

Abstract
Fibroblast growth factors (FGFs) play diverse roles in several developmental processes. Mutations leading to deregulated FGF signaling can cause human skeletal dysplasias and cancer.1,2 Here we report a missense mutation (Ser99Asp) in exon 2 of FGF9 in 12 patients with multiple synostoses syndrome (SYNS) in a large Chinese family. In vitro studies demonstrate that FGF9S99N is expressed and secreted as efficiently as wild-type FGF9 in transfected cells. However, FGF9S99N induces compromised chondrocyte proliferation and differentiation, which is accompanied by enhanced osteogenic differentiation and matrix mineralization of bone marrow-derived mesenchymal stem cells (BMSCs). Biochemical analysis reveals that S99N mutation in FGF9 leads to significantly impaired FGF signaling, as evidenced by diminished activity of Erk1/2 pathway and decreased β-catenin and c-Myc expression when compared with wild-type FGF9. Importantly, the binding of FGF9S99N to its receptor is severely impaired although the dimerization ability of mutant FGF9 itself or with wild-type FGF9 is not detectably affected, providing a basis for the defective FGFR signaling. Collectively, our data demonstrate a previously uncharacterized mutation in FGF9 as one of the causes of SYNS, implicating an important role of FGF9 in normal joint development.
Introduction
Multiple synostoses syndrome (SYNS) is an autosomal-dominant disorder characterized by the joint fusions that commonly involve proximal interphalangeal, carpal-tarsal, humeroradial, and cervical spine joints. A subpopulation of patients also manifests characteristic facies and progressive conductive deafness due to fusion of the ossicles. Previous studies revealed that the mutations in NOG (MIM 602991) on chromosome 17q22 and growth differentiation factor-5 (GDF5 [MIM 601146]) on chromosome 20q11.2 are responsible for multiple synostoses syndrome (SYNS1 [MIM 186500], SYNS2 [MIM 610017], respectively).3,4 NOG encodes a secreted protein, Noggin, that is involved in developmental processes, including the regulation of chondrogenesis in somites and limb buds, where it acts as an antagonist to the bone morphogenetic proteins (BMPs).5–7 It has been shown that mutations in NOG not only cause SYNS1 but also lead to proximal symphalangism (SYM1 [MIM 185800]) and tarsal-carpal coalition syndrome (TCC [MIM 186570]),3,8,9 diseases with clinical features similar to SYNS1. GDF5, also known as cartilage-derived morphogenetic protein 1 (CDMP1), is a secreted growth factor expressed during the course of skeletal development and belongs to TGF-β superfamilies. The mutations in GDF5 in both mouse and human are known to cause abnormal joint development.10–12
Here, we report a five-generation family with common features of SYNS from Qinghai Province, China. The 12 affected individuals in this pedigree display the fusions of proximal interphalangeal, carpal-tarsal, and humeroradial joints. Unexpectedly, linkage analysis and sequencing results indicated that the disease phenotype was associated with neither NOG nor GDF5 mutations. Instead, a missense mutation in fibroblast growth factor 9 was identified in all affected individuals. Thus, our study identified FGF9 as the third disease-causing gene for SYNS phenotype, implicating a crucial role of FGF9 in human joint development.
Subjects and Methods
Human Subjects
A five-generation family with autosomal-dominant SYNS was ascertained through initial identification of a proband (IV-9 in Figure 1) from Qinghai Province, China. Approval for this study was obtained from the committee of Shanghai Second Medical University (SHSMU) charged with reviewing ethics. After informed consent was signed, all affected individuals and family members who agreed to participate in this study were evaluated by experienced orthopedists and radiologists. The diagnosis was made on the basis of family history and physical and radiographic examinations on elbows, hands, feet, and cervical and lumbar spines. The conductive hearing was evaluated by tuning fork test. Peripheral blood samples were collected and genomic DNA was extracted from all family members with QIAamp DNA blood mini kit (QIAGEN). The control DNA samples used for assessing the frequency of DNA sequence alterations in the population were obtained from 250 normal, unrelated, ethnically matched individuals.
Figure 1.
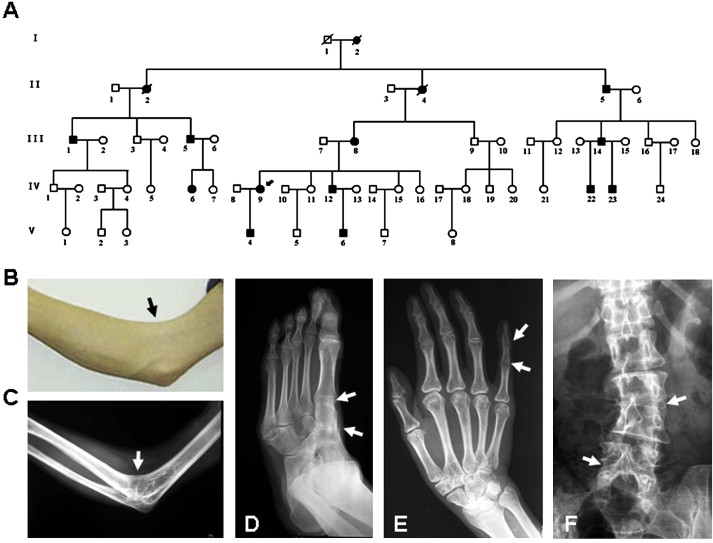
A Chinese Pedigree with Autosomal-Dominant SYNS
(A) SYNS pedigree. Squares, circles, and slashed-through symbols indicate males, females, and individuals known to be deceased, respectively. Affected individuals are indicated by black symbols. Arrow marks the proband with SYNS in this pedigree (IV-9).
(B and C) Right elbow photograph (B) and radiograph (C) of individual IV-12 show typical humeroradial synostoses. Arrows indicate the inability to flex the right elbow joint.
(D) Left-foot radiograph of individual IV-12 shows the synostoses among first metatarsal, cuneiform, and navicular (arrows).
(E) Right-hand radiograph of individual IV-9 displays the synostoses of interphalangeal joints in the fifth digit (arrows).
(F) Lumbar-spine radiograph of individual IV-9 shows the synostoses of lumbar joints at L2-3 and L4-5 (arrows).
Genome-wide Scan and Mutation Analysis
The genotypes of the family members were determined by using the microsatellites from Applied Biosystems Prism Linkage Mapping Set Version 2 according to the manufacturer's instructions on a MegaBACE sequencer. Genetic Profiler (version 1.1) was used for genotype assignment, and MLINK program from the LINKAGE 5.1 computer program package was used for two-point linkage analysis.13 Cyrillic (version 2.0) was used for haplotype analysis. For mutation detection, we performed PCR with the PCR product presequencing kit (USB), purified appropriate PCR products, and sequenced them with dye-primer chemistry (Applied Biosystems) on an ABI 377 automated sequence analyzer (Applied Biosystems). The resulting sequences were analyzed with the Sequencher sequence alignment program (ACGT codes). The primers used for mutation detection are listed in the Supplemental Data available online.
Cell Culture and Transfection
COS-7 or 293T cell lines used for expression and secretion tests were grown in Dulbecco's modified Eagle's medium (DMEM), supplemented with 10% heat-inactivated fetal calf serum, 100 units/ml penicillin, and 100 μg/ml streptomycin. Cells were transfected with Flag-tagged plasmids encoding either wild-type or mutant FGF9, and empty vector by using LipofectAMINE 2000 (Invitrogen). Cell lysates were prepared and culture media were collected 48 hr after transfection. The conditional media composed of normal complete medium and transfected cell cultured medium (volume 1:1) were used for subsequent experiments.
Cell Differentiation Assays
Rat calvaria chondrogenic cell line (RCJ 3.1 C5.18 cells) was maintained in α-MEM supplemented with 15% heat-inactivated fetal calf serum, 100 units/ml penicillin, 100 μg/ml streptomycin, 2 mM sodium pyruvate (GIBCO BRL), and 10−7 M dexamethasone. The confluenced cells were further cultured with conditional medium containing FGF9 or FGF9S99N in the presence of 50 μg/ml ascorbic acid phosphate, 10 mM β-glycerophosphate (Sigma), and heparin (2 μg/ml). Chondrocyte differentiation was assessed by alcian blue staining as previously reported.14 For quantification of the chondrocyte staining, cells were solubilized in 1% SDS by shaking for 30 min at room temperature. Absorbances were taken at 605 nm for triplicate samples. Mouse bone marrow-derived mesenchymal stem cells (BMSCs) were induced to osteoblastic differentiation under the same condition as mentioned above. The osteoblastic phenotype was assessed by alkaline phosphatase (ALP) staining. Cells were fixed with 4% paraformaldehyde for 10 min at 4°C, washed with 0.056 M 2-amino-2-methylpropanol (AMP; Sigma) buffer and stained with substrate solution (0.5 mg/ml naphthol AS-MX phosphate and 0.5 mg/ml Fast Red Violet LB salt in 0.056 M AMP buffer) for 5 min, and the reaction was stopped with TE buffer. The matrix mineralization was visualized by alizarin red staining.15 Cells were washed and fixed as mentioned above. After fixation, the cell layers were washed with PBS and stained with 1% alizarin red S (Sigma) in 0.1% NH4OH (pH 6.5) for 2 min. The cell layers were washed with H2O and then observed and photographed with an Olympus CKX41 microscope.
mRNA and Protein Assays
Total RNA was extracted from the cultures at the indicated time, with the Trizol reagent RNA extraction protocol according to the manufacturer's instructions (Invitrogen). Semiquantitative RT-PCR was performed with cDNA reverse transcribed from 1 μg of total RNA. PCR cycles varied from 20 to 37 cycles. For protein-blot analysis, cell cultures were lysed in RIPA buffer containing a protease inhibitor cocktail (Sigma). Culture media of DNA-transfected cells were incubated with heparin-sepharose beads overnight at 4°C. The beads were washed twice with phosphate-buffered saline (PBS) and suspended in Laemmli buffer. The eluted or cell lysate proteins were separated by SDS-PAGE (12.5% gels) and blotted with the antibodies indicated.
GST Pull-Down Assay
GST-FGF9 fusion proteins were extracted and immobilized on glutathione-sepharose beads by incubation of the purified GST-protein fusions with glutathione-sepharose beads. 293T cells were transiently transfected with Flag-FGF9, Flag-FGF9S99N, and empty vector. Cell lysates and culture media were subjected to incubation with GST-FGF9 sepharose for 4 hr at 4°C. Precipitates from lysates and media were separated by SDS-PAGE, transferred to nitrocellulose membranes, and probed with the indicated antibodies. Whole-cell lysates and media were blotted with Flag and α-tubulin antibodies as the input controls. All antibodies used in this study are listed in the Supplemental Data.
Statistics
Results are presented as means ± standard deviation (SD). The statistical differences were analyzed with Student's t test. Statistical significance was assumed for p < 0.05.
Results
Clinical Phenotype in Patients with SYNS in a Qinghai Family
Twelve affected individuals in this pedigree were diagnosed as SYNS on the basis of their clinical history and physical and radiographic findings (Figure 1A). The individual IV-12 manifested typical humeroradial synostoses at the right elbow joint (Figures 1B and 1C) and semidislocation at the left elbow joint, which was accompanied by the synostoses among first metatarsal, cuneiform, and navicular (Figure 1D). The proband (IV-9, female, 37 years old) showed the typical SYNS features such as humeroradial synostoses and cubitus valgus at the elbows, coupled with the synostoses at the interphalangeal joints in the fifth digit, fusions among first metatarsal, cuneiform, and navicular, and the fusions of lumbar joints at L2-3 and L4-5 (Figures 1E and 1F). The other affected individuals (age 5–68, 8 men and 2 women) showed variable clinical features. The individuals II-5, III-1, III-5, III-8, III-14, and IV-6 manifested semidislocation and cubitus valgus at the elbows. They also had synostoses between first metacarpal and trapezium, or synostoses among first metatarsal, cuneiform, and navicular. The individuals IV-22, IV-23, V-4, and V-6 at the age of 11 or below displayed mild semidislocation or cubitus valgus at elbow joints and limitation of finger joint flexion, suggesting that the SYNS phenotype is age dependent. In addition to the proband IV-9, individuals IV-6 and V-6 had the synostoses of lumbar joints at L4-5 and L3-4, respectively. The joint abnormalities in 12 affected individuals are summarized in Table 1. The hearing, stature, and intelligence were normal in all affected individuals.
Table 1.
Phenotype Variability of Individuals Affected with SYNS by X-Ray Examination
| Affected Individual | Age (years) | Sex | Anomaly at Elbow Jointa | Synostosis at Handsb | Synostosis at Feetc | Synostosis at Lumbar |
|---|---|---|---|---|---|---|
| II-5 | 68 | M | + | + | ++ | − |
| III-1 | 54 | M | + | − | ++ | − |
| III-5 | 38 | M | + | + | ++ | − |
| III-8 | 62 | F | + | ++ | ++ | − |
| III-14 | 37 | M | + | + | ++ | − |
| IV-6 | 16 | F | + | + | ++ | L4-5 |
| IV-9 | 37 | F | ++ | + | ++ | L2-3, L4-5 |
| IV-12 | 29 | M | ++ | + | ++ | − |
| IV-22 | 11 | M | + | +/− | − | − |
| IV-23 | 6 | M | + | +/− | − | − |
| V-4 | 11 | M | +/− | +/− | − | − |
| V-6 | 5 | M | + | − | − | L3-4 |
+, mild elbow abnormalities, e.g., cubitus valgus and semi-dislocation. ++, severe elbow abnormality, e.g., humeroradial synostosis. +/−, cubitus valgus or other elbow abnormalities.
+, the synostosis involved first metacarpal and trapezium or fifth interphalangeal joints. ++, the synostosis involved first metacarpal and trapezium, plus fifth interphalangeal joints. +/−, the synostosis trend involved fifth interphangeal joints.
++, the synostosis involved first metatarsal, cuneiform, and navicular. −, no synostosis or other abnormalities.
Association of SYNS Phenotype in This Pedigree with neither NOG nor GDF5 Mutation
To examine whether SYNS in this large family was due to the mutations in either NOG or GDF5, we performed linkage analysis by using genetic markers from chromosome 17q22 and 20q11.2, respectively. Unexpectedly, no linkage with either of the two genes was detected (Table S1). To exclude the possibility that SYNS in this family is caused by two independently segregating mutations in NOG and GDF5, we also sequenced the coding regions and flanking introns of these two genes in three affected individuals and did not find any mutations. These data suggest that a mutation in a gene other than NOG or GDF5 is responsible for the SYNS observed in this family.
FGF9 S99N Mutation as a Genetic Alteration in SYNS
To search for such genetic alterations, we performed a genome-wide screen and identified a single locus on chromosome 13q11-q12 that cosegregated with the disease. The maximum two-point LOD score was 3.7 at D13S1236 (θ = 0.0). Fine mapping and haplotype analysis narrowed the critical interval to 8.6 Mb between D13S175 and D13S221 (Table 2 and Figure S1).
Table 2.
The LOD Scores of Two-Point Linkage Analysis from Fine Mapping on Chromosome 13
| Lod Score (Z) at Recombination Fraction (θ) | ||||||
|---|---|---|---|---|---|---|
| Marker | 0.0 | 0.1 | 0.2 | 0.3 | 0.4 | 0.5 |
| D13S175 | 1.464375 | 3.107013 | 2.612414 | 1.857932 | 0.919577 | 0.000000 |
| D13S1236 | 3.700658 | 3.873736 | 3.184450 | 2.259354 | 1.158830 | 0.000000 |
| D13S1275 | 0.543834 | 2.798210 | 2.485964 | 1.817558 | 0.921112 | 0.000000 |
| D13S1243 | −0.362458 | 2.059668 | 1.913292 | 1.429418 | 0.760590 | 0.000000 |
| D13S221 | 0.429933 | 3.706325 | 3.166717 | 2.255349 | 1.121422 | 0.000000 |
| D13S1254 | −2.907339 | 0.116978 | 0.340966 | 0.286847 | 0.113492 | 0.000000 |
| D13S217 | −9.250479 | −0.011491 | 0.578008 | 0.609531 | 0.379275 | 0.000000 |
Based on a human genome database, it is known that 22 candidate genes are located in this region. Among them, fibroblast growth factor 9 (FGF9, [MIM 600921]) seems to be one of the most attractive candidate genes because available information indicates that mammalian FGFs control a wide spectrum of biological functions during development and adult life,16 and deregulated FGF signaling is closely associated with human diseases including craniosynostoses and skeletal dysplasia syndromes.17 Moreover, previous studies have showed that FGF9 can induce endochondral ossification in cranial mesenchyme and regulate early hypertrophic chondrocyte differentiation and skeletal vascularization in the developing stylopod.18 Thus, we focused on FGF9 to search for potential mutations. FGF9 localizes at chromosome 13q12, the region between D13S175 and D13S1236 (Figure 2A). It has three exons with an open reading frame of 624 bp, encoding a peptide of 208 amino acids (Figure 2B). By analyzing the sequences of PCR products amplified from each FGF9 exon, we were able to identify a heterozygous mutation of G-to-A transition at nucleotide position 296 within exon 2, leading to an amino acid change from serine (AGT) to asparagine (AAT) at the 99th amino acid residue (Figures 2C and 2D). Significantly, the exact G-to-A mutation was detected in all 12 affected, but not in unaffected, members in the family. In addition, we screened 250 unrelated, ethnically matched individuals (i.e., 500 chromosomes) and found no such mutation in FGF9 exon 2 (data not shown). Thus, our study has identified the FGF9 S99N mutation as a genetic alteration that cosegregates with multiple synostoses syndrome.
Figure 2.
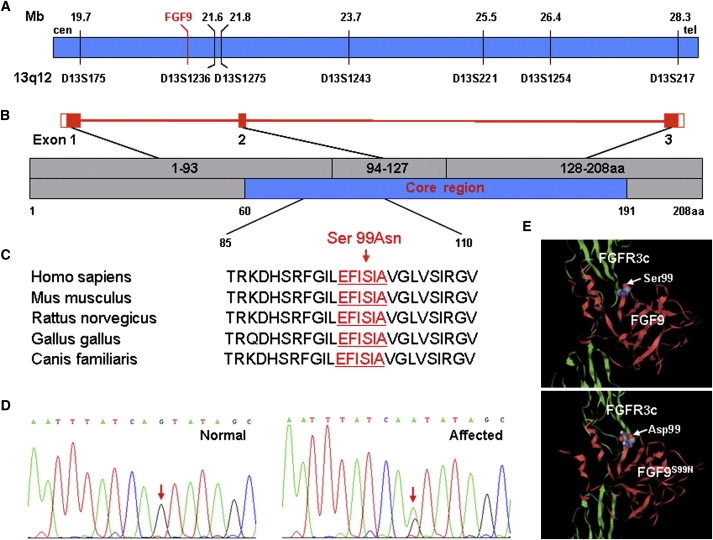
Identification of S99N Mutation in the Core Region of FGF9
(A) Physical map of the region between genetic marker D13S175 and D13S217. The red line indicates FGF9 locus. The black lines show the physical distances (upper) and genetic markers (lower). This map is based on human genomic sequence NM_002010.1.
(B) Illustration of the exon-intron structure of FGF9. The exons marked as red boxes are numbered as exon 1, 2, and 3 and encode amino acids 1–93, 94–127, and 128–208 (gray, upper), respectively. The core region of FGF9 marked as blue (lower) spans from amino acid 60 to 191.
(C) Amino acid sequence from amino acid 85 to 110 in homologs of FGF9 aligned by BLAST. Red characters indicate the highly conserved motif (EFISIA) between amino acid 96 and 101 of FGF9. Ser99Asn mutation is indicated by an arrow.
(D) Sequence chromatograms of FGF9 mutation. Left and right diagrams show normal and mutation sequence, respectively. The mutation site (G > A) is indicated (arrows).
(E) 3D structure of FGF9-FGFR3c complex modeled with Molscript software. Red and green ribbons indicate FGF9 and FGFR3c, respectively. Upper diagram shows wild-type FGF9 binding to FGFR3c and lower diagram shows potential binding and structural alteration of mutant FGF9 and FGFR3c. The substituted residue is indicated by arrows.
To gain insight into this FGF9 mutation, we performed a computer simulation of the structure of the FGF9 protein with Molscript software. The result predicts that FGF9 3D structure and its binding to the receptor may be altered because of S99N mutation (Figure 2E), suggesting it as a consequential mutation.
Normal Expression and Secretion of FGF9S99N When Expressed in Cells
Structurally, FGF9 does not have a typical secretory signal peptide. Its secretion is known to be mediated via a hydrophobic region containing a highly conserved EFISIA motif and a noncleaved signal sequence consisting of the first 33 residues.19,20 A previous report showed that artificial I98N, I100N, and A101N mutations in the FGF9 EFISIA motif can significantly change local hydrophobicity and interfere with FGF9 secretion.21 These findings raised the possibility that S99N mutation could interfere with FGF9 secretion. Therefore, we transfected COS-7 or 293T cells with Flag-tagged wild-type (WT) or mutant FGF9-expressing construct. The expression and secretion of WT or mutant FGF9 in cell lysate and culture medium were measured by western blotting with an anti-Flag antibody. There were no detectable differences in the amount of WT and mutant FGF9 proteins found in cell lysate and medium, respectively (Figure 3A), indicative of normal expression and secretion of mutant protein. The result is in line with the fact that substitution of serine (−0.80) with asparagine (−3.50) in FGF9 does not significantly alter its hydrophobicity in Kyte-Doolittle scale (Figure S2),22 which is critical for FGF9 secretion.21
Figure 3.
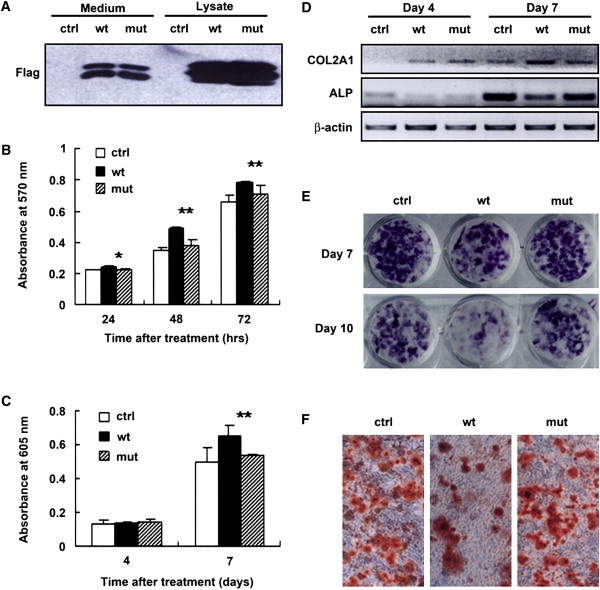
Abnormal Cell Proliferation and Differentiation Induced by FGF9S99N
(A) Expression and secretion of FGF9S99N by transfected cells. FGF9S99N in medium was detected at the level comparable to wild-type FGF9.
(B) RCJ cell proliferation was assayed by MTT colorimetric method in quadruplicate at the time points indicated. The data shown are representative of at least four independent experiments. ∗ indicates p < 0.05; ∗∗ indicates p < 0.01 (WT versus mutant).
(C) RCJ cells were differentiated to chondrocytes in conditional medium for the indicated time. The chondrocytes were stained with alcian blue and, after solubilization, absorbances were taken at 605 nm in triplicate. The representative of three independent experiments is shown. ∗∗ indicates p < 0.01 (WT versus mutant).
(D) RCJ cells were differentiated to chondrocytes in conditional medium and cell cultures were lysed and total RNA was extracted at the indicated time. The expression levels of COL2A1 (for chondrocytes) and ALP (for osteoblasts) were assayed by semiquantitative RT-PCR. β-actin was shown as inner control. For osteoblastic differentiation, BMSCs were differentiated to osteoblasts in conditional medium for 7 days and 10 days.
(E and F) Cell cultures were stained with naphthol AS-MX phosphate for ALP activity (E) and alizarin red S for matrix mineralization (F, images of day 7). The photographs were taken under Olympus CKX41 microscope (magnification, 10×).
Abnormal Cell Proliferation and Differentiation Associated with FGF9S99N
To examine a potential functional consequence to this FGF9 mutation, we determined FGF9-mediated cellular effects. FGF signaling is involved in multiple cellular processes, including cell growth, differentiation, migration, and survival depending on the cellular context.23 We collected the culture medium from cells expressing either WT or mutant FGF9 and assessed their activity on chondrocyte proliferation and differentiation. Consistent with previously reports,14 conditioned medium containing FGF9 promoted cell proliferation of rat calvaria chondrogenic cell line (RCJ 3.1 C5.18 cells) (Figure 3B), as well as mouse primary chondrocytes (Figure S3A). In contrast, no such effect was observed when FGF9S99N-containing medium was used, suggesting that S99N mutation compromised FGF9 to promote chondrogenic cell proliferation. By using in vitro RCJ cell culture model for chondrocyte differentiation, we also evaluated the ability of mutant FGF9 to influence cell differentiation. We found that WT FGF9 induced chondrocyte differentiation, evidenced by increased alcian blue staining after 7 day culture. However, chondrocyte differentiation induced by FGF9S99N remained at a comparable level with control cells (Figure 3C). The expression levels of type II collagen 1 (COL2A1) gene, a well-characterized, specific marker of commitment to the chondrogenic lineage, and ALP, the most widely recognized biochemical marker for osteoblast activity, were analyzed by semiquantitative RT-PCR. The alteration of COL2A1 expression was found to be consistent with the result of alcian blue staining (Figure 3D). Meanwhile, a decreased expression upon WT FGF9 treatment and unchanged expression of ALP were observed when compared to control (Figure 3D), suggesting that FGF9 mutant is unable to exert an inhibitory effect on osteoblastic differentiation as WT FGF9 does. For further evaluation of the effect of FGF9 on osteogenic differentiation, BMSCs were used as a model system because it is well documented that BMSCs can be directed toward the osteogenic lineage in vitro when they are treated with dexamethasone, β-glycerophosphate, and ascorbic acid. With cell differentiation in the presence of either WT FGF9 or FGF9S99N, osteoblast activity was visualized by ALP substrate staining. The results indicated that WT FGF9 exerts an inhibitory effect on osteogenic differentiation, whereas FGF9S99N has no such effect. ALP activity remains unchanged when compared to control cells (Figure 3E). Furthermore, extracellular matrix calcification was detected by alizarin red S staining at days 7 and 10. As a consequence of changes in ALP expression and activity, similar results were observed in the extracellular matrix calcification upon WT FGF9 or FGF9S99N treatment (Figure 3F and Figure S3B). These observations suggest that S99N mutation of FGF9 causes impaired chondrogenesis and aberrantly enhanced osteogenesis.
Diminished Activation of the MAPK as a Result of FGF9 S99N Mutation
To explore the molecular mechanism by which FGF9S99N causes impaired chondrocyte proliferation and differentiation, we probed the signaling pathways downstream of the FGFR activation by using the PathScan Multiplex Western Cocktail I (Detection Kit #7100, New England BioLab), which allows simultaneous detection of Phospho-p90RSK, Phospho-Akt, Phospho-p44/42 MAPK, and Phospho-S6 Ribosomal Proteins. Early passages of NIH 3T3 cells, which express FGFR3, were used as target cells to assess the activity of FGF9. Incubation of conditioned medium collected from WT FGF9-expressing cells was associated with elevated phosphorylation of p44/42 (ERK1/2) and p90RSK, but not Akt or S6 ribosomal protein (data not shown). Given the fact that p90RSK is a downstream target of p44/42 MAPK, the result indicates that binding of FGF9 to its receptor results in activation of the MAPK pathway. With that, we compared FGF9S99N with WT FGF9 for their ability to activate the MAPK pathway. Incubation of the conditioned medium collected from cells expressing S99N FGF9 mutant failed to simulate p44/42 and p90RSK phosphorylation (Figure 4A), indicating an inability of the FGF9 mutant to activate Erk1/2 signaling.
Figure 4.
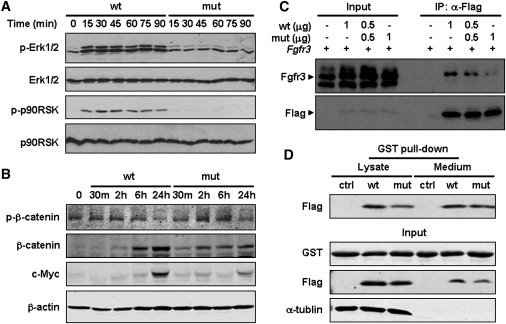
Compromised Activation of Erk1/2 and Wnt Signaling Pathways as a Result of Decreased Receptor Binding of FGF9S99N
(A) Loss of activation of Erk1/2 signaling by FGF9S99N. NIH 3T3 cells treated with the conditional medium for the indicated time were lysed and subjected to western blotting with the indicated antibodies. Compared to WT FGF9, phosphorylation of Erk1/2 and its downstream effector p90RSK induced by FGF9S99N was significantly impaired.
(B) Abnormal activation of Wnt signaling by FGF9S99N. RCJ cells were treated with the same medium as in (A) for the indicated time. Cell lysates were immunoblotted with phospho-β-catenin, β-catenin, c-Myc, and β-actin antibodies.
(C) Compromised receptor-binding capacity of FGF9S99N. 293T cells were transiently cotransfected with Fgfr3 and Flag-FGF9 or Flag-FGF9S99N, or with Fgfr3 and both at the indicated amount of expressing vectors. Cell lysates were immunoprecipitated with anti-Flag and then immunoblotted with the indicated antibodies. Total cell lysates were included as input controls.
(D) FGF9S99N is able to form dimer with FGF9. 293T cells transiently transfected with Flag-FGF9, Flag-FGF9S99N, or empty vector were lysed after 48 hr. The cell lysates and media were subjected to incubation with GST-FGF9 beads. The precipitates were immunoblotted with Flag and GST antibodies. The quantitative analyses for all blots are shown as in Figure S5 on line.
Impaired Wnt/β-Catenin Signaling Pathway as a Result of S99N Mutation
WNT and FGF signaling pathways crosstalk during a variety of cellular processes, such as human colorectal carcinogenesis, mouse mammary tumor virus (MMTV)-induced carcinogenesis, E2A-Pbx-induced leukemogenesis, early embryogenesis, body-axis formation, limb-bud formation, and neurogenesis.24 Both WNT and FGF signals downregulate GSK3β (GSK3B) activity, resulting in the stabilization and the nuclear accumulation of β-catenin, which plays a crucial role in multiple steps during chondrogenesis and chondrocyte maturation.25 In light of the cross-talk between the Wnt and FGF pathways, we further investigated the effect of S99N mutation of FGF9 on the Wnt/β-catenin signaling pathway. As expected, phosphorylation of β-catenin (Ser33/37/Thr41) by GSK3β decreased when RCJ cells were treated with WT FGF9 for 24 hr, and subsequently total β-catenin and its downstream target c-Myc were significantly induced. However, S99N mutation of FGF9 attenuated β-catenin accumulation and c-Myc expression, accompanied by an increase in phospho-β-catenin level (Figure 4B), indicating an impaired ability of FGF9 to activate the Wnt/β-catenin pathway because of S99N mutation.
Compromised Binding of FGF9S99N to Its Receptor
To understand S99N mutation-associated deregulation of cellular signaling, we examined the binding of this mutant to its receptor, fibroblast growth factor receptor 3 (FGFR3; [MIM 134934]). 293T cells were transiently cotransfected with Fgfr3 and Flag-FGF9 or Flag-FGF9S99N, or both constructs at a ratio of 1:1. Cell lysates were immunoprecipitated with an anti-Flag antibody and blotted with an anti-Fgfr3 antibody. Consistent with the prediction by computer simulation (Figure 2E), S99N mutation causes severe defects in receptor binding of FGF9, evidenced by the finding that a significantly decreased amount of Fgfr3 was immunoprecipitated from the lysate containing Flag-FGF9S99N. Of note is the observation that the amount of Fgfr3 precipitated from the lysate containing Flag-FGF9 and Flag-FGF9S99N (half of each) was in between WT and mutant FGF9 alone (Figure 4C). The data indicates that the FGF9 mutation leads to an impaired binding to its receptor.
Dimerization of FGF9S99N Itself or with WT FGF9
It has been well known that the formation of a ternary complex between the FGF ligand, FGFR, and heparan sulfate (HS) is crucial for FGF signaling. Previous work demonstrated that ligand dimerization can significantly enhance the binding of FGF2 to FGFR1, the dimerization of the receptor, and the induction of downstream signal-transduction pathways.26 We thus assessed the effect of S99N mutation on FGF9 dimerization. GST-FGF9 on Sepharose beads were subjected to incubation with either Flag-FGF9 or Flag-FGF9S99N; Flag-tagged proteins precipitated by GST-FGF9 were analyzed by immunoblotting. As shown in Figure 4D, the amount of WT FGF9 or FGF9S99N precipitated by GST-FGF9 Sepharose was comparable in the cell lysate and medium. Furthermore, we performed a chemical cross-linking assay, showing that FGF9S99N is able to form dimer or tetramer as WT FGF9 does (Figure S4). These findings suggested that there was little effect of S99N mutation on the dimerization itself or with wild-type FGF9.
Discussion
SYNS was first reported by Maroteaux et al.,27 and thereafter many other designations were used to describe this disorder, including WL syndrome, symphalangism-brachydactyly syndrome, deafness-symphalangism syndrome of Herrmann, and facioaudiosymphalangism syndrome.28,29 Available evidence indicates that the variation of clinical features correlates with genetic heterogeneity in SYNS. Two disease-causing genes, NOG and GDF5, have been identified to be responsible for SYNS1 and SYNS2, respectively.30,31 SYNS1 is caused by a NOG mutation that usually displays synostoses at the elbow joint, proximal symphalangism, carpal and tarsal fusion, and progressive conduction deafness. Other features include a broad hemicylindrical nose without alar flare and cervical vertebral fusions. SYNS2 resulting from GDF5 mutation was first described by Akarsu et al.31 and validated by Dawson et al.4 The major clinical features were tarsal-carpal coalition, humeroradial synostoses, brachydactyly, proximal symphalangism, a broad hemicylindrical nose, and vertebral fusion. However, conductive deafness usually does not appear. The patients harboring FGF9 S99N mutation in this large pedigree share most clinical features with the SYNS cases reported previously, including humeroradial synostoses or semidislocation at elbow joint, synostoses of the interphalangeal joints, carpal fusion occurring at metacarpal and trapezium, tarsal fusion occurring between cuneiform and navicular, and lumbar vertebral fusion. It was noted that only mild semidislocation or cubitus valgus at elbow joints, or limitation of finger joint flexion, was found in 4 patients at the age of 11 or below, implicating that SYNS phenotype is age dependent. On the basis of the facts that no mutations in either NOG or GDF5 and the tight cosegregation of FGF9 S99N mutation with SYNS phenotype were detected in this pedigree, we believe that FGF9 could be a third locus for SYNS phenotype.
It should be noted that the three different gene mutations have been identified as SYNS disease-causative genes. It may implicate that the mutant genes cause the disease phenotype through a similar pathway or mechanism. NOG encodes a secreted protein, Noggin, which was shown to bind to BMP7 (MIM 112267) and inhibit BMP signaling. The BMP binding affinity of noggin variants is highly correlated with alterations in bone formation and apoptosis in chick limb development.32 It was also shown that the requirement of Noggin during skeletogenesis differs between species and between specific skeletal elements within species.3 GDF5 is a secreted growth factor expressed during several steps in skeletal development, including the formation of the cartilage anlagen (chondrogenesis), chondrocyte differentiation, and joint morphogenesis.33,34 A missense mutation (R438L) in GDF5 was reported to be responsible for abnormal joint development, although the mutant protein encoded by the SYNS allele was secreted as a mature GDF5 dimer.4
FGF9 (glia-activating factor) belongs to the FGF superfamily, which consists of 22 members involved in various developmental processes including the formation of mesoderm during gastrulation, patterning during early postimplantation, and development of various tissues such as limb, nervous, and skeletal systems in vertebrates.35,36 FGF9 was originally purified as a heparin-binding, secreted glycoprotein from cultured human glioma cells.37 Whereas mutations in FGF9 were identified in 10 out of 203 tumor tissues and cell lines,2 little is known about the association of germline mutation of FGF9 with other human pathologies. In fact, a similar phenotype to SYNS in a spontaneous dominant mouse mutant was reported previously, showing elbow and knee joint synostosis (Eks).38 Most recently, a missense mutation that replaces Asn143 with threonine in the Fgf9 gene was identified in Eks mutant mice.39 Combined with these findings in mice, our data indicate that mutations in FGF9, in addition to those in NOG and GDF5, are responsible for SYNS.
FGF9 and its high-affinity receptor FGFR3 had similar developmental expression patterns, particularly in areas of active chondrogenesis.40 Targeted overexpression of FGF9 to cartilage of transgenic mice leads to reduced proliferation and terminal differentiation of chondrocytes similar to that observed in the human disorders, such as achondroplasia and thanatophoric dysplasia caused by activation mutations in FGFR3.40 Consistent with these findings, loss of Fgfr3 function in mice resulted in a phenotype that is the opposite of that observed in achondroplasia and thanatophoric dwarfism.41,42 It was reported that Fgf9-deficient mice exhibited a disproportionate shortening of proximal skeletal elements, which was a prominent feature of patients with FGFR3-induced chondrodysplasia syndromes, and that the Fgf9−/− limb phenotype resulted from loss of Fgf9 functions after formation of the mesenchymal condensation.18 These findings collectively suggest that although the biological function of FGF9-FGFR3 may vary with the gene dosage and developmental stage or cell types, FGFR3-mediated signaling plays an important role in skeletal development. Of note, FGF9-associated SYNS features multiple joint fusions, implicating a role for FGF9 in joint development. Together with the data derived from mouse models, the compromised FGFR3 binding of the FGF9 mutant found in SYNS patients suggests that an adequate strength of FGF9-FGFR3 signaling is crucial for normal skeletal and joint development.
It has been well documented that FGF signaling is involved in multiple cellular processes, including cell growth, differentiation, migration, and survival depending on the cellular context.42 Three major intracellular pathways—ERK1/2, PI3K, and PLCγ—have been shown to mediate the activation of FGFRs.43 Among them, the ERK pathway regulates cell proliferation, chondrocyte differentiation, and synthesis of extracellular matrix components.44 We showed that in contrast to wild-type FGF9—which through binding to FGFR345,46 induced a rapid increase in phosphorylated ERK1/2 and consequently increased phosphorylation of its downstream effector, p90RSK—the ERK pathway was not detectably stimulated by FGF9S99N, suggesting a functional defect of the FGF9 mutant. Indeed, biochemical analysis indicates that S99N mutation in FGF9 resulted in an impaired FGFR3 binding, an outcome predicted by computation simulation.
It was shown that conditional deletion of the β-catenin gene in chondrocytes causes decreased chondrocyte proliferation and delayed chondrocyte maturation,47 and overexpression of inhibitor of β-catenin and TCF (ICAT) in chondrocytes resulted in suppression of chondrocyte proliferation and differentiation in transgenic mice.48 We showed that the FGF9 mutation resulted in an increased phospho-β-catenin level and subsequently deregulated expression of β-catenin and c-Myc, consistent with the active cross-talk between Wnt and FGF signaling pathways that has been shown to take place during a variety of cellular processes.
Taken together, our data indicate that as a result of the S99N mutation, the ability of FGF9 to bind its receptor is severely impaired, leading to the defective ERK signaling and deregulated WNT/β-catenin pathways. Cell-signaling abnormalities due to FGF9 S99N mutation result in an impaired chondrogenesis and aberrantly enhanced osteogenesis, leading to joint fusions during development. Thus, we demonstrate, for the first time, that S99N mutation in FGF9 is the third cause of SYNS, implicating a crucial role of FGF9 in human joint development.
Supplemental Data
Supplemental Data include a list of primers, five figures, and one table and can be found with this article online at http://www.ajhg.org/.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
GenBank, http://www.ncbi.nlm.nih.gov/Genbank/
HUGO Gene Nomenclature Committee, http://www.genenames.org/
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/
Acknowledgments
We thank all members of SYNS family for their selfless contribution to scientific discovery. We thank Prof. Yong-jun Wang (RCJ3.1C5.18 cell line), Prof. Qi-qun Tang (MC3T3E1 cell line), and Prof. Lin Chen (mouse Fgfr3-expressing vector) for their kind help in the critical materials mentioned above. We thank Prof. Dong-qing Wei and Huai-dong Song for the technical assistance. This work is partially supported by grants from the Chinese National Science Fund for Distinguished Young Scholars (39925023 to Z.-g.W., 30625019 to W.H.), National Natural Science Foundation of China (30470951 to M.-m.G., 30530390 to Z.-g.W.), Ministry of Science and Technology of China (2006BAI23B02), the Science and Technology Commission of Shanghai Municipality (07DZ22929, 06DZ05907), and the E-Institutes of Shanghai Municipal Education Commission (E03003).
References
- 1.Eswarakumar V.P., Lax I., Schlessinger J. Cellular signaling by fibroblast growth factor receptors. Cytokine Growth Factor Rev. 2005;16:139–149. doi: 10.1016/j.cytogfr.2005.01.001. [DOI] [PubMed] [Google Scholar]
- 2.Abdel-Rahman W.M., Kalinina J., Shoman S., Eissa S., Ollikainen M., Elomaa O., Eliseenkova A.V., Bützow R., Mohammadi M., Peltomäki P. Somatic FGF9 mutations in colorectal and endometrial carcinomas associated with membranous beta-catenin. Hum. Mutat. 2008;29:390–397. doi: 10.1002/humu.20653. [DOI] [PubMed] [Google Scholar]
- 3.Gong Y., Krakow D., Marcelino J., Wilkin D., Chitayat D., Babul-Hirji R., Hudgins L., Cremers C.W., Cremers F.P.M., Brunner H.G. Heterozygous mutations in the gene encoding noggin affect human joint morphogenesis. Nat. Genet. 1999;21:302–304. doi: 10.1038/6821. [DOI] [PubMed] [Google Scholar]
- 4.Dawson K., Seeman P., Sebald E., King L., Edwards M., Williams J., Mundlos S., Krakow D. GDF5 is a second locus for multiple-synostosis syndrome. Am. J. Hum. Genet. 2006;78:708–712. doi: 10.1086/503204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zimmerman L.B., De Jesus-Escobar J.M., Harland R.M. The Spemann organizer signal noggin binds and inactivates bone morphogenetic protein 4. Cell. 1996;86:599–606. doi: 10.1016/s0092-8674(00)80133-6. [DOI] [PubMed] [Google Scholar]
- 6.Brunet L.J., McMahon J.A., McMahon A.P., Harland R.M. Noggin, cartilage morphogenesis, and joint formation in the mammalian skeleton. Science. 1998;280:1455–1457. doi: 10.1126/science.280.5368.1455. [DOI] [PubMed] [Google Scholar]
- 7.McMahon J.A., Takada S., Zimmerman L.B., Fan C.M., Harland R.M., McMahon A.P. Noggin-mediated antagonism of BMP signaling is required for growth and patterning of the neural tube and somite. Genes Dev. 1998;12:1438–1452. doi: 10.1101/gad.12.10.1438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dixon M.E., Armstrong P., Stevens D.B., Bamshad M. Identical mutations in NOG can cause either tarsal/carpal coalition syndrome or proximal symphalangism. Genet. Med. 2001;3:349–353. doi: 10.1097/00125817-200109000-00004. [DOI] [PubMed] [Google Scholar]
- 9.Brown D.J., Kim T.B., Petty E.M., Downs C.A., Martin D.M., Strouse P.J., Moroi S.E., Milunsky J.M., Lesperance M.M. Autosomal dominant stapes ankylosis with broad thumbs and toes, hyperopia, and skeletal anomalies is caused by heterozygous nonsense and frameshift mutations in NOG, the gene encoding noggin. Am. J. Hum. Genet. 2002;71:618–624. doi: 10.1086/342067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Storm E.E., Huynh T.V., Copeland N.G., Jenkins N.A., Kingsley D.M., Lee S.J. Limb alterations in brachypodism mice due to mutations in a new member of the TGF beta-superfamily. Nature. 1994;368:639–643. doi: 10.1038/368639a0. [DOI] [PubMed] [Google Scholar]
- 11.Thomas J.T., Lin K., Nandedkar M., Camargo M., Cervenka J., Luyten F.P. A human chondrodysplasia due to a mutation in a TGF-beta superfamily member. Nat. Genet. 1996;12:315–317. doi: 10.1038/ng0396-315. [DOI] [PubMed] [Google Scholar]
- 12.Thomas J.T., Kilpatrick M.W., Lin K., Erlacher L., Lembessis P., Costa T., Tsipouras P., Luyten F.P. Disruption of human limb morphogenesis by a dominant negative mutation in CDMP1. Nat. Genet. 1997;17:58–64. doi: 10.1038/ng0997-58. [DOI] [PubMed] [Google Scholar]
- 13.Kruglyak L., Daly M.J., Reeve-Daly M.P., Lander E.S. Parametric and nonparametric linkage analysis: Unified multipoint approach. Am. J. Hum. Genet. 1996;58:1347–1363. [PMC free article] [PubMed] [Google Scholar]
- 14.Weksler N.B., Reid E.S., Horton W.A., Lunstrum G.P. Differential effects of fibroblast growth factor (FGF) 9 and FGF2 on proliferation, differentiation and terminal differentiation of chondrocytic cells in vitro. Biochem. J. 1999;342:677–682. [PMC free article] [PubMed] [Google Scholar]
- 15.Shimoaka T., Ogasawara T., Yonamine A., Chikazu D., Kawano H., Nakamura K., Itoh N., Kawaguchi H. Regulation of osteoblast, chondrocyte, and osteoclast functions by fibroblast growth factor (FGF)-18 in comparison with FGF-2 and FGF-10. J. Biol. Chem. 2002;277:7493–7500. doi: 10.1074/jbc.M108653200. [DOI] [PubMed] [Google Scholar]
- 16.Ornitz D.M., Marie P.J. FGF signaling pathways in endochondral and intramembranous bone development and human genetic disease. Genes Dev. 2002;16:1446–1465. doi: 10.1101/gad.990702. [DOI] [PubMed] [Google Scholar]
- 17.Govindarajan V., Overbeek P.A. FGF9 can induce endochondral ossification in cranial mesenchyme. BMC Dev. Biol. 2006;6 doi: 10.1186/1471-213X-6-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hung I.H., Yu K., Lavine K.J., Ornitz D.M. FGF9 regulates early hypertrophic chondrocyte differentiation and skeletal vascularization in the developing stylopod. Dev. Biol. 2007;307:300–313. doi: 10.1016/j.ydbio.2007.04.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ornitz D.M. FGF signaling in the developing endochondral skeleton. Cytokine Growth Factor Rev. 2005;16:205–213. doi: 10.1016/j.cytogfr.2005.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Revest J.M., DeMoerlooze L., Dickson C. Fibroblast growth factor 9 secretion is mediated by a non-cleaved amino-terminal signal sequence. J. Biol. Chem. 2000;275:8083–8090. doi: 10.1074/jbc.275.11.8083. [DOI] [PubMed] [Google Scholar]
- 21.Miyakawa K., Hatsuzawa K., Kurokawa T. A hydrophobic region locating at the center of fibroblast growth factor-9 is crucial for its secretion. J. Biol. Chem. 1999;274:29352–29357. doi: 10.1074/jbc.274.41.29352. [DOI] [PubMed] [Google Scholar]
- 22.Kyte J., Doolittle R.F. A simple method for displaying the hydropathic character of a protein. J. Mol. Biol. 1982;157:105–132. doi: 10.1016/0022-2836(82)90515-0. [DOI] [PubMed] [Google Scholar]
- 23.Böttcher R.T., Niehrs C. Fibroblast growth factor signaling during early vertebrate development. Endocr. Rev. 2005;26:63–77. doi: 10.1210/er.2003-0040. [DOI] [PubMed] [Google Scholar]
- 24.Katoh M., Katoh M. Cross-talk of WNT and FGF signaling pathways at GSK3beta to regulate beta-catenin and SNAIL signaling cascades. Cancer Biol. Ther. 2006;5:1059–1064. doi: 10.4161/cbt.5.9.3151. [DOI] [PubMed] [Google Scholar]
- 25.Macsai C.E., Foster B.K., Xian C.J. Roles of Wnt signalling in bone growth, remodelling, skeletal disorders and fracture repair. J. Cell. Physiol. 2008;215:578–587. doi: 10.1002/jcp.21342. [DOI] [PubMed] [Google Scholar]
- 26.Safran M., Eisenstein M., Aviezer D., Yayon A. Oligomerization reduces heparin affinity but enhances receptor binding of fibroblast growth factor 2. Biochem. J. 2000;345:107–113. [PMC free article] [PubMed] [Google Scholar]
- 27.Maroteaux P., Bouvet J.P., Briard M.L. La maladie des synostoses multiples. Nouv. Presse Med. 1972;1:3041–3047. [PubMed] [Google Scholar]
- 28.Herrmann J. Symphalangism and brachydactyly syndrome: Report of the WL symphalangism-brachydactyly syndrome: Review of literature and classification. Birth Defects Orig. Artic. Ser. 1974;10:23–53. [PubMed] [Google Scholar]
- 29.Hurvitz S.A., Goodman R.M., Hertz M., Katznelson M.B.-M., Sack Y. The facio-audio-symphalangism syndrome: Report of a case and review of the literature. Clin. Genet. 1985;28:61–68. doi: 10.1111/j.1399-0004.1985.tb01219.x. [DOI] [PubMed] [Google Scholar]
- 30.Krakow D., Reinker K., Powell B., Cantor R., Priore M.A., Garber A., Lachman R.S., Rimoin D.L., Cohn D.H. Localization of a multiple synostoses-syndrome disease gene to chromosome 17q21–22. Am. J. Hum. Genet. 1998;63:120–124. doi: 10.1086/301921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Akarsu A.N., Rezaie T., Demirtas M., Farhud D.D., Sarfarazi M. Multiple synostosis type 2 (SYNS2) maps to 20q11.2 and caused by a missense mutation in the growth/differentiation factor 5 (GDF5) Am. J. Hum. Genet. 1999;65(Suppl):A281. [Google Scholar]
- 32.Groppe J., Greenwald J., Wiater E., Rodriguez-Leon J., Economides A.N., Kwiatkowski W., Affolter M., Vale W.W., Belmonte J.C.I., Choe S. Structural basis of BMP signalling inhibition by the cystine knot protein Noggin. Nature. 2002;420:636–642. doi: 10.1038/nature01245. [DOI] [PubMed] [Google Scholar]
- 33.Chang S.C., Hoang B., Thomas J.T., Vukicevic S., Luyten F.P., Ryba N.J., Kozak C.A., Reddi A.H., Moos M. Cartilage-derived morphogenetic proteins: New members of the transforming growth factor-beta superfamily predominantly expressed in long bones during human embryonic development. J. Biol. Chem. 1994;269:28227–28234. [PubMed] [Google Scholar]
- 34.Storm E.E., Kingsley D.M. Joint patterning defects caused by single and double mutations in members of the bone morphogenetic protein (BMP) family. Development. 1996;122:3969–3979. doi: 10.1242/dev.122.12.3969. [DOI] [PubMed] [Google Scholar]
- 35.Popovici C., Conchonaud F., Birnbaum D. Functional phylogeny relates LET-756 to fibroblast growth factor 9. J. Biol. Chem. 2004;279:40146–40152. doi: 10.1074/jbc.M405795200. [DOI] [PubMed] [Google Scholar]
- 36.Santos-Ocampo S., Colvin J.S., Chellaiah A., Ornitz D.M. Expression and biological activity of mouse fibroblast growth factor-9. J. Biol. Chem. 1996;271:1726–1731. doi: 10.1074/jbc.271.3.1726. [DOI] [PubMed] [Google Scholar]
- 37.Miyamoto M., Naruo K., Seko C., Matsumoto S., Kondo T., Kurokawa T. Molecular cloning of a novel cytokine cDNA encoding the ninth member of the fibroblast growth factor family, which has a unique secretion property. Mol. Cell. Biol. 1993;13:4251–4259. doi: 10.1128/mcb.13.7.4251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Murakami H., Okawa A., Yoshida H., Nishikawa S., Moriya H., Koseki H. Elbow knee synostosis (Eks): A new mutation on mouse Chromosome 14. Mamm. Genome. 2002;13:341–344. doi: 10.1007/s00335-001-2143-6. [DOI] [PubMed] [Google Scholar]
- 39.Harada M., Murakami H., Okawa A., Okimoto N., Hiraoka S., Nakahara T., Akasaka R., Shiraishi Y., Futatsugi N., Mizutani-Koseki Y. FGF9 monomer-dimer equilibrium regulates extracellular matrix affinity and tissue diffusion. Nat. Genet. 2009;41:289–298. doi: 10.1038/ng.316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Garofalo S., Kliger-Spatz M., Cooke J.L., Wolstin O., Lunstrum G.P., Moshkovitz S.M., Horton W.A., Yayon A. Skeletal dysplasia and defective chondrocyte differentiation by targeted overexpression of fibroblast growth factor 9 in transgenic mice. J. Bone Miner. Res. 1999;14:1909–1915. doi: 10.1359/jbmr.1999.14.11.1909. [DOI] [PubMed] [Google Scholar]
- 41.Deng C., Wynshaw-Boris A., Zhou F., Kuo A., Leder P. Fibroblast growth factor receptor 3 is a negative regulator of bone growth. Cell. 1996;84:911–921. doi: 10.1016/s0092-8674(00)81069-7. [DOI] [PubMed] [Google Scholar]
- 42.Dailey L., Ambrosetti D., Mansukhani A., Basilico C. Mechanisms underlying differential responses to FGF signaling. Cytokine Growth Factor Rev. 2005;16:233–247. doi: 10.1016/j.cytogfr.2005.01.007. [DOI] [PubMed] [Google Scholar]
- 43.Schönwasser D.C., Marais R.M., Marshall C.J., Parker P.J. Activation of the mitogen-activated protein kinase/extracellular signal-regulated kinase pathway by conventional, novel, and atypical protein kinase C isotypes. Mol. Cell. Biol. 1998;18:790–798. doi: 10.1128/mcb.18.2.790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Stanton L.A., Michael Underhilla T., Beier F. MAP kinases in chondrocyte differentiation. Dev. Biol. 2003;263:165–175. doi: 10.1016/s0012-1606(03)00321-x. [DOI] [PubMed] [Google Scholar]
- 45.Kang S., Dong S., Gu T., Guo A., Cohen M., Lonial S., Khoury H., Fabbro D., Gilliland D., Bergsagel P. FGFR3 activates RSK2 to mediate hematopoietic transformation through tyrosine phosphorylation of RSK2 and activation of the MEK/ERK pathway. Cancer Cell. 2007;12:201–214. doi: 10.1016/j.ccr.2007.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Stavridis M.P., Lunn J.S., Collins B.J., Storey K.G. A discrete period of FGF-induced Erk1/2 signalling is required for vertebrate neural specification. Development. 2007;134:2889–2894. doi: 10.1242/dev.02858. [DOI] [PubMed] [Google Scholar]
- 47.Akiyama H., Lyons J.P., Mori-Akiyama Y., Yang X., Zhang R., Zhang Z., Deng J.M., Taketo M.M., Nakamura T., Behringer R.R. Interactions between Sox9 and beta-catenin control chondrocyte differentiation. Genes Dev. 2004;18:1072–1087. doi: 10.1101/gad.1171104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chen M., Zhu M., Awad H., Li T.F., Sheu T.J., Boyce B.F., Chen D., O'Keefe R.J. Inhibition of beta-catenin signaling causes defects in postnatal cartilage development. J. Cell Sci. 2008;121:1455–1465. doi: 10.1242/jcs.020362. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.