

Abstract
Odonto-onycho-dermal dysplasia (OODD), a rare autosomal-recessive inherited form of ectodermal dysplasia including severe oligodontia, nail dystrophy, palmoplantar hyperkeratosis, and hyperhidrosis, was recently shown to be caused by a homozygous nonsense WNT10A mutation in three consanguineous Lebanese families. Here, we report on 12 patients, from 11 unrelated families, with ectodermal dysplasia caused by five previously undescribed WNT10A mutations. In this study, we show that (1) WNT10A mutations cause not only OODD but also other forms of ectodermal dysplasia, reaching from apparently monosymptomatic severe oligodontia to Schöpf-Schulz-Passarge syndrome, which is so far considered a unique entity by the findings of numerous cysts along eyelid margins and the increased risk of benign and malignant skin tumors; (2) WNT10A mutations are a frequent cause of ectodermal dysplasia and were found in about 9% of an unselected patient cohort; (3) about half of the heterozygotes (53.8%) show a phenotype manifestation, including mainly tooth and nail anomalies, which was not reported before in OODD; and (4) heterozygotes show a sex-biased manifestation pattern, with a significantly higher proportion of tooth anomalies in males than in females, which may implicate gender-specific differences of WNT10A expression.
Main Text
The ectodermal dysplasias (EDs) are a large, heterogeneous, and growing group of disorders characterized by defects in morphogenesis of skin, hair, nails, teeth, and sweat, sebaceous, submucous, and mammary glands. Numerous, more or less distinct entities of ectodermal dysplasia, including syndromal forms and monosymptomatic oligodontia as well, have been reported. Most of them are very rare, and their cause is often unknown.
Recently, Adaimy et al.1 identified a homozygous nonsense WNT10A (MIM 606268) mutation (c.697G>T [p.E233X]) as the cause of autosomal-recessive odonto-onycho-dermal dysplasia (OODD [MIM 257980]) in three consanguineous Lebanese families. OODD was first described by Fadhil et al.2 and was further delineated by others3–5 as a distinct rare condition characterized by severe oligodontia, nail dystrophy, hypotrichosis, erythematous lesions of face, smooth tongue with reduced fungiform and filiform papillae, and palmoplantar hyperkeratosis with increased sweating. Additional single-case reports of apparently different forms of EDs, either with a quite similar phenotype but no distinct symptoms distinguishing them from OODD6,7 or with additional unique symptoms of numerous cysts along eyelid margins in Schöpf-Schulz-Passarge syndrome (SSPS [MIM 224750]),8–16 were published. Until now, the cause of SSPS was unknown and genetic heterogeneity was discussed on the basis of an apparent autosomal-recessive as well as autosomal-dominant mode of inheritance.
OODD is considered a rare condition, and no reports were published of WNT10A-related ED except the one by Adaimy et al.1 However, because phenotypic variability was observed in these families, we wondered whether WNT10A mutations might explain a broader spectrum of EDs. Therefore, we screened the WNT10A gene in patients with clinical findings resembling OODD or SSPS or with unclassified forms of ED. All patients and their relatives who were genotyped consented to participate in our study on ED and oligodontia, which was sanctioned by an independent institutional ethics committee in accordance with the national regulations and GCP/ICH guidelines. All index patients and their relatives were ascertained by physician-initiated referral or had contacted us independently via the German ED support group. Clinical data were obtained by an examination performed by one of us, through questionnaires, or from medical records. All index patients were prescreened for the absence of EDA (MIM 300451) mutations (data not shown). DNA was isolated from peripheral blood leukocytes via standard procedures. PCR was performed with specific primer pairs (Table S1, available online) selected with Primer3 and checked with MFOLD and SNPCheck. The PCR products were treated with ExoSAP-IT (USB Corporation, Cleveland, OH, USA), in accordance with the manufacture's instructions, and sequenced with the BigDye Terminator v3.1 Cycle Sequencing Kit (Applied Biosystems, Carlsbad, CA, USA). Mutation screening was performed on a 3730 DNA Analyzer (Applied Biosystems).
The WNT10A gene spans approximately 13.4 kb on human chromosome 2q35, contains four exons, and encodes a messenger RNA of 2.4 kb. The protein consists of two domains: a signal peptide (amino acid position 1-35) and the Wnt domain (amino acid position 60-417). Five previously undescribed nucleotide substitutions within the coding region of the WNT10A gene were identified as causing different manifestations of ED in 12 patients from 11 unrelated families of German and Turkish origin (for pedigree details, see Figure 1). Three of these (c.27G>A [p.W9X], c.321C>A [p.C107X], and c.1128C>A [p.C376X]) predicted premature termination of translation and were found in index patients in either homozygous or compound heterozygous state together with a missense mutation. Two nucleotide substitutions (c.383G>A [p.R128Q] and c.682T>A [p.F228I]) were shown to affect amino acids from evolutionary highly conserved protein regions (NCBI Protein Database) and were identified as disease causing, because they were found to be associated recurrently with a severe ED phenotype when occurring in homozygous or compound heterozygous state together with a nonsense mutation (Table 1; Figures S1 and S2). Another previously undescribed nucleotide substitution (c.493G>A [p.G165R]) (refSNP ID: ss131007345; dbSNP database) probably represents a rare polymorphism, given that it was found in trans position with the considered pathogenetic mutation c.682T>A in two completely unaffected individuals of family 123 (II:2 and II:3). In addition, we found the already known SNP c.1087A>C (p.N363H) (refSNP ID: rs34972707; dbSNP Database) in heterozygous state in one unaffected relative of family 071 (III:3).
Figure 1.
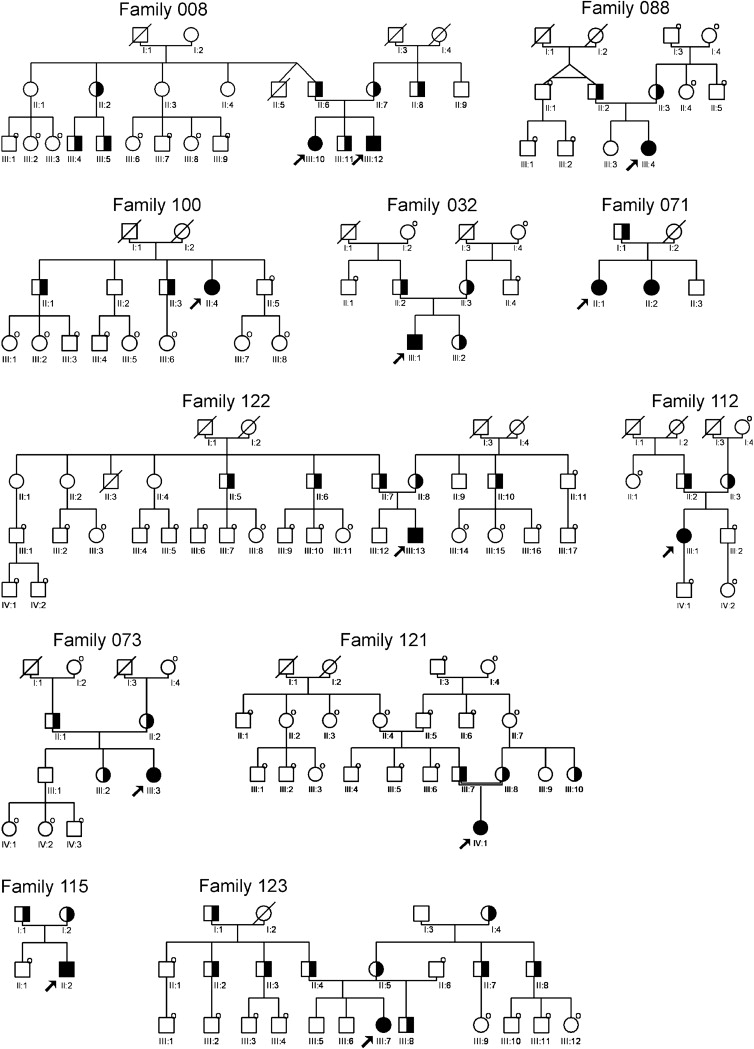
Pedigrees of 12 Patients with WNT10A-Related ED
Filled symbols indicate homozygous or compound heterozygous mutations; half-filled symbols indicate heterozygous mutations in individuals. “O” at the upper right part of a symbol indicates that the individual was not tested.
Table 1.
Clinical Symptoms and Mutational Results in 12 Patients with WNT10A Mutations
| Patient | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 |
| Family ID | 008 | 008 | 088 | 100 | 032 | 071 | 122 | 112 | 073 | 121 | 115 | 123 |
| Pedigree position | III:10 | III:12 | III:4 | II:4 | III:1 | II:1 | III:13 | III:1 | III:3 | IV:1 | II:2 | III:7 |
| Clinical Symptoms | ||||||||||||
| Eruption of first teeth at age | 5mo. | 5mo. | 6mo. | n.d. | 7mo. | n.d. | n.d. | 5mo. | 4mo. | 9mo. | 10mo. | n.d. |
| Primary teeth abnormal | − | − | + | + | − | + | + | − | − | + | + | − |
| Permanent teeth missing | + | + | + | + | + | + | + | + | + | n.d. | + | + |
| Sparse scalp hair | − | − | + | + | − | + | − | + | + | + | + | − |
| Sparse body hair | + | + | − | + | + | + | − | + | − | n.d. | n.d. | n.d. |
| Sparse eyebrows | + | + | + | − | − | + | − | + | − | n.d. | + | + |
| Short eyelashes | − | − | + | − | − | + | − | − | − | n.d. | + | + |
| Hypohidrosis | − | − | − | − | + | + | − | − | − | n.d. | − | + |
| Hyperhidrosis | − | − | − | + | + | − | + | − | − | n.d. | − | − |
| Dry skin | − | − | + | + | − | + | − | + | + | n.d. | + | + |
| Soft, thin skin | − | − | − | + | + | − | − | + | n.d. | n.d. | n.d. | |
| Palmar hyperkeratosis | − | − | (+) | + | − | (+) | − | − | − | n.d. | − | (+) |
| Hyperkeratosis on dorsal hands | − | − | − | + | − | + | − | − | − | n.d. | n.d. | − |
| Plantar hyperkeratosis | − | − | + | + | + | + | − | − | − | n.d. | − | − |
| Palmoplantar sudation | − | − | − | + | − | + | − | − | − | n.d. | + | − |
| Dyshidrotic blistering | − | − | n.d. | + | − | n.d. | − | − | − | n.d. | + | − |
| Dystrophic fingernails | − | − | + | + | + | (+) | − | − | + | + | − | − |
| Dystrophic toenails | − | − | + | + | + | + | + | − | + | + | + | − |
| Photophobia | − | − | − | − | − | + | − | + | + | n.d. | − | − |
| Lid cysts | − | − | − | + | − | − | − | − | − | n.d. | − | − |
| Mutational Results | ||||||||||||
| Nucleotide substitution (first allele)a | c.321C>A | c.321C>A | c.321C>A | c.321C>A | c.321C>A | c.1128C>A | c.321C>A | c.321C>A | c.682T>A | c.27G>A | c.321C>A | c.682T>A |
| Nucleotide substitution (second allele)a | c.383G>A | c.383G>A | c.321C>A | c.321C>A | c.321C>A | c.1128C>A | c.682T>A | c.682T>A | c.682T>A | c.27G>A | c.321C>A | c.682T>A |
| Amino acid substitutions | p.C107X p.R128Q | p.C107X p.R128Q | p.C107X p.C107X | p.C107X p.C107X | p.C107X p.C107X | p.C376X p.C376X | p.C107X p.F228I | p.C107X p.F228I | p.F228I p.F228I | p.W9X p.W9X | p.C107X p.C107X | p.F228I p.F228I |
+: present; −: not present; (+): mild; n.d.: no data.
According to GenBank NM_025216.2.
At least 200 control chromosomes of healthy, anonymized individuals from our institute's registry (Münster controls) were tested for each identified previously undescribed nucleotide substitution by restriction enzyme analysis, high-resolution melting (HRM) analysis, or sequencing (Table S2). For mutation restriction analysis, the PCR products were digested with the specific restriction endonuclease (New England Biolabs, Frankfurt, Germany) at the recommended conditions. For HRM analysis, primers for amplifying an approximately 200 bp region surrounding the different mutations were designed (Table S3). The PCR and HRM analyses were performed on a LightCycler 480 (Roche Diagnostics, Penzberg, Germany) with the LightCycler 480 High Resolution Melting Master Kit (Roche Diagnostics), in accordance with the manufacture's instructions. DNA fragments with altered mobility were sequenced. For each of the three nonsense mutations and the missense mutation c.383G>A, no carriers were found within the control group. The missense mutation c.682T>A, which is considered disease causing by phenotypic consequences, was found in two of 396 control chromosomes (∼0.5%). The considered SNP c.493G>A (refSNP ID: ss131007345; dbSNP Database) was found in 0.7% of the control chromosomes.
WNT10A belongs to a highly conserved gene family encoding secreted signaling molecules. In general, Wnt proteins regulate cell-to-cell interactions and are implicated in multiple developmental processes during embryogenesis, as well as in homeostasis in adult tissues by inhibiting the β-catenin degradation complex and allowing interaction with nuclear transcription factors LEF/TCF and regulation of target gene expression17,18 (canonical Wnt signaling pathway). In mouse and chicken embryos, Wnt10a was shown to be upregulated in skin,19 in placodes at the onset of hair follicle morphogenesis, and in the oral epithelium at the first steps of tooth morphogenesis20–22 (mouse) and was identified to be involved in the formation of the apical ectodermal ridge during limb development23 (chicken).
The phenotypic expression in our patients with WNT10A mutations shows a high degree of variability (Table 1 and Figure S3); however, it is so far without recognizable phenotype-genotype correlation. Hypohidrosis (two patients) and hyperhidrosis (two patients) were reported, and a change in sudation pattern from increased to decreased could be observed in another patient with puberty. Differences in sweat secretion in skin, palms, and soles was described, including normal or decreased ability to sweat in general but increased palmoplantar sudation. Increased sweating only at night and discomfort at temperatures above 25°C were reported in one patient. Nevertheless, most patients reported dry skin, which is most likely caused by a decreased number of sebaceous glands, as reported by Burket et al.9 and also confirmed by microscopic evaluation of a skin biopsy in one of our patients. Nails may be normal, flat, convex, thin, soft, splitting, slow growing, minimized, or apparently absent at birth but starting to grow later in childhood, and they may be differently affected regarding finger- and toenails in the same patient. Palmoplantar skin showed dyshidrotic blistering and peeling, hyperkeratotic plaques (which were also reported on the dorsal fingers), or severe hyperkeratosis with painful lacerations, usually milder on palms than on soles, in six patients but was normal in five patients. The tongue was not studied in detail in our patients. However, in photographs, the filiform and fungiform papillae appear to be reduced in at least two cases (Figure S4).
Overall, WNT10A mutations were found to cause a broad continuum of phenotypes, ranging from apparently isolated severe oligodontia with only very mild other ED symptoms (patients 1 and 2), to several “variants” of OODD with or without facial skin erythema, palmoplantar hyperkeratosis, or nail dysplasia, to SSPS in one patient (patient 4), who also showed the typical finding of numerous cysts along the upper and lower eyelid margins. Her brother was reported to have similar symptoms but a milder skin manifestation.
Photophobia, reported by Zirbel et al.4 as an unusual symptom in one OODD patient, was also found in three of our patients. It might be caused by primary disturbed retinal pigment epithelium (RPE) morphogenesis or failure in photoreceptor protection in the degenerating retina. There are some reasons to consider Wnt/β-catenin signaling as an important pathway in RPE morphogenesis via direct target genes such as Mitf, a key regulator of the formation of pigment cells that transactivates promoters of the tyrosinase gene family controlling pigment synthesis. It is interesting to note that in the human MITF gene (MIM 156845), mutations produce Waardenburg syndrome (MIM 193510), which is characterized by hypoplasia of iris stroma and hypopigmented ocular fundus, inter alia. Decreased pigment synthesis might also be the cause of the pale skin that was described as a translucent appearance in patient 9 and the premature graying of hair at age 14 in patient 1. A further indication for the role of Wnt/β-catenin signaling in RPE morphogenesis is the reverse observation that some variants of familial adenomatous polyposis caused by APC gene (MIM 611731) mutations show congenital nodular hyperpigmented RPE lesions, indicating increased availability of β-catenin for WNT signaling, given that the APC protein is a component of the β-catenin APC-GSK3β-Axin destruction complex24,25. Another explanation for the photophobia, however, may come from the in vivo experiments in mice performed by Yi et al.,26 who demonstrated Wnt10a expression during cone photoreceptor degeneration induced by oxidative stress, indicating prosurvival protection activity, a function that should get lost when mutated. However, none of the patients was examined by an ophthalmologist for iris pigmentation or had an electroretinogram for cone dystrophy. Thus, we suggest further studies of the eyes in patients with WNT10A mutations in order to possibly discover supportive data for one of these hypotheses.
Eccrine poromas, basal cell carcinomas, and squamous cell carcinoma were described in SSPS.10–12 In our patient with SSPS (family 100-II:4), a porocarcinoma at the left heel was also diagnosed. Because we identified SSPS as part of the phenotypic spectrum, follow-up studies are needed to find out whether, like in SSPS, other phenotypes with WNT10A mutations also have an increased skin tumor risk.
The most consistent symptom in all cases, and, in our opinion, the most specific diagnostic criterion, was the severe oligodontia concerning the permanent teeth (Table 2 and Figure S4), with apparently normal (six patients) or comparatively less disturbed deciduous dentition with conical frontal teeth or agenesis of the upper lateral or central incisors (patients 3, 4, 6, 7, 10, and 11). These findings well reflect the importance of Wnt/β-catenin signaling in tooth formation, especially the formation of succedaneous teeth.27
Table 2.
Tooth Findings in 11 Patients with WNT10A Mutations
| Patient | Family ID | Pedigree Position | Age (yrs) |
Upper Right |
Upper Left |
||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 18 | 17 | 16 | 15 | 14 | 13 | 12 | 11 | 21 | 22 | 23 | 24 | 25 | 26 | 27 | 28 | ||||
| 1 | 008 | III:10 | 15 | ■ | ■ | + | ■ | [■] | + | ■ | + | + | ■ | [■] | [■] | [■] | + | ■ | ■ |
| 2 | 008 | III:12 | 10 | ■ | ■ | + | [g] | + | [■] | ▾ | ▾ | ▾ | ▾ | [■] | + | [g] | ■ | ■ | ■ |
| 3 | 088 | III:4 | 11 | ■ | ■ | ■ | [■] | [■] | [■] | ■ | [■] | [■] | ■ | [■] | [■] | [■] | ■ | ■ | ■ |
| 4 | 100 | II:4 | 25 | ■ | ■ | ■ | [■] | [■] | ■ | ■ | ■ | ■ | ■ | [■] | [■] | + | ■ | ■ | ■ |
| 5 | 032 | III:1 | 10 | ■ | ■ | + | [g] | ■ | [■] | ■ | ▾ | ▾ | ■ | [■] | ■ | [g] | + | ■ | ■ |
| 6 | 071 | II:1 | 28 | Implants | |||||||||||||||
| 7 | 122 | III:13 | 12 | ■ | ■ | + | [■] | [■] | [■] | ■ | + | + | ■ | [■] | [■] | [■] | + | ■ | ■ |
| 8 | 112 | III:1 | 8 | ■ | + | ■ | [g] | [■] | [g] | [■] | + | + | [■] | [g] | [■] | [■] | + | ■ | ■ |
| 9 | 073 | III:3 | 18 | ■ | + | + | [■] | [■] | ■ | ▾ | + | + | ▾ | ■ | [■] | [■] | + | + | ■ |
| 11 | 115 | II:2 | 8 | ■ | ■ | ■ | [■] | [■] | [■] | [■] | [■] | [■] | [■] | [■] | [■] | [■] | ■ | ■ | ■ |
| 12 | 123 | III:7 | 12 | ■ | + | + | [■] | + | ■ | ▾ | + | + | [■] | [■] | [■] | [■] | + | + | ■ |
| 12 | 123 | III:7 | 12 | ■ | + | + | [■] | + | + | + | [■] | [■] | + | [■] | [■] | [■] | + | + | g |
| 11 | 115 | II:2 | 8 | ■ | ■ | ■ | [■] | [■] | [■] | [■] | [■] | [■] | [■] | [■] | [■] | [■] | ■ | ■ | ■ |
| 9 | 073 | III:3 | 18 | ■ | ■ | + | [■] | + | + | + | ■ | ■ | + | + | + | + | + | + | ■ |
| 8 | 112 | III:1 | 8 | ■ | ■ | + | [■] | [■] | [g] | [■] | [■] | [■] | [■] | [g] | [■] | [■] | + | ■ | ■ |
| 7 | 122 | III:13 | 12 | ■ | ■ | + | [■] | + | + | + | + | + | + | + | [■] | [■] | + | ■ | ■ |
| 6 | 071 | II:1 | 28 | + | Implants | + | |||||||||||||
| 5 | 032 | III:1 | 10 | ■ | ■ | + | [■] | ■ | [■] | [■] | [■] | [■] | [■] | [■] | ■ | [g] | + | g | ■ |
| 4 | 100 | II:4 | 25 | ■ | ■ | ■ | + | [■] | [■] | [■] | [■] | [■] | [■] | [■] | [■] | + | ■ | ■ | ■ |
| 3 | 088 | III:4 | 11 | ■ | ■ | ■ | [■] | + | [■] | [■] | [■] | [■] | [■] | [■] | + | [■] | ■ | ■ | ■ |
| 2 | 008 | III:12 | 10 | ■ | + | ■ | [■] | + | [■] | [■] | [■] | [■] | [■] | + | + | [■] | ■ | + | ■ |
| 1 | 008 | III:10 | 15 | ■ | ■ | + | [■] | + | [■] | [■] | [■] | [■] | [■] | [■] | + | + | + | ■ | ■ |
| Family ID | Pedigree Position | Age (yrs) |
48 |
47 |
46 |
45 |
44 |
43 |
42 |
41 |
31 |
32 |
33 |
34 |
35 |
36 |
37 |
38 |
|
| Lower Right | Lower Left | ||||||||||||||||||
“Age” indicates age of patient when panoramic radiograph was taken.
18–11, 12–28, 48–41, and 31–38: tooth position according to the World Dental Federation [FDI] notation (ISO-3950 notation). In this two-digit numbering, the first number represents a tooth's quadrant (1, upper right; 2, upper left; 3, lower left; 4, lower right) and the second number represents the number of the tooth from the midline of the face (1, central incisor; 2, lateral incisor; 3, canine; 4, first premolar; 5, second premolar; 6, first molar; 7, second molar; 8, wisdom tooth).
■: absent permanent tooth and tooth germ; [■]: deciduous tooth with absent permanent tooth germ; [g]: deciduous tooth with underlying permanent tooth germ; g: permanent tooth germ;▾: conical tooth; +: permanent tooth present.
A blank entry indicates that there was no information available.
In SSPS, genetic heterogeneity was suggested on the basis of observed both autosomal-recessive and autosomal-dominant modes of inheritance in different families.8,13 In our opinion, however, mild symptoms, even if multifocal, reflect heterozygous manifestations rather than genetic heterogeneity. In fact, 21 out of 39 heterozygous mutated relatives (53.8%) had minor disease-associated symptoms, such as abnormal shape or agenesis of one or two (rarely up to six) permanent teeth, usually upper lateral incisors, upper canines, or lower lateral or central incisors, nail dystrophy, dry skin, palmoplantar hyperkeratosis, sparce scalp hair, sparse eyelashes, or sparse eyebrows (Table 3). The total frequency of heterozygotes with any phenotype manifestation differs nonsignificantly between females (8/14; 57%) and males (13/25; 52%). In all relatives with minor symptoms, the entire WNT10A gene was sequenced, and in completely unaffected relatives, a targeting mutational analysis was performed. All individuals with minor symptoms were heterozygous for one of the mutations described above, and none of the tested relatives with two wild-type alleles had any symptoms. Phenotype manifestation in heterozygotes was not described in the families reported by Adaimy et al.1 However, because the mutational spectrum in our patients differs from the one mutation found by Adaimy et al.,1 a mutation-related phenotype in heterozygotes can not be excluded.
Table 3.
Clinical Manifestation in Heterozygotes
| Substitution | Sex | Family ID | Pedigree Position |
Affected Structures |
|||
|---|---|---|---|---|---|---|---|
| Teeth | Nails | Skin | Hair | ||||
| p.W6X | Male | 121 | III:7 | - | - | - | S |
| p.W6X | Female | 121 | III:8 | - | Y | - | L |
| p.W6X | Female | 121 | III:10 | A | - | - | - |
| p.C107X | Male | 008 | II:6 | - | Y | H,D | - |
| p.C107X | Male | 008 | III:4 | A | Y | K,P | - |
| p.C107X | Male | 008 | III:5 | - | - | - | - |
| p.C107X | Male | 008 | III:11 | - | - | - | - |
| p.C107X | Male | 088 | II:2 | A | - | - | - |
| p.C107X | Male | 100 | II:1 | B2 | - | - | - |
| p.C107X | Male | 100 | II:3 | A | - | - | - |
| p.C107X | Male | 032 | II:2 | G | - | - | - |
| p.C107X | Male | 122 | II:5 | - | - | - | - |
| p.C107X | Male | 122 | II:6 | C | - | - | - |
| p.C107X | Male | 122 | II:7 | A | - | - | - |
| p.C107X | Male | 115 | I:1 | - | - | - | - |
| p.C107X | Female | 008 | II:2 | - | - | K | - |
| p.C107X | Female | 088 | II:3 | - | Y | - | - |
| p.C107X | Female | 032 | II:3 | - | - | - | - |
| p.C107X | Female | 032 | III:2 | - | - | - | - |
| p.C107X | Female | 112 | II:3 | - | - | - | - |
| p.C107X | Female | 115 | I:2 | - | - | - | - |
| p.R128Q | Male | 008 | II:8 | - | - | - | - |
| p.R128Q | Female | 008 | II:7 | - | - | - | E |
| p.F228I | Male | 122 | II:10 | A | - | - | - |
| p.F228I | Male | 112 | II:2 | - | - | - | - |
| p.F228I | Male | 073 | II:1 | - | - | K | - |
| p.F228I | Male | 123 | I:1 | - | - | - | - |
| p.F228I | Male | 123 | II:2 | - | - | - | - |
| p.F228I | Male | 123 | II:3 | - | - | - | - |
| p.F228I | Male | 123 | II:4 | B1 | - | - | - |
| p.F228I | Male | 123 | II:7 | M | - | - | - |
| p.F228I | Male | 123 | II:8 | - | - | - | - |
| p.F228I | Male | 123 | III:8 | - | - | - | - |
| p.F228I | Female | 122 | II:8 | - | - | - | - |
| p.F228I | Female | 073 | II:2 | - | Y | - | S |
| p.F228I | Female | 073 | III:2 | - | - | K | - |
| p.F228I | Female | 123 | I:4 | M | - | - | - |
| p.F228I | Female | 123 | II:5 | - | - | - | - |
| p.C376X | Male | 071 | I:1 | - | - | - | - |
The use of a dash indicates that no abnormalities are present.
Teeth: A, markedly small, conical, sharp, or missing upper lateral permanent incisors; G, all permanent teeth are markedly small; C, agenesis of upper permanent canines; B1, agenesis of lower right central incisor; B2, agenesis of both lower central incisors; M, agenesis of 2 to 6 permanent teeth except third molars without further information available.
Nails: Y, nail dystrophy.
Skin: H, hypohidrosis; D, dry skin; K, hyperkeratosis of palms and/or soles; P, laceration of finger tips.
Hair: S, sparse scalp hair; L, sparse eyelashes; E, sparse eyebrows.
Phenotypic manifestation in heterozygotes of an autosomal-recessive disorder is known from other conditions, such as some metabolic diseases (e.g., congenital adrenal hyperplasia [MIM 201910]28 and alpha-1-antitrypsin deficiency [MIM 107400])29,30 or bone dysplasias (e.g., otospondylomegaepiphyseal dysplasia [OSMED] [MIM 215150]/Stickler syndrome type III [MIM 184840]).31 The phenomenon was also described recently in EDAR [MIM 604095]-related hypohidrotic ED [MIM 224900], in which “some presumably recessive mutations may show phenotypic expression in carriers.”32 Because of the high frequency of clinical manifestation in heterozygotes seen in our study, this possibility should also be conveyed during counseling of individuals or families with WNT10A-related ED.
Finally, another surprising result, which may provide new insights into pattern formation in humans, came to our attention: Whereas in homozygous or compound heterozygous patients, no phenotypic differences between males and females were evident, we found a tendency of a sex-biased manifestation pattern in heterozygous individuals. Thus, heterozygous males predominantly showed agenesis or hypoplasia of permanent upper lateral incisors (rarely canines or lower central incisors) and less nail or hair manifestation than females. This observation seems to be supported by family histories of additional male relatives with tooth anomalies (family 088 [II:1]; family 100 [I:2]; family 121 [III:5 and III:6]) and females with nail and hair anomalies (family 071 [I:1]; family 073 [I:4]) and by the literature.8,13 Thus, data were analyzed by Fisher's exact test with SPSS software version 17.0 for Windows (SPSS, Chicago, IL, USA). Accordingly, in heterozygous individuals with phenotypic manifestation, teeth of males were significant more frequently affected (10/13; 77%) than those of females (2/8; 25%) (p = 0.029) (Figure 2). Further studies are necessary for the verification of any sex-biased phenotypic expression in heterozygotes that would indicate that haploinsufficiency in males and females has different consequences as a result of tissue-specific sex-dependent differences in WNT10A activity in the sense of a gender-dependent micromodification of the phenotype. Increasing evidence of the important functions of fine-tuning of signaling in tooth development comes from the observation that premolar-like teeth in half of the animals are present in mouse lines overexpressing ectodysplasin.33 Moreover, it was very recently shown that the number of teeth, as well as the molar cusp pattern, can be modified by modulation of several different signal pathways34 and that the stimulation of the Wnt pathway in the oral epithelium leads to abundant de novo tooth formation in transgenic mice.27 These findings support the hypothesis that the diversity of tooth types and numbers may have resulted from modifications of gene expression within evolutionary conserved signal pathways, and one may also suspect that micromodulation of gene expression is also responsible for many kinds of sexual dimorphism. Thus, a higher concentration of an mRNA in one sex, observed when the same tissue is compared between the sexes, was indeed designated as a major contributor to gender-specific differences in gene expression.35,36 Conversely, it can be assumed that the phenotypic effect of loss-of-function mutations of certain genes with different gender-specific expression levels may vary and would explain our findings of tooth abnormalities occurring predominantly in heterozygous males and affecting mainly the upper lateral incisors.
Figure 2.
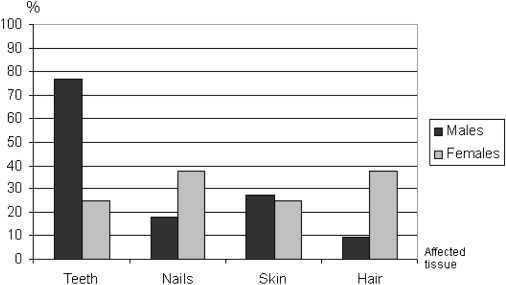
Distribution of Involved Ectodermal Structures in Heterozygous Mutated Males and Females with Phenotypic Manifestation
Sexual dimorphism for human tooth size has been reported in the general population,37,38 but although the mechanism has become more obvious,34 the contributing genes are still unknown. Our data may suggest WNT10A as a good candidate for further research in this field.
Agenesis or hypoplasia of permanent upper lateral incisors is frequent in the population. Thus, Witkop39 stated that the prevalence in the U.S. population is approximately 1:67 (1.5%). In at least part of the cases, an autosomal-dominant trait with reduced penetrance and variable expression (selective tooth agenesis type 4; STHAG4 [MIM 150400]) was suggested, and in the supposed homozygous state by parental consanguinity, adontia of permanent teeth (MIM 206780) but unaffected primary dentition was observed.39–44 Altogether, this pattern of tooth anomalies is comparable to that which we found in the patients and their relatives reported here. Thus, testing for heterozygous WNT10A mutations in STHAG4 might be worthwhile.
The frequency of WNT10A-related ED in our cohort was higher than expected from the rarely reported cases in the literature. Although in our clientele of unselected patients from 123 families with ED or isolated severe oligodontia, mutational proven Christ-Siemens-Touraine syndrome (CST [MIM 305100]) was the by far most common diagnosis (63/123, approximately 51%), homozygous or compound heterozygous WNT10A mutations were identified in patients from 11 of these 123 families (approximately 9%) representing 25% (11/44) of the genotyped subgroup of index patients remaining after exclusion of CST and other easily recognizable types of ED.
In conclusion, WNT10A mutation analysis might become an important diagnostic test in many types of ED with severe oligodontia concerning the permanent teeth, with or without sweating problems, palmoplantar hyperkeratosis, or nail and hair anomalies, that are now difficult to classify, as well as in isolated oligodontia.
Acknowledgments
We are grateful for the generosity of the individuals with ectodermal dysplasia and their families for their participation in this work, as well as for the German support group Selbsthilfegruppe Ektodermale Dysplasie e.V. We also would like to thank dentists and orthodontists Thomas Henning, Androniki-M. Matzke, Carola Zeitz, Johannes Röhling, Rainer Schmelzeisen, Ingrid Rudzki-Janson, Richard Meissen, Rainer Ehring, Carolin Ehring, Gudbjartur Holm, Günther Kramer, and Anette Thieme for providing us with panoramic radiographs and dental records. The technical assistance of Ursula Antkoviak, Silvia Fleige-Menzen, Sabine von Rüden, and Britta Hitschfeld is gratefully acknowledged.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
dbSNP Database, http://www.ncbi.nlm.nih.gov/SNP/
GenBank, http://www.ncbi.nlm.nih.gov/sites/entrez?db=nucleotide
MFOLD, http://mobyle.pasteur.fr/cgi-bin/MobylePortal/portal.py?form=mfold
NCBI Protein Database, http://www.ncbi.nlm.nih.gov/protein/
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/
References
- 1.Adaimy L., Chouery E., Mégarbané H., Mroueh S., Delague V., Nicolas E., Belguith H., de Mazancourt P., Mégarbané A. Mutation in WNT10A is associated with an autosomal recessive ectodermal dysplasia: the odonto-onycho-dermal dysplasia. Am. J. Hum. Genet. 2007;81:821–828. doi: 10.1086/520064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fadhil M., Ghabra T.A., Deeb M., Der Kaloustian V.M. Odontoonychodermal dysplasia: a previously apparently undescribed ectodermal dysplasia. Am. J. Med. Genet. 1983;14:335–346. doi: 10.1002/ajmg.1320140213. [DOI] [PubMed] [Google Scholar]
- 3.Arnold W.P., Merkx M.A.W., Steijlen P.M. Variant of odontoonychodermal dysplasia? Am. J. Med. Genet. 1995;59:242–244. doi: 10.1002/ajmg.1320590224. [DOI] [PubMed] [Google Scholar]
- 4.Zirbel G.M., Ruttum M.S., Post A.C., Esterly N.B. Odonto-onycho-dermal dysplasia. Brit J Derm. 1995;133:797–800. doi: 10.1111/j.1365-2133.1995.tb02761.x. [DOI] [PubMed] [Google Scholar]
- 5.Mégarbané H., Haddad M., Delague V., Renoux J., Boehm N., Mégarbané A. Further delineation of the odonto-onycho-dermal dysplasia syndrome. Am. J. Med. Genet. 2004;129A:193–197. doi: 10.1002/ajmg.a.30188. [DOI] [PubMed] [Google Scholar]
- 6.Fried K. Autosomal recessive hydrotic ectodermal dysplasia. J. Med. Genet. 1977;14:137–139. doi: 10.1136/jmg.14.2.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mégarbané A., Noujeim Z., Fabre M., Der Kaloustian V.M. New form of hidrotic ectodermal dysplasia in a Lebanese family. Am. J. Med. Genet. 1998;75:196–199. [PubMed] [Google Scholar]
- 8.Schöpf E., Schulz H.J., Passarge E. Syndrome of cystic eyelids, palmo-plantar keratosis, hypodontia and hypotrichosis as a possible autosomal recessive trait. Birth Defects Orig. Artic. Ser. 1971;VII:219–221. [PubMed] [Google Scholar]
- 9.Burket J.M., Burket B.J., Burket D.A. Eyelid cysts, hypodontia, and hypotrichosis. J. Am. Acad. Dermatol. 1984;10:922–925. doi: 10.1016/s0190-9622(84)80448-x. [DOI] [PubMed] [Google Scholar]
- 10.Font R.L., Stone M.S., Schanzer M.C., Lewis R.A. Apocrine hidrocystomas of the lids, hypodontia, palmar-plantar hyperkeratosis, and onychodystrophy: a new variant of ectodermal dysplasia. Arch. Ophthal. 1986;104:1811–1813. doi: 10.1001/archopht.1986.01050240085045. [DOI] [PubMed] [Google Scholar]
- 11.Nordin H., Mansson T., Svensson A. Familial occurrence of eccrine tumours in a family with ectodermal dysplasia. Acta Derm. Venereol. 1988;68:523–530. [PubMed] [Google Scholar]
- 12.Perret C. Schöpf syndrome. Brit J Derm. 1989;120:131–132. [Google Scholar]
- 13.Küster W., Hammerstein W. Das Schöpf-Syndrom. Hautarzt. 1992;43:763–766. [PubMed] [Google Scholar]
- 14.Monk B.E., Pieris S., Soni V. Schöpf-Schulz-Passarge syndrome. Brit J Derm. 1992;127:33–35. doi: 10.1111/j.1365-2133.1992.tb14822.x. [DOI] [PubMed] [Google Scholar]
- 15.Craigen W.J., Levy M.L., Lewis R.A. Schöpf-Schulz-Passarge syndrome with an unusual pattern of inheritance. Am. J. Med. Genet. 1997;71:186–188. doi: 10.1002/(sici)1096-8628(19970808)71:2<186::aid-ajmg12>3.0.co;2-a. [DOI] [PubMed] [Google Scholar]
- 16.Castori M., Ruggieri S., Giannetti L., Annessi G., Zambruno G. Schöpf-Schulz-Passarge syndrome: further delineation of the phenotype and genetic considerations. Acta Derm. Venereol. 2008;88:607–612. doi: 10.2340/00015555-0547. [DOI] [PubMed] [Google Scholar]
- 17.Logan C.Y., Nusse R. The Wnt signaling pathway in development and disease. Annu. Rev. Cell Dev. Biol. 2004;20:781–810. doi: 10.1146/annurev.cellbio.20.010403.113126. [DOI] [PubMed] [Google Scholar]
- 18.Gordon M.D., Nusse R. Wnt signaling: multiple pathways, multiple receptors, and multiple transcription factors. J. Biol. Chem. 2006;281:22429–22433. doi: 10.1074/jbc.R600015200. [DOI] [PubMed] [Google Scholar]
- 19.Wang J., Shackleford G.M. Murine Wnt10a and Wnt10b: cloning and expression in developing limbs, face and skin of embryos and in adults. Oncogene. 1996;13:1537–1544. [PubMed] [Google Scholar]
- 20.Reddy S., Andl T., Bagasra A., Lu M.M., Epstein D.J., Morrisey E.E., Millar S.E. Characterization of Wnt gene expression in developing and postnatal hair follicles and identification of Wnt5a as a target of Sonic hedgehog in hair follicles morphogenesis. Mech. Dev. 2001;107:69–82. doi: 10.1016/s0925-4773(01)00452-x. [DOI] [PubMed] [Google Scholar]
- 21.Dassule H.R., McMahon A.P. Analysis of epithelial-mesenchymal interactions in the initial morphogenesis of the mammalian tooth. Dev. Biol. 1998;202:215–227. doi: 10.1006/dbio.1998.8992. [DOI] [PubMed] [Google Scholar]
- 22.Miletich I., Sharpe P.T. Normal and abnormal dental development. Hum. Mol. Genet. 2003;12 doi: 10.1093/hmg/ddg085. Spec No 1:R69–R73. [DOI] [PubMed] [Google Scholar]
- 23.Narita T., Sasaoka S., Udagawa K., Ohyama T., Wada N., Nishimatsu S., Takada S., Nohno T. Wnt10a is involved in AER formation during chick limb development. Dev. Dyn. 2005;233:282–287. doi: 10.1002/dvdy.20321. [DOI] [PubMed] [Google Scholar]
- 24.Tiret A., Parc C. Fundus lesions of adenomatous polyposis. Curr. Opin. Ophthalmol. 1999;10:168–172. doi: 10.1097/00055735-199906000-00003. [DOI] [PubMed] [Google Scholar]
- 25.Burke J.M. Epithelial phenotype and the RPE: is the answer blowing in the Wnt? Prog. Retin. Eye Res. 2008;27:579–595. doi: 10.1016/j.preteyeres.2008.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yi H., Nakamura R.E., Mohamed O., Dufort D., Hackam A.S. Characterization of Wnt signaling during photoreceptor degeneration. Invest. Ophthalmol. Vis. Sci. 2007;48:5733–5741. doi: 10.1167/iovs.07-0097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Järvinen E., Salazar-Ciudad I., Birchmeier W., Taketo M.M., Jernvall J., Thesleff I. Continuous tooth generation in mouse is induced by activated epithelial Wnt/beta-catenin signaling. Proc. Natl. Acad. Sci. USA. 2006;103:18627–18632. doi: 10.1073/pnas.0607289103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Blanché H., Vexiau P., Clauin S., Le Gall I., Fiet J., Mornet E., Dausset J., Bellanné-Chantelot C. Exhaustive screening of the 21-hydroxylase gene in a population of hyperandrogenic women. Hum. Genet. 1997;101:56–60. doi: 10.1007/s004390050586. [DOI] [PubMed] [Google Scholar]
- 29.Hersh C.P., Dahl M., Ly N.P., Berkey C.S., Nordestgaard B.G., Silverman E.K. Chronic obstructive pulmonary disease in alpha1-antitrypsin PI MZ heterozygotes: a meta-analysis. Thorax. 2004;59:843–849. doi: 10.1136/thx.2004.022541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fischer H.P., Ortiz-Pallardo M.E., Ko Y., Esch C., Zhou H. Chronic liver disease in heterozygous alpha1-antitrypsin deficiency PiZ. J. Hepatol. 2000;33:883–892. doi: 10.1016/s0168-8278(00)80119-1. [DOI] [PubMed] [Google Scholar]
- 31.Vikkula M., Mariman E.C.M., Lui V.C.H., Zhidkova N.I., Tiller G.E., Goldring M.B., van Beersum S.E.C., de Waal Malefijt M.C., van den Hoogen F.H.J., Ropers H.H. Autosomal dominant and recessive osteochondrodysplasias associated with the COL11A2 locus. Cell. 1995;80:431–437. doi: 10.1016/0092-8674(95)90493-x. [DOI] [PubMed] [Google Scholar]
- 32.Van der Hout A.H., Oudesluijs G.G., Venema A., Verheij J.B.G.M., Mol B.G.J., Rump P., Brunner H.G., Vos Y.J., van Essen A.J. Mutation screening of the ectodysplasin-A receptor gene EDAR in hypohidrotic ectodermal dysplasia. Eur. J. Hum. Genet. 2008;16:673–679. doi: 10.1038/sj.ejhg.5202012. [DOI] [PubMed] [Google Scholar]
- 33.Kangas A.T., Evans A.R., Thesleff I., Jernvall I. Nonindependence of mammalian dental characters. Nature. 2004;432:211–214. doi: 10.1038/nature02927. [DOI] [PubMed] [Google Scholar]
- 34.Tummers M., Thesleff I. The importance of signal pathway modulation in all aspects of tooth development. J. Exp. Zoolog. B Mol. Dev. Evol. 2009;312B:309–319. doi: 10.1002/jez.b.21280. [DOI] [PubMed] [Google Scholar]
- 35.Ellegren H., Parsch J. The evolution of sex-biased genes and sex-biased gene expression. Nat. Rev. Genet. 2007;8:689–698. doi: 10.1038/nrg2167. [DOI] [PubMed] [Google Scholar]
- 36.Yang X., Schadt E.E., Wang S., Wang H., Arnold A.P., Ingram-Drake L., Drake T.A., Lusis A.J. Tissue-specific expression and regulation of sexually dimorphic genes in mice. Genome Res. 2006;16:995–1004. doi: 10.1101/gr.5217506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Acharya A.B., Mainali S. Sex discrimination potential of buccolingual and mesiodistal tooth dimensions. Forensic Sci. Int. 2008;173:47–56. doi: 10.1111/j.1556-4029.2008.00778.x. [DOI] [PubMed] [Google Scholar]
- 38.Guatelli-Steinberg D., Sciulli P.W., Betsinger T.K. Dental crown size and sex hormone concentrations: another look at the development of sexual dimorphism. Am. J. Phys. Anthropol. 2008;137:324–333. doi: 10.1002/ajpa.20878. [DOI] [PubMed] [Google Scholar]
- 39.Witkop C.J., Jr. Agenesis of succedaneous teeth: an expression of the homozygous state of the gene for the pegged or missing maxillary lateral incisor trait. Am. J. Med. Genet. 1987;26:431–436. doi: 10.1002/ajmg.1320260222. [DOI] [PubMed] [Google Scholar]
- 40.Woolf C.M. Missing maxillary lateral incisors: a genetic study. Am. J. Hum. Genet. 1971;23:289–296. [PMC free article] [PubMed] [Google Scholar]
- 41.Hoo J.J. Anodontia of permanent teeth (OMIM #206780) and pegged/missing maxillary lateral incisors (OMIM #150400) in the same family. Am. J. Med. Genet. 2000;90:326–327. doi: 10.1002/(sici)1096-8628(20000214)90:4<326::aid-ajmg12>3.0.co;2-o. [DOI] [PubMed] [Google Scholar]
- 42.Cramer M. Case report of complete anodontia of the permanent dentition. Am. J. Orthod. 1947;33:760–764. doi: 10.1016/s0096-6347(47)90074-4. [DOI] [PubMed] [Google Scholar]
- 43.Gorlin R.J., Herman N.G., Moss S.J. Complete absence of the permanent dentition: an autosomal recessive disorder. Am. J. Med. Genet. 1980;5:207–209. doi: 10.1002/ajmg.1320050215. [DOI] [PubMed] [Google Scholar]
- 44.Burzynski N., Escobar V. Classification genetics of numeric anomalies of dentition. Birth Defects. 1983;13:95–106. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.