

Abstract
Endothelial cell migration and proliferation, central steps in both physiologic and pathologic angiogenesis, require cytoskeletal-dependent remodeling, which is, in large part, achieved by the dynamic regulation of the β-actin network. Specifically, the β-actin network has previously been shown to be (i) enriched in regions of highly motile cytoplasm, and (ii) modulated by its isoactin-specific barbed-end capping protein, β cap73. We hypothesize that regulated over-expression of β cap73 could disrupt angiogenesis by capping β-actin-filament assembly thus inhibiting the incipient cellular migration and microvascular morphogenesis that ensues. Indeed, upon infection of capillary endothelial cells (cEC) with an adenovirus encoding the full-length βcap73 (Ad-βcap73), there is a robust cellular rounding response that occurs concomitantly with cytoskeletal disruption, as visualized with immunofluorescence microscopy. Further, we demonstrate that over-expression of Ad-βcap73 inhibits cEC migration in wound healing studies. Quantitative in vitro angiogenesis assays reveal that Ad-βcap73 not only prevents endothelial cells from forming capillary-like networks, but also induces the collapse of preformed endothelial tubes. In testing whether Ad-βcap73 impairs angiogenic events by inducing anoikis/apoptosis, we demonstrate that β cap73 infection activates a caspase-3-mediated cell death response as observed by quantitative Western blotting and immunofluorescence analyses. Altogether, these findings suggest that endothelial-specific targeting and βcap73 over-expression may represent an innovative therapeutic approach capable of abrogating pathologic angiogenesis.
Keywords: βcap73, adenovirus, endothelium, neovascularization, wound healing, anoikis
Introduction
Angiogenesis, the sprouting and generation of nascent capillaries from pre-existing microvessels, occurs throughout life (Lamalice et al., 2007). Upon angiogenic stimulation, local endothelial cells are activated to remodel their extracellular matrix via secretion of various proteases; subsequently, endothelial cell migration and proliferation occurs. Vessel remodeling and stabilization occurs upon recruitment of smooth muscle cells/pericytes, while tube maturation culminates with the initiation of blood flow (Lamalice et al. 2007).
Microvascular morphogenesis is likely regulated by the dynamic remodeling of the endothelial cytoskeleton; moreover, it is becoming evident that diverse angiogenic processes including migration, proliferation, and differentiation may be isoactin-dependent. For example, in cells expressing both muscle and non-muscle isoforms, α-smooth muscle actin is largely restricted to areas of non-motile cytoplasm, i.e. stress fibers, while non-muscle β-actin is specifically localized to domains of highly motile and actively remodeling cytoplasm, such as the leading edge ([DeNofrio et al., 1989], [Herman and D’Amore, 1985], [Bassell et al., 1998], and [Hoock et al., 1991]). In neuronal pathfinding, growth factor stimulation induces the specific localization of β-actin mRNA and protein to highly motile growth cones and filopodia, thus driving local β-actin polymerization in areas of active cell extension (Zhang at al., 1999). Also, fibroblasts that recruit β-actin mRNA using isoform-specific 3′-UTR sequences to actively remodeling lamellopodia show an increase in cell migration when compared to fibroblasts that are devoid of β-actin mRNA at the leading edge (Kislauskis et al., 1999). Thus, stationary and stable cytoplasm is linked with α- and γ-actin isoforms, while transient and remodeling cellular structures are associated with the β-actin cytoskeleton.
Specific spatial, temporal and cell-type restricted patterns suggest that, although highly homologous, actin isoforms have individual and non-overlapping roles in governing cellular functions. Indeed, upon differentiation, non-muscle precursors replace cytoplasmic β- and γ-actins for their skeletal, cardiac, or smooth muscle cell-specific equivalents upon maturation (Khaitlina, 2007). Additionally, in Drosophila flightless mutants, the human β-actin isoform is unable functionally compensate for the endogenous flight muscle specific actin despite remarkable sequence conservation, with only 15 amino acid divergences (Brault et al., 1999). Although considerable data suggest that actin isoforms perform specialized subcellular roles, the molecular mechanisms connecting microenvironmental signaling to isoactin-dependent cytoskeletal mobilization during angiogenesis remain equivocal.
While investigating β-actin filament dynamics during angiogenesis, our laboratory identified a novel β-actin specific capping molecule, β cap73 ([Shuster and Herman, 1995], [Shuster et al., 1996], and [Welch and Herman 2002]). Specifically, βcap73 binds to the barbed ends of β, but not α-, actin filaments thus inhibiting actin polymerization (Welch and Herman 2002). Additionally, βcap73 co-localizes with β-actin in the forward aspects of motile cytoplasm (Shuster and Herman, 1995). In motility and spreading-impaired cells, β cap73 levels are up-regulated ([Welch and Herman, 2002] and [Potter et al., 1998]). Therefore, we predict that a tightly-regulated signaling complex of membrane and isoactin associating proteins function to and through β cap73, thus modulating β-actin mediated processes and leading edge dynamics during endothelial motility.
In our continued efforts aimed at revealing the mechanisms directing isoactin-dependent cell migration and in our pursuit of innovative therapeutic solutions for pathologic angiogenesis, we have performed a series of experiments focused on curtailing endothelial migration and morphogenesis by specifically targeting the β-actin network. As such, we interrogated the potential of β cap73 over-expression in regulating β-actin-mediated cytoskeletal remodeling during capillary endothelial cell (cEC) function in angiogenic processes. In order to induce the regulated over-expression of β cap73, we generated an adenoviral construct expressing the full-length β cap73 (Ad-βcap73), which readily transduces our cEC cultures. Strikingly, we demonstrate that Ad-βcap73 markedly disrupts angiogenic processes, including migration and morphogenesis, in part by inducing apoptosis/anoikis. Importantly, our findings implicate that targeting cytoskeletal remodeling by manipulating endogenous isoactin-specific control proteins, such as βcap73, could offer unique opportunities for anti-angiogenesis therapeutics.
Materials and Methods
Adenovirus construction
βcap73 was cloned from a bovine retinal capillary endothelial cell (cEC) library as previously published (Welch and Herman, 2002). The full length β cap73 (NCIB accession number AY152693, nucleotide start 2300, stop 4596) was subcloned from the pEF-BOS vector into pAdTrack-CMV, in a strategy to generate an N-terminal myc tagged fusion product. The forward primer contained a KpnI site, a Kozak consensus site, and a region targeting the myc sequence (5′GCGGTTACCACCACCATGGAACAAAAACTCATCTCAGAA-3′). The reverse primer contained an XbaI restriction site, and a region complementary to the β cap73 3′ sequence (5′GTATCTAGACTAGCACACGAGCCCCTGCCG-3′). The βcap73 -pAdTrack-CMV construct, confirmed by restriction digestion and DNA sequencing, was linerarized with PmeI and co-transformed with the pAdEasy-1 adenoviral backbone into E. coli BJ5183 and selected for kanamycin resistance. The recombinant plasmid was transfected into the 293 packaging line. The resultant Ad-βcap73, an Ad5 serotype deleted for E1 and E3, was amplified and purified using CsCl density gradient centrifugation. Adenoviral titer was determined by using the Adeno-X Rapid Titer Kit according to the manufacturer’s instructions (Clontech).
Western Blotting
2×105 cEC were infected with 500 MOI of either Ad-βcap73 or Ad-GFP. After 24 hours (h), both floating and attached cells were pelleted, washed with PBS, and treated with lysis buffer (0.1% Triton-X100, 40mM HEPES, 50mM PIPES, 75mM NaCl, 1mM MgCl2, 0.5mM EGTA, 1mM PMSF, and Sigma protease inhibitor cocktail at 1:100). Equal cell numbers were run using SDS-PAGE electrophoresis. Proteins were transferred to nitrocellulose membranes and blocked with 3% BSA/TBST for 1h at room temperature (RT), incubated with primary antibodies for 2h RT, washed in TBST, and incubated with secondary antibodies for 1h RT. Chemiluminescence was detected using a CCD detection system (UVP) and WesternC reagent (Bio Rad). Antibodies were used at 1:1000 and include c-myc (9E10, Santa Cruz), GFP (I-19, Santa Cruz), β-actin (Hoock et al. 1991), anti-βcap73 (Covance custom sera), and activated caspase-3 (Sigma). All secondary antibodies were HRP-conjugated and used at 1:10,000.
Immunofluorescence
2×105 cECs were plated on 12mm glass coverslips in 24 well plates, allowed to settle for 2–3h, and infected with 500 MOI of either Ad-βcap73 or Ad-GFP. After approximately 20h of infection, monolayers were mechanically injured. After 1–2h, cells were fixed with 4%formaldehyde/1X DMEM and permeabilized with lysis buffer (0.1% Triton-X100, 50mM HEPES, 50mM PIPES, 1mM MgCl2, 0.1mM EGTA, and 75mM KCl). Cells were washed with PBS/0.02% azide, and incubated with primary antibodies (1:100: β-actin (Hoock et al., 1991), activated caspase-3 (Sigma)) for 45 minutes at RT and secondary antibodies (1:200: Alexa-488 or 546 conjugated) for 30 minutes RT. Alexa488-Phalloidin was incubated with the secondary antibodies and used at a concentration of 0.25U. For DNA staining, Hoechst dye (Molecular Probes) was added to the mounting media (9:1 glycerol to PBS/0.02%azide mixture) at a 1:1000 dilution. Images were obtained with a Zeiss Axiovert 200M equipped with a Hamamatsu (Orca ER) camera and a mercury fluorescence light source (X-Cite).
Matrigel Tube Formation/Collapse Assays
For the prevention of tube formation studies, 250μL of growth factor reduced Matrigel (BD Biosciences) was polymerized for 1h at 37°C in 8-well chamberslides. Subsequently, 5×104 cECs were plated per well in 200uL 5% BCS/DMEM and let settle for 30 min at 37°C. Then, 500 MOI of experimental/control viruses were added to the wells. Tube formation was observed at 18h post-plating. Images were taken using a Zeiss Axiovert 200M microscope equipped with a Hamamatsu (Orca ER) camera and a mercury fluorescence lamp (X-Cite) and analyzed using ImageJ. For tube-collapse studies, Matrigel and cells were prepared as above. After tube structures started to appear approximately 5h post plating, 500 MOI of experimental/control virus was applied. After 22h, images were captured using a Zeiss Axiovert 200M microscope equipped with a Hamamatsu (Orca ER) camera and a mercury fluorescence lamp (X-Cite) and analyzed using ImageJ.
Wound Healing Assays
2×105 cECs were plated into 24-well plates, and infected with 500 MOI of either adenovirus. At 24h post infection, monolayers were mechanically injured. Time-lapse microscopy was employed using a motorized stage (Ludl) and captured using Metamorph software (Molecular Devices) and a Zeiss Axiovert 200M microscope equipped with a Hamamatsu (Orca ER) camera and a mercury fluorescence lamp (X-Cite). Images were captured every 10 minutes using phase contrast and GFP settings for four hours. Wound area was measured using ImageJ and percent wound closure over times was calculated.
Results
Generation of Ad-βcap73
Our laboratory has previously identified and cloned β cap73, a β-actin specific capping protein that serves as the molecular link between the β-actin filament network and the plasma membrane-cytoskeletal crosslinker, ezrin ([Shuster and Herman, 1995], [Shuster et al., 1996], and [Welch and Herman 2002]). As such, we sought to learn whether targeting this critical link would impair the endothelial migration and morphogenesis that is required for angiogenesis. We generated an adenoviral construct to over-express the full length β-actin specific capping protein, βcap73. This adenovirus has two separate expression cassettes: (1) CMV-driven expression of βcap73, and (2) CMV-driven expression of GFP as a vital marker (Fig. 1A). In control treatments, the Ad-GFP is employed, which is a single expression cassette that drives GFP expression via a CMV promoter (Fig. 1A). When cEC are infected with adenoviruses, while attached to Matrigel, we observe a rapid viral transduction; protein product is transcribed as early as 6 hours post-infection with an increase in both percentage of cells infected and in GFP fluorescence intensity over time (Fig. 1B). 24 hours post-infection, cells are then harvested for Western blotting. We detect the exogenously expressed βcap73 (an 110kD myc-tagged species) as probed with anti-myc IgG, and the over-expressed and endogenous β cap73 using a rabbit polyclonal antibody, which is specific for β cap73 (Fig. 1C and data not shown). The GFP expression levels are similar in both experimental and control treatments. Thus, our adenoviral-βcap73 model is not only able to effectively drive the over-expression of this molecule in vitro.
Figure 1. Generation of Adenoviral-βcap73 (Ad- βcap73).

(A) Map of the viral expression cassettes of Ad-βcap73 (top) and Ad-GFP (bottom). CMV promoter, PA: poly-adenylation sequence.(B) cEC are plated on Matrigel, and infected with Ad-βcap73 or Ad-GFP. Infected cultures are analyzed for onset of viral protein expression at 4, 6, 8, and 10h post-infection. The appearance of GFP-positive cells starting at 6 hours post-infection reveals that viral proteins are actively being transcribed in these cells. (C) cEC lysates, generated after infection with either Ad-βcap73 or Ad-GFP, were probed for the myc-tagged βcap73 (110kD), the over-expressed βcap73 recognized by βcap73-specific IgGs (110kD), GFP (27kD), and β-actin (42kD).
Ad-βcap73 inhibits the endothelial migratory response to injury
As cell motility is mainly driven by the reorganization and polymerization of β-actin filaments, we hypothesized that over-expression of the β-actin specific capping molecule, β cap73, would inhibit endothelial migration post-injury. Endothelial monolayers infected with either Ad-GFP or Ad-βcap73 are mechanically injured, and time lapse imaging is employed to document wound closure over time. All GFP-infected cEC monolayers steadily migrate over time, with wound closure completed by 4 hours post-injury (Fig. 2A). However, in the β cap73 over-expressing cells, normal wound healing is severely impaired: cell monolayers display perturbed cell-cell and/or cell-matrix contacts, characterized by numerous contracted cells and acellular gaps behind the wound edge (Fig. 2A). As quantified in percent wound closure over time, all control GFP-infected wounds heal at consistent rates over time; however, there is a marked inhibitory effect of Ad-βcap73 on cEC motility, causing a 50% impairment in wound closure (Fig. 2B). Since βcap73-infected cEC demonstrate an abnormal wound healing response, we performed immunofluorescent analyses on adenoviral-treated, migrating endothelial monolayers. In high resolution, GFP-infected cEC uniformly generate a polarized leading edge, rich in β-actin; additionally, upon phalloidin-labeling, cells display intact and intricate actin stress fiber organization (Fig. 2C). However, infection with βcap73 markedly alters normal endothelial cell structure and function: cEC are unable to spread and generate a proper monolayer formation (Fig. 2C). Also, lamellopodia that mark polarized and actively migrating cells are absent from βcap73-infected cultures. Instead, cells appear to contract, with long, slender, β-actin rich filopodia that do not appear to preferentially localize into the wound (Fig. 2C). Notably, phalloidin staining reveals a reduction in stress fibers and a diminution in global actin cytoskeletal organization in Ad-βcap73 treated cEC (Fig. 2C). Taken together, these results suggest that over-expression of βcap73 causes a disorganization in cytoskeletal architecture linked with impaired cell migration.
Figure 2. Ad-βcap73 inhibits the endothelial migratory response to injury.

(A) cEC monolayers infected with Ad-GFP or Ad-βcap73 were mechanically injured and migration was captured and quantified using digital imaging microscopy. In GFP-infected monolayers, post-injury migration persists unimpeded and wound closure is complete within four hours (injured space highlighted with dashed lines). However, in the βcap73 -overexpressing endothelial cultures, the wound remains unclosed due to defective cell shape and motility, which results in an imperfect monolayer where post-injury migration, cell-cell and cell-matrix contacts are perturbed (solid enclosed lines). Scale bar: 100μm (B) Quantitative analysis demonstrates that Ad-GFP wounds close steadily over the four hour experimental time period while the Ad-βcap73 wounds are impaired by approximately 50% in their healing rates. (C) cEC monolayers infected with either Ad-GFP or Ad-βcap73 were mechanically injured. Two hours post-injury, cells were processed for fluorescence imaging. In GFP-infected monolayers, cells polarize immediately post injury and establish a leading edge adjacent to the wound (dashed line). However, in βcap73 -infected cultures the contiguous monolayer is disrupted following perturbations in cell shape, motility, cell-cell and cell-matrix adhesions, which is apparent with differential interference contrast (DIC) imaging (gaps behind wound edge demarcated by dashed line), and following anti-β-actin IgG and fluorescent phalloidin localization. Note βcap73 -infected cells detaching from the matrix (arrowhead). Also, phalloidin-stress fiber staining is highly diminished in βcap73 -infected cells (asterisks) and β-actin rich filopodia (blunt arrowhead) are prevalent within the βcap73 -infected, but not control populations. Scale bar: 25μm
Ad-βcap73 inhibits capillary morphogenesis
During angiogenesis, the endothelium markedly undergoes cell shape changes in generating functional tube structures. We postulated that increased β cap73 levels would impair β-actin mediated cEC shape changes that occur during in vitro morphogenesis. Immediately post-seeding equal cell numbers on Matrigel, cEC are infected with either Ad-GFP or Ad-βcap73. Approximately 18 hours after infection, cEC are analyzed for tube formation. Extensively organized endothelial architecture, i.e. nodes, ringlets, and tube structure, is observed subsequent to GFP infection (Fig. 3A). However, upon elevating levels of β cap73, there is a marked alteration of in vitro morphogenesis: cEC cluster in abnormal node alignments, are devoid of internodal connections, and remain as single cells, unincorporated into any tubular or nodal structures (Fig. 3A). Compared to control populations, β cap73 over-expressing cEC exhibit a significant 62% decrease in both tube number and in total tube length (Fig. 3B). Therefore, we posit that Ad-βcap73 inhibits normal angiogenic processes, including tube formation.
Figure 3. Ad-βcap73 inhibits capillary morphogenesis.
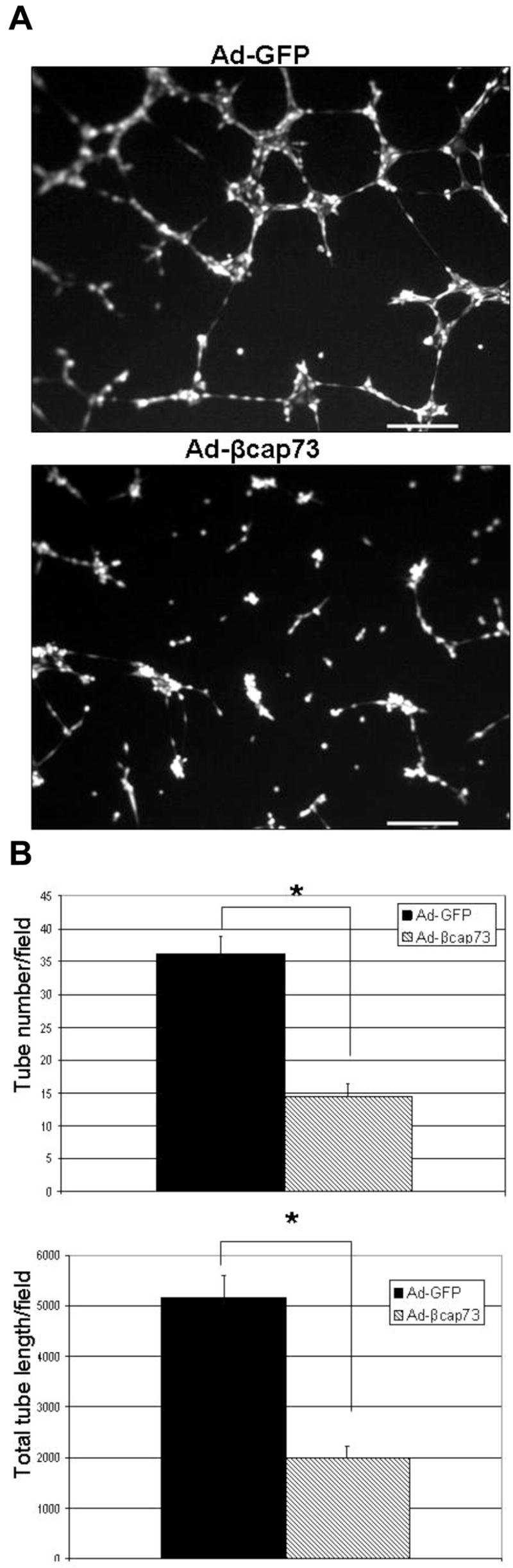
(A) cEC were plated on growth factor reduced Matrigel, infected with Ad-GFP or Ad-βcap73 infection, and observed using phase contrast (data not shown) and GFP fluorescence microscopy. Fluorescence imaging reveals that β cap73 over-expressing cEC are significantly impaired in their ability to undergo in vitro angiogenesis: there is a noticeable absence of endothelial-lined tubules with a definitive lack of inter-connectedness when compared to GFP-infected control cultures. Scale bar: 250μm (B) Quantitative image analysis reveals that compared to Ad-GFP, both tube number (left) and total tube length (right) is reduced by 62% in Ad-βcap73 infected populations (p<0.0001).
Ad-βcap73 causes collapse of capillary endothelial cell-derived tubes in vitro
With the finding that β cap73 is able to prevent cEC morphogenesis, we tested whether Ad-βcap73 could collapse newly establishing vessels. As in vitro tube formation commences in the absence of viral infection, cEC coalesce into nodes, align into tube-like precursors, and generate cellular projections towards adjacent nodes (data not shown). Once capillary formation is established, at approximately 5 hours post seeding upon Matrigel, cEC are infected with either Ad-GFP or Ad-βcap73; thus, at the time of analysis (22 hours post plating) cEC express viral protein for approximately 11 hours. Compared with GFP-infected populations, which contained numerous nodes, ringlets, and tubes, the βcap73 - infected populations are unable to maintain capillary endothelial structure and exhibit altered cell shape, with heavily contracted/poorly spread cells present within any remaining structure (Fig. 4A). In comparison to controls, βcap73 over-expressing cEC demonstrate a 55 and 61% decrease in tube number and total tube length, respectively (Fig. 4B). Therefore, Ad-βcap73 may have the potential to regress preformed vessels in pathologic angiogenesis.
Figure 4. Ad-βcap73 causes collapse of capillary endothelial cell-derived tubes in vitro.
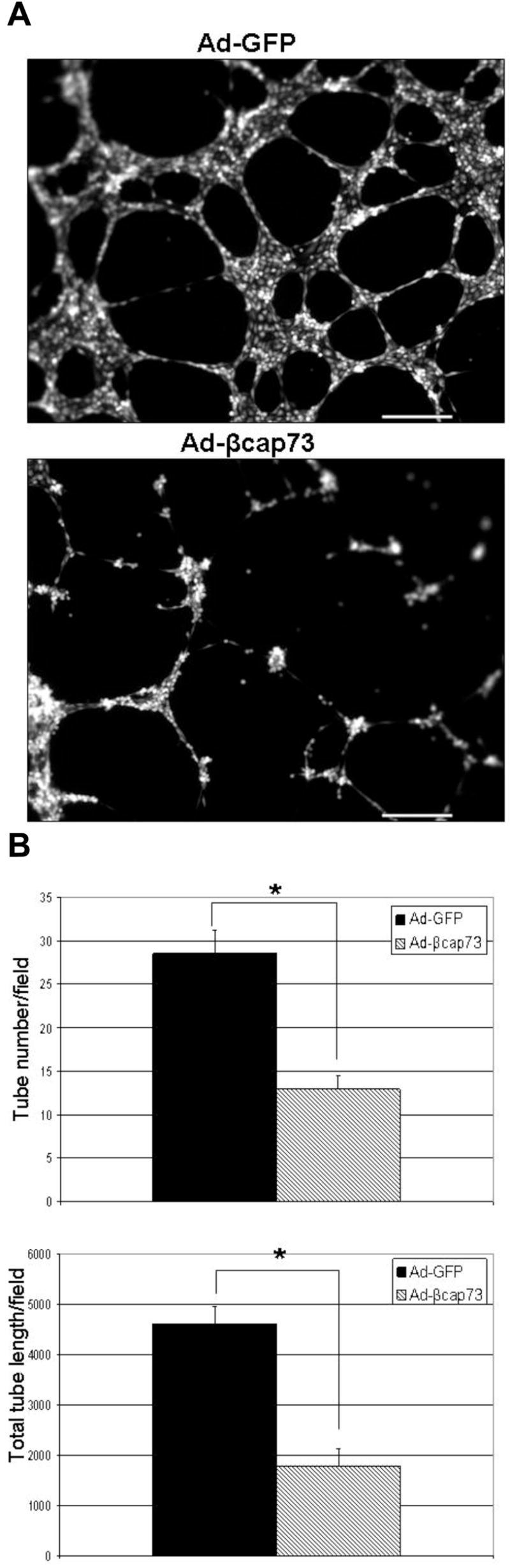
(A) In vitro angiogenesis was enabled on growth factor reduced Matrigel. Subsequently, cEC present within nascent pre-formed capillaries were infected with either Ad-GFP or Ad-βcap73. In contrast to the GFP-infected cEC, which continue to remodel in vitro assembled capillaries, βcap73-overexpressing cEC are unable to sustain angiogenesis. Indeed, βcap73-infected cEC present within angiogenic sprouts begin a catastrophic disassembly, typified by a noticeable absence of extended tip cells, disruption of inter-connected nodal arrays, and accumulation of retracted endothelial mounds, which lack an angiogenic phenotype. Scale bar: 250μm (B) Quantitative analysis reveals that compared to Ad-GFP, tube number (left) is reduced by 55% and total tube length (right) is reduced by 61% in Ad-βcap73 infected populations (p<0.002, and p<0.00002, respectively).
Ad-βcap73 induces anoikis/apoptosis in cEC
Cells that are unable to maintain cell-cell or cell-matrix connections are described to initiate a specialized form of apoptosis known as anoikis ([Gilmore, 2005], [Grossman, 2002], and [Reddig and Juliano, 2005]). With the observation that βcap73 -over-expressing cEC actively round and detach (Fig. 5A), we hypothesized that over-expression of βcap73 prevents cells from generating and/or maintaining their cell-cell and cell-matrix interactions that are dependent on actin cytoskeletal remodeling; therefore, βcap73-infected cells may undergo anoikis. Firstly, we analyzed nuclear architecture in endothelial monolayers and at the wound edge. While normal monolayer organization and nuclear integrity is observed in GFP-infected cEC, strikingly, there is marked DNA fragmentation that coincides with cellular contraction in βcap73 -infected cells (Fig. 5B). Additionally, we interrogated endothelial tube structures for markers of apoptosis, including induction of a key initiator of the cell death cascade, caspase-3 and nuclear fragmentation. Although there is a slight detection of caspase-3 in GFP-infected endothelial tubes, there is extensive expression of activated caspase-3 in β cap73 over-expressing cultures (Fig. 5C). A substantial population of βcap73 -infected cEC expresses activated caspase-3 with prominent localization in remaining nodes, tubes, and regressed/collapsed structures; however, in control treatments, activated caspase-3 is restricted to cells within large nodes, a feature that is representative of the normal cell death that occurs during this model of in vitro angiogenesis. Also, fragmented nuclei, a late stage marker of apoptosis, are present in high numbers throughout β cap73 over-expressing cEC. Finally, we performed Western blot analyses on infected cell lysates for both activated caspase-3 and for its target protein, PARP. A significant increase in the 17kD species representing activated caspase-3 and cleaved PARP is observed in the βcap73 over-expressing cells (Fig. 5D). Thus, we propose that, in part, Ad-βcap73 exerts its inhibitory effects on normal angiogenic processes, including cEC migration and morphological changes, by inducing apoptosis/anoikis.
Figure 5. Ad-βcap73 induces anoikis/apoptosis in cEC.

(A) Live, infected cEC cultures in phase contrast (top panels) and GFP fluorescence (bottom panels). Note the rounded and detached cells, which are present in the Ad-βcap73 infected cultures (arrowheads). Scale bar: 250μm (B) Confluent cEC cultures were infected with Ad-GFP or Ad-βcap73 for 20 hours prior to fixation, permeabilization, and imaging for DIC, nuclei (Hoechst) and F-actin (phalloidin). Note contraction of βcap73 -infected cells, fragmentation of nuclei, diminished phalloidin staining, and perturbation of monolayer formation as compared with Ad-GFP infected cells. Scale bar: 25μm (C) In vitro angiogenesis was enabled on growth factor reduced Matrigel. cEC within nascent pre-formed capillary-like structures were treated with either Ad-GFP or Ad-βcap73. Matured tubes were fixed, permeabilized, and visualized using phase contrast, Hoechst-labeled nuclei, and active caspase-3 fluorescent IgGs. Note the abnormal cell structure/alignment in phase contrast, the intense caspase-3 staining, and the fragmented nuclei in Ad-βcap73-infected endothelial tubes. Scale bar: 100μm (D) cEC were infected for 24 hours with either Ad-GFP or Ad-βcap73 viruses at identical MOI prior to lysis in SDS gel sample buffer, electrophoresis and Western blotting. Ad-GFP and Ad-βcap73 over-expressing cell lysates were subsequently probed for activated caspase-3, a central effector caspase (17kD), and cleaved PARP, a substrate of activated caspase-3 (89kD). In βcap73 -overexpressing cells, steady state levels of activated caspase-3 and cleaved PARP levels are significantly elevated.
Discussion
Ad-βcap73 antagonizes vital angiogenic processes
Controlling pathologic angiogenesis will likely require continued development of innovative strategies and novel tools that disrupt endothelial migration and morphogenesis. Because of this, we sought to further delineate the molecular mechanisms controlling endothelial β-actin dynamics during cell migration and angiogenesis in vitro by adenoviral-mediated over-expression of the β-actin specific capping molecule, β cap73. Here, we demonstrate Ad-βcap73 delivery to cEC derived from mammalian retina affects in vitro capillary morphogenesis via altering actin-dependent cytoskeletal dynamics. Ad- βcap73 infected endothelial cell cultures exhibit impaired cell migration, enhanced cellular rounding and detachment from growth substrates. Concomitantly, there is marked increase in detachment-dependent cell death, or anoikis.
Ad-βcap73 inhibits cellular responses to injury and motility by cytoskeletal disorganization
Notably, β-actin has been functionally linked with areas of actively remodeling cytoplasm, including filopods, fan lamellae, and growth cones ([Bassell et al., 1998], [Hoock et al., 1991], and [Herman, 1993]). Indeed, elevated synthesis of β-actin in vitro causes a marked increase in cellular spreading (Schevzov et al., 1992). On the other hand, increased βcap73 protein levels are linked with impaired cell spreading (Welch and Herman, 2002). As postulated, we find that βcap73 over-expression leads to defects in cell polarization, migration, and wound healing. These observations are in stark contrast to previous studies in which over-expression of gelsolin or CapG, actin capping proteins with isoform-independent capping activities, elevates fibroblast motility ([Cunningham et al., 1991] and [Sun et al., 1995]). Moreover, CapG expression levels, endogenously up-regulated when conditioned with unidirectional flow, coincide with increased endothelial cell migration and faster wound healing (Pellieux et al., 2003). Also, gelsolin and CapG null mice experience lengthened bleeding times associated with alterations in platelet function, dampened inflammation due to decreased neutrophil migration, and slower wound healing caused by impaired fibroblast motility (Witke et al., 1995). Notably, gelsolin and CapG have no strict preference for any specific actin isoform, while βcap73 binds and caps filaments of β-actin, exclusively. Here, we report that β cap73 elevation in cEC promotes cytoskeletal collapse and β-actin mislocalization. These combined results suggest that an intact and dynamically reorganizing β-actin network is integral for normal cell function.
βcap73 over-expression impairs capillary morphogenesisin vitro
During angiogenesis, endothelial remodeling is integral to capillary formation and function (Lamalice, 2007). During in vitro tube formation, uninfected control cultures or cells infected with Ad-GFP assemble into highly organized arrays of tube-like structures. However, upon increased expression of βcap73, the inter-connected network of capillary-like tubes is strikingly absent. Interestingly, after allowing capillary morphogenesis to occur for several hours in the absence of virus, β cap73 over-expression induces the collapse of tube-like structure. In contrast to our findings, other work suggests that during active capillary formation, endothelial cells embedded in a collagen matrix significantly up-regulate gelsolin levels, and these morphology-differentiated endothelial cells have higher spontaneous motility (Salazar et al., 1999). Our results continue to support the notion that distinct actin-capping proteins govern unique cellular functions, since elevated levels of β cap73 are inhibitory to capillary morphogenesis while elevated levels of gelsolin are essential for normal tube formation.
βcap73 over-expression induces detachment-mediated cell death
For adherent cells, it is critical to preserve attachments to the growth substrate; once detached, cells undergo a specialized form of apoptosis known as anoikis ([Gilmore, 2005], [Grossman, 2002], and [Reddig and Juliano, 2005]). Indeed, modulation of cell shape through specialized micropatterned surfaces reveals that cell morphology translates to cell function: cellular spreading stimulates growth while cellular rounding induces apoptosis (Chen et al., 1998). As such, with βcap73 over-expression leading to cellular contraction and detachment, we predicted that Ad-βcap73 may induce anoikis/apoptosis. Indeed, in endothelial monolayers, activation of a caspase-3-mediated apoptotic cascade coincides with a dramatic reduction in actin stress fiber assembly. Also, our results reveal that the inhibition of in vitro angiogenesis is partially mediated by apoptosis, as activated capsase-3 is highly expressed in βcap73 -overexpressing capillary endothelial tubes. Western blot analysis confirms that apoptosis occurs in βcap73-overexpressing cells, as indicated by activated caspase-3 and cleaved PARP. Interestingly, during apoptosis, activated caspase-3 cleaves gelsolin; this truncated actin binding protein no longer requires activation by calcium for its actin-filament severing activity (Cooper and Schafer, 2000). Presumably, apoptotic progression is propagated by gelsolin-mediated severing of actin filaments. Indeed, over-expression of gelsolin prevents caspase-3 activation and loss of mitochondrial membrane potential, each reflective of early events in apoptosis (Silacci et al., 2004). Since we demonstrate that βcap73 over-expression activates caspase-3 and others have shown that gelsolin is a caspase-3 target, it is possible that βcap73 over-expressing cEC activate gelsolin, which enables actin filament severing, leading to cytoskeletal collapse and a commitment to apoptosis. If valid, this hypothesis may shed light on the molecular mechanisms leading to the extensive cytoskeletal disruption, observed at the level of β-actin and total actin networks, when βcap73 is over-expressed.
There is a dynamic reciprocity between cytoskeletal integrity and cellular adhesion, each playing a crucial role in determining cell survival (Reddig and Juliano 2005). Interestingly, the perturbations in the cytoskeleton that are induced by the actin stabilizing drug, jasplakinolide, or the actin depolymerizing drug, cytochalasin D, which both solely do not induce cell death, are able to enhance the commitment to the apoptosis that is induced by cytokine-withdrawl (Posey and Bierer, 1999). These data may shed light on our results where a detachment-induced apoptotic phenotype arises in Ad-βcap73 infected cells. Perhaps, perturbation of cytoskeletal dynamics, either with actin-stabilizing drugs, actin-depolymerizing agents, or isoactin-specific capping activities initiates a signaling cascade that terminates in detachment-dependent cell death. If so, this insight might prove useful as we prepare to develop strategies capable of acutely disrupting invasive migratory or unwanted proliferative behaviors.
βcap73-mediated control of endothelial cytoskeletal dynamics
Paradoxically, actin capping proteins that limit filament growth in vitro have been shown to stimulate actin-based cell motility; an effect that has been attributed to a downstream stimulation of Arp2/3-dependent dendritic nucleation upon Arp2/3 complex binding sides of tightly capped actin filaments ([Bear, 2008] and [Akin and Mullins, 2008]). This work reveals that elevation in capping protein levels does not alter actin assembly kinetics but stimulates a dynamic reorganization of the underlying cytoskeletal network (Akin and Mullins, 2008). These new findings may help to explain the motility-stimulating phenomena of other actin capping proteins, which are not selective in their isoactin-interacting activity, but these data do not adequately account for the regulatory role that isoactin-specific capping proteins play in modulating cell shape and motility. We demonstrate, here, that over-expression of an isoactin-specific capping protein fosters cytoskeletal reorganization, while inhibiting cell motility, promoting cell rounding, and interfering with in vitro angiogenesis. These findings are consistent with earlier reports that β-actin filament assembly drives cell migration and spreading, while γ- and α-actins are largely involved in controlling cell shape. The regulated expression of β cap73 is likely to ensure that β-actin filament assembly is tightly coordinated and localized to actively remodeling cytoplasm at the plasma membrane and that actin filament branching occurs secondary to β-actin filament elongation at the membrane interface. Future experiments are aimed at delineating the molecular mechanisms the control βcap73 -dependent inhibition of cEC migration and angiogenesis, when endogenous protein levels are up-regulated and cytoskeletal protein-protein interactions are dis-equilibrated. Finally, we predict that regulated, endothelial-specific over-expression of β cap73 via use of an endothelial-specific promoter, should prove to be an innovative anti-angiogenic therapeutic approach for the abrogation of pathologic angiogenesis in vivo.
Acknowledgments
This work was supported by the NIH grants T32 DK07542 (to J.T.D.) and NIH EY 15125 (to I.M.H.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Akin O, Mullins RD. Capping protein increases the rate of actin-based motility by promoting filament nucleation by the Arp2/3 complex. Cell. 2008;133:841–51. doi: 10.1016/j.cell.2008.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bassell GJ, et al. Sorting of beta-actin mRNA and protein to neurites and growth cones in culture. J Neurosci. 1998;18:251–65. doi: 10.1523/JNEUROSCI.18-01-00251.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bear JE. Follow the monomer. Cell. 2008;133:765–7. doi: 10.1016/j.cell.2008.05.012. [DOI] [PubMed] [Google Scholar]
- Brault V, et al. Substitution of flight muscle-specific actin by human (beta)-cytoplasmic actin in the indirect flight muscle of Drosophila. J Cell Sci. 1999;112:3627–39. doi: 10.1242/jcs.112.21.3627. [DOI] [PubMed] [Google Scholar]
- Chen CS, et al. Micropatterned surfaces for control of cell shape, position, and function. Biotechnol Prog. 1998;14:356–63. doi: 10.1021/bp980031m. [DOI] [PubMed] [Google Scholar]
- Cooper JA, Schafer DA. Control of actin assembly and disassembly at filament ends. Curr Opin Cell Biol. 2000;12:97–103. doi: 10.1016/s0955-0674(99)00062-9. [DOI] [PubMed] [Google Scholar]
- Cunningham CC, Stossel TP, Kwiatkowski DJ. Enhanced motility in NIH 3T3 fibroblasts that overexpress gelsolin. Science. 1991;251:1233–6. doi: 10.1126/science.1848726. [DOI] [PubMed] [Google Scholar]
- DeNofrio D, Hoock TC, Herman IM. Functional sorting of actin isoforms in microvascular pericytes. J Cell Biol. 1989;109:191–202. doi: 10.1083/jcb.109.1.191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilmore AP. Anoikis. Cell Death Differ. 2005;12:1473–7. doi: 10.1038/sj.cdd.4401723. [DOI] [PubMed] [Google Scholar]
- Grossmann J. Molecular mechanisms of “detachment-induced apoptosis: Anoikis. Apoptosis. 2002;7:247–60. doi: 10.1023/a:1015312119693. [DOI] [PubMed] [Google Scholar]
- Herman IM. Functional sorting of actin isoforms. Curr Opin Cell Biol. 1993;5:48–55. doi: 10.1016/s0955-0674(05)80007-9. [DOI] [PubMed] [Google Scholar]
- Herman IM, D’Amore PA. Microvascular pericytes contain muscle and nonmuscle actins. J Cell Biol. 1985;101:43–52. doi: 10.1083/jcb.101.1.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoock TC, Newcomb PM, Herman IM. Beta actin and its mRNA are localized at the plasma membrane and the regions of moving cytoplasm during the cellular response to injury. J Cell Biol. 1991;112:653–64. doi: 10.1083/jcb.112.4.653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khaitlina S. Mechanisms of spatial segregation of actin isoforms. Tsitologiia. 2007;49:345–54. [PubMed] [Google Scholar]
- Kislauskis EH, Zhu X, Singer RH. beta-Actin messenger RNA localization and protein synthesis augment cell motility. J Cell Biol. 1997;136:1263–70. doi: 10.1083/jcb.136.6.1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamalice LF, LeBoeuf F, Huot J. Endothelial cell migration during angiogenesis. Circ Res. 2007;100:782–94. doi: 10.1161/01.RES.0000259593.07661.1e. [DOI] [PubMed] [Google Scholar]
- Pellieux C, et al. Cap G, a gelsolin family protein modulating protective effects of unidirectional shear stress. J Biol Chem. 2003;278:29136–44. doi: 10.1074/jbc.M300598200. [DOI] [PubMed] [Google Scholar]
- Posey SC, Bierer BE. Actin stabilization by jasplakinolide enhances apoptosis induced by cytokine deprivation. 1999;274:4259–65. doi: 10.1074/jbc.274.7.4259. [DOI] [PubMed] [Google Scholar]
- Potter DA, et al. Calpain regulated actin remodeling during cell spreading. J Cell Biol. 1998;141:647–62. doi: 10.1083/jcb.141.3.647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddig PJ, Juliano RL. Clinging to life: cell to matrix adhesions and cell survival. Cancer Metastasis Rev. 2005;24:425–39. doi: 10.1007/s10555-005-5134-3. [DOI] [PubMed] [Google Scholar]
- Salazar R, Bell SE, Davis GE. Coordinate induction of the actin cytoskeletal regulatory proteins gelsolin, vasodilator-stimulated phosphoprotein, and profilin during capillary morphogenesis in vitro. Exp Cell Res. 1999;249:22–32. doi: 10.1006/excr.1999.4460. [DOI] [PubMed] [Google Scholar]
- Schevzov G, Llyod C, Gunning P. High level expression of transfected beta- and gamma-actin genes differentially impacts on myoblast cytoarchitecture. J Cell Biol. 1992;117:775–85. doi: 10.1083/jcb.117.4.775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shuster CB, Herman IM. Indirect association of ezrin with F-actin: isoform specificity and calcium sensitivity. J Cell Biol. 128(5):837–48. doi: 10.1083/jcb.128.5.837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shuster CB, et al. Beta cap73: a novel beta actin-specific binding protein. Cell Motil Cytoskeleton. 1996;35:175–87. doi: 10.1002/(SICI)1097-0169(1996)35:3<175::AID-CM1>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- Silacci P, et al. Gelsolin superfamily proteins: key regulators of cellular functions. Cell Mol Life Sci. 2004;61:2614–23. doi: 10.1007/s00018-004-4225-6. [DOI] [PubMed] [Google Scholar]
- Sun HQ, et al. Effects of CapG overespression on agonist-induced motility and second messenger generation. J Cell Biol. 1995;129:147–56. doi: 10.1083/jcb.129.1.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welch AY, Herman IM. Cloning and characterization of betaCAP73, a novel regulator of beta-actin assembly. Int J Biochem Cell Biol. 2002;34:864–81. doi: 10.1016/s1357-2725(01)00159-5. [DOI] [PubMed] [Google Scholar]
- Witke W, et al. Hemostatic, inflammaroty, and fibroblast responses are blunted in mice lacking gelsolin. Cell. 1995;81:41–51. doi: 10.1016/0092-8674(95)90369-0. [DOI] [PubMed] [Google Scholar]
- Zhang HL, Singer RH, Bassell GJ. Neurotrophin regulation of beta-actin mRNA and protein localization within growth cones. J Cell Biol. 1999;147(1):59–70. doi: 10.1083/jcb.147.1.59. [DOI] [PMC free article] [PubMed] [Google Scholar]