

Abstract
We examined the pattern of nucleotide substitution in 4933 conserved single-copy orthologous protein-coding genes of human, rhesus, mouse, and rat. Consistent with previous studies, the median ratio of the number of nonsynonymous substitutions per nonsynonymous site (dN) to the number of synonymous substitutions per synonymous site (dS) was significantly higher in the comparison between the two primates than in the comparison between the two rodents. This pattern was particularly strong in the case of genes expressed in the immune system, but also occurred in other genes, including a set of highly conserved genes involved in the regulation of transcription. Both synonymous and nonsynonymous differences occurred independently in the same codons in the primates and in the rodents to a greater extent than expected by chance, but the extent of the deviation from random expectation was much greater in the case of nonsynonymous differences. Parallel amino acid replacements occurred at the same sites in the primates and rodents far more frequently than expected by chance, but tended to involve very conservative amino acid changes. Divergent amino acid changes involved more chemically different amino acids than parallel changes, and divergent amino acid replacements between the primates were significantly more radical than those between the rodents. These results are most easily explained on the hypothesis that the evolution of these genes has been shaped largely by purifying selection, which has been less effective in primates than in rodents, presumably as a consequence of lower long-term effective population sizes in the former.
Keywords: purifying selection, nearly neutral theory, parallel evolution, slightly deleterious mutation
1. Introduction
Abundant evidence suggests that natural selection at the molecular level predominantly takes the form of purifying selection; that is, selection against deleterious mutations. In most protein-coding genes, the number of synonymous of synonymous substitutions per synonymous site (dS) exceeds the number of nonsynonymous (amino acid-altering) substitutions per nonsynonymous site (dN), indicating that numerous nonsynonymous mutations have been eliminated by purifying selection (Nei 1987). A number of recent studies have reported a surprisingly high prevalence of parallel or convergent amino acid replacements (i.e., homoplasy at the amino acid sequence level; Bazykin et al. 2007; Rogozin et al. 2008; Rokas and Carroll 2008). One possible explanation of this phenomenon is that it is a consequence of purifying selection acting to constrain the set of permissible amino acid replacements (Rokas and Carroll 2008). In addition, the evolutionary conservation of numerous sequences outside of protein-coding genes implies that many such motifs are also subject to purifying selection (Bejerano et al. 2004; Casillas et al. 2007; Sakuraba et al. 2008). Moreover, in populations of a wide variety of organisms, nonsynonymous polymorphisms tend to be rare in comparison to synonymous polymorphisms in the same genes, indicating that purifying selection against slightly deleterious variants is an ongoing process (Hughes et al. 2003, 2008; Hughes 2005, 2007;Hughes and Hughes 2007a; Irausquin and Hughes 2008).
According to the “nearly neutral” theory of molecular evolution, the efficiency with which slightly deleterious variants can be removed from populations is predicted to depend on the extent of recombination and on effective population size (Ohta 1973, 2002; Lynch 2007; Hughes 2008). Genomes or genomic regions with low recombination rates are expected to show elevated accumulation of nonsynonymous substitutions, a prediction supported by data from sex chromosomes (Berlin and Ellegren 2006; Wykoff et al. 2002), mitochondrial genomes (Nachman et al. 1994, 1996; Rand and Kann 1996), selfing and asexual organisms (Barraclough et al. 2007; Bustamante et al. 2002), and the comparison of genomic regions with different recombination rates (Haddrill et al. 2007). Likewise, there is evidence that the accumulation of nonsynonymous mutations is accelerated in species with low long-term effective population sizes due to a bottlenecked population history (Ohta 1993a; Hughes and Hughes 2007b).
As evidence for the accumulation of nonsynonymous mutations in species with low effective population size, Ohta (1993b) pointed to the observation of a higher dN/dS in primates than in rodents or artiodactyls. This observation, which was originally based on just 17 gene loci, is consistent with the nearly neutral theory if one assumes that effective population sizes in primates have been smaller on average than in the other two orders of mammals studied (Ohta1993b). Comparison of thousands of putative orthologs among complete genomes confirmed Ohta’s (1993b) original observation of a higher dN /dS in primates than in rodents (Chimpanzee Sequencing and Analysis Consortium 2005; Rhesus Macaque Genome Sequencing and Analysis Consortium 2007; Ellegren 2008).
Here we analyze a set of nearly 5000 conserved single-copy orthologs from the genomes of two primates (human and rhesus macaque) and two rodents (rat and mouse). We analyze only single copy genes to avoid possibly confounding effects of non-reciprocal inter-locus recombination (“gene conversion”) in the case of multi-gene families (Teshima and Innan 2004). Indeed, there is evidence that biased gene conversion can be a major source of slightly deleterious nonsynonymous substitutions in primates (Galtier et al. 2008). We test whether the tendency toward increased dN /dS in primates remains constant across different functional categories of genes. In particular, we compare dN /dS in genes involved in the immune response with those involved in the regulation of transcription because of previous evidence that the former are relatively unconstrained at the amino acid level in mammals while the latter are highly constrained (Murphy 1993; Hughes 1997; Hughes and Friedman 2008).
In addition, examining individual orthologous codons, we test the extent to which the same codons show differences both between the two primate species and between the two rodent species. We compare amino acid difference that occurred divergently with those that occurred in parallel in the two lineages (Figure 1) in order to examine the kinds of amino acid replacements involved. We test the hypothesis that purifying selection promotes parallel amino acid replacements, by limiting acceptable replacements at many sites to a small set of chemically similar amino acids (Rokas and Carroll 2008). Then, using parallel amino acid replacements as a basis for comparison, we test the prediction that primate-specific divergent amino acid replacement should show a greater tendency to introduce chemically dissimilar amino acids than do rodent-specific divergent replacements, as expected if the former include a higher proportion of slightly deleterious replacement mutations.
Figure 1.
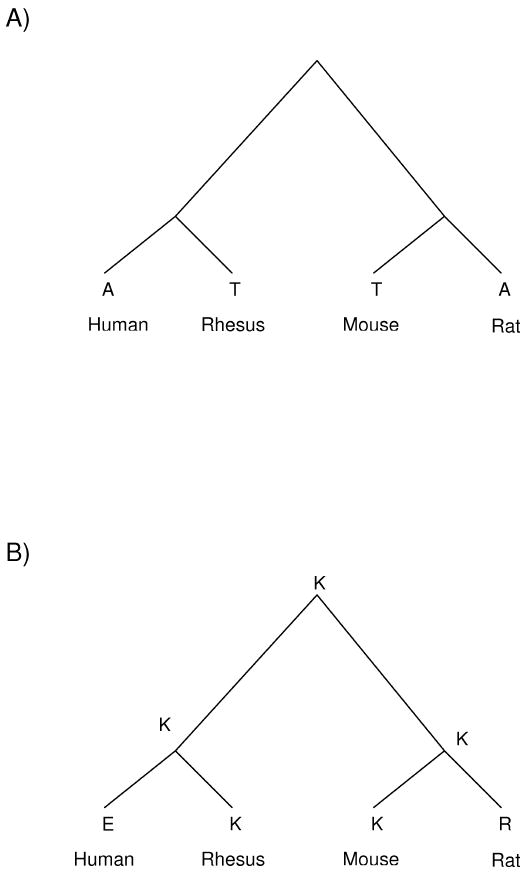
Examples of parallel (A) and divergent (B) patterns of amino acid replacement, illustrated by aligned amino acid sites (A) 147 and (B) 453 of 78Kd centrosomal protein (encoded by CEP78). Amino acids are indicated by the single-letter code.
2. Methods
2.1. Sequences Analyzed
The following mammalian complete genomes were obtained from Ensembl version 49 (Hubbard et al. 2007; assembly version and number of loci in parentheses): Homo sapiens (NCBI36; 19,902 transcripts), Macaca mulatta (MMUL1; 20,258), Mus musculus (NCBIM37, 21,424), Rattus norvegicus (RGSC3.4; 20,828). The protein-coding sequences (the shortest predicted sequence per locus) were compiled from the above set and formatted into a sequence database. To find sets of orthologs, the Blastclust software (Altschul et al. 1997) was employed which searches for all homologous sequences in a database and then organizes the homologs into families by a single-linkage method. This ensures that each gene is assigned to only one family. This step was performed using the following criteria to establish homology (Hughes et al. 2005a): a minimum E-value of 10-6 and at least 70 percent similarity across at least 85 percent of the sequence lengths. Gene families were retained which had a single putative ortholog in each of the four mammalian genomes.
This set of orthologs was then further edited to exclude a small number of genes for which dS (see below) was very high or undefined in either the human-rhesus or mouse-rat comparison, indicating the probable lack of a true orthologous relationship. In order to include only a set of genes that have been subject largely to purifying selection on the amino acid sequence, we excluded a small number of genes for which dN exceeded dS (see below) in either the human-rhesus comparison or the mouse-rat comparison. To minimize possible effects of misalignment, we included in the final data set only genes for which the coefficient of variation (CV) in length (number of codons) across the four species was less than 10%. Genes in these excluded categories amounted to only 1.6% of the original set of putative orthologs. The final data set consisted of 4933 putative single copy orthologs (Supplementary Table S1), with a mean CV in length across species of 1.60% (± 0.03 S.E.) and a median CV in length of 0.62%.
Each ortholog was scored for its role in biological processes using information from the Gene Ontology (GO) project (Gene Ontology Consortium 2000). Genes were categorized as having a role in the immune system if the GO annotations included the biological processes immune response and/or inflammatory response. Similarly, genes were categorized as having a role in the regulation of transcription if the GO annotations for biological processes included regulation of transcription. We chose to analyze these two functional categories because there were numerous genes in our data set belonging to each category and because, on the basis of previous literature (e.g., Murphy 1993; Hughes 1997; Hughes and Friedman 2008), we expected immune system genes to be relatively unconstrained and genes involved in the regulation of transcription to be subject to strong functional constraint.
2.2. Data Analysis
Members of each ortholog set were globally aligned at the amino acid level using ClustalW (Thompson et al. 1994), and the alignment was imposed on the corresponding nucleotide sequence. The number of synonymous of synonymous substitutions per synonymous site (dS) and the number of nonsynonymous substitutions per nonsynonymous site (dN) were estimated by Yang and Nielsen’s (2000) method and by the Nei and Gojobori method (1986), using the PAML software (Yang 1997). The correlation between dN estimates by the two methods was > 0.99 in both primates and rodents, while the correlation between the estimates of dN /dS by the two methods was 0.945 in primates and 0.967 in rodents. Because the results of the two methods were very similar, only those of Yang and Nielsen’s (2000) method are reported below.
Synonymous and nonsynonymous nucleotide differences between human and rhesus and between mouse and rat were counted at each individual aligned codon using Nei and Gojobori’s (1986) method. The latter simple method was used in this case because more complicated methods estimate parameters (such as nucleotide content) from the data, which is not applicable in the case of a single codon (Hughes and Friedman 2005). In addition, Nei and Gojobori’s (1986) method averages across evolutionary pathways, thereby providing a conservative count for the occurrence of synonymous and nonsynonymous differences.
At codons that showed an amino acid difference both between human and rhesus and between mouse and rat, we focused on two specific patterns of amino acid change (Figure 1). In parallel change, the same two amino acid residues occurred in the two primate species and in the two rodent species (Figure 1A). With sequences of just these four species it was not possible to determine which of the two residues was ancestral, it could be inferred that amino acid replacements had occurred in parallel (Figure 1A). In divergent change, the same amino acid residue was found in one of the two primate species and in one of the two rodent species, while two different residues were found in the other primate and in the other rodent (Figure 1B). In the latter case, the principle of parsimony leads to the inference that the ancestral residue was the one shared now by one primate and one rodent (Figure 1B). Thus, at such a site, it can be inferred that independent divergent changes (away from the ancestral state) have occurred in both the primate and rodent lineages.
The chemical similarity between pairs of amino acid was measured by the chemical distances of Miyata, Miyazawa, and Yasunaga (1979) and of Xia and Xie (2002). Since the results using the two distances were essentially identical, only the former is reported here (designated MMY distance in the following). The MMY distance has an arbitrary scale ranging from 0.06 to 5.23, with a median value for the 190 possible amino acid pairs of 2.365. Because the variables analyzed were generally not normally distributed, we report statistical tests using nonparametric methods; however, in every case, analogous parametric tests yielded essentially identical results (not shown).
3. Results
3.1. Synonymous and Nonsynonymous Substitutions
In comparisons of 4933 single-copy orthologous genes, the mean dS between human and rhesus was 0.0900 ± 0.0009 S.E., with a median value of 0.0733 and a range of 0.0075 to 0.4957; and mean dN was 0.0181 ± 0.004, with a median of 0.0102 and a range of 0.0000 to 0.2666. When the same set of orthologs were compared between mouse and rat, mean dS was 0.1955 ± 0.0010 (median = 0.1885; range = 0.0187 to 0.7112); and mean dN was 0.0263 ± 0.004 (median = 0.0191; range = 0.0000 to 0.2742). In spite of the overall higher levels of both synonymous and nonsynonymous substitution in the mouse-rat comparison than in the human-rhesus comparison, both the mean (0.196 ± 0.003) and median (0.138) of the ratio dN /dS were higher in the case of the human-rhesus comparison than in the rat-mouse comparison (0.134 ± 0.002 and 0.102, respectively). Figure 2A illustrates the distribution of the difference between dN /dS for the two primate species and dN /dS for the two rodent species. Both mean and median values of the difference were substantially greater than zero (Figure 2A). Overall, dN /dS for the primates was greater than that for the rodents in 3355 genes (68.0%), while the reverse was true in 1578 genes (32.0%). The difference between median dN /dS between primates and rodents was highly significant (Sign test; P < 0.001).
Figure 2.
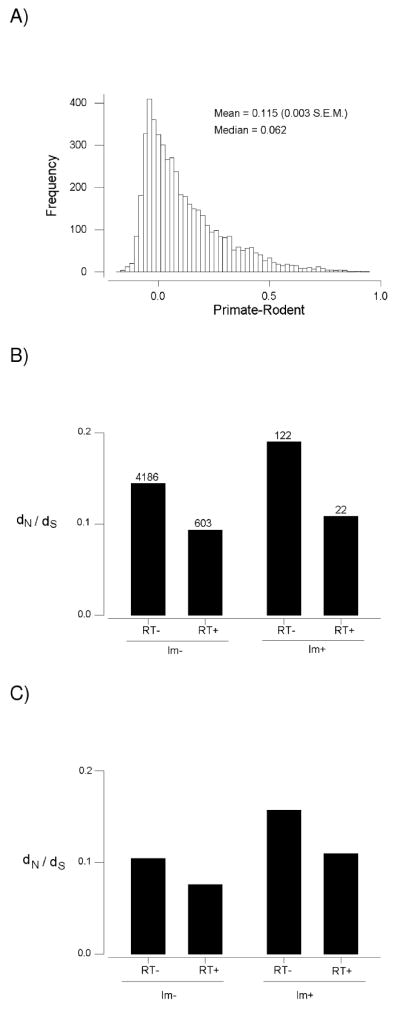
(A) Frequency distribution of the difference between dN /dS in the primates and dN /dS in the rodents for 4933 orthologous single-copy genes from human, rhesus, mouse, and rat. (B) Median dN /dS between human and rhesus in orthologous genes categorized by presence (Im+) or absence (Im-) of immune system function and by presence (RT+) or absence (RT-) of function in regulation of transcription. Numbers of genes in each category are shown. The difference among categories with respect to median dN /dS was highly significant (P < 0.001; Kruskal-Wallis test). (C) Median dN /dS between mouse and rat in orthologous genes categorized by presence (Im+) or absence (Im-) of immune system function and by presence (RT+) or absence (RT-) of function in regulation of transcription. Numbers of genes in each category are as in Figure 2B. The difference among categories with respect to median dN /dS was highly significant (P < 0.001; Kruskal-Wallis test).
Genes were classified with regard to their function in the immune system and in regulation of transcription; 4186 genes had neither of these functions, 603 functioned in regulation of transcription but not in the immune system, 122 functioned in the immune system but not in regulation of transcription, and 22 had both functions (Figure 2B). In comparisons between the two primate species, median dN /dS differed significantly among the four categories of genes (P < 0.001; Kruskal-Wallis test), with the highest median value (0.190) occurring in genes with immune system function only and the lowest median value (0.094) occurring in genes that function in regulation of transcription but not in the immune system (Figure 2B). Likewise, in comparisons between the two rodent species, median dN /dS differed significantly among the four categories of genes (P < 0.001; Kruskal-Wallis test), with the highest median value (0.157) occurring in genes with immune system function only and the lowest median value (0.076) occurring in genes functioning in the regulation of transcription but not in the immune system (Figure 2C).
When each of the four categories was analyzed separately, median dN /dS in primates was significantly greater than that in rodents (P < 0.001 for each categories except that of genes functioning in both the immune system and regulation of transcription, where P = 0.017; Sign tests). The median difference between dN /dS in primates and dN /dS in rodents also differed significantly among the four categories (P < 0.001; Kruskal-Wallis test). The median difference was highest (0.106) in genes with immune system function only and lowest (0.024) in genes functioning in regulation of transcription but not in the immune system.
Across all genes, there were 2,592,614 aligned codons at which both synonymous and nonsynonymous differences were possible (i.e., excluding stop codons and methionine and tryptophan codons). At 81,283 of these codons (3.1%), there was a nonsynonymous difference between human and rhesus, while at 124,540 (4.8%), there was a nonsynonymous difference between mouse and rat. If nonsynonymous differences occurred independently in the primates and the rodents, one would expect 3904.6 codons to show nonsynonymous differences both between human and rhesus and mouse and rat. In fact, there were 9837 codons which showed nonsynonymous differences both between human and rhesus and mouse and rat, about 2.52 times as many as expected. The deviation from independence was highly significant (χ2 = 9774.9; 1 d.f.; P < 0.001).
Synonymous differences occurred at 159,690 (6.2%) codons between human and rhesus and at 359,071 (13.8%) codons between mouse and rat. There were 28,402 codons showing synonymous differences both between the two primates and between the two rodents. This value was again greater than that (22,116.7) expected under the hypothesis of independence, and the deviation from independence was highly significant (χ2 = 2209.5; 1 d.f.; P < 0.001). However, the ratio of observed to expected numbers was much lower in the case of synonymous differences (1.28) than in the case of nonsynonymous differences (2.52).
3.2. Amino Acid Replacements
Among the codon sites at which nonsynonymous differences occurred both between the two primates and between the two rodents, 2198 showed a parallel pattern of amino acid replacement (Figure 1A), whereas 5303 showed a divergent pattern of amino acid replacement (Figure 1B). Thus, divergent amino acid replacements occurred 2.41 times as frequently as parallel amino acid replacements, implying that the latter have occurred at a far higher level than expected. On the unrealistic assumption that all amino acid changes are equally likely, one would expect divergent changes to outnumber parallel changes by 18:1. We used the frequency of occurrence of different amino acid replacements in the set of divergent changes to construct a more realistic null hypothesis. On this basis we predict, that divergent changes should outnumber parallel changes by about 4.08:1. The difference between observed an expected numbers was highly significant (χ2 = 439.5; 1 d.f.; P < 0.001), indicating a significant excess of parallel amino acid changes in comparison to divergent changes.
When genes were categorized by function in the immune system and in regulation of transcription, there was a significant difference among categories with respect to the frequency of occurrence of genes with one or more parallel changes (χ2 = 27.5; 3 d.f.; P < 0.001; Figure 3A). The highest percentage of genes with one or more parallel changes (39.3%) occurred in genes functioning in the immune system but not in regulation of transcription, while the lowest percentage (19.9%) occurred in genes functioning in regulation of transcription but not in the immune system (Figure 3A).
Figure 3.
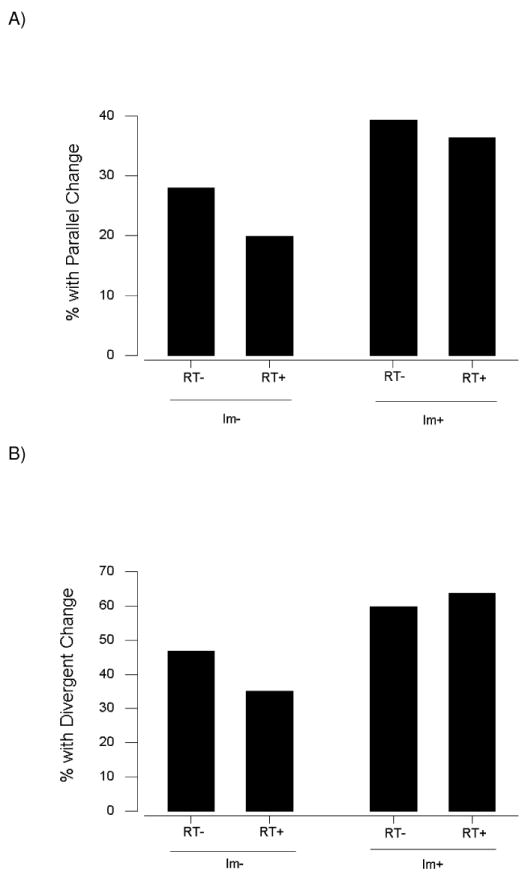
Percentages of genes with parallel (A) and divergent (B) amino acid replacements in orthologous genes categorized by presence (Im+) or absence (Im-) of immune system function and by presence (RT+) or absence (RT-) of function in regulation of transcription. In each case, there was a significant difference among categories: (A) χ2 = 27.5; 3 d.f.; P < 0.001; (B) χ2 = 41.9; 3 d.f.; P < 0.001.
Likewise, there was a significant difference among categories with respect to the frequency of occurrence of genes with one or more divergent changes (χ2 = 41.9; 3 d.f.; P < 0.001; Figure 3B). The percentage of genes with one or more divergent changes was highest in genes functioning in both the immune system and in the regulation of transcription (63.6%) and almost as high in genes functioning in the immune system but not in regulation of transcription (59.8%; Figure 3B). By contrast, the lowest percentage of genes with one or more divergent changes (35.2%) was seen in genes functioning in regulation of transcription but not in the immune system (Figure 3B).
Overall, 2260 genes showed at least one divergent amino acid change, and 926 of these (41.0%) showed at least one parallel change as well. By contrast, one or more parallel changes were observed in only 422 (15.8%) of the 2673 genes lacking a divergent change. The difference in the proportions was highly significant (χ2 = 391.2; 1 d.f.; P < 0.001), indicating a positive association between the occurrence of parallel and divergent changes in the same gene.
Of the 3355 genes having dN /dS greater in the primates than in the rodents, 1931 (57.6%) had at least one divergent amino acid change. By contrast, one or more divergent amino acid changes occurred in only 329 of 1578 (20.8%) genes having dN /dS greater in the rodents than in the primates. The difference in proportions was highly significant (χ2 = 582.5; 1 d.f.; P < 0.001). Similarly, at least one parallel change occurred in 1144 (34.1%) of genes having dN /dS greater in the primates than in the rodents but in only 204 (12.9%) of the remaining genes. The difference in proportions was highly significant (χ2 = 242.2; 1 d.f.; P < 0.001).
3.3. Amino Acid Chemical Distances
The pairs of amino acids involved in cases of parallel amino acid replacement included 137 (72.1%) of the 190 possible amino acid pairs. Yet certain amino acid pairs occurred with much greater frequency than others. Of the 2198 cases of parallel amino acid replacement, 28.5% were due to just three amino acid pairs: I-V (272 cases or 12.4%), A-T (199 cases or 9.1%), and N-S (155 cases or 7.1%). Using the pairs of amino acids involved in divergent changes as a basis for comparison, we used χ2 tests of independence to test for over-representation of each amino acid pair in the set of parallel amino acid replacements. Six amino acid pairs showed significant over-representation (at the 5% level or better, Bonferroni-corrected for multiple testing; Figure 4A). These six pairs all involved relatively conservative changes from the point of view of the chemical distances between amino acids (MMY distances in parentheses): I-V (0.85), A-T (0.90), N-S (1.31), Q-R (1.13), K-R (0.40), and H-R (0.82; Figure 4A). The median MMY distance of the 2198 parallel amino acid changes was 0.90. This value was significantly lower than the median MMY distance for divergent amino acid changes either in rodents (1.13) or in primates (1.14; P < 0.001 in each case; Mann-Whitney test; Figure 4B).
Figure 4.
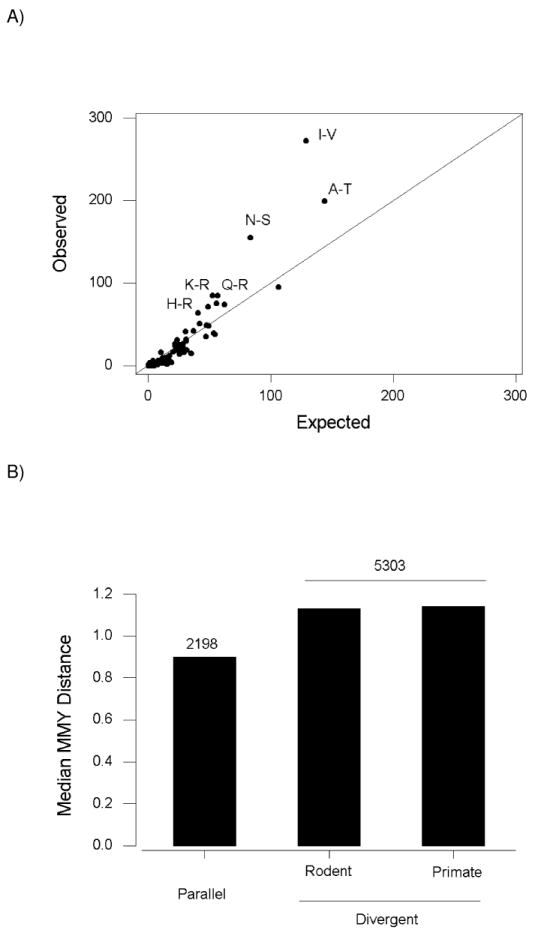
(A) Observed vs. expected numbers of amino acid pairs involved in parallel amino acid replacements. Expected numbers were based on divergent amino acid replacements. The six amino acid pairs (single-letter code) are shown for which the observed number significantly (P < 0.05; Bonferroni-corrected) exceeded the expected number. The line is a 45° line. (B) Median MMY chemical distance between amino acids in parallel and divergent amino acid replacements. The median distance for parallel replacements was significantly different from that for divergent amino acid replacements either in rodents or in primates (Mann-Whitney test; P < 0.001 in each case). The median distance for divergent amino acid replacements in primates was significantly different from that in rodents (Sign test; P < 0.001).
In the case of divergent amino acid replacements, although the median MMY distance in the primates (1.14) was only slightly higher than that in the rodents (1.13), the difference was highly significant statistically (P < 0.001; Sign test; Figure 4B). Of the 5303 sites with divergent amino acid replacements, the MMY distance for the primates was greater than that for the rodents in 2784 (52.5%), whereas the MMY distance for rodents was greater than that for primates at 2510 sites (47.3%) and equal to that for primates at 9 sites (0.2%). The median MMY distance for divergent amino acid replacements in primates was nonetheless significantly lower than the median value (2.365) for the 190 possible amino acid pairs (Sign test; P < 0.001). Of the 5303 sites with divergent amino acid replacements, at 4048 (76.3%) the MMY distance was less than 2.365.
We further analyzed MMY at divergently evolving amino acid sites by looking separately at sites (N = 4806) in genes for which dN /dS was greater in primates than in rodents and at sites (N = 497) in genes for which dN /dS was greater in rodents than in primates. In the former set of sites, there was a highly significant difference in median MMY distance between primates (1.31) and rodents (1.13; Sign test; P < 0.001). By contrast, in the latter sites, there was no significant difference in median MMY distance between primates (0.91) and rodents (1.13; Sign test; n.s.). Thus, the effect of greater MMY distance in the case of divergent amino acid replacements in primates was essentially a feature of the subset of genes where dN /dS was greater in primates than in rodents.
4. Discussion
Examination of the pattern of nucleotide substitution in 4933 conserved single-copy orthologous protein-coding genes of human, rhesus, mouse, and rat showed significantly higher median dN /dS in the comparison between the two primates than in the comparison between the two rodents, consistent with previous analyses (Ohta 1993d; Chimpanzee Sequencing and Analysis Consortium 2005; Rhesus Macaque Genome Sequencing and Analysis Consortium 2007; Ellegren 2008). On the assumption that effective population sizes have generally been larger in the rodents than in the primates, this result supports the prediction of the “nearly neutral” theory that nonsynonymous substitution will be elevated in species with small effective population sizes (Ohta 1993b). This effect is expected if many nonsynonymous mutations are slightly deleterious, since the efficiency of purifying selection in removing slightly deleterious mutations is reduced when effective population size is low (Ohta 1993b).
Other interpretations of our results are possible, including the hypothesis that positive selection on amino acid replacements has occurred more frequently between the two primate species than between the two rodent species. However, it is hard to imagine what sort of selection would occur broadly at isolated codons across a large number of single-copy genes in primates but not in rodents. Note also that the genes in question are generally conserved at the amino acid sequence level, with dN less than dS in every case. Moreover, there were several lines of evidence that argued against the hypothesis of positive selection and favored that of inefficient purifying selection. This evidence involved the patterns of nucleotide substitution in genes of different functional categories and the patterns of parallel and divergent amino acid replacement. Taken together, the overall pattern suggested a predominant role for purifying selection in the evolution of nonsynonymous sites in these genes, with differences in evolutionary rate attributable mainly to differences in the strength and/or effectiveness of purifying selection.
Consistent with previous analyses (Murphy 1993; Hughes 1997; Hughes and Friedman 2008), we found that, in both primates and rodents, median dN /dS was elevated in genes with immune system function, particularly in comparison to a set of highly conserved genes involved in the regulation of transcription. Again, this pattern also might be explained either by positive selection on immune system genes or by relaxation or inefficiency of purifying selection on immune system genes. But, whatever process explains the elevated dN /dS in the immune system genes, it is worth noting that the dN /dS ratio was greater in primates than in rodents even in the conserved genes involved in the regulation of transcription. Even if there were certain instances of positive selection on the immune system genes, it seems unlikely that the same type of selection could be involved in the case of the conserved genes functioning in the regulation of transcription. Thus, it seems unlikely that positive selection can account for the elevated dN /dS in primates across functional categories of genes.
It is important to note that the immune system genes involved in the present analyses did not include the highly polymorphic genes of the major histocompatibility complex, which are subject to a form of balancing selection that accelerates the rate of amino acid substitution in the peptide-binding region of the molecule (Hughes and Nei 1988) nor the other multi-gene families of immune system effectors for which there is evidence of past diversifying selection (Hughes 2002). In the human population, immune system genes show significantly reduced evidence of ongoing purifying selection on nonsynonymous variants in comparison to other genes (Hughes et al. 2005b), consistent with the hypothesis that the elevated values of dN /dS in immune system genes occurs mainly as a result of reduction in the stringency of purifying selection.
We found that both synonymous and nonsynonymous changes occurred at the same codon independently in the primate and rodent lineages significantly more frequently than expected by chance. As regards synonymous changes, this effect may reflect differences in mutability of certain codons. However, the effect was much more pronounced in the case of nonsynonymous changes than in the case of synonymous changes, with nonsynonymous changes occurring in the same codon independently in the two lineages over two and a half times as frequently as expected by chance. The distinctive pattern at nonsynonymous sites implicates purifying selection, suggesting that certain codon positions are more likely to undergo amino acid replacements because they are less subject to purifying selection.
Analysis of parallel amino acid changes in the two lineages further supported the hypothesis that purifying selection plays a key role in shaping evolution of these genes. Parallel amino acid change has sometimes been taken as a sign of positive selection (Zhang and Kumar 1997), but studies showing the wide prevalence of parallel amino acid changes provide evidence against this interpretation (Bazykin et al. 2007; Rogozin et al. 2008; Rokas and Carroll 2008). In our data set, parallel amino acid changes disproportionately involved a small set of very conservative amino acid replacements, and the median chemical distance was significantly lower in parallel changes than in divergent changes. Thus, by permitting only certain conservative changes at certain sites, purifying selection seems to increase the likelihood of parallel amino acid changes at those sites. These results support the hypothesis that parallel amino acid changes occur mainly as a result of purifying selection, rather than positive selection (Rokas and Carroll 2008). In our data set, parallel amino acid replacements occurred in the two lineages at a far higher level than expected, based on the patterns of amino acid replacement that occurred in the case of divergent changes. This high level of parallel change suggests that sites there are numerous sites where a limited set of conservative amino acid changes are permitted.
Both divergent and parallel amino acid changes were disproportionately likely to occur in genes for which dN /dS was greater in primates than in rodents. Moreover, in the latter genes, divergent changes tended to introduce more divergent amino acids in the primate lineage than in the rodent lineage, as indicated by significantly greater median MMY distance in the primates than in the rodents. Note that, even though the median MMY distance at these sites in primates (1.31) was higher than that in rodents, it was still significantly lower than the median MMY distance for all possible amino acid pairs (2.365). In fact, 1.31 corresponds to the N-S pair, at about the 22nd percentile of the distribution of MMY distance values. This finding is consistent with the hypothesis that the elevated dN /dS in primates is due mainly to the fixation of slightly deleterious mutations, since slightly deleterious mutations are likely to involve greater chemical dissimilarity of amino acid residues than those that are strictly neutral, yet are less likely to involve extremely radical changes.
In summary, our results showed evidence of elevated dN /dS in primates in comparison to rodents, and this pattern was particularly enhanced in genes with immune system function. Moreover, elevated dN /dS was statistically associated with increased frequencies of both convergent and divergent amino acid replacements; and with larger chemical distances in the case of divergent amino acid changes. These patterns are most easily explained by a greater rate of fixation of slightly deleterious mutations in primates than in rodents as a consequence of lower effective population sizes in the former. Thus our results provide support for the nearly-neutral theory of molecular evolution.
Supplementary Material
Acknowledgments
This research was supported by grant GM43940 from the National Institutes of Health.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barraclough TG, Fontaneto D, Ricci C, Hernious EA. Evidence for inefficient selection against deleterious mutations in cytochrome oxidase I of asexual bdelloid rotifers. Mol Biol Evol. 2007;24:1952–1962. doi: 10.1093/molbev/msm123. [DOI] [PubMed] [Google Scholar]
- Bazykin GA, Kondrashov FA, Brudno M, Poliakov A, Dubchak I, Kondrashov AS. Extensive parallelism in protein evolution. Biology Direct 2007. 2007;2:20. doi: 10.1186/1745-6150-2-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bejerano G, Pheasant M, Makunin I, Stephen S, Kent WJ, Mattick JS, Haussler D. Ultraconserved elements in the human genome. Science. 2004;304:1321–1325. doi: 10.1126/science.1098119. [DOI] [PubMed] [Google Scholar]
- Berlin S, Ellegren H. Fast accumulation of nonsynonymous mutations on the female-specific W chromosome in birds. J Mol Evol. 2006;62:66–72. doi: 10.1007/s00239-005-0067-6. [DOI] [PubMed] [Google Scholar]
- Bustamante CD, Nielsen R, Sawyer SA, Olsen KM, Purugganan MD, Hartl DL. The cost of inbreeding in Arabidopsis. Nature. 2002;416:531–534. doi: 10.1038/416531a. [DOI] [PubMed] [Google Scholar]
- Casillas S, Barbadilla A, Bergman CM. Purifying selection maintains highly conserved noncoding sequences in Drosophila. Mol Biol Evol. 2007;24:2222–2234. doi: 10.1093/molbev/msm150. [DOI] [PubMed] [Google Scholar]
- Chimpanzee Sequencing and Analysis Consortium. Initial sequence of the chimpanzee genome and comparison with the human genome. Nature. 2005;437:69–87. doi: 10.1038/nature04072. [DOI] [PubMed] [Google Scholar]
- Ellegren H. Comparative genomics and the study of evolution by natural selection. Mol Ecol. 2008;17:4586–4596. doi: 10.1111/j.1365-294X.2008.03954.x. [DOI] [PubMed] [Google Scholar]
- Galtier N, Duret L, Glémin S, Ranwez V. GC-biased gene conversion promotes the fixation of deleterious amino acid changes in primates. Trends Genet. 2008;25:1–5. doi: 10.1016/j.tig.2008.10.011. [DOI] [PubMed] [Google Scholar]
- Gene Ontology Consortium. Gene Ontology: tool for the unification of biology. Nature Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haddrill PR, Halligan DL, Tomaras D, Charlesworth B. Reduced efficacy of selection in regions of the Drosophila genome that lack crossing over. Genome Biol 2007. 2007;8:R18. doi: 10.1186/gb-2007-8-2-r18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubbard TJ, Aken BL, Beal K, Ballester B, Caccamo M, Chen Y, Clarke L, Coates G, Cunningham F, Cutts T, Down T, Dyer SC, Fitzgerald S, Fernandez-Banet J, Graf S, Haider S, Hammond M, Herrero J, Holland R, Howe K, Howe K, Johnson N, Kahari A, Keefe D, Kokocinski F, Kulesha E, Lawson D, Longden I, Melsopp C, Megy K, Meidl P, Ouverdin B, Parker A, Prlic A, Rice S, Rios D, Schuster M, Sealy I, Severin J, Slater G, Smedley D, Spudich G, Trevanion S, Vilella A, Vogel J, White S, Wood M, Cox T, Curwen V, Durbin R, Fernandez-Suarez XM, Flicek P, Kasprzyk A, Proctor G, Searle S, Smith J, Ureta-Vidal A, Birney E. Ensembl 2007. Nucleic Acids Res. 2007;35:D610–D617. doi: 10.1093/nar/gkl996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes AL. Rapid evolution of immunoglobulin superfamily C2 domains expressed in immune system cells. Mol Biol Evol. 1997;14:1–5. doi: 10.1093/oxfordjournals.molbev.a025694. [DOI] [PubMed] [Google Scholar]
- Hughes AL. Natural selection and diversification of vertebrate immune effectors. Immunol Rev. 2002;190:161–168. doi: 10.1034/j.1600-065x.2002.19012.x. [DOI] [PubMed] [Google Scholar]
- Hughes AL. Evidence for abundant slightly deleterious polymorphisms in bacterial populations. Genetics. 2005;169:533–538. doi: 10.1534/genetics.104.036939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes AL. Micro-scale signature of purifying selection in Marburg virus genomes. Gene. 2007;392:266–272. doi: 10.1016/j.gene.2006.12.038. [DOI] [PubMed] [Google Scholar]
- Hughes AL. Near neutrality: leading edge of the neutral theory of molecular evolution. Ann NY Acad Sci. 2008;1133:162–179. doi: 10.1196/annals.1438.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes AL, Friedman R. Variation in the pattern of synonymous and nonsynonymous difference between two fungal genomes. Mol Biol Evol. 2005;22:1320–1324. doi: 10.1093/molbev/msi120. [DOI] [PubMed] [Google Scholar]
- Hughes AL, Friedman R. Codon-based tests of positive selection, branch lengths, and the evolution of mammalian immune system genes. Immunogenetics. 2008;60:495–506. doi: 10.1007/s00251-008-0304-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes AL, Hughes MA. More effective purifying selection in RNA viruses than in DNA viruses. Gene. 2007a;404:117–125. doi: 10.1016/j.gene.2007.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes AL, Hughes MA. Coding sequence polymorphism in avian mitochondrial genomes reflects population histories. Mol Ecol. 2007b;16:1369–1376. doi: 10.1111/j.1365-294X.2007.03242.x. [DOI] [PubMed] [Google Scholar]
- Hughes AL, Nei M. Pattern of nucleotide substitution at MHC class I loci reveals overdominant selection. Nature. 1988;335:167–170. doi: 10.1038/335167a0. [DOI] [PubMed] [Google Scholar]
- Hughes AL, Packer B, Welch R, Bergen AW, Chanock SJ, Yeager M. Widespread purifying selection at polymorphic sites in human protein-coding loci. Proc Natl Acad Sci USA. 2003;100:15754–15757. doi: 10.1073/pnas.2536718100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes AL, Ekollu V, Friedman R, Rose JR. Gene family content-based phylogeny of prokaryotes: the effect of search criteria. Syst Biol. 2005a;54:268–276. doi: 10.1080/10635150590923335. [DOI] [PubMed] [Google Scholar]
- Hughes AL, Packer B, Welsch R, Chanock SJ, Yeager M. High level of functional polymorphism indicates a unique role of natural selection at human immune system loci. Immunogenetics. 2005b;57:821–827. doi: 10.1007/s00251-005-0052-7. [DOI] [PubMed] [Google Scholar]
- Hughes AL, Friedman R, Rivailler P, French JO. Synonymous and nonsynonymous polymorphisms and divergences in bacterial genomes. Mol Biol Evol. 2008;25:2199–2209. doi: 10.1093/molbev/msn166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irausquin SJ, Hughes AL. Distinctive pattern of sequence polymorphism in the NS3 protein of hepatitis C virus type 1b reflects conflicting evolutionary pressures. J Gen Virol. 2008;89:1921–1929. doi: 10.1099/vir.0.2008/000992-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch M. The origins of genomic architecture. Sunderland MA: Sinauer; 2007. [Google Scholar]
- Miyata T, Miyazawa S, Yasunaga T. Two types of amino acid substitutions in protein evolution. J Mol Evol. 1979;12:219–236. doi: 10.1007/BF01732340. [DOI] [PubMed] [Google Scholar]
- Murphy PM. Molecular mimicry and the generation of host defense protein diversity. Cell. 1993;72:823–826. doi: 10.1016/0092-8674(93)90571-7. [DOI] [PubMed] [Google Scholar]
- Nachman MW, Boyer SN, Aquadro CF. Nonneutral evolution at the mitochondrial NADH dehydrogenase subunit 3 gene in mice. Proc Natl Acad Sci USA. 1994;91:6364–6368. doi: 10.1073/pnas.91.14.6364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nachman MW, Brown WM, Stoneking M, Aquadro CF. Nonneutral mitochondrial DNA variation in humans and chimpanzees. Genetics. 1996;142:953–963. doi: 10.1093/genetics/142.3.953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nei M. Molecular evolutionary genetics. New York: Columbia University Press; 1987. [Google Scholar]
- Nei M, Gojobori T. Simple methods for estimating the numbers of synonymous and nonsynonymous nucleotide substitutions. Mol Biol Evol. 1986;3:418–426. doi: 10.1093/oxfordjournals.molbev.a040410. [DOI] [PubMed] [Google Scholar]
- Ohta T. Slightly deleterious mutant substitutions in evolution. Nature. 1973;246:96–98. doi: 10.1038/246096a0. [DOI] [PubMed] [Google Scholar]
- Ohta T. Amino acid substitution at the Adh locus of Drosophila is facilitated by small population size. Proc Natl Acad Sci USA. 1993a;90:4548–4551. doi: 10.1073/pnas.90.10.4548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohta T. An examination of the generation-time effect on molecular evolution. Proc Natl Acad Sci USA. 1993b;90:10676–10680. doi: 10.1073/pnas.90.22.10676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohta T. Near-neutrality in evolution of genes and gene regulation. Proc Natl Acad Sci USA. 2002;99:16134–16137. doi: 10.1073/pnas.252626899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rand DM, Kann LM. Excess amino acid polymorphism in mitochondrial DNA: contrasts among genes from Drosophila, mice, and humans. Mol Biol Evol. 1996;13:735–748. doi: 10.1093/oxfordjournals.molbev.a025634. [DOI] [PubMed] [Google Scholar]
- Rhesus Macaque Genome Sequencing and Analysis Consortium. Evolutionary and biomedical insights from the rhesus macaque genome. Science. 2007;316:222–234. doi: 10.1126/science.1139247. [DOI] [PubMed] [Google Scholar]
- Rogozin IB, Thomson K, Csürös M, Carmel L, Koonin EV. Homoplasy in genome-wide analysis of rare amino acid replacements: the molecular-evolutionary basis for Vavilov’s law of homologous series. Biology Direct 2008. 2008;3:7. doi: 10.1186/1745-6150-3-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rokas A, Carroll SB. Frequent and widespread parallel evolution of protein sequences. Mol Biol Evol. 2008;25:1943–1953. doi: 10.1093/molbev/msn143. [DOI] [PubMed] [Google Scholar]
- Sakuraba Y, Kimura T, Masuya H, Noguchi H, Sezutsu H, Takahasi KR, Toyoda A, Fukumura R, Murata T, Sakaki Y, Yamamura M, Wakana S, Noda T, Shiroishi T, Gondo Y. Identification and characterization of new long conserved noncoding sequences in vertebrates. Mamm Genome. 2008;19:703–712. doi: 10.1007/s00335-008-9152-7. [DOI] [PubMed] [Google Scholar]
- Teshima KM, Innan H. The effect of gene conversion on the divergence between duplicated genes. Genetics. 2004;166:1553–1560. doi: 10.1534/genetics.166.3.1553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson JD, Higgins DG, Gibson TJ. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, positions-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22:4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyckoff GJ, Joyce L, Wu C-I. Molecular evolution of functional genes on the mammalian Y chromosome. Mol Biol Evol. 2002;19:1633–1636. doi: 10.1093/oxfordjournals.molbev.a004226. [DOI] [PubMed] [Google Scholar]
- Xia X, Xie Z. Protein structures, neighbor effect, and a new index of amino acid similarities. Mol Biol Evol. 2002;19:58–67. doi: 10.1093/oxfordjournals.molbev.a003982. [DOI] [PubMed] [Google Scholar]
- Yang Z. PAML: a program package for phylogenetic analysis by maximum likelihood. Comput Appl BioSci. 1997;13:555–556. doi: 10.1093/bioinformatics/13.5.555. [DOI] [PubMed] [Google Scholar]
- Yang Z, Nielsen R. Estimating synonymous and nonsynonymous substitution rates under realistic evolutionary models. Mol Biol Evol. 2000;17:32–43. doi: 10.1093/oxfordjournals.molbev.a026236. [DOI] [PubMed] [Google Scholar]
- Zhang J, Kumar S. Detection of convergent and parallel evolution at the amino acid sequence level. Mol Biol Evol. 1997;14:527–536. doi: 10.1093/oxfordjournals.molbev.a025789. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.