

Abstract
Pemetrexed represents the first antifolate cancer drug to be approved by the FDA in 20 years; it is currently in widespread use for first line therapy of mesothelioma and non- small cell lung cancer. Pemetrexed has more than one site of action; the primary site is thymidylate synthase. We now report that the secondary target is the downstream folate-dependent enzyme in de novo purine synthesis, aminoimidazolecarboxamide ribonucleotide formyltransferase (AICART). The substrate of the AICART reaction, ZMP, accumulated in intact pemetrexed-inhibited tumor cells, identifying AICART as the step in purine synthesis which becomes rate-limiting after drug treatment. The accumulating ZMP causes an activation of AMP-activated protein kinase with subsequent inhibition of the mammalian target of rapamycin (mTOR) and hypophosphorylation of the downstream targets of mTOR that control initiation of protein synthesis and cell growth. We suggest that the activity of pemetrexed against human cancers is a reflection of its direct inhibition of folate-dependent target proteins combined with prolonged inhibition of the mTOR pathway secondary to accumulation of ZMP.
Keywords: Purine synthesis, ZMP, AMP-activated protein kinase, mammalian target of rapamycin, pemetrexed
Introduction
The antifolate pemetrexed is a potent inhibitor of thymidylate synthase and a very efficient substrate for human folylpolyglutamate synthetase, creating anabolites that are retained intracellularly after drug exposure (1,2). However, pemetrexed also had at least one other target that became apparent from a continued antiproliferative effect in cell cultures exposed to exogenous thymidine, which prevents the cytotoxic effects of thymidylate synthase inhibition (3,4). A study of the activity of pemetrexed against several recombinant mouse and human enzymes in vitro led to the conclusion that both glycinamide ribonucleotide transformylase (GART) and dihydrofolate reductase (DHFR) were potential secondary targets for the polyglutamate forms of pemetrexed (4). The secondary target for pemetrexed is generally taken as GART (5), which was suggested, but not proven, by the in vitro enzyme kinetic data (4). The question of the identity of any secondary targets for pemetrexed has become of substantial interest given the clinical responses to the drug in lung cancers (6) an unusual activity for folate antimetabolites.
In a series of cell culture experiments, we have developed evidence that the target for pemetrexed secondary to thymidylate synthase is the second folate-dependent enzyme in purine synthesis, aminoimidazolecarboxamide ribonucleotide formyltransferase (AICART), not GART, the target for lometrexol (6R-DDATHF). The significance of this is substantial, since the substrate for GART is very labile and is probably inert due to this lability, whereas the substrate for AICART is ZMP, a known activator of the AMP-activated protein kinase, AMPK (7), a key controlling element in the mTOR pathway (8).
A therapeutic approach built around reactivation of control on a pathway often dormant in tumor cells due to loss of tumor suppressor gene product function or constitutive activation of c-oncogenes would be very attractive. The accumulation of ZMP offers such an approach, due to the placement of AMPK in the mTOR pathway. Such accumulation of a regulatory intermediate behind a metabolic block had been predicted to be a theoretical avenue for therapeutic drug design, but this case represents a rare instance in which this theory has been reduced to practice. Hence, pemetrexed is indeed a multi-targeted inhibitor whose activity appears to extend beyond the traditional targets of antifolates.
Materials and Methods
Cell Culture maintenance and reagents
CCRF-CEM human lymphoblastic leukemia cells were maintained at a density between 105–106 cells/ml in RPMI 1640 medium supplemented with 10% dialyzed fetal bovine serum. Reversal experiments were performed in media supplemented with 20 mM HEPES and 40 mM MOPS. Pemetrexed and (6R)-DDATHF were obtained from Eli Lilly and Co. and were dissolved in PBS. All other culture reagents were from Sigma Aldrich and were of the highest available quality. 6-[4-(2-piperidin-1-yl-etoxy)-phenyl)]-3-pyridin-4-yl-pyrrazolo [1,5-a] pyrimidine, (compound C) was also obtained from Sigma-Aldrich. Stock concentrations were determined spectrophotometrically. In studies using modifying agents 5.6 μM of thymidine, 32 μM hypoxanthine, 320 μM AICA were used, unless otherwise noted. Cell counts were obtained using a Beckman Coulter Counter. When CEM cells were treated for longer than 48 hours, fresh drug-containing media was applied at 48 hours.
N-Formyl glycinamide ribonucleotide (FGAR) synthesis from 14C-glycine
Exponentially-growing CEM cells were harvested and resuspended in RPMI 1640 medium without glutamine or serum and azaserine was added to 10 μM for 30 min at 37 °C. Glutamine at 2 μM and 0.25 μCi/ml of 14C-glycine were added and incubation was continued at 37 °C for 1 h. Nucleotides were extracted and radioactivity in the FGAR peak was measured by chromatography on AG1 resin as previously described (9).
HPLC analysis of ZMP
Following drug exposure, culture densities were determined electronically. Cell pellets were washed with PBS, resuspended in ice-cold 5% trichloroacetic acid at a density of 106 cells/200 ul, vortexed vigorously, and held on ice for 5 min. Debris was removed by centrifugation at 5,000 × g for 10 min, and the acid-soluble fractions were neutralized by two extractions with diethyl ether. Samples were passed through a 0.45 uM syringe filter before being injected onto a Phenosphere SAX 250 × 2.0 mm HPLC column (Phenomenex). Absorbance was monitored at 280 nm using a linear gradient of 5 mM NH4H2PO4, pH 2.8 to 750 mM NH4H2PO4, pH 3.9 over 25 min at a flow rate of 0.2 ml/min. ZMP concentration was determined by fitting peak absorbance to a standard curve generated from synthetic ZMP. The detection limit of this assay was < 60 pmol/106 cells.
Immunoblot analysis
Cells were lysed in lysis buffer containing 62.5mM Tris, pH 6.5, 5% glycerol, 2% SDS, 5% 2-mercapothethanol, 50mM NaF, 0.2 mM Na3VO4 and 1× protease inhibitor complete mixture (Roche Applied Science). Protein concentrations were determined using Bio-Rad assay, against a standard of BSA. Total cellular protein (40 μg) were resolved on 7.5%, 4–15%, and 15% SDS-polyacrylamide gels and were transferred to an Immobilon-P polyvinylidene fluoride membrane (Millipore). Membranes were blocked with either 5% milk or Starting Block Buffer (Pierce), washed, and probed with antibodies against AMPKα, phospho-AMPKα (T172) (1:250), 4EBP1 (1:1000), phospho-4EBP1 (T70) (1:1000), S6 kinase (1:1000), phospho-S6 kinase (T389) (1:500), acetyl CoA carboxylase (1:1000), Raptor and phosphoRaptor (S792)(1:1000), phosphoacetyl-CoA carboxylase (S79) (1:1000) at 4 °C overnight. All antibodies were purchased from Cell Signaling. Total and phospho-S6 Kinase, 4EBP1, and total and phosphoRaptor antibodies were diluted in Starting Block Buffer (Pierce), all other antibodies were diluted in 5% BSA in TBST. Membranes were incubated with anti-rabbit secondary antibody with horseradish peroxidase conjugate for 1hr at room temperature (Pierce). Chemiluminescence was detected using the SuperSignal West Pico and West Dura Chemiluminescent Substrate Kits (Pierce).
Results
Reversal of the growth inhibition of pemetrexed by purines and thymidine
The growth of human CEM leukemia cells is inhibited by pemetrexed at tens of nanomolar concentrations. This growth inhibition is not reversed by preformed purines, such as hypoxanthine (Hx), but inclusion of thymidine in the culture medium does reverse the growth inhibitory effects of pemetrexed, shifting the concentrations of drug needed to affect growth by about 12-fold, as shown for the CEM human leukemia cell line in Fig. 1A. Inclusion of both thymidine and hypoxanthine reverses the effects of pemetrexed at even high concentrations (Fig. 1A), in agreement with previous literature (3,4). These observations have previously been interpreted to mean that pemetrexed inhibits thymidylate synthase as its primary target, and has a second site of action within the folate pathways, presumably on purine synthesis, that is only affected at higher drug concentrations (3,4). In order to determine whether the first or second folate dependent enzyme of de novo purine synthesis, GART or AICART, respectively (Fig. 1C), is affected by drug, we tested the effects of aminoimidazolecarboxamide (AICA) on the inhibition of growth of CEM cells by pemetrexed. AICA is metabolized to the corresponding ribonucleotide, AICAR monophosphate (also known as ZMP) by adenine phosphoribosyltransferase (APRT) and, thus, inclusion of AICA in growth medium introduces a purine pathway intermediate into the cell that is downstream of GART, but upstream of AICART (Fig. 1C). If the secondary target of pemetrexed were GART, inclusion of thymidine and AICA in the medium should completely reverse growth inhibition (9,10) whereas, if AICART were inhibited, either no effect or a mild exacerbation of growth inhibition would be expected. Others have shown that AICA by itself at 300 μM did not change the potency of pemetrexed against CEM cells (4). However, AICA (320 μM) mildly enhanced the growth inhibition by pemetrexed in the presence of thymidine (Fig. 1A). In contrast, AICA shifted the sensitivity of CEM cells to the GART inhibitor 6R-DDATHF (Fig. 1B), similarly to that seen in mouse cells (9,10). We concluded that the secondary target of pemetrexed was not GART, and was most likely AICART.
Figure 1. Reversal of CEM cell growth inhibition by AICA indicated that the second target of pemetrexed is AICART, not GART.

CEM cells were treated with the indicated concentrations of pemetrexed alone (A, no add) or 6R-DDATHF (B, no add) or in the presence of TdR (5.6 μM), Hx (32 μM), AICA (320 μM), or a combination of TdR with either Hx or AICA. Drug and modifying agents were added simultaneously and drug-containing medium was changed at 48 hours. Cell growth was determined after 96 hours and cell number is expressed relative to controls without drug. Panel C depicts the folate-dependent steps of de novo purine synthesis and the site of entry of AICA and AICAR into the pathway. De novo purine synthesis consists of ten sequential enzymatic reactions starting with 1-phosphoribosyl-5-pyrophosphate (PRPP) of which two, GART and AICART, are folate-dependent. The figure depicts the substrate of the GART reaction, glycinamide ribonucleotide (GAR), and the product of this reaction formylglycinamide ribonucleotide (FGAR), as well as the salvage of AICA, AICAR, and hypoxanthine catalyzed by adenine phosphoribosyltransferase (APRT), adenosine kinase (AK), and hypoxanthine-guanine phosphoribosyltransferase (HGPRT), respectively.
Effects of pemetrexed on GART and AICART in intact cells
In the original determination of the site of action of lometrexol within the purine pathway in mouse cells (9), we had used the facts that the enzyme downstream of GART was sensitive to the glutamine analog azaserine, and the enzyme immediately preceding GART in this pathway incorporated glycine into the purine skeleton (Fig. 1C). We previously used these metabolic aspects to demonstrate that DDATHF inhibited the accumulation of 14C-glycine into N-formylglycinamide ribonucleotide (FGAR) in azaserine-treated mouse leukemic cells and, hence, had GART as a primary target (9). This is also the case in intact CEM cells for 6R-DDATHF (Fig. 2A) over the concentration range that is growth-inhibitory (Fig 1B). In contrast, pemetrexed gradually inhibited 14C-FGAR accumulation (Fig. 2A) to a lower extent that did not reach 50 % over the range of concentrations associated with drug action. Hence, any effect of pemetrexed directly or indirectly on GART in intact cells appears to be limited. When the metabolic fate of the intermediates of this pathway were followed further downstream, it was found that the ZMP pool expanded in pemetrexed-treated cells, indicating an effect of pemetrexed or its metabolites predominantly on AICART (Fig. 2B). If inhibition of GART played a role in the secondary effects of pemetrexed, ZMP accumulation would not have been observed (Fig. 1C). To this point, ZMP accumulation did not occur in 6R-DDATHF-treated CEM cells (Fig. 2B). When CEM cells were treated with AICA in the presence of pemetrexed, the accumulation of ZMP was exacerbated, implying that blockage of de novo purine synthesis at the AICART step was restricting the flow of intermediates through this pathway (Fig. 1C and see below). The accumulation of ZMP was progressive after drug treatment (Fig. 2C), occurred over the range of concentrations at which pemetrexed was growth-inhibitory, and became more intense at concentrations causing inhibition of a secondary target reversible by hypoxanthine (Figs. 1A and 2B).
Figure 2. Substrate accumulation studies support an effect of pemetrexed on AICART in intact CEM cells.

(A.) Accumulation of 14C-glycine as FGAR in pemetrexed and (6R)-DDATHF-treated CEM cells. Cells were exposed to either (6R)-DDATHF or pemetrexed plus 5.6 μM thymidine for 24 hours, nucleotides extracted and FGAR separated by anion-exchange chromatography. (B.) Increased cellular ZMP pools in pemetrexed-treated but not (6R)-DDATHF-treated CEM cells. Cells were exposed to drugs for 48 hours, nucleotides extracted and ZMP separated by hplc. (C.) CEM cells were exposed to 1μM pemetrexed for the indicated periods and ZMP analyzed as in (B.) The bar shown at zero time indicated the limit of sensitivity of the assay.
The question arises whether the levels of ZMP seen in pemetrexed-treated tumor cells are sufficient to play a role in the inhibitory effects of pemetrexed. One can approach this question by comparing the levels of ZMP that accumulate in pemetrexed-treated cells with those in cells whose growth is inhibited by the ribonucleoside analog of AICA, AICAR, whose substantial cytotoxicity is generally taken to be solely due to ZMP accumulation (11). At growth-inhibitory concentrations of AICAR, cellular levels of ZMP range from 0.4–2 mM (Fig. 3). Hence, the ZMP levels seen in pemetrexed-treated cells are sufficient to be at least partially causal of the growth-inhibition seen with pemetrexed. However, one should note that higher concentrations of ZMP accumulate in high (≥500 μM) AICAR concentrations than in pemetrexed.
Figure 3. Growth inhibition by ZMP.

(A.) CEM cells were exposed to the indicated concentrations of AICAR for 24 hours, and ZMP (open circles) was then analyzed as in Fig. 2B. Cell number (filled circles) was measured electronically after 24 hours. Panel B correlates the cell number after 24 hours of growth with the intracellular ZMP pool from panel A.
Effects of accumulating ZMP on AMP-activated protein kinase and the mTOR pathway
The accumulation of ZMP in cells treated with AICAR and the substantial cytotoxicity of AICAR is thought due to the activation of AMPK by ZMP leading, in turn, to an inhibition of mTOR (11). These effects of AICAR were also seen in CEM cells (Figs. 3 and 4B,C). We studied the activation of AMPK by pemetrexed and thymidine. The phosphorylation of the α-subunit of AMPK enhances the kinase activity of this protein, but the phosphorylation of a direct target of AMPK, acetyl-CoA carboxylase (ACC), is a useful and direct indicator of the activity of AMPK in intact cells (12). A 1μM concentration of pemetrexed promoted activation of AMPK as demonstrated by both direct phosphorylation at T172 and by an enhanced phosphorylation of ACC at residue S79 (Fig. 4B). Activation of AMPK is known to cause inhibition of mTOR by phosphorylation of TSC2, upstream of mTOR, as well as a direct phosphorylation of the Raptor component of the mTORC1 complex (Fig. 4A) (13,14). Accordingly, a robust hypophosphorylation of two key mTOR targets, 4EBP1 and S6K1 was seen in pemetrexed-treated CEM cells (Fig 4C), as well as an increase in phosphorylation of S792 of Raptor (Fig. 4B). The phosphorylation of S6K1 by mTOR is confined to a single residue (T389)(15), and this phosphorylation was eliminated by pemetrexed (Fig. 4C), whereas four residues on 4EBP1 are phosphorylated by mTOR (16); the broader migration pattern seen in pemetrexed-treated cells with the pan-4EBP1 antibody (Fig 3C) suggests that several of these phosphorylation sites are affected by drug. Thymidine was included in these experiments to separate the effects of inhibition of thymidylate synthase from any effect caused by accumulating ZMP. However, phosphorylation of AMPK and hypophosphorylation of 4EBP1 were also observed in cells treated with pemetrexed for 48 hours at doses as low as 0.1 μM in the absence of thymidine. The effects of pemetrexed on activation of AMPK, as observed by the phosphorylation of the AMPK substrates ACC and Raptor, and the hypophosphorylation of the downstream targets of mTOR were exacerbated in the presence of AICA (Fig. 5B), in concert with the enhanced accumulation of ZMP seen under these conditions (Fig. 5A).
Figure 4. Effects of pemetrexed on activation of AMPK and inhibition of mTOR.
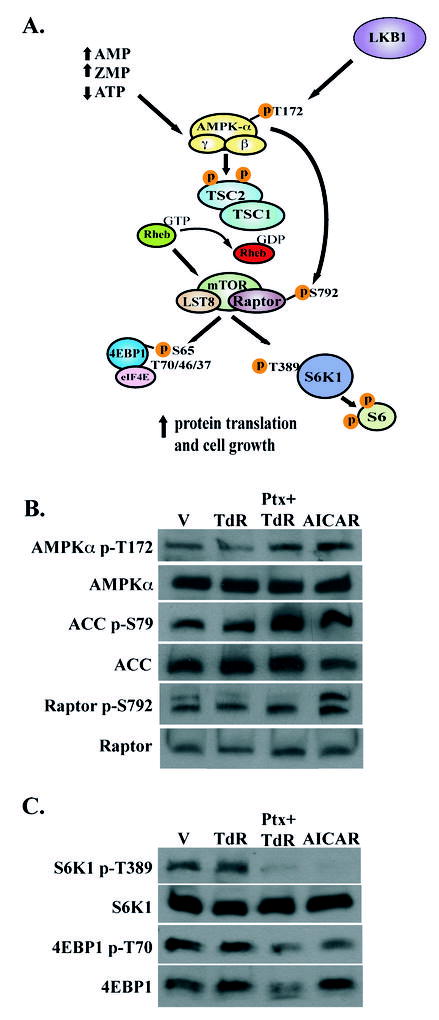
(A.) Schematic diagram showing activation of AMPK by either AMP or ZMP that results in inhibition of mTOR and its downstream targets. (B,C.) Western blot analysis of total and phosphorylated AMPK, ACC, Raptor, S6K1, and 4EBP1. The molecular masses of these bands were 62 kD (AMPK), 280 kD (ACC), 150 kDa (Raptor), S6K1 (70 kD), and 15–20 kD (4EBP1). Equal levels of total protein (40 μg) was loaded in each lane; use of actin as a control demonstrated equal loading between lanes in this and the following figures (not shown). Vehicle was PBS, pemetrexed was used at 1 μM, AICAR was at 250 μM, and TdR was 5.6μM; drug exposure was 48 hours.
Figure 5. Expansion of the ZMP pool by AICA enhances the effect of pemetrexed on the AMPK-mTOR pathway.
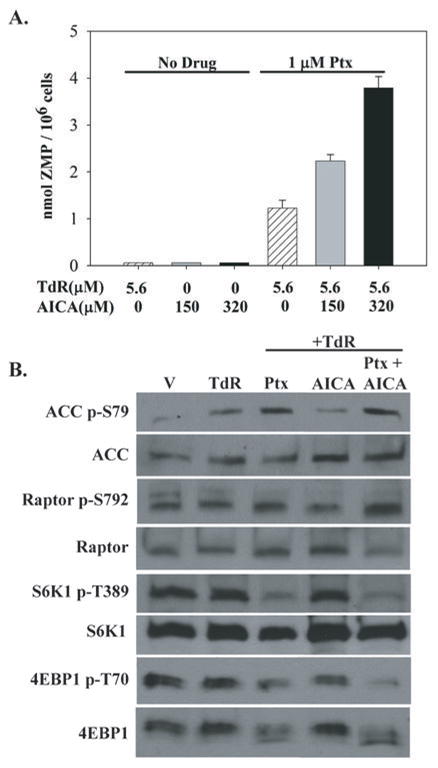
Exposure to AICA for 48 hours increased cellular levels of ZMP in a dose-related manner in pemetrexed-treated but not untreated CEM cells (A). In panel B, western blot analysis and drug exposures were as in Fig. 4B; AICA was used at 320 μM.
Reversal of the effects of pemetrexed by hypoxanthine
It was clear that a source of preformed purines, such as hypoxanthine, reversed the effects of pemetrexed on its secondary target (Fig. 1A). Initially, we thought this effect indicated a role for a diminution of purine nucleotide pools by pemetrexed inhibition of AICART, but direct measurement of ATP pools by hplc indicated that ATP levels not only did not decrease, but also increased somewhat in pemetrexed-treated cells in the absence of hypoxanthine (data not shown) 6. Rather, the mechanism of this hypoxanthine reversal of the secondary target of pemetrexed appeared to be a prevention of the accumulation of ZMP (Fig. 6A), presumably due to the known feedback effects of an expanded purine pool on early steps in purine synthesis (18,19). As would be predicted, the inclusion of hypoxanthine in the medium also decreased the phosphorylation of ACC and Raptor as well as the hypophosphorylation of S6K1 and 4EBP1 caused by pemetrexed (Fig. 6B).
Figure 6. Prevention of the secondary effects of pemetrexed by purines and by compound C.

(A.) When CEM cells were exposed to 1 μM pemetrexed and 5.6 μM thymidine for 48 hours, hypoxanthine in the medium prevented the accumulation of ZMP, in a dose-dependent manner that correlated with protection from growth inhibition. Cell growth (filled circles) and ZMP levels (open circles) were determined as in Fig. 3. (B.) Cellular activity of AMPK was assessed by western blot analysis of phosphorylation of ACC and Raptor, and the activity of mTOR was indicated by the phosphorylation of S6K1 and 4EBP1. CEM cells were exposed to drug and modifying agents for 48 hours as in Fig. 4 and Hx was used at 32 μM; conditions for western blots were as in Fig. 4. (C.) Compound C (1 μM, 48 hour exposure) prevented the cellular activity of AMPK by pemetrexed without interfering with the phosphorylation of AMPK. Vehicle was DMSO in PBS; all other conditions were as in Fig. 4.
Compound C effects support the role of AMPK activation in the pemetrexed-induced mTOR inhibition
The accumulation of ZMP in pemetrexed-treated cells and the activating effects of ZMP on AMPK suggest that the inhibition of mTOR by pemetrexed is mediated by a ZMP-dependent activation of AMPK. To determine the intermediacy of activation of AMPK in this mechanism, the effects of an inhibitor of AMPK, compound C (20), on the pemetrexed-induced mTOR inhibition was studied. As expected, compound C inhibited the enhanced AMPK activity in cells treated with pemetrexed and thymidine, as judged by an inhibition of the phosphorylation of ACC (Fig. 6C). The effects of 1 μM compound C on AMPK blocked the inhibition of mTOR by pemetrexed, as judged by the hyperphosphorylation of S6K1 in the presence of pemetrexed and compound C (Fig. 6C), a striking contrast to the marked hypophosphorylation of S6K1 caused by pemetrexed. Interestingly, the phosphorylation of AMPK in the presence of pemetrexed and compound C was greater than that seen with pemetrexed, in spite of the fact that compound C diminished the level of ACC phosphorylation; this suggested that compound C was blocking the activity of AMPK while allowing phosphorylation of the α-subunit of AMPK. Overall, we concluded that the effects of pemetrexed on the mTOR signaling pathway were caused by the activation of AMPK by accumulation of ZMP.
Discussion
The search for new therapeutic agents useful against cancer has focused on molecularly-targeted small molecules. In ideal cases, these would be agents inhibitory to steps unique to oncogenically transformed cells that were not shared with normal stem cells required for host survival. Such steps have rarely been identified, although the Gleevec-sensitive BCR-Abl and Iressa-selective mutations in the EGFR protein are two well-studied cases (21,22). One of the most promising approaches seems to be the design of therapeutic agents that affect pathways dependent on tumor suppressor genes, whose function is often eliminated or dramatically altered during transformation. The mTOR pathway responsible for balance of energy metabolism, protein and lipid synthesis, and growth involves a series of upstream controlling proteins recognized as tumor suppressor proteins, including LKB1, PTEN, TSC1 and 2, and others recognized as cellular oncogenes, such as AKT and PI3 kinase (8). Perhaps the central element in this pathway that controls the influence of the upstream tumor suppressor gene products/oncogenes on the downstream direct activator of protein synthesis, mTORC1, is AMPK, a protein kinase that activates the TSC1/TSC2 complex by phosphorylation (13) and inhibits the mTORC1 complex by phosphorylation of the Raptor component of this complex (14). The phosphorylated TSC1/TSC2 complex inactivates the Rheb GTP component needed for mTOR signaling. AMPK is composed of three subunits, and activation of the catalytic α-subunits is facilitated by binding of AMP to the γ-subunit; ZMP is thought to mimic the effect of AMP on the γ-subunit (7,23). In this study, we make the case that the accumulation of ZMP behind a block of AICART will activate AMPK and inhibit the downstream activity of mTORC1.
The concept that the accumulation of a toxic substrate behind a metabolic block could be used for therapeutics dates back to the classic treatise by Webb on enzyme inhibition in vivo (24). It is difficult to find examples in the chemotherapeutic literature, but the accumulation of ZMP behind an AICART block that we describe here makes a case for the utility and effectiveness of metabolite accumulation as a mechanism of anticancer agents. A metabolic inhibitor of any linear metabolic pathway would be expected to cause the accumulation of substrate behind the block, at least transiently, and such accumulation could result in a limitation of the effects of the inhibitor, e.g., the excessive accumulation of dUMP in cells treated with precursors of FdUMP (25), or enhancement of an enzyme inhibitor, e.g., raltitrexid (26).
Why has inhibition of AICART by pemetrexed or its metabolites not been found during prior surveys of folate enzymes for sensitivity to this drug? Earlier in vitro analysis of the kinetics of inhibition of several folate-dependent enzymes by pemetrexed and its polyglutamates led to the conclusion that inhibition of AICART was unlikely to contribute to the biological effects of this drug. Thus, the Ki measured for recombinant human AICART for pemetrexed itself was 3 μM, and for the more potent long-chain polyglutamate metabolites of pemetrexed was 0.26 μM when measured at a fixed concentration of ZMP (50 μM), whereas the activity of these compounds against GART were more favorable but still not very high (Ki = 65 nM). However, we herein demonstrate that any effects of pemetrexed on de novo purine synthesis in intact tumor cells are apparently due to effects on AICART, not GART. The solution to this apparent dilemma may be in the binding mechanisms of these two enzymes: GART can bind its two substrates, glycinamide ribonucleotide and 10-formyltetrahydrofolate polyglutamates in a random manner (27), with either substrate binding first, whereas AICART obeys an ordered sequential binding mechanism, with 10-formyltetrahydrofolate polyglutamates binding first before ZMP can bind (27,28).
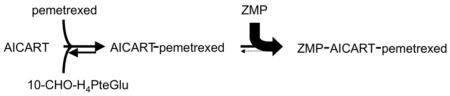
Hence, for AICART, when ZMP accumulates behind an initial blockade of the pathway, the expanded ZMP pool would promote the reformation of inhibited ternary complex whenever this complex dissociates to binary AICART-pemetrexed complex. Hence, the likelihood that the bound pemetrexed or its polyglutamates can leave the enzyme is greatly diminished, an example of metabolic trapping of an inhibitor on the enzyme active site by an expanded pool of second substrate. This mechanism is formally identical to the metabolic trapping of FdUMP on the surface of thymidylate synthase responsible for the stabilization of TS ternary complexes by the expanded pool of 5,10-methylenetetrahydrofolate polyglutamates induced by high doses of folinic acid (5-formyltetrahydrofolate) (29,30).
In theory, the secondary effect of pemetrexed reversible by a combination of thymidine and purine (Fig. 1) might be due to an inhibition of DHFR causing a cellular deficiency of 10-formyltetrahydrofolate. There are four pieces of evidence that argue against this interpretation: 1. The potency of pemetrexed and its metabolites against DHFR in vitro is quite low (72 nM) compared to the activity of pemetrexed polyglutamates against thymidylate synthase (1.3 nM)(4). 2. The substantial excess of DHFR over thymidylate synthase in leukemic cells implies that DHRF must be almost completely inhibited before any inhibition of cell growth can occur (31). 3. The primary inhibition of thymidylate synthase by pemetrexed polyglutamates would eliminate the importance of any inhibition of DHFR, given evidence that DHFR becomes irrelevant to cell survival or growth in the absence of thymidylate synthase activity (32,33). That is, substantial levels of a dihydrofolate reductase inhibitor as potent as methotrexate are without effect on the growth of leukemic cells exposed to a complete pharmacologic or genetic block of thymidylate synthase. And 4. Accumulation of ZMP has been sought in MTX-treated tumor cells and does not occur, although MTX treatment does enhance the ZMP accumulation in cells treated with AICAR (34). This enhanced accumulation of ZMP made from AICAR in methotrexate-treated cells is consistent with reports that methotrexate results in inhibition of AICART both by depletion of 10-formyltetrahydrofolate and by a direct inhibition of AICART by methotrexate polyglutamates (35).
It was appreciated that pemetrexed targets more than one site since early in the development of this compound, but the multiple targets were thought to all involve other enzymes on the folate pathways (1,3). We now present evidence that inhibition of AICART by pemetrexed causes a mechanistically important but indirect effect on the AMP-dependent kinase-mTOR pathway. We note that this should be the case for any drug targeted against AICART, a concept that suggests the development of drugs specifically against AICART but without activity against thymidylate synthase. Pharmacological inhibition of mTOR with direct inhibitors is currently of substantial interest for cancer therapeutics, and current mTOR inhibitors have shown activity against human lung cancers (36,37). Perhaps this spillover of the effects of pemetrexed to the mTOR pathway explains the activity of pemetrexed in lung cancers, an unusual pattern for classical antifolates.
Footnotes
This work was supported in part by grant CA 27605 from the National Institutes of Health, DHHS.
The authors declare no conflict of interest.
Author contributions: ACR and RGM designed the research; ACR, CLH, RGM, and SBR performed the experiments; RGM wrote the manuscript.
Abbreviations: AMPK, AMP-activated protein kinase; mTOR, mammalian target of rapamycin; AICA, 5-Amino-4-imidazolecarboxamide; AICAR, 1-β-D-ribofuranosyl-5-aminoimidazole-4-carboxamide; ZMP, 1-β-D-ribofuranosyl-5-Aminoimidazole-4-carboxamide-5′-phosphate; AICART, aminoimidazolecarboxamide ribonucleotide formyltransferase; GART, glycinamide ribonucleotide formyltransferase; DHFR, dihydrofolate reductase; mTOR, mammalian target of rapamycin; FGAR, N-formylglycinamide ribonucleotide; ACC, acetylCoA carboxylase.
Others had previously shown that ATP pools were not diminished by pemetrexed and, in fact, increased by about the factor we observed (17). We would suggest that this effect is due to the block in cell cycle progression with pemetrexed which prevents the loss of purine nucleotides caused by DNA synthesis, which would negate the effect of a de novo block of purine synthesis on ATP and GTP pools.
References
- 1.Habeck LL, Mendelsohn LG, Shih C, et al. Substrate specificity of mammalian folylpolyglutamate synthetase for 5,10-dideazatetrahydrofolate analogs. Mol Pharmacol. 1995;48:326–33. [PubMed] [Google Scholar]
- 2.Chattopadhyay S, Moran RG, Goldman ID. Pemetrexed: biochemical and cellular pharmacology, mechanisms, and clinical applications. Mol Cancer Ther. 2007;6:404–17. doi: 10.1158/1535-7163.MCT-06-0343. [DOI] [PubMed] [Google Scholar]
- 3.Taylor EC, Hamby JM, Shih C, et al. Synthesis and antitumor activity of 5-deaza-5,6,7,8-tetrahydrofolic acid and its N10-substituted analogues. J Med Chem. 1989;32:1517–22. doi: 10.1021/jm00127a019. [DOI] [PubMed] [Google Scholar]
- 4.Shih C, Chen VJ, Gossett LS, et al. LY231514, a pyrrolo[2,3-d]pyrimidine-based antifolate that inhibits multiple folate-requiring enzymes. Cancer Res. 1997;57:1116–23. [PubMed] [Google Scholar]
- 5.Chattopadhyay S, Zhao R, Krupenko SA, Krupenko N, Goldman ID. The inverse relationship between reduced folate carrier function and pemetrexed activity in a human colon cancer cell line. Mol Cancer Ther. 2006;5:438–49. doi: 10.1158/1535-7163.MCT-05-0243. [DOI] [PubMed] [Google Scholar]
- 6.Scagliotti GV, Parikh P, von Pawel J, et al. Phase III study comparing cisplatin plus gemcitabine with cisplatin plus pemetrexed in chemotherapy-naive patients with advanced-stage non-small-cell lung cancer. J Clin Oncol. 2008;26:3543–51. doi: 10.1200/JCO.2007.15.0375. [DOI] [PubMed] [Google Scholar]
- 7.Davies SP, Helps NR, Cohen PT, Hardie DG. 5′-AMP inhibits dephosphorylation, as well as promoting phosphorylation, of the AMP-activated protein kinase. Studies using bacterially expressed human protein phosphatase-2C alpha and native bovine protein phosphatase-2AC. FEBS Lett. 1995;377:421–5. doi: 10.1016/0014-5793(95)01368-7. [DOI] [PubMed] [Google Scholar]
- 8.Corradetti MN, Guan KL. Upstream of the mammalian target of rapamycin: do all roads pass through mTOR? Oncogene. 2006;25:6347–60. doi: 10.1038/sj.onc.1209885. [DOI] [PubMed] [Google Scholar]
- 9.Beardsley GP, Moroson BA, Taylor EC, Moran RG. A new folate antimetabolite, 5,10-dideaza-5,6,7,8-tetrahydrofolate is a potent inhibitor of de novo purine synthesis. J Biol Chem. 1989;264:328–33. [PubMed] [Google Scholar]
- 10.Moran RG, Baldwin SW, Taylor EC, Shih C. The 6S- and 6R-diastereomers of 5, 10-dideaza-5, 6, 7, 8-tetrahydrofolate are equiactive inhibitors of de novo purine synthesis. J Biol Chem. 1989;264:21047–51. [PubMed] [Google Scholar]
- 11.Rattan R, Giri S, Singh AK, Singh I. 5-Aminoimidazole-4-carboxamide-1-beta-D-ribofuranoside inhibits cancer cell proliferation in vitro and in vivo via AMP-activated protein kinase. Journal of Biological Chemistry. 2005;280:39582–93. doi: 10.1074/jbc.M507443200. [DOI] [PubMed] [Google Scholar]
- 12.Winder WW, Hardie DG. Inactivation of acetyl-CoA carboxylase and activation of AMP-activated protein kinase in muscle during exercise. Am J Physiol. 1996;270:E299–304. doi: 10.1152/ajpendo.1996.270.2.E299. [DOI] [PubMed] [Google Scholar]
- 13.Inoki K, Zhu T, Guan KL. TSC2 mediates cellular energy response to control cell growth and survival. Cell. 2003;115:577–90. doi: 10.1016/s0092-8674(03)00929-2. [DOI] [PubMed] [Google Scholar]
- 14.Gwinn DM, Shackelford DB, Egan DF, Mihaylova MM, Mery A, Vasquez DS, Turk BE, Shaw RJ. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Molecular Cell. 2008;30:214–26. doi: 10.1016/j.molcel.2008.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kimura N, Tokunaga C, Dalal S, et al. A possible linkage between AMP-activated protein kinase (AMPK) and mammalian target of rapamycin (mTOR) signalling pathway. Genes Cells. 2003;8:65–79. doi: 10.1046/j.1365-2443.2003.00615.x. [DOI] [PubMed] [Google Scholar]
- 16.Fadden P, Haystead TA, Lawrence JC., Jr Identification of phosphorylation sites in the translational regulator, PHAS-I, that are controlled by insulin and rapamycin in rat adipocytes. J Biol Chem. 1997;272:10240–7. doi: 10.1074/jbc.272.15.10240. [DOI] [PubMed] [Google Scholar]
- 17.Chen VJ, Bewley JR, Andis SL, et al. Preclinical cellular pharmacology of LY231514 (MTA): a comparison with methotrexate, LY309887 and raltitrexed for their effects on intracellular folate and nucleoside triphosphate pools in CCRF-CEM cells. Br J Cancer. 1998;78 (Suppl 3):27–34. doi: 10.1038/bjc.1998.751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wyngaarden JB, Ashton DM. Feedback control of purine biosynthesis by purine ribonucleotides. Nature. 1959;183:747–8. doi: 10.1038/183747a0. [DOI] [PubMed] [Google Scholar]
- 19.Caskey CT, Ashton DM, Wyngaarden JB. The Enzymology Of Feedback Inhibition Of Glutamine Phosphoribosylpyrophosphate Amidotransferase By Purine Ribonucleotides. J Biol Chem. 1964;239:2570–9. [PubMed] [Google Scholar]
- 20.Zhou G, Myers R, Li Y, et al. Role of AMP-activated protein kinase in mechanism of metformin action. J Clin Invest. 2001;108:1167–74. doi: 10.1172/JCI13505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Druker BJ, Tamura S, Buchdunger E, et al. Effects of a selective inhibitor of the Abl tyrosine kinase on the growth of Bcr-Abl positive cells. Nat Med. 1996;2:561–6. doi: 10.1038/nm0596-561. [DOI] [PubMed] [Google Scholar]
- 22.Pao W, Miller V, Zakowski M, et al. EGF receptor gene mutations are common in lung cancers from “never smokers” and are associated with sensitivity of tumors to gefitinib and erlotinib. Proc Natl Acad Sci U S A. 2004;101:13306–11. doi: 10.1073/pnas.0405220101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hardie DG. AMP-activated/SNF1 protein kinases: conserved guardians of cellular energy. Nat Rev Mol Cell Biol. 2007;8:774–85. doi: 10.1038/nrm2249. [DOI] [PubMed] [Google Scholar]
- 24.Webb J. Enzyme and Metabolic Inhibitors. New York: Academic Press; 1963. [Google Scholar]
- 25.Moran RG, Spears CP, Heidelberger C. Biochemical determinants of tumor sensitivity to 5-fluorouracil: ultrasensitive methods for the determination of 5-fluoro-2′-deoxyuridylate, 2′-deoxyuridylate, and thymidylate synthetase. Proc Natl Acad Sci U S A. 1979;76:1456–60. doi: 10.1073/pnas.76.3.1456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rutenber EE, Stroud RM. Binding of the anticancer drug ZD1694 to E. coli thymidylate synthase: assessing specificity and affinity. Structure. 1996;4:1317–24. doi: 10.1016/s0969-2126(96)00139-6. [DOI] [PubMed] [Google Scholar]
- 27.Sanghani SP, Moran RG. Tight binding of folate substrates and inhibitors to recombinant mouse glycinamide ribonucleotide formyltransferase. Biochemistry. 1997;36:10506–16. doi: 10.1021/bi970825u. [DOI] [PubMed] [Google Scholar]
- 28.Vergis JM, Beardsley GP. Catalytic mechanism of the cyclohydrolase activity of human aminoimidazole carboxamide ribonucleotide formyltransferase/inosine monophosphate cyclohydrolase. Biochemistry. 2004;43:1184–92. doi: 10.1021/bi035139b. [DOI] [PubMed] [Google Scholar]
- 29.Danenberg PV, Danenberg KD. Effect of 5, 10-methylenetetrahydrofolate on the dissociation of 5-fluoro-2′-deoxyuridylate from thymidylate synthetase: evidence for an ordered mechanism. Biochemistry. 1978;17:4018–24. doi: 10.1021/bi00612a022. [DOI] [PubMed] [Google Scholar]
- 30.Keyomarsi K, Moran RG. Mechanism of the cytotoxic synergism of fluoropyrimidines and folinic acid in mouse leukemic cells. J Biol Chem. 1988;263:14402–9. [PubMed] [Google Scholar]
- 31.Jackson RC, Hart LI, Harrap KR. Intrinsic resistance to methotrexate of cultured mammalian cells in relation to the inhibition kinetics of their dihydrololate reductases. Cancer Res. 1976;36:1991–7. [PubMed] [Google Scholar]
- 32.Moran RG, Mulkins M, Heidelberger C. Role of thymidylate synthetase activity in development of methotrexate cytotoxicity. Proc Natl Acad Sci U S A. 1979;76:5924–8. doi: 10.1073/pnas.76.11.5924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ayusawa D, Koyama H, Seno T. Resistance to methotrexate in thymidylate synthetase-deficient mutants of cultured mouse mammary tumor FM3A cells. Cancer Res. 1981;41:1497–501. [PubMed] [Google Scholar]
- 34.Beckers A, Organe S, Timmermans L, et al. Methotrexate enhances the antianabolic and antiproliferative effects of 5-aminoimidazole-4-carboxamide riboside. Mol Cancer Ther. 2006;5:2211–7. doi: 10.1158/1535-7163.MCT-06-0001. [DOI] [PubMed] [Google Scholar]
- 35.Allegra CJ, Drake JC, Jolivet J, Chabner BA. Inhibition of phosphoribosylaminoimidazolecarboxamide transformylase by methotrexate and dihydrofolic acid polyglutamates. Proc Natl Acad Sci U S A. 1985;82:4881–5. doi: 10.1073/pnas.82.15.4881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gridelli C, Rossi A, Morgillo F, et al. A randomized phase II study of pemetrexed or RAD001 as second-line treatment of advanced non-small-cell lung cancer in elderly patients: treatment rationale and protocol dynamics. Clin Lung Cancer. 2007;8:568–71. doi: 10.3816/CLC.2007.n.045. [DOI] [PubMed] [Google Scholar]
- 37.Pandya KJ, Dahlberg S, Hidalgo M, et al. A randomized, phase II trial of two dose levels of temsirolimus (CCI-779) in patients with extensive-stage small-cell lung cancer who have responding or stable disease after induction chemotherapy: a trial of the Eastern Cooperative Oncology Group (E1500) J Thorac Oncol. 2007;2:1036–41. doi: 10.1097/JTO.0b013e318155a439. [DOI] [PubMed] [Google Scholar]