

Abstract
Cannabinoid agonists might serve as neuroprotective agents in neurodegenerative disorders. Here, we examined this hypothesis in a rat model of Huntington’s disease (HD) generated by intrastriatal injection of the mitochondrial complex II inhibitor malonate. Our results showed that only compounds able to activate CB2 receptors were capable of protecting striatal projection neurons from malonate-induced death. That CB2 receptor agonists are neuroprotective was confirmed by using the selective CB2 receptor antagonist, SR144528, and by the observation that mice deficient in CB2 receptor were more sensitive to malonate than wild-type animals. CB2 receptors are scarce in the striatum in healthy conditions but they are markedly up-regulated after the lesion with malonate. Studies of double immunostaining revealed a significant presence of CB2 receptors in cells labelled with the marker of reactive microglia OX-42, and also in cells labelled with GFAP (a marker of astrocytes). We further showed that the activation of CB2 receptors significantly reduced the levels of tumor necrosis factor-α (TNF-α) that had been increased by the lesion with malonate. In summary, our results demonstrate that stimulation of CB2 receptors protect the striatum against malonate toxicity, likely through a mechanism involving glial cells, in particular reactive microglial cells in which CB2 receptors would be up-regulated in response to the lesion. Activation of these receptors would reduce the generation of proinflammatory molecules like TNF-α. Altogether our results support the hypothesis that CB2 receptors could constitute a therapeutic target to slowdown neurodegeneration in HD.
Keywords: cannabinoids, CB1 and CB2 receptors, Huntington’s disease, malonate, neuroprotection
Introduction
In addition to well-described pharmacological actions in the brain, such as analgesia, hypokinesia, antiemesis, hypothermia, and orexigenic effects, cannabinoids have been recently associated with the control of the cell survival/death decision, being able to behave as antiproliferative agents but also to protect neurons from different types of insults (for review, see Fernández-Ruiz et al., 2005 and 2007). The neuroprotectant effect seems to be mediated by the activation of the cannabinoid type-1 (CB1) receptor and its well-described effects on glutamate release or calcium influx, although the contribution of alternative mechanisms (i.e., antioxidant and/or antiinflammatory properties of cannabinoids) can not be ruled out (Fernández-Ruiz et al., 2005 and 2007, for recent reviews). This neuroprotectant potential has been examined in conditions of acute neurodegeneration, e.g. hypoxia-ischemia (Nagayama et al., 1999; Sinor et al., 2000; Fernández-López et al., 2006) and brain trauma (Panikashvili et al., 2001), and also in chronic neurodegenerative disorders including Alzheimer’s disease (Ramírez et al., 2005; Esposito et al., 2007), Parkinson’s disease (Lastres-Becker et al., 2005; García-Arencibia et al., 2007), multiple sclerosis (Pryce et al., 2003; Arévalo-Martín et al., 2003), amyotrophic lateral sclerosis (Kim et al., 2006; Shoemaker et al., 2007), and others (see Fernández-Ruiz et al., 2005, for review).
Cannabinoids may also offer neuroprotection in Huntington’s disease (HD) (see Fernández-Ruiz et al., 2005 and 2007; Sagredo et al., 2007a, for recent reviews). HD is an adult-onset and autosomal-dominant neurodegenerative disorder characterized by progressive neuronal death that is associated with abnormal movements (chorea) and cognitive decline (Walker, 2007). The disease is caused by an expansion in a CAG triplet repeat in the gene coding the protein huntingtin (Cattaneo et al., 2005), which results toxic for neurons located mainly in the striatum and, to a lesser extent, in several cortical areas. In the striatum, toxicity is mainly limited to striatal projection neurons, but scarcely affects striatal interneurons and dopaminergic afferents (Brouillet et al., 2005; Cattaneo et al., 2005). The mechanism(s) responsible for this particular preference in neuronal degeneration remain(s) a major uncertainty in HD, together with the development of efficient therapies to halt or slowdown disease progression (McMurray, 2001; Wright and Barker, 2007, for review). Studies using a medium-throughput cell-based assay to screen a compound library against the in vitro toxicity of mutated huntingtin found neuroprotection with several plant-derived cannabinoids (Aiken et al., 2004), although a further study did not replicate these results (Wang et al., 2005). The issue has been also examined in vivo, although the results proved that the type of cannabinoid agonist able to provide neuroprotection was different depending on the type of animal model used for this disease (see Sagredo et al., 2007a, for review). This variability might be related to the fact that these models only reproduce few of the pathogenic mechanisms that cooperate to kill the striatal projection neurons in HD patients, e.g. protein misfolding, failed proteolysis, mitochondrial dysfunction, excitotoxicity, inflammation and oxidative stress. For example, Pintor et al. (2006) demonstrated that CB1 receptors can be a target to reduce excitotoxic death in HD, since only agonists of this receptor type reduced the striatal degeneration typical of rats lesioned with the excitotoxin quinolinate. However, CB1 receptor agonists failed to provide neuroprotection when the striatal atrophy was generated with mitochondrial toxins like 3-nitropropionic acid (3-NP) (Brouillet et al., 2005), and the same occurs with cannabinoid type-2 (CB2) receptor agonists (Sagredo et al., 2007b). The use of this neurotoxin replicates the mitochondrial defects found in HD patients and related genetic models (Gu et al., 1996), which originates the generation of reactive oxygen species (Alexi et al., 1998, for review). This possibly explains why, in this model, neuroprotection was attained exclusively with antioxidant cannabinoids like Δ9-tetrahydrocannabinol (Δ9-THC; Lastres-Becker et al., 2004) or cannabidiol (CBD; Sagredo et al., 2007b). The neuroprotective potential of Δ9-THC has also been examined in rats with striatal degeneration generated by malonate (Lastres-Becker et al., 2003), another complex II inhibitor, which is known to kill striatal neurons through mechanisms involving the apoptotic machinery (Toulmond et al., 2004). However, results in the malonate model were poorly informative, presumably because of the overlap between multiple mechanisms activated by this plant-derived cannabinoid, and suggested that more selective cannabinoid agonists should be examined in this model (Lastres-Becker et al., 2003). This has been carried out in the present study using the CB1 receptor agonist arachidonyl-2-chloroethylamide (ACEA) (Hillard et al., 1999), the CB2 receptor agonist HU-308 (Hanus et al., 1999), and the plant-derived cannabinoid CBD, a compound with low affinity for cannabinoid receptors but having notable antioxidant properties (Mechoulam et al., 2002). The degree of GABA depletion, the levels of transcripts for neuron-specific enolase (NSE), and the number of apoptotic nuclei measured by TUNEL staining, were the variables used to determine the toxicity of malonate and the potential neuroprotective effect of these cannabinoids. Additional pharmacological, biochemical and histological experiments, aimed at determining the importance of CB2 receptors in malonate-lesioned animals, were conducted in the second part of this study.
Materials and Methods
Animals, treatments and sampling
Animals
Male Sprague-Dawley rats, or CB2-/- mice and their respective wild-type littermates (bred in our animal facilities from a donation by Dr. Nancy Buckley, California State Polytechnic University, Pomona, CA, U.S.A.; see Buckley et al., 2000; Buckley, 2008), were housed in a room with controlled photoperiod (08:00-20:00 light) and temperature (22 ± 1°C). They had free access to standard food and water and were used at adult age (3 month-old; 300-400 g weight, for rats; and 3 month-old; 20-25 g weight, for mice) for experimental purposes, all conducted according to European rules (directive 86/609/EEC). In the case of mice, their genetic profile (CB2+/+, CB2+/- and CB2-/-) was identified by PCR analysis, as described by Buckley et al. (2000), using DNA extracted from a piece of tail of each mouse. Only homozygous animals (CB2+/+ or CB2-/-) were used in these experiments.
Intrastriatal injection of malonate
Rats were injected stereotaxically (coordinates: +0.8 mm anterior, +2.9 mm lateral from the bregma, -4.5 mm ventral from the dura mater) into the left striatum with 2 M malonate in a volume of 1 μl or were sham-operated in the same left striatum but with no injection of the toxin. The contralateral striatum of each animal remained always unaffected. Animals were used for experimental analysis 48 h later (and at shorter times for qRT-PCR analysis). The same procedure was used in the case of mice with the only difference of coordinates used that were: +1 mm anterior, +2.1 lateral and -2.8 ventral.
Cannabinoid treatments
ACEA was purchased from Tocris (Biogen, Madrid, Spain), HU-308 was kindly provided by Pharmos (Israel), CBD was synthesized as previously described (Gaoni and Mechoulam, 1971), and SR144528 was kindly provided by Sanofi-Aventis (Montpellier, France). All compounds were prepared in Tween 80-saline solution (1/16 v/v) for i.p. administration (volume dosage: 2 ml/kg weight). The doses used were based on previous pharmacological studies (Rinaldi-Carmona et al., 1998; Hillard et al., 1999; Hanus et al., 1999; Mechoulam et al., 2002). In a first series of experiments, animals (n=6-8 individuals per group) were i.p. administered with ACEA (3 mg/kg); HU-308 (5 mg/kg), CBD (5 mg/kg), or vehicle 30 min before and 2 hours after the intrastriatal injection of malonate. Animals were killed 46 hours after the second cannabinoid injection and their brains were rapidly removed and frozen in 2-methylbutane cooled in dry ice, and stored until evaluation for the degree of malonate-induced striatal injury. In a second set of experiments, SR144528 was injected i.p. at a dose of 1 mg/kg according to the same schedule, with or without another injection of HU-308 (5 mg/kg). Animals were also killed 46 hours after the second SR144528 and/or HU-308 injection and their brains were collected and processed as described above. The same procedure for killing the animals and collecting the brains was used in the case of the experiments conducted with CB2+/+ or CB2-/- mice lesioned with malonate or sham-operated. In a third group of experiments, malonate-injected and sham-operated rats were decapitated at different times after the lesion (0, 24 and 48 h). Their brains were quickly and carefully removed, the striata dissected and frozen for qRT-PCR analysis. A group of brains of 48 h after the lesion, and their corresponding sham-operated controls, were also removed and fixed in 4% paraformaldehyde in PBS for 24 h. Finally, the last series of experiments repeated the same treatment schedule with HU-308 (and, in some cases, CBD) used in the first series, although animals were used to collect brains for analysis of molecular mechanisms potentially involved in the neuroprotective effect of HU-308.
HPLC determination
Brain coronal slices (around 500 μm thick) were made at levels containing the substantia nigra, the globus pallidus and the caudate-putamen, according to Palkovits and Brownstein (1988). Subsequently, the three structures were dissected and homogenized in 20-40 volumes of cold 150 mM potassium phosphate buffer, pH 6.8. Each homogenate was distributed in aliquots, one to be used for the analysis of protein concentration (Lowry et al., 1951). The other aliquots were used for the measurement of neurotransmitter contents (GABA, dopamine and its major metabolite, DOPAC) by HPLC coupled to electrochemical detection, according to our previously described methods (Lastres-Becker et al., 2005; Sagredo et al., 2007b). They were diluted (½) with 0.4 N perchloric acid containing 0.4 mM sodium disulfite, 0.90 mM EDTA and the corresponding internal standard (β-aminobutirate for GABA and dihydroxybenzylamine for dopamine and DOPAC). Afterwards, samples were centrifuged for 3 min (15000 g) and the supernatants directly injected into the HPLC system, except in the case of those samples for the determination of GABA that were subjected to a previous derivatization process with o-phthaldehide (OPA)-sulfite solution (14.9 mM OPA, 45.4 mM sodium sulfite and 4.5% ethanol in 327 mM borate buffer, pH 10.4; see details in Sagredo et al., 2007b). HPLC system consisted of the following elements. The pump was an isocratic Spectra-Physics 8810. The column was a RP-18 (Spherisorb ODS-2; 150 mm, 4.6 mm, 5 μm particle size; Waters, Massachusetts, USA). The mobile phase, previously filtered and degassed, consisted of: (i) 0.06 M sodium dihydrogen phosphate, 0.06 mM EDTA and 20-30% methanol (pH 4.4), for the determination of GABA, and (ii) 100 mM citric acid, 100 mM sodium acetate, 1.2 mM heptane sulphonate, 1 mM EDTA and 7% methanol (pH 3.9), for the determination of dopamine and DOPAC. The flow rate was 0.8 ml/min. The effluent was monitored with a Metrohm bioanalytical system amperometric detector using a glassy carbon electrode. The potential was 0.85V relative to an Ag/AgCl reference electrode with a sensitivity of 50 nA (approx. 2 ng per sample). In the case of the determination of dopamine and DOPAC, the effluent was monitored with a coulochemical detector (Coulochem II, ESA) using a procedure of oxidation/reduction (conditioning cell: +360 mV; analytical cell #1: +50 mV; analytical cell #2: -340 mV), procedure that reaches a sensitivity of 50 nA (10 pg per sample). In both cases, the signal was recorded on a Spectra-Physics 4290 integrator. The results were obtained from the peaks and calculated by comparison with the area under the corresponding internal standard peak. Values were expressed as ng or μg/mg of protein.
In situ hybridization techniques
Brain slicing
Coronal sections, 40 μm-thick, were cut in a cryostat, according to the Paxinos and Watson atlas (1986). Sections were thaw-mounted onto Superfrost Plus glass slides and dried briefly at 30°C and stored at -80°C until used.
Analysis of mRNA levels for NSE and SOD-1 and -2
Briefly, sections were fixed in 4% paraformaldehyde for 5 min and, after rinsing twice in PBS, were acetylated by incubation in 0.25% acetic anhydride, prepared in 0.1M triethanolamine/0.15M sodium chloride (pH 8.0), for 10 min. Sections were rinsed in 0.3M sodium chloride/0.03M sodium citrate, pH 7.0, dehydrated and delipidated by ethanol/chloroform series. For hybridization, we used a synthetic 45-base probe, whose sequence was selected from the previously-published complete sequence for rat NSE (5’-TCT GGG TGA CTT GGG GCT CAA GGT ATC AAG GTA ACT ATG GCG GGT-3’; Katagiri et al., 1993). The oligonucleotide probe was 3’-end labelled with [35S]-dATP using terminal deoxynucleotidyl-transferase. Sections were, then, hybridized with [35S]-labelled oligonucleotide probes (7.5 × 105 dpm per section), washed and exposed to X-ray film (βmax, Amersham) for 10 days, and developed (D-19, Kodak) for 6 min at 20°C. The intensity of the hybridization signal was assessed by measuring the grey levels in the films with a computer-assisted videodensitometer. Adjacent brain sections were co-hybridized with a 100-fold excess of cold probe or with RNAse to assert the specificity of the signal (data not shown). Similar procedures were used for the analysis of mRNA levels of SOD-1 and SOD-2. We used synthetic 45-base probes, selected from the previously-published sequences for SOD-1 (5’-TCC AGT CTT TGT ACT TTC TTC ATT TCC ACC TTT GCC CAA GTC ATC-3’) and SOD-2 (5’-TGA TCT GCG CGT TAA TGT GCG GCT CCA GCG CGC CAT AGT-3’) (Kunikowska and Jenner, 2001).
Real time qRT-PCR analysis
Total RNA was extracted from lesioned or non-lesioned rat striata using RNATidy reagent (AppliChem. Inc., Cheshire, CT, U.S.A.), and, after RNA extraction, samples were treated with DNAse I (Roche Diagnostics, Barcelona, Spain) in order to eliminate possible DNA contamination. The total amount of RNA extracted was quantitated by spectrometry at 260 nm and its integrity was evaluated in agarose geles. In each case, 1 μg of total RNA was transcribed into complementary DNA using the First Strand cDNA Synthesis kit for RT-PCR (AMV, Roche Diagnostics, Barcelona, Spain). The reaction mixture was kept frozen at -80°C until enzymatic amplification. Quantitative real-time PCR assays were established in order to quantify the CB2 receptor mRNA expression and the housekeeping gene glyceraldehyde-3-phosphate dehydrogenase (GAPDH) using the LightCycler (LC) technology (Roche Diagnostics, Barcelona, Spain). It is important to remark that malonate lesion did not affect the expression of GAPDH (CT values: control: 20.5 ± 0.5, n=7; malonate-lesioned: 20.86 ± 0.49, n=7; t=0.47, df=12, p=0.646) allowing that it can be used as a housekeeping gene. The LC FastStart DNA Master SybrGreen (Roche Diagnostics, Barcelona, Spain) kit was used for amplifications. The following primers were used: CB2 receptor forward: 5’-CGGCTTGGAGTTCAACCCTA-3’; CB2 receptor reverse: 5’-ACAACAAGTCCACCCCAT GAG-3’; GAPDH forward: 5’-GAAGGGCTCATGACCACAGT-3’; GAPDH reverse: 5’-GAACACAGACCATGTCA-3’. The concentration of primers were 0.5 μM. The PCR assays were performed using 2 μl of the 20 μl cDNA reaction. All assays were carried out twice as independent PCR runs for each cDNA sample. Mean values were used for further calculations. A negative (no template) control was measured in each of the PCR runs. Standard curves were calculated for quantification purposes using ten-fold serial dilutions of cDNA from spleen rat. The transcript amounts were calculated using the second derivate maximum mode of the LC-software version 1.01. The specific transcript quantities were normalized to the transcript amounts of the reference gene GAPDH. All further calculations and statistical analyses were carried out with these values referred to as relative expression ratios.
Histochemical techniques
FluoroJade B staining
This staining was used to compare the magnitude of the damage generated by the intrastriatal application of malonate in CB2+/+ and CB2-/- mice. The procedure used was previously described by Schmued et al. (1997) and was used here with a few minor modifications. Briefly, brains were sliced in a cryostat to obtain serial coronal sections (20 μm thick) as described for in situ hybridization techniques, but using an Atlas of mouse brain (Lehmann and Gautier, 1974). Sections were collected on gelatine-coated slices. They were immersed in a xilol solution for 5 min and then hydrated in graded ethanol (100, 96 and 70%; 5 min each) and distilled water. Sections were then immersed in 0.06% potassium permanganate for 15 min at room temperature, and then placed into distilled water for 2 min. Following this, sections were immersed in a solution of 0.001% FluoroJade B (Chemicon International, Temecula, CA, USA) in 0.1% where they were maintained for 30 min in the darkness and at room temperature. Afterwards, sections were rinsed in distilled water and dehydrated in ethanol (96% and 100%; 5 min each) and then passed to xilol and mounted on coverslips with EUKITT mounting medium (Panlab, Barcelona, Spain). All sections were observed and photographed under a fluorescence microscope with blue (450-490 nm) excitation light. FluoroJade B-positive cells were counted in the lesioned and non-lesioned striatum using four randomly selected, non-overlapping fields and using a 20X objective. The final number of FluoroJade B-positive cells was the mean of the four fields of view used in each section. Analysis was always conducted by experimenters who were blinded to all animal characteristics.
TUNEL staining
TUNEL (TdTmediated dUTP nick-end labeling) reaction was developed in striatal sections (20 μm thick) obtained from lesioned or non-lesioned brains and collected on gelatine-coated slides. They were first permeabilized for 30 min in PBS containing 0.25% Triton X-100. Then, they were incubated in 1×TdT buffer containing biotinylated dUTP (200 pM; Roche Diagnostic, Barcelona, Spain) and TdT enzyme (30 units; Roche Diagnostic, Barcelona, Spain), for 2h at 37°C in a humidified chamber. To stop the reaction, sections were rinsed in PBS for 5 min and then reacted with streptavidin-conjugated Alexa Fluor 488 (1 mg/ml, Molecular Probes, Eugene, OR, USA) for 2 h to detect and quantify the fluorescein-16-dUTP-labelled DNA by light microscopy using a procedure of quantification similar to FluoroJade B staining.
Immunohistochemistry
The protocol used for the immunohistochemical staining is basically the same as previously described (Benito et al, 2003) with slight modifications. Fixed brains were embedded in paraffin. Sections (4 μm-thick) were obtained with a Leica microtome and mounted on glass slides. Then, tissue sections were deparaffinizied and extensively washed in potassium phosphate-buffered saline (KPBS) (50 mM) and endogenous peroxidase was blocked by incubation in peroxidase-blocking solution (Dako, Denmark) for 20 min, at room temperature. In order to obtain a more efficient immunostaining, sections were subjected to an antigen retrieval procedure (Shi et al., 2001). Briefly, sections were placed in a stainless steel pressure cooker containing a boiling solution (sodium citrate 0.01M, pH 10). After heating under pressure for 2 min, samples were removed and extensively washed in KPBS. Tissue sections were then incubated with the primary antibody (polyclonal anti-CB2 receptor, 1:1500 dilution in KPBS, Affinity Bioreagents, USA). After 24h incubation at 4°C, sections were washed in 50 mM KPBS and incubated with biotinylated goat anti-rabbit antibody (1:200), at room temperature for 1h followed by avidin-biotin complex (Vector Elite, Burlingame, CA, U.S.A.), according to the manufacturer’s instructions. Visible reaction product was produced by treating the sections with 0.04% diaminobenzidine (DAB, Dako, Glostrup. Denmark), 2.5% nickel sulfate and 0.01% H2O2, dissolved in 0.1 M sodium acetate. Sections were then dehydrated and sealed with cover slips. The observations and photography of the slides were done using a Nikon Eclipse 90i microscope and a Nikon DXM 1200F camera. Controls for the immunohistochemistry included the preabsorption and co-incubation of the antibodies with the corresponding immunogenic proteins (CB2, fusion protein against amino acids 1-33 of human-CB2 at 5 μg/ml) and incubation in the absence of primary antibody. Adjacent sections to those employed in the immunohistochemical studies were used for hematoxilineosin and Nissl stainings.
Immunofluorescence
For double-labelling studies, fixed brains were sliced in a vibratome. Floating sections were sequentially incubated with polyclonal anti-CB2 receptor (1:100; Affinity Bioreagents, Golden, CO, U.S.A.) followed by incubation with monoclonal anti-OX-42 (CD11b, 1:50; Chemicon International, Temecula, CA, U.S.A.). After incubation with the corresponding primary antibody, the sections were washed in Tris-buffered saline and incubated (at room temperature for 1 hour) with an Alexa 488 anti-rabbit antibody conjugate made in goat (1:200; Molecular Probes, Eugene, OR, U.S.A.), rendering green fluorescence for anti-CB2 receptors, or with Alexa 568 anti-mouse antibody conjugate made in goat (1:200; Molecular Probes, Eugene, OR, U.S.A.) rendering red fluorescence for anti-OX-42. To obtain complete identification of the cellular types exhibiting CB2 receptor-positive immunostaining, additional double-labelling studies were performed using a monoclonal antibody against the marker of astrocytes GFAP (1:50, Sigma-Aldrich, Madrid, Spain). The protocol included two consecutive steps: immunofluorescence and incubation with an Alexa 568 anti-mouse antibody conjugate, followed by immunofluorescence with anti-CB2 receptor antibody, as described above. A Nikon Eclipse 90i microscope and a Nikon DXM 1200F camera were used for slide observation and photography. Lastly, immunofluorescence staining was also used to detect increase in caspase 3 after the malonate lesion. In this case, a polyclonal antibody against caspase 3 (1:250; Promega Biotech Ibérica, Madrid, Spain) was used and the analysis was carried out following the same procedure described above.
Analysis of TNF-α levels
The levels of TNF-α in the striatum of rats of the different groups of interest under examination were analyzed using the Rat Tumor Necrosis Factor-Alpha Ultrasensitive (Rt TNF-α US) ELISA kit (Invitrogen Corporation, Carlstad, CA, U.S.A.). Values were expressed as pg/mg of protein.
Statistics
qRT-PCR and FluoroJade B data were assessed by Student’s t-test, whereas the remaining data were assessed by ANOVA followed by the Student-Newman-Keuls test. In both cases, we used the GraphPad software (version 4.0). One-way ANOVA was used for the data expressed as % of the lesioned side over the non-lesioned side, exactly the data that are presented in figures and tables, whereas two-way ANOVA (treatment × brain side) was used to assess the raw data with the values of the lesioned and non-lesioned sides separately (mentioned in the text).
Results
Experiment I: Treatment with ACEA, HU-308 or CBD in malonate-lesioned rats
Malonate injection produced a marked reduction in GABA contents (expressed as % in the lesioned side over the non-lesioned side for each subject) in the caudate-putamen (Figure 1). It also decreased GABA concentrations in the globus pallidus, which receives GABA terminals from the striatum (Table 1). Other nuclei also receiving GABA terminals, such as the substantia nigra, showed however slight and non-significant reduction in GABA contents, but this can be explained by the presence in this structure of other GABA-containing neurons that were not affected by malonate (Table 1). GABA depletion likely reflects the death of striatal projection neurons caused by this mitochondrial toxin, as previously described (Zeevalk et al., 2002) and confirmed here by the following observations: (i) the high number of apoptotic nuclei measured by the TUNEL staining (Figure 2); (ii) the increase in caspase-3 immunoreactivity (Figure 2); and (iii) the loss of mRNA levels for NSE (Figure 3). Malonate also reduced the contents of dopamine and its metabolite DOPAC in the caudate-putamen (Table 1). This might reflect either: (i) a dysfunction of nigrostriatal dopaminergic neurons that are under the influence of striatal output neurons (Calabresi et al., 2000), or (ii) a loss of striatal dopamine terminals due to a direct effect of malonate (Alfinito et al., 2003).
Figure 1.
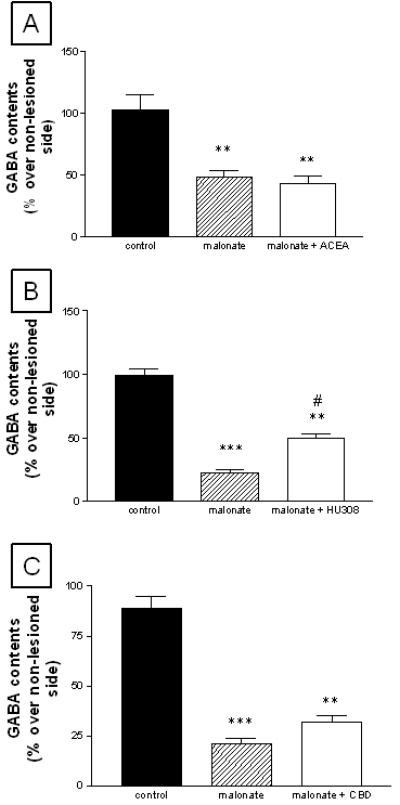
GABA contents in the striatum of rats subjected to unilateral injections of malonate and treated with ACEA (panel A), HU-308 (panel B) or CBD (panel C), and their respective controls. Details in the text. Values correspond to % of the lesioned side over the non-lesioned one for each subject, and are presented as means ± SEM of 6-8 subjects per group. Data were assessed by one-way analysis of variance followed by the Student-Newman-Keuls test (*p<0.05, **p<0.005, ***p<0.0005 vs the controls; #p<0.05 vs the malonate group)
Table 1.
GABA, dopamine and DOPAC contents in different basal ganglia of rats subjected to unilateral injections of malonate and treated with ACEA, HU-308 or CBD, and their respective controls. Details in the text. Values correspond to % of the lesioned side over the non-lesioned one for each subject, and are presented as means ± SEM of 6-8 subjects per group. Data were assessed by one-way analysis of variance followed by the Student-Newman-Keuls test (*p<0.05, **p<0.005, ***p<0.0005 vs the controls; #p<0.05 vs the malonate group)
| Treatments | GABA contents | DA contents | DOPAC contents | |
|---|---|---|---|---|
| Globus pallidus | Substantia nigra | Caudate-putamen | Caudate-putamen | |
| Control | 97.2 ± 5.6 | 81.0 ± 9.2 | 87.5 ± 12.3 | 91.7 ± 9.5 |
| Malonate | 68.3 ± 12.7* | 78.5 ± 9.3 | 27.0 ± 4.6** | 41.6 ± 4.7** |
| Malonate + ACEA | 61.1 ± 12.2* | 92.1 ± 5.2 | 23.8 ± 5.4** | 44.9 ± 9.7** |
| Control | 112.0 ± 15.6 | 97.5 ± 7.2 | 104.3 ± 11.7 | 98.2 ± 12.6 |
| Malonate | 48.0 ± 18.8* | 89.8 ± 7.2 | 11.6 ± 7.6*** | 36.6 ± 9.1** |
| Malonate + HU-308 | 90.1 ± 13.4# | 93.8 ± 7.7 | 34.1 ± 5.2**# | 53.9 ± 5.7* |
| Control | 106.3 ± 11.9 | 104.2 ± 5.4 | 96.0 ± 7.9 | 96.2 ± 3.8 |
| Malonate | 62.0 ± 9.4* | 98.7 ± 8.4 | 7.6 ± 1.8*** | 14.0 ± 3.3*** |
| Malonate + CBD | 54.4 ± 8.2* | 86.3 ± 8.6 | 12.6 ± 4.9*** | 17.3 ± 5.5*** |
Figure 2.
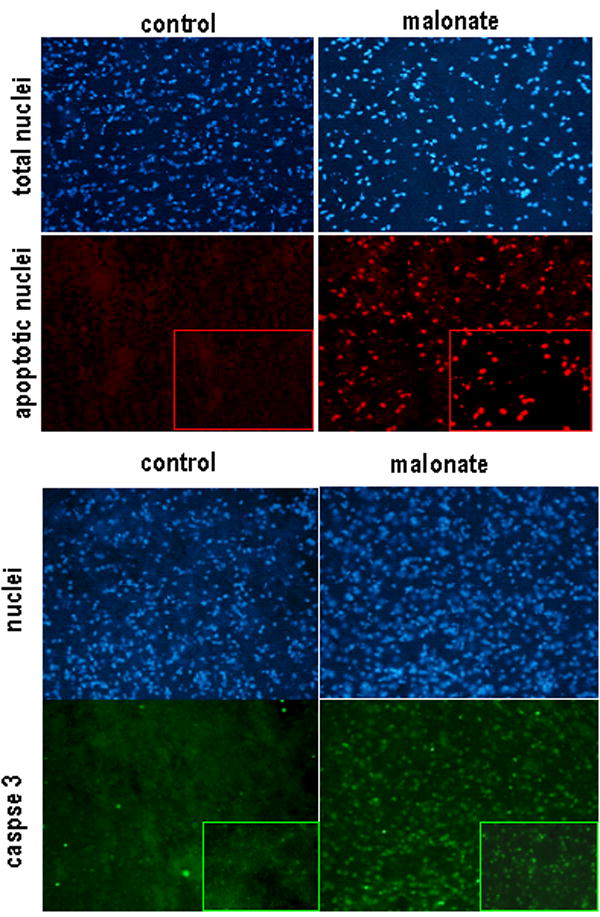
TUNEL staining and caspase-3 immunohistochemistry in the striatum of rats subjected to unilateral injections of malonate and their respective controls. Details in the text. For both cases, upper panels correspond to Hoescht staining for identifying total nuclei. Magnification was 20x.
Figure 3.
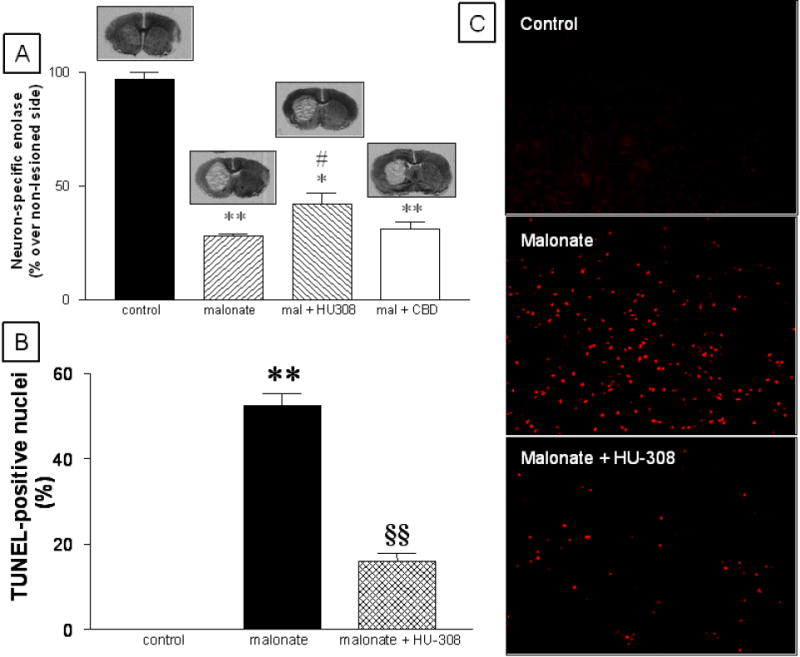
mRNA levels for neuron-specific enolase (panel A) and number of apoptotic nuclei (panel B) in the striatum of rats subjected to unilateral injections of malonate and treated with HU-308 (and CBD in the case of neuron-specific enolase-mRNA levels), and their respective controls. Representative images of TUNEL staining for each experimental group are shown in panels C (magnification was 10x). Details in the text. Values correspond to % of the lesioned side over the non-lesioned one for each subject, and are presented as means ± SEM of 5-8 subjects per group. Data were assessed by one-way analysis of variance followed by the Student-Newman-Keuls test (*p<0.005, **p<0.0005 vs the controls; #p<0.05 vs the malonate group or CBD groups; §§p<0.005 vs the malonate group).
The treatment with HU-308 partially reduced malonate-induced GABA deficit in the striatum (F(2,17)=94.21, p<0.0001; Figure 1) and the globus pallidus (F(2,18)=4.83, p<0.05; Table 1). HU-308 modified GABA contents only in the lesioned side (control: 1.91 ± 0.13 μg/mg of protein; malonate: 0.76 ± 0.07, p<0.001 vs controls; malonate + HU-308: 1.12 ± 0.08, p<0.05 vs the other two groups; F(2,17)=36.81, p<0.0001), but not in the non-lesioned side (control: 1.90 ± 0.06; malonate: 1.95 ± 0.11; malonate + HU-308: 1.96 ± 0.11; F(2,17)=0.112, ns), indicating that its effects were neuroprotective (visible only in the lesioned side) rather than up-regulatory (visible in both sides). The same observations were made for the remaining regions and parameters (data not shown). The treatment with HU-308 also attenuated the reduction in mRNA levels for NSE (F(3,21)=85.73, p<0.0001; Figure 3) and the increase in the number of apoptotic nuclei (F(2,14)=188.9, p<0.0001; Figure 3) caused by the administration of malonate, confirming the neuroprotective properties of this CB2 receptor agonist in this model. HU-308 also reduced the malonate-induced dopamine deficit, as reflected by a partial recovery in the contents for this neurotransmitter (F(2,18)=31,98, p<0.0001) and its metabolite DOPAC (F(2,19)=11.35, p<0.005) (Table 1). By contrast, the treatment with ACEA did not attenuate the neurochemical deficits caused by the administration of malonate (Table 1 and Figure 1), thus indicating that the activation of CB1 receptors does not protect striatal projection neurons from death in this model. We found the same result when the cannabinoid used was CBD (Table 1 and Figures 1 and 3).
Experiment II: Importance of CB2 receptors in malonate-lesioned rats
In the second part of this study, we first examined whether co-administration of HU-308 with the selective CB2 receptor antagonist SR144528 could reverse the neuroprotective effects of HU-308 injected alone. As shown in Figure 4, malonate-induced GABA depletion in animals co-injected with HU-308 and SR144528 was similar to the value measured in animals receiving vehicle and significantly different to the case of animals receiving HU-308 alone (F(4,29)=15.78, p<0.0001). The administration of SR144528 alone was without effect (Figure 4). We also conducted some experiments to compare the magnitude of striatal lesion caused by malonate in CB2 receptor-deficient and wild-type mice. The number of degenerating cells measured by FluoroJade B staining was 2-fold higher in the striatal parenchyma of CB2-/- mice compared to the case of CB2+/+ animals (Figure 4). Degenerating cells were not detectable in the respective controls for both CB2+/+ and CB2-/- mice (data not shown).
Figure 4.
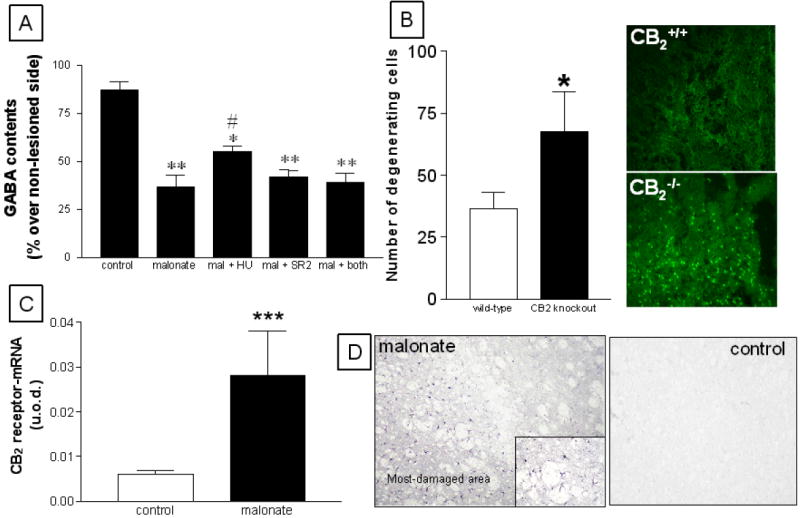
Panel A: GABA contents in the striatum of rats subjected to unilateral injections of malonate and treated with HU-308, SR144528, or both, and their respective controls. Details in the text. Values correspond to % of the lesioned side over the non-lesioned one for each subject, and are presented as means ± SEM of 5-7 subjects per group. Data were assessed by one-way analysis of variance followed by the Student-Newman-Keuls test (*p<0.05, **p<0.005 vs the controls; #p<0.05 vs the other malonate groups). Panel B: Number of degenerating cells measured in FluoroJade B stained-sections (representative images are 20x) corresponding to the striatum of CB2+/+ and CB2-/- mice subjected to unilateral injections of malonate. Details in the text. Values are means ± SEM of 5 subjects per group. Data were assessed by the Student’s t-test (*p<0.005 vs the wild-type group). Panel C: mRNA levels for CB2 receptors (measured by RT-PCR) in the striatum of rats subjected to unilateral injections of malonate and their respective controls. Details in the text. Values are means ± SEM of 5-7 subjects per group. Data were assessed by the Student’s t-test (***p<0.005 vs the controls). Panel D: Immunostaining corresponding to CB2 receptors in the striatum of rats subjected to unilateral injections of malonate and their respective controls. Note the glial-like appearance of CB2 positive cells in the inset in the panel corresponding to malonate-lesioned animals, as well as the spatial segregation within the lesioned striatum. Magnification was 20x.
CB2 receptors are thought to be absent (or scarcely present) in the striatum of adult mammals. We have re-examined this issue here and found near to non-detectable immunoreactivity for this receptor type in the non-lesioned striatum using classic immunohistochemical staining methods (Figure 4). Further data obtained using qRT-PCR demonstrated that CB2 receptor-mRNA transcripts may be in fact present in the intact striatum (Figure 4) in concordance with data described in other brain structures (reviewed in Fernández-Ruiz et al., 2007), although their levels were very modest and close to the non-specific signals. However, the injection of malonate within the striatum produced a marked increase (approximately 5-fold) in the levels of CB2 receptor transcripts (Figure 4), supporting the idea that these receptors are significantly up-regulated in response to lesion. This response was already evident at 24 hours after the lesion and remains stable up to 48 hours (Figure 5). The up-regulation of CB2 receptors was also evident using classic immunohistochemical staining methods, which showed a clear spatial segregation in CB2 receptor immunostaining within the lesioned striatum, with areas affected by malonate administration being strongly positive for this receptor (Figure 4). A preliminary examination of morphological characteristics of cells expressing CB2 receptors indicated that they could be glial cells, either reactive microglia and activated astrocytes. To confirm this hypothesis, we conducted double labelling experiments with selective markers of glial cells and found co-localization of CB2 receptors with the marker of reactive microglia OX-42, and also with the marker of astrocytes GFAP (Figure 6). In the case of microglial cells, CB2 receptors are located in many but not all activated cells (Figure 6), whereas the astrocytes showed CB2 receptor staining in cell bodies and proximal portions of cellular processes (Figure 6). By contrast, the same type of double-labelling analyses proved a poor signal for the CB2 receptor in the non-lesioned contralateral striatum (data not shown), in concordance with the data obtained with DAB immunostaining (Figure 4).
Figure 5.
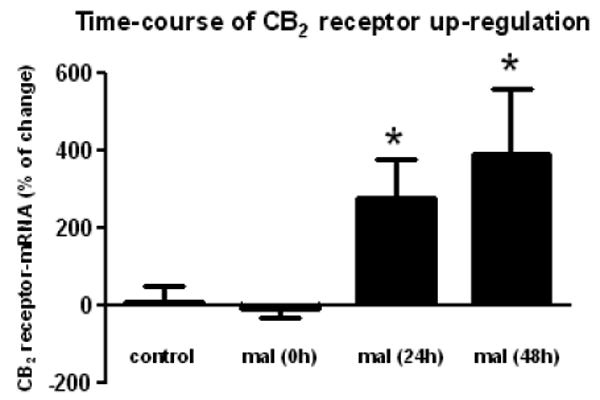
Time-course of CB2 receptor up-regulation (measured by RT-PCR) in the striatum of rats subjected to unilateral injections of malonate and their respective controls. Details in the text. Values correspond to % of change in the lesioned side over the non-lesioned one for each subject, and are presented as means ± SEM of 5-7 subjects per group. Data were assessed by the one-way analysis of variance followed by the Student-Newman-Keuls test [*p<0.05 vs controls and malonate (0h)].
Figure 6.
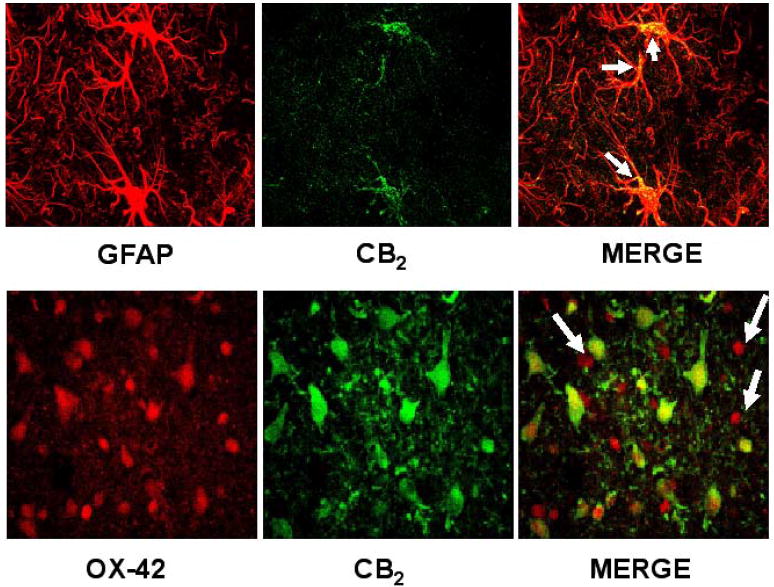
Cannabinoid CB2 receptors are expressed by astrocytes, as revealed by colocalization with GFAP (upper panels), and reactive microglia, as revealed by colocalization with OX-42 (bottom panels). Upper panels: 100x magnification photographs of the striatum of lesioned animals. CB2 receptors are located in cell bodies and proximal portions of cellular processes of astrocytes (marked with arrows). Bottom panels: 100x magnification photographs of the striatum of lesioned animals. CB2 receptors are abundantly expressed in areas of intense microgliosis, although not all reactive microglial cells are CB2 receptor-positive (marked with arrows). Additional non-microglial cells (mainly astrocytes, as previously shown in upper panels) are also CB2-positive.
Experiment III: Cellular events linked to the toxicity of malonate that are controlled by CB2 receptors
Here we wanted to determine the type of cytotoxic events elicited by malonate that might be limited by the activation of CB2 receptors. Given the role played by reactive microglia in the generation of reactive oxygen species (Fernández-Ruiz et al., 2007, for review), we first studied whether CB2 receptors might enhance the endogenous antioxidant mechanisms. Our results proved that HU-308 was unable to restore the malonate-induced reduction of mRNA levels for SOD-1 (F(3,21)=26.32, p<0.0001; see Figure 7) and SOD-2 (F(3,22)=4.13, p<0.05; see Figure 7). We then evaluated whether CB2 receptors might play a role in the control of proinflammatory factors that, released by reactive microglia, become toxic for neurons (McCarty, 2006, for review). We focused on TNF-α and found that its levels significantly increased in the striatum after the malonate injection (F(2,14)=7.546, p<0.01; see Figure 7), whereas the treatment with HU-308 reduced the levels of TNF-α back to values similar to those found in control animals (Figure 8).
Figure 7.
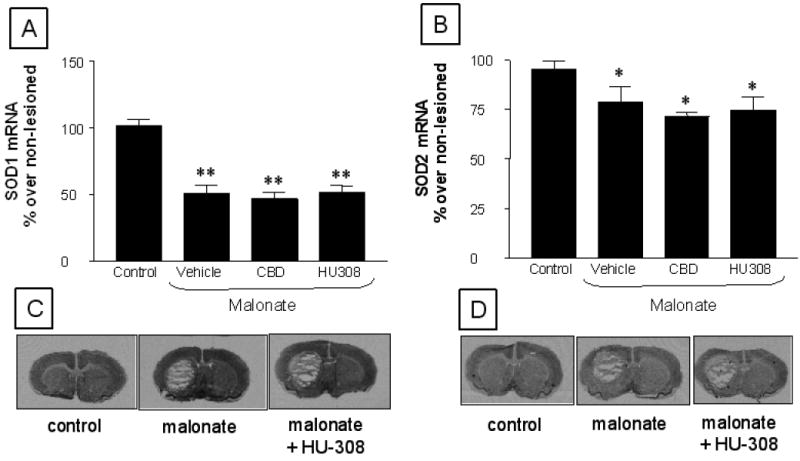
mRNA levels for SOD-1 (panel A and representative images in panel C) and SOD-2 (panel B and representative images in panel D) in the striatum of rats subjected to unilateral injections of malonate treated with HU-308 or CBD, and their respective controls. Details in the text. Values correspond to % of the lesioned side over the non-lesioned one for each subject, and are presented as means ± SEM of 5-6 subjects per group. Data were assessed by one-way analysis of variance followed by the Student-Newman-Keuls test (*p<0.05, **p<0.001 vs the controls)
Figure 8.
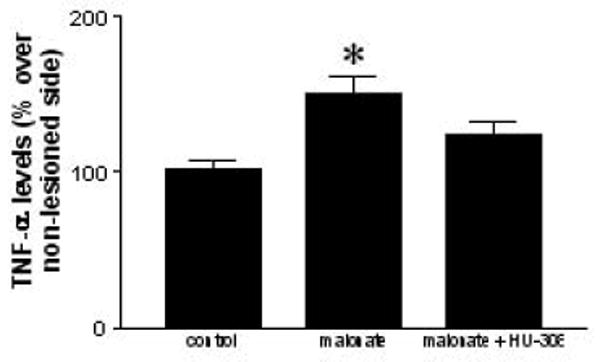
TNF-α levels in the striatum of rats subjected to unilateral injections of malonate treated with HU-308, and their respective controls. Details in the text. Values correspond to % of the lesioned side over the non-lesioned one for each subject, and are presented as means ± SEM of 6-8 subjects per group. Data were assessed by one-way analysis of variance followed by the Student-Newman-Keuls test (*p<0.01 vs the controls).
Discussion
Cannabinoid agonists might serve to delay/arrest disease progression in HD given their well-demonstrated neuroprotectant properties (for review, see Fernández-Ruiz et al., 2005 and 2007; Sagredo et al., 2007a). This has been recently evaluated in animal models of striatal injury generated by the excitotoxin quinolinate (Pintor et al., 2006) or the mitochondrial complex II inhibitors malonate or 3-NP (Lastres-Becker et al., 2003 and 2004; Sagredo et al., 2007b). Certain cannabinoid agonists are effective against the toxicity caused by quinolinate (Pintor et al., 2006) or 3-NP (Lastres-Becker et al., 2004; Sagredo et al., 2007b). However, the results obtained in rats lesioned with malonate were inconclusive (Lastres-Becker et al., 2003) and demanded a further evaluation with compounds having more selectivity for the different targets of the cannabinoid signaling. We have carried out this re-evaluation in the present study, obtaining strong evidence that activation of CB2 receptors is the key mechanism for cannabinoids to protect striatal neurons from death in this HD model. This conclusion is based on the following observations: (i) the unique cannabinoid that reduced the magnitude of striatal lesion caused with malonate was HU-308, and this compound has well-described CB2 receptor agonist properties, with low affinities for other known receptors (Hanus et al., 1999), (ii) the neuroprotective effect of HU-308 was reversed by a selective CB2 receptor antagonist, (iii) the intrastriatal injection of malonate in CB2 receptor knockout mice led to a lesion of greater magnitude than in the case of wild-type animals, and (iv) an increased expression of CB2 receptor was observed to be associated with ongoing striatal degeneration. These observations, together with the lack of neuroprotective effects of ACEA or CBD, supports a major role for CB2 receptors in detriment of additional mechanisms like the activation of CB1 receptors or the antioxidant and receptor-independent properties of certain cannabinoid agonists. The absence of neuroprotective effect is notably important in the case of CBD because this plant-derived cannabinoid protected striatal neurons from death in another model of HD, rats lesioned with 3-NP (Sagredo et al., 2007b). This would possibly indicate that oxidative injury is not a relevant mechanism in malonate toxicity, but it seems to be crucial in 3-NP toxicity (Alexi et al., 1998, for review). In addition, the death of striatal neurons caused by malonate is mainly apoptotic (Toulmond et al., 2004), a fact confirmed here by the data of TUNEL staining and caspase-3 immunohistochemistry, whereas the death caused by 3-NP has been reported to progress by mechanisms other than apoptosis (Brouillet et al., 2005). Therefore, both models present substantial differences and this possibly explains why the type of cannabinoid agonist(s) that provides neuroprotection against the striatal damage caused by each neurotoxin may be different (reviewed in Sagredo et al., 2007a).
Our study suggests that CB2 receptors are up-regulated in response to malonate-induced striatal damage. We believe that this up-regulatory response contributes to a more general response of the cannabinoid signaling against different types of stimuli that may damage the brain (see Fernández-Ruiz et al., 2005 and 2007, for review). For example, Panikashvili et al. (2001) observed a 10-fold increase in the generation of the endocannabinoid 2-arachidonoylglycerol after closed head injury. Jin et al. (2000) described that CB1 receptors are induced in neuronal cells after experimental stroke, whereas an increase of these receptors in response to excitotoxic stimuli in neonatal rats was reported by Hansen et al. (2001). As regards to CB2 receptors, our data suggest that this receptor is poorly expressed in striatal neurons and glial cells (e.g. quiescent microglial cells and astrocytes) in healthy conditions, in concordance with previous studies (for review, see Walter and Stella, 2004). However, the lesion of the striatum with malonate caused a marked elevation of CB2 receptor levels. We have provided evidence that this up-regulatory response occurred in astrocytes and reactive microglial cells, as previously observed in other neuroinflammatory/neurodegenerative disorders (Walter and Stella, 2004; Ehrhart et al., 2005; Fernández-Ruiz et al., 2007). Importantly, the activation of these receptors served to reduce the neuronal damage in those disorders where CB2 receptors were up-regulated in glial elements, in particular in reactive microglial cells recruited at the lesioned structures. This was the case of Alzheimer’s disease (Benito et al., 2003; Ramírez et al., 2005; Esposito et al., 2007), multiple sclerosis (Pryce et al., 2003; Arévalo-Martín et al., 2003; Maresz et al., 2005; Yiangou et al., 2006; Benito et al., 2007), cerebral ischemia (Ashton et al., 2007; Zhang et al., 2007), Down’s syndrome (Nuñez et al., 2008), peripheral nerve injury (Zhang et al., 2003), and amyotrophic lateral sclerosis (Yiangou et al., 2006; Kim et al., 2006; Shoemaker et al., 2007). Our present data, showing up-regulation of CB2 receptors in glial elements and protection of striatal neurons after the activation of this receptor in a model of striatal damage, support that this might be the case of HD too. In the case of astrocytes, we hypothesize that CB2 receptors might facilitate the trophic role exerted by these glial cells on neuronal survival (e.g. by providing energy substrates or prosurvival factors; see Fernández-Ruiz et al., 2007, for review), although this possibility has not been examined yet. By contrast, in the case of reactive microglia, CB2 receptors might play a key role by reducing the toxicity exerted by these cells on neurons. This effect could be mediated by inhibiting the production of cytotoxic factors, such as nitric oxide, reactive oxygen species or proinflammatory cytokines, that are released by activated microglial cells when these cells are recruited and migrate at lesioned sites (Carrier et al., 2004; Witting and Stella, 2004; Walter and Stella, 2004; Ehrhart et al., 2005). We had preliminary evidence that one key factor might be TNF-α, whose production increased rapidly after malonate application in a series of time-course experiments (data not shown). The administration of the CB2 receptor agonist HU-308 significantly attenuated the malonate-induced increase in TNF-α levels at the time-point at which the elevation of this cytokine was maximal. This observation agrees with previous studies showing: (i) reduction of TNF-α levels and/or TNF-α-triggered signaling by CB2 receptor agonists in different inflammatory conditions in the brain (Klegeris et al., 2003; Ehrhart et al., 2005) and also in the periphery (Gallily et al., 2000; Rajesh et al., 2007); and (ii) an enhanced generation of TNF-α and other proinflammatory cytokines after the CB2 receptor blockade (Puffenbarger et al., 2000). On the other hand, CB2 receptors do not appear to be involved in the control of oxidative injury because their activation did not restore the malonate-induced decrease in mRNA levels of SOD-1 and -2, which agrees with the lack of protective effect of CBD in this model.
In summary, our data show that CB2 receptor expression in the striatum increased in response to neurodegeneration produced by a mitochondrial toxin, and that activation of these receptors produces significant neuroprotective effects, as indicated by both selective CB2 receptors agonists and mice lacking this cannabinoid receptor type. These neuroprotective effects are likely exerted through a mechanism involving glial cells, in particular reactive microglial cells. In these cells, the activation of CB2 receptors would reduce the generation of proinflammatory molecules like TNF-α. Collectively, these data suggest that CB2 receptors represent a potential therapeutic target to slow the progression of degeneration in HD and other neurodegenerative disorders.
Acknowledgments
This work was supported by grants from CIBERNED (CB06/05/0089 to OS, MRP and JFR, and CB06/05/1109 to CB, RMT and JR), Santander-UCM (PRR27/05-13975 to JFR), MEC (SAF2006/11333 to OS, MRP and JFR; and SAF2007/61565 to CB, RMT and JR), CAM (S-SAL-0261/2006 to OS, MRP, CB, RMT, JR and JFR). and the US National Institute on Drug Abuse and the Israel Science Foundation to RM. Ilia Aroyo is an undergraduate student supported by the Socrates-Erasmus Programme, while María Ruth Pazos is a predoctoral fellow supported by the “Programa FPI-Ministerio de Educación y Ciencia”. Authors are indebted to Pharmos and Sanofi-Aventis for the gift of HU-308 and SR144528, respectively, to Dr. Nancy Buckley (California State Polytechnic University, Pomona, CA, U.S.A.) for the donation of CB2+/- mice, and to Patricia Rodríguez Valsero and Yolanda García Movellán for their technical and administrative assistance.
References
- Aiken CT, Tobin AJ, Schweitzer ES. A cell-based screen for drugs to treat Huntington’s disease. Neurobiol Dis. 2004;16:546–555. doi: 10.1016/j.nbd.2004.04.001. [DOI] [PubMed] [Google Scholar]
- Alexi T, Hughes PE, Faull RL, Williams CE. 3-Nitropropionic acid’s lethal triplet: cooperative pathways of neurodegeneration. Neuroreport. 1998;9:R57–R64. doi: 10.1097/00001756-199808030-00001. [DOI] [PubMed] [Google Scholar]
- Alfinito PD, Wang SP, Manzino L, Rijhsinghani S, Zeevalk GD, Sonsalla PK. Adenosinergic protection of dopaminergic and GABAergic neurons against mitochondrial inhibition through receptors located in the substantia nigra and striatum, respectively. J Neurosci. 2003;23:10982–10987. doi: 10.1523/JNEUROSCI.23-34-10982.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arévalo-Martin A, Vela JM, Molina-Holgado E, Borrell J, Guaza C. Therapeutic action of cannabinoids in a murine model of multiple sclerosis. J Neurosci. 2003;23:2511–2516. doi: 10.1523/JNEUROSCI.23-07-02511.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashton JC, Rahman RM, Nair SM, Sutherland BA, Glass M, Appleton I. Cerebral hypoxia-ischemia and middle cerebral artery occlusion induce expression of the cannabinoid CB2 receptor in the brain. Neurosci Lett. 2007;412:114–117. doi: 10.1016/j.neulet.2006.10.053. [DOI] [PubMed] [Google Scholar]
- Benito C, Nuñez E, Tolon RM, Carrier EJ, Rabano A, Hillard CJ, Romero J. Cannabinoid CB2 receptors and fatty acid amide hydrolase are selectively overexpressed in neuritic plaque-associated glia in Alzheimer’s disease brains. J Neurosci. 2003;23:11136–11141. doi: 10.1523/JNEUROSCI.23-35-11136.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benito C, Romero JP, Tolon RM, Clemente D, Docagne F, Hillard CJ, Guaza C, Romero J. Cannabinoid CB1 and CB2 receptors and fatty acid amide hydrolase are specific markers of plaque cell subtypes in human multiple sclerosis. J Neurosci. 2007;27:2396–2402. doi: 10.1523/JNEUROSCI.4814-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brouillet E, Jacquard C, Bizat N, Blum D. 3-Nitropropionic acid: a mitochondrial toxin to uncover physiopathological mechanisms underlying striatal degeneration in Huntington’s disease. J Neurochem. 2005;95:1521–1540. doi: 10.1111/j.1471-4159.2005.03515.x. [DOI] [PubMed] [Google Scholar]
- Buckley NE, McCoy KL, Mezey E, Bonner T, Zimmer A, Felder CC, Glass M, Zimmer A. Immunomodulation by cannabinoids is absent in mice deficient for the cannabinoid CB2 receptor. Eur J Pharmacol. 2000;396:141–149. doi: 10.1016/s0014-2999(00)00211-9. [DOI] [PubMed] [Google Scholar]
- Buckley NE. The peripheral cannabinoid receptor knockout mice: an update. Br J Pharmacol. 2008;153:309–318. doi: 10.1038/sj.bjp.0707527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calabresi P, Centonze D, Gubellini P, Marfia GA, Pisani A, Sancesario G, Bernardi G. Synaptic transmission in the striatum: from plasticity to neurodegeneration. Prog Neurobiol. 2000;61:231–265. doi: 10.1016/s0301-0082(99)00030-1. [DOI] [PubMed] [Google Scholar]
- Carrier EJ, Kearn CS, Barkmeier AJ, Breese NM, Yang W, Nithipatikom K, Pfister SL, Campbell WB, Hillard CJ. Cultured rat microglial cells synthesize the endocannabinoid 2-arachidonylglycerol, which increases proliferation via a CB2 receptor-dependent mechanism. Mol Pharmacol. 2004;65:999–1007. doi: 10.1124/mol.65.4.999. [DOI] [PubMed] [Google Scholar]
- Cattaneo E, Zuccato C, Tartari M. Normal huntingtin function: an alternative approach to Huntington’s disease. Nat Rev Neurosci. 2005;6:919–930. doi: 10.1038/nrn1806. [DOI] [PubMed] [Google Scholar]
- Ehrhart J, Obregon D, Mori T, Hou H, Sun N, Bai Y, Klein T, Fernandez F, Tan J, Shytle RD. Stimulation of cannabinoid receptor 2 (CB2) suppresses microglial activation. J Neuroinflammation. 2005;2:29. doi: 10.1186/1742-2094-2-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esposito G, Scuderi C, Savani C, Steardo L, Jr, De Filippis D, Cottone P, Iuvone T, Cuomo V, Steardo L. Cannabidiol in vivo blunts beta-amyloid induced neuroinflammation by suppressing IL-1beta and iNOS expression. Br J Pharmacol. 2007;151:1272–1279. doi: 10.1038/sj.bjp.0707337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernández-López D, Pazos MR, Tolón RM, Moro MA, Romero J, Lizasoain I, Martínez-Orgado J. The cannabinoid agonist WIN55212 reduces brain damage in an in vivo model of hypoxic-ischemic encephalopathy in newborn rats. Pediatr Res. 2007;62:255–260. doi: 10.1203/PDR.0b013e318123fbb8. [DOI] [PubMed] [Google Scholar]
- Fernández-Ruiz J, González S, Romero J, Ramos JA. Cannabinoids in Neurodegeneration and Neuroprotection. In: Mechoulam R, editor. Cannabinoids as Therapeutics (MDT) Birkhaüser Verlag; Switzerland: 2005. pp. 79–109. [Google Scholar]
- Fernández-Ruiz J, Romero J, Velasco G, Tolón RM, Ramos JA, Guzmán M. Cannabinoid CB2 receptor: a new target for the control of neural cell survival? Trends Pharmacol Sci. 2007;28:39–45. doi: 10.1016/j.tips.2006.11.001. [DOI] [PubMed] [Google Scholar]
- Gallily R, Breuer A, Mechoulam R. 2-Arachidonylglycerol, an endogenous cannabinoid, inhibits tumor necrosis factor-alpha production in murine macrophages, and in mice. Eur J Pharmacol. 2000;406:R5–R7. doi: 10.1016/s0014-2999(00)00653-1. [DOI] [PubMed] [Google Scholar]
- Gaoni Y, Mechoulam R. The isolation and structure of delta-1-tetrahydrocannabinol and other neutral cannabinoids from hashish. J Am Chem Soc. 1971;93:217–224. doi: 10.1021/ja00730a036. [DOI] [PubMed] [Google Scholar]
- García-Arencibia M, González S, de Lago E, Ramos JA, Mechoulam R, Fernández-Ruiz J. Evaluation of the neuroprotective effect of cannabinoids in a rat model of Parkinson’s disease: importance of antioxidant and cannabinoid receptor-independent properties. Brain Res. 2007;1134:162–170. doi: 10.1016/j.brainres.2006.11.063. [DOI] [PubMed] [Google Scholar]
- Gu M, Gash MT, Mann VM, Javoy-Agid F, Cooper JM, Schapira AH. Mitochondrial defect in Huntington’s disease caudate nucleus. Ann Neurol. 1996;39:385–389. doi: 10.1002/ana.410390317. [DOI] [PubMed] [Google Scholar]
- Hansen HH, Schmid PC, Bittigau P, Lastres-Becker I, Berrendero F, Manzanares J, Ikonomidou C, Schmid HH, Fernández-Ruiz JJ, Hansen HS. Anandamide, but not 2-arachidonoylglycerol, accumulates during in vivo neurodegeneration. J Neurochem. 2001;78:1415–1427. doi: 10.1046/j.1471-4159.2001.00542.x. [DOI] [PubMed] [Google Scholar]
- Hanus L, Breuer A, Tchilibon S, Shiloah S, Goldenberg D, Horowitz M, Pertwee RG, Ross RA, Mechoulam R, Fride E. HU-308: a specific agonist for CB2, a peripheral cannabinoid receptor. Proc Natl Acad Sci U S A. 1999;96:14228–14233. doi: 10.1073/pnas.96.25.14228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hillard CJ, Manna S, Greenberg MJ, DiCamelli R, Ross RA, Stevenson LA, Murphy V, Pertwee RG, Campbell WB. Synthesis and characterization of potent and selective agonists of the neuronal cannabinoid receptor (CB1) J Pharmacol Exp Ther. 1999;289:1427–1433. [PubMed] [Google Scholar]
- Jin KL, Mao XO, Goldsmith PC, Greenberg DA. CB1 cannabinoid receptor induction in experimental stroke. Ann Neurol. 2000;48:257–261. [PubMed] [Google Scholar]
- Katagiri T, Feng X, Ichikawa T, Usui H, Takahashi Y, Kumanishi T. Neuron-specific enolase (NSE) and non-neuronal enolase (NNE) mRNAs are co-expressed in neurons of the rat cerebellum: in situ hybridization histochemistry. Mol Brain Res. 1993;19:1–8. doi: 10.1016/0169-328x(93)90142-c. [DOI] [PubMed] [Google Scholar]
- Kim K, Moore DH, Makriyannis A, Abood ME. AM1241, a cannabinoid CB2 receptor selective compound, delays disease progression in a mouse model of amyotrophic lateral sclerosis. Eur J Pharmacol. 2006;542:100–105. doi: 10.1016/j.ejphar.2006.05.025. [DOI] [PubMed] [Google Scholar]
- Klegeris A, Bissonnette CJ, McGeer PL. Reduction of human monocytic cell neurotoxicity and cytokine secretion by ligands of the cannabinoid-type CB2 receptor. Br J Pharmacol. 2003;139:775–786. doi: 10.1038/sj.bjp.0705304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunikowska G, Jenner P. 6-Hydroxydopamine-lesioning of the nigrostriatal pathway in rats alters basal ganglia mRNA for copper, zinc- and manganese-superoxide dismutase, but not glutathione peroxidase. Brain Res. 2001;922:51–64. doi: 10.1016/s0006-8993(01)03149-3. [DOI] [PubMed] [Google Scholar]
- Lastres-Becker I, Bizat N, Boyer F, Hantraye P, Brouillet E, Fernandez-Ruiz J. Effects of cannabinoids in the rat model of Huntington’s disease generated by an intrastriatal injection of malonate. Neuroreport. 2003;14:813–816. doi: 10.1097/00001756-200305060-00007. [DOI] [PubMed] [Google Scholar]
- Lastres-Becker I, Bizat N, Boyer F, Hantraye P, Fernández-Ruiz J, Brouillet E. Potential involvement of cannabinoid receptors in 3-nitropropionic acid toxicity in vivo. Neuroreport. 2004;15:2375–2379. doi: 10.1097/00001756-200410250-00015. [DOI] [PubMed] [Google Scholar]
- Lastres-Becker I, Molina-Holgado F, Ramos JA, Mechoulam R, Fernández-Ruiz J. Cannabinoids provide neuroprotection against 6-hydroxydopamine toxicity in vivo and in vitro: relevance to Parkinson’s disease. Neurobiol Dis. 2005;19:96–107. doi: 10.1016/j.nbd.2004.11.009. [DOI] [PubMed] [Google Scholar]
- Lehmann A, Gautier M. Stereotaxic Atlas of the Brain of the Mouse. Paris, France: 1974. pp. 1–68. Book. [Google Scholar]
- Lowry OH, Rosebrough NJ, Farr AL, Randall RJ. Protein measurement with the Folin phenol reagent. J Biol Chem. 1951;193:265–275. [PubMed] [Google Scholar]
- Maresz K, Carrier EJ, Ponomarev ED, Hillard CJ, Dittel BN. Modulation of the cannabinoid CB2 receptor in microglial cells in response to inflammatory stimuli. J Neurochem. 2005;95:437–445. doi: 10.1111/j.1471-4159.2005.03380.x. [DOI] [PubMed] [Google Scholar]
- McMurray CT. Huntington’s disease: new hope for therapeutics. Trends Neurosci. 2001;24:S32–S38. doi: 10.1016/s0166-2236(00)01997-4. [DOI] [PubMed] [Google Scholar]
- Mechoulam R, Parker LA, Gallily R. Cannabidiol: an overview of some pharmacological aspects. J Clin Pharmacol. 2002;42:11S–19S. doi: 10.1002/j.1552-4604.2002.tb05998.x. [DOI] [PubMed] [Google Scholar]
- Nagayama T, Sinor AD, Simon RP, Chen J, Graham SH, Jin KL, Greenberg DA. Cannabinoids and neuroprotection in global and focal cerebral ischemia and in neuronal cultures. J Neurosci. 1999;19:2987–2995. doi: 10.1523/JNEUROSCI.19-08-02987.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Núñez E, Benito C, Tolón RM, Hillard CJ, Griffin WS, Romero J. Glial expression of cannabinoid CB2 receptors and fatty acid amide hydrolase are beta amyloid-linked events in Down’s syndrome. Neuroscience. 2008;151:104–110. doi: 10.1016/j.neuroscience.2007.10.029. [DOI] [PubMed] [Google Scholar]
- Palkovits M, Brownstein J. Maps and Guide to Microdissection of the Rat Brain. Elsevier; 1988. [Google Scholar]
- Panikashvili D, Simeonidou C, Ben-Shabat S, Hanus L, Breuer A, Mechoulam R, Shohami E. An endogenous cannabinoid (2-AG) is neuroprotective after brain injury. Nature. 2001;413:527–531. doi: 10.1038/35097089. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Watson C. The rat brain in stereotaxic coordinates. Academic Press; London: 1986. [DOI] [PubMed] [Google Scholar]
- Pintor A, Tebano MT, Martire A, Grieco R, Galluzzo M, Scattoni ML, Pezzola A, Coccurello R, Felici F, Cuomo V, Piomelli D, Calamandrei G, Popoli P. The cannabinoid receptor agonist WIN 55,212-2 attenuates the effects induced by quinolinic acid in the rat striatum. Neuropharmacology. 2006;51:1004–1012. doi: 10.1016/j.neuropharm.2006.06.013. [DOI] [PubMed] [Google Scholar]
- Pryce G, Ahmed Z, Hankey DJ, Jackson SJ, Croxford JL, Pocock JM, Ledent C, Petzold A, Thompson AJ, Giovannoni G, Cuzner ML, Baker D. Cannabinoids inhibit neurodegeneration in models of multiple sclerosis. Brain. 2003;126:2191–2202. doi: 10.1093/brain/awg224. [DOI] [PubMed] [Google Scholar]
- Puffenbarger RA, Boothe AC, Cabral GA. Cannabinoids inhibit LPS-inducible cytokine mRNA expression in rat microglial cells. Glia. 2000;29:58–69. [PubMed] [Google Scholar]
- Rajesh M, Mukhopadhyay P, Bátkai S, Haskó G, Liaudet L, Huffman JW, Csiszar A, Ungvari Z, Mackie K, Chatterjee S, Pacher P. CB2-receptor stimulation attenuates TNF-alpha-induced human endothelial cell activation, transendothelial migration of monocytes, and monocyte-endothelial adhesion. Am J Physiol Heart Circ Physiol. 2007;293:H2210–H2218. doi: 10.1152/ajpheart.00688.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramirez BG, Blazquez C, Gomez del Pulgar T, Guzman M, de Ceballos ML. Prevention of Alzheimer’s disease pathology by cannabinoids: neuroprotection mediated by blockade of microglial activation. J Neurosci. 2005;25:1904–1913. doi: 10.1523/JNEUROSCI.4540-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rinaldi-Carmona M, Barth F, Millan J, Derocq JM, Casellas P, Congy C, Oustric D, Sarran M, Bouaboula M, Calandra B, Portier M, Shire D, Breliere JC, Le Fur GL. SR 144528, the first potent and selective antagonist of the CB2 cannabinoid receptor. J Pharmacol Exp Ther. 1998;284:644–650. [PubMed] [Google Scholar]
- Sagredo O, García-Arencibia M, de Lago E, Finetti S, Decio A, Fernández-Ruiz J. Cannabinoids and neuroprotection in basal ganglia disorders. Mol Neurobiol. 2007a;36:82–91. doi: 10.1007/s12035-007-0004-3. [DOI] [PubMed] [Google Scholar]
- Sagredo O, Ramos JA, Decio A, Mechoulam R, Fernández-Ruiz J. Cannabidiol reduced the striatal atrophy caused by 3-nitropropionic acid in vivo by mechanisms independent of the activation of cannabinoid, vanilloid TRPV1 and adenosine A2A receptors. Eur J Neurosci. 2007b;26:843–851. doi: 10.1111/j.1460-9568.2007.05717.x. [DOI] [PubMed] [Google Scholar]
- Schmued LC, Albertson C, Slikker W., Jr Fluoro-Jade: a novel fluorochrome for the sensitive and reliable histochemical localization of neuronal degeneration. Brain Res. 1997;751:37–46. doi: 10.1016/s0006-8993(96)01387-x. [DOI] [PubMed] [Google Scholar]
- Shi SR, Cote RJ, Taylor CR. Antigen retrieval techniques: current perspectives. J Histochem Cytochem. 2001;49:931–937. doi: 10.1177/002215540104900801. [DOI] [PubMed] [Google Scholar]
- Shoemaker JL, Seely KA, Reed RL, Crow JP, Prather PL. The CB2 cannabinoid agonist AM-1241 prolongs survival in a transgenic mouse model of amyotrophic lateral sclerosis when initiated at symptom onset. J Neurochem. 2007;101:87–98. doi: 10.1111/j.1471-4159.2006.04346.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinor AD, Irvin SM, Greenberg DA. Endocannabinoids protect cerebral cortical neurons from in vitro ischemia in rats. Neurosci Lett. 2000;278:157–160. doi: 10.1016/s0304-3940(99)00922-2. [DOI] [PubMed] [Google Scholar]
- Toulmond S, Tang K, Bureau Y, Ashdown H, Degen S, O’Donnell R, Tam J, Han Y, Colucci J, Giroux A, Zhu Y, Boucher M, Pikounis B, Xanthoudakis S, Roy S, Rigby M, Zamboni R, Robertson GS, Ng GY, Nicholson DW, Fluckiger JP. Neuroprotective effects of M826, a reversible caspase-3 inhibitor, in the rat malonate model of Huntington’s disease. Br J Pharmacol. 2004;141:689–697. doi: 10.1038/sj.bjp.0705662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker FO. Huntington’s disease. Lancet. 2007;369:218–228. doi: 10.1016/S0140-6736(07)60111-1. [DOI] [PubMed] [Google Scholar]
- Walter L, Stella N. Cannabinoids and neuroinflammation. Br J Pharmacol. 2004;141:775–785. doi: 10.1038/sj.bjp.0705667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, Duan W, Igarashi S, Morita H, Nakamura M, Ross CA. Compounds blocking mutant huntingtin toxicity identified using a Huntington’s disease neuronal cell model. Neurobiol Dis. 2005;20:500–508. doi: 10.1016/j.nbd.2005.03.026. [DOI] [PubMed] [Google Scholar]
- Witting A, Stella N. Cannabinoid signaling in glial cells in health and disease. Curr Neuropharmacol. 2004;2:115–124. [Google Scholar]
- Wright BL, Barker RA. Established and emerging therapies for Huntington’s disease. Curr Mol Med. 2007;7:579–587. doi: 10.2174/156652407781695738. [DOI] [PubMed] [Google Scholar]
- Yiangou Y, Facer P, Durrenberger P, Chessell IP, Naylor A, Bountra C, Banati RR, Anand P. COX-2, CB2 and P2X7-immunoreactivities are increased in activated microglial cells/macrophages of multiple sclerosis and amyotrophic lateral sclerosis spinal cord. BMC Neurol. 2006;6:12. doi: 10.1186/1471-2377-6-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeevalk GD, Manzino L, Sonsalla PK. Protection of malonate-induced GABA but not dopamine loss by GABA transporter blockade in rat striatum. Exp Neurol. 2002;176:193–202. doi: 10.1006/exnr.2002.7917. [DOI] [PubMed] [Google Scholar]
- Zhang J, Hoffert C, Vu HK, Groblewski T, Ahmad S, O’Donnell D. Induction of CB2 receptor expression in the rat spinal cord of neuropathic but not inflammatory chronic pain models. Eur J Neurosci. 2003;17:2750–2754. doi: 10.1046/j.1460-9568.2003.02704.x. [DOI] [PubMed] [Google Scholar]
- Zhang M, Martin BR, Adler MW, Razdan RK, Jallo JI, Tuma RF. Cannabinoid CB2 receptor activation decreases cerebral infarction in a mouse focal ischemia/reperfusion model. J Cereb Blood Flow Metab. 2007;27:1387–1396. doi: 10.1038/sj.jcbfm.9600447. [DOI] [PMC free article] [PubMed] [Google Scholar]