

Abstract
Environment-induced relapse is a major concern in drug addiction because of the strong associations formed between drug reward and environment. Cocaine-conditioned place preference is an ideal experimental tool to examine adaptations in the molecular pathways that are activated upon re-exposure to an environment previously paired with drug reward. To better understand the mechanism of cocaine-conditioned place preference we have used western blot analysis to examine changes in phosphorylation of cAMP-response element binding protein (CREB), dopamine- and cyclic AMP-regulated phosphoprotein 32 (DARPP-32), extra-cellular signal-regulated kinase (ERK) and GluR1, key molecular substrates altered by cocaine, in the nucleus accumbens (NAc) and dorsal hippocampus (DHC) of C57BL/6 mice. Our studies revealed that re-exposing mice to an environment in which they were previously given cocaine resulted in increased levels of Ser133 phospho-CREB and Thr34 phospho-DARPP-32 with a corresponding decrease in Thr75 phospho-DARPP-32 in the NAc. In DHC there were increased levels of phospho-CREB, Thr183/Tyr185 phospho-ERK, and Ser845 phospho-GluR1. These data suggest that the formation of contextual drug reward associations involves recruitment of the DHC-NAc circuit with activation of the DARPP-32/CREB pathway in the NAc and the ERK/CREB pathway in the DHC.
Keywords: cocaine, cocaine-conditioned place preference, cyclic AMP-response element binding protein, dopamine-and cyclic AMP-regulated phosphoprotein 32, dorsal hippocampus, nucleus accumbens
Exposure to drugs of abuse usurp the normal reward and learning mechanisms of the brain causing maladaptive changes in molecular signal transduction pathways leading to the addictive state. Addiction is characterized by compulsive drug use, and carries a high risk of relapse following cessation (Hyman 2005; Hyman et al. 2006). Mesolimbic dopamine neurons from the ventral tegmental area (VTA) projecting to the nucleus accumbens (NAc) serve as the primary reward circuitry of the brain, via connections with the striatum, medial prefrontal cortex and hippocampus, which mediates learning and memory and goal directed behaviors (Kelley 2004). A widely used behavioral assay in preclinical drug abuse research is cocaine-conditioned place preference (CPP). CPP utilizes the natural reward pathways of the NAc and pathways of learning and memory in the dorsal hippocampus (DHC) allowing the ability to learn associations between the rewarding, euphoric effects of drugs of abuse with a specific environment through repeated pairing of the two (Bardo 1998; Bardo and Bevins 2000; Meyers et al. 2003).
Changes in the function and expression of specific genes and proteins shown to underlie the persistent and pervasive neurobiological and behavioral changes that characterize addiction have provided insight into the mechanisms of the addictive properties of drugs of abuse. Of particular interest is the transcription factor cAMP-response element binding protein (CREB), which, upon phosphorylation at Ser133, activates gene expression by binding CRE elements in promoter regions of other proteins and transcription factors that mediate neural plasticity (Berke and Hyman 2000; Hyman and Malenka 2001). Cocaine and other abused drugs increase CREB phosphorylation in the NAc (Brami-Cherrier et al. 2005) and hippocampus (Kuo et al. 2007) after acute exposure, suggesting CREB-mediated pathways have a role in the altered response to subsequent drug exposure. Furthermore, CREB in the NAc has been directly implicated in the rewarding properties of cocaine (Carlezon et al. 1998; Walters and Blendy 2001). In the DHC increased CREB activity is observed following training tasks and memory provoking stimuli (Taubenfeld et al. 1999) akin to the CPP assay, and increased DHC CREB phosphorylation facilitates long-term associative learning (Brightwell et al. 2007).
The molecular pathways contributing to the extent of phosphorylation of CREB at Ser133 are diverse and controlled by a number of kinases and phosphatases that vary with brain region. In the NAc and hippocampus CREB is directly phosphorylated at Ser133 by cAMP activation of protein kinase A (PKA) (Gonzalez and Montminy 1989), and ERK activation of p90 ribosomal kinase (RSK) and mitogen-and stress-activated protein kinase pathways (MSK) (Xing et al. 1996; Deak et al. 1998; Mayr and Montminy 2001), kinases implicated in reward-related learning such as CPP (Valjent et al. 2000; Self 2004; Miller and Marshall 2005; Ferguson et al. 2006). In the NAc, acute cocaine exposure activates D1 dopamine receptors that induce CREB phosphorylation directly by PKA activation (Hyman and Malenka 2001; Nestler 2001). Acute cocaine exposure also activates ERK pathways leading to CREB phosphorylation (Valjent et al. 2000; Brami-Cherrier et al. 2005).
Another important regulator of CREB phosphorylation is dopamine- and cyclic AMP-regulated phosphoprotein 32 (DARPP-32). In the NAc DARPP-32 is regulated by the PKA and Ca2+ signaling pathways (Halpain et al. 1990; Svenningsson et al. 2004). PKA-mediated phosphorylation at Thr34 results in increases in Ser133 CREB phosphorylation via inhibition of protein phosphatase 1 whereas phosphorylation at Thr75 converts DARPP-32 into an inhibitor of PKA decreasing levels of Ser133 CREB phosphorylation (Nairn et al. 2004).
The current study explored the molecular substrates activated in the NAc and DHC of cocaine-treated mice that were reintroduced to an environment in which they were previously given cocaine (cocaine-paired) or saline (cocaine-unpaired) 2 days after CPP testing. We found that cocaine-paired mice had higher levels of Ser133 phospho-CREB (P-CREB) in both NAc and DHC compared with cocaine-unpaired mice and also compared with saline-treated mice reintroduced into the environment in which they were given saline (saline-paired). Furthermore, cocaine-paired mice displayed decreased Thr75 phospho-DARPP-32 (P-DAR-PP-32) and increased Thr34 P-DARPP-32 in the NAc with no difference in levels of Thr202/Tyr204 phospho-ERK2 (P-ERK2) or Ser845 GluR1 phosphorylation (phospho-GluR1), a PKA target and an important mediator of synaptic plasticity (Wolf et al. 2004; Wang et al. 2005). In the DHC, cocaine-paired mice exhibited higher levels of P-ERK2 and phospho-GluR1. These results provide evidence of increased PKA signaling in the NAc and increased PKA and ERK signaling in the DHC as persistent drug-related changes induced and triggered by contextual association, which provide insight into the mechanism of drug reward and relapse.
Materials and methods
Animals
Eight- to 10-week-old C57BL/6 mice (Jackson Labs, Bar Harbor, ME, USA) were used in this study. Animals were handled and weighed each day prior to testing, and allowed to habituate to the testing room for 1 h prior to each day’s experiment.
Drugs and antibodies
Cocaine HCl was dissolved in physiological saline at a concentration of 1 mg/mL, and injected at 0.01 mL/g body weight at a dose of 10 mg/kg. Antibodies for Ser133 P-CREB, CREB, Thr34 P-DARPP-32, Thr75 P-DARPP-32, Thr183/Tyr 185 P-ERK1/2, and ERK were obtained from Cell Signaling Technology (Danvers, MA, USA), GluR1 and Ser845 phospho-GluR1 were obtained from Abcam (Cambridge, MA, USA) and actin was obtained from Chemicon (Temecula, CA, USA).
Conditioned place preference
Our model employed a 5-day CPP protocol shown to be effective for CPP (Carlezon et al. 1998). Because preliminary studies found a more substantial change in preference when conditioned in the morning (data not shown), we utilized a semi-biased design during which all animals were conditioned in the morning. To have an operational definition of reward with high face validity we used a method to screen mice that had an initial bias, and defined reward as the difference in time spent in the chamber where drug was given on the test day minus the time spent in the same chamber on the preconditioning day. This allowed us to control for as many biasing variables as possible (Roma and Riley 2005).
Specifically, we used a three-chamber place preference apparatus (Med Associates Inc., St. Albans, VT, USA) made of two equal-sized (16.8 × 12 cm) preference chambers connected by a central chamber (7.2 × 12 cm), outfitted with sliding guillotine-style doors between each chamber, and photo-beams wired to a computer to record animal location and activity. The central chamber had a smooth floor and was colored gray, while each preference chamber was either white with a floor made of a fine metal mesh or black with a floor made of equally spaced metal bars.
On the first day of experimentation (preconditioning; Fig. 1a) mice were placed in the central chamber for a 1-min (60 s) habituation period with the sliding doors closed, followed by a 20-min (1200 s) period of free exploration throughout the whole apparatus. Time spent in each chamber was recorded, and mice were returned to their cage. To control for apparatus bias, a set of criteria were used to exclude some mice from the study. First, any mouse that spent more than 600 s of the 1200-s pre-conditioning period in the central gray chamber was excluded. Of the remaining mice, the time spent in the gray chamber was subtracted from 1200 (the total test time), and divided by two to give a ‘half-time’ (equal to the amount of time the mouse would spend in each chamber, black and white, if it demonstrated no innate preference). The half-time was multiplied by 0.2 and this value was added or subtracted to the half-time to give a range of allowable time a mouse can spend in the black or white chamber. If the time the animal spent in one chamber exceeded the upper or lower limits of the range then the animal was assumed to have an apparatus bias and was removed from the study.
Fig. 1.

Schematic of cocaine-conditioned place preference protocol and behavioral results. (a) Following a pre-conditioning assessment of initial preference (day 1) mice were randomly assigned to saline-paired (SAL; n = 11), cocaine-paired (COC; n = 10) or cocaine-unpaired (UNP; n = 6) treatment groups, and received saline or cocaine (10 mg/kg, i.p.), respectively on days 2–4 in the morning (AM) and were confined to the black or white chamber for 20 min. Four hours later all mice were injected with saline and confined in the opposite chamber for 20 min. On day 5 (test) mice were allowed free access to both chambers. (b) The mice given cocaine spent significantly greater time in the cocaine-paired chamber (COC and UNP) while saline-paired mice showed no change in chamber preference. Difference score equals difference in time spent in chamber from preconditioning to test. *p < 0.01 versus SAL.
The remaining mice were then randomly assigned to either cocaine or saline-paired groups. On days 2–4 (conditioning; Fig. 1a) mice were given an injection of cocaine or saline and confined randomly to either the black or white chamber for 20 min (AM session). Mice were then returned to their home cages. After 4 h a saline injection (0.01 mL/g body weight) was given to all mice and they were confined to the opposite side to which they received saline or cocaine in the AM session for 20 min (PM session). On the final day (test; Fig. 1a) mice were again placed in the central chamber with the doors closed for a 1-min habituation period. The doors were raised and the mice were allowed to walk freely about the chamber. Time spent in each chamber was recorded.
Preference was defined as the difference in time spent in the chamber where drug was given on the test day minus the time spent in the same chamber on the pre-conditioning day and is reported as difference score in Fig. 1b. Data obtained from pairing in black or white chambers were collapsed because initial experiments showed that the preference obtained by pairing in the black or pairing in the white was indistinguishable (data not shown).
Western immunoblotting
Forty-eight hours following the test day, mice that had been given saline were confined to the chamber in which they were given saline (saline-paired group) and mice that had been given cocaine were confined to either the chamber in which they were given cocaine (cocaine-paired group) or saline (cocaine-unpaired group) for a 20-min period followed immediately by rapid decapitation, whole brain dissection and freezing in isopentane at −40°C. Brains were sectioned in a cryostat in the coronal plane up to the rostral end of the NAc (shell and core) and 0.5 mm deep bilateral tissue punches spanning approximately +1.7 to +1.2 mm relative to bregma (Paxinos and Franklin 2003), were obtained with a 17-gauge stainless steel stylet. The same brain was sectioned to the rostral end of the hippocampus and 0.5 mm deep bilateral tissue punches of the DHC containing the dentate gyrus and CA1 regions spanning approximately −2.70 to −3.20 mm relative to bregma (Paxinos and Franklin 2003), were obtained with a 17-gauge stainless steel stylet. Tissue was sonicated in 1% sodium dodecyl sulfate buffer in Tris–EDTA, pH 7.4, containing 1x protease inhibitor cocktail (Sigma, St Louis, MO, USA), 5 mM NaF, and 1x phosphatase inhibitor cocktail (Sigma). Samples were boiled for 5 min and centrifuged at 16 100 g for 10 min. Supernatants were collected and protein concentration determined by bicinchoninic acid assay (Pierce, Rockford, IL, USA); 30 μg of protein were loaded on a 12% sodium dodecyl sulfate polyacrylamide gel, transferred to polyvinylidene fluoride membranes (Immuno-Blot polyvinylidene fluoride, 0.2 μm; Bio-Rad Laboratories, Hercules, CA, USA) and blocked in blocking buffer (5% non-fat dried milk in 0.25 M Tris–HCl, pH 7.6, 1.37 M NaCl, 0.1% Tween) for 60 min. Membranes were probed with primary antibody to Ser133 P-CREB (1:850), CREB (1:850), Thr34 P-DARPP-32 (1:500), Thr75 P-DARPP-32 (1::500), DARPP-32 (1:1000), Thr183/Tyr185 P-ERK1/2 (1:850), ERK (1:1000), Ser845 phospho-GluR1 (1:850), GluR1 (1:5000), or β-actin (1:20 000) for 12–48 h at 4°C. They were then washed four times for 5 min each in blocking buffer and exposed to goat anti-rabbit (1:5000 for P-CREB, CREB, P-ERK1/2, ERK, phospho-GluR1, GluR1, Thr34 P-DARPP-32, Thr75 P-DARPP-32, and DARPP-32) or horse anti-mouse (1:30 000 for β-actin) horseradish peroxidase-linked IgG (Vector Laboratories, Burlingame, CA, USA) for 1 h at 22.5°C. Blots were washed once for 15 min and four times for 5 min each in Tris-buffered saline. Protein bands were detected by chemiluminescence (Western Lightning; Perkin Elmer Life Sciences, Boston, MA, USA) and exposed on X-Omat Blue autoradiographic film (Kodak, Rochester, NY, USA). Films were scanned and optical density determined using the NIH Image program (NIH, Bethesda, MD, USA).
Statistics
For conditioned place preference, difference score data and for western immunoblots, the optical density data were analyzed by one-way ANOVA followed by post hoc comparisons (Fisher’s probability of least significant difference) between treatment groups using Statview software (SAS Institute Inc., Cary, NC, USA).
Results
Prior to western blot analysis described in Figs 2–5, mice were assessed for conditioned place preference behavior as outlined in Fig. 1(a). Mice were randomly assigned to three treatment groups. These included mice treated with saline (days 2–4, Fig. 1a) and reintroduced to the saline-paired chamber 48 h following behavioral testing on day 5 (saline-paired) (Fig. 1a) and mice that were treated with cocaine and reintroduced to the cocaine-paired chamber (cocaine-paired) or reintroduced to the saline-paired chamber (cocaine-unpaired). To evaluate preference for cocaine, difference score was calculated as time spent in the cocaine-paired chamber on the test day minus time spent in the cocaine-paired chamber on the pre-conditioning day (Fig. 1a).
Fig. 2.

Quantitative analysis of protein levels in the nucleus accumbens (NAc) of saline-paired, cocaine-paired, and cocaine-unpaired mice. (a) Punches of NAc tissue (shell and core, +1.7 mm relative to bregma) (Paxinos and Franklin 2003), were obtained from mice from saline-paired (SAL), cocaine-paired (COC), and cocaine-unpaired (UNP) treatment groups. (b) Analysis of protein levels of CREB, DARPP-32, ERK2, and GluR1 in the NAc of mice using western blot analysis from the above three treatment groups. Protein levels were normalized to actin and values are shown as fold-induction (mean optical density ± SEM) compared with saline-paired controls. No differences were observed between any of the treatment conditions; n = 6–9 mice per treatment group for each protein examined.
Fig. 5.

Quantitative analysis of phosphorylated protein levels in the dorsal hippocampus (DHC) of saline-paired, cocaine-paired, and cocaine-unpaired mice. Phosphorylation levels of Ser133 P-CREB, Tyr185 P-ERK2, and Ser845 P-GluR1 in saline-paired (SAL), cocaine-paired (COC), and cocaine-unpaired (UNP) mice were detected by western blot analysis. Protein phosphorylation levels were normalized to their respective total protein levels and values are shown as fold-induction (mean optical density ± SEM) compared with saline-paired controls. *p < 0.05 versus saline-paired and cocaine-unpaired groups; n = 6–9 mice per group for each protein examined.
Conditioned place preference
A comparison of the difference in time spent on the drug paired side revealed a main effect of drug treatment (F2,23 = 9.526, p = 0.001). Post hoc analysis revealed that mice given cocaine had significantly higher difference scores compared to mice in the saline-paired group (p < 0.01; Fig. 1b). Data for the saline-paired group were collapsed for both time and chamber color because mice received saline in the AM and PM sessions and in both black and white chambers; however, they did not reveal main effects of either variable (i.e. no difference for AM vs. PM, nor black chamber vs. white chamber respectively). As no significant differences in time spent in a chamber were observed in any of the analyses performed for the saline-paired mice, only data for AM pairing (same time as cocaine pairing for the cocaine group) is presented in Fig. 1 (PM pairing data is not shown).
Western immunoblotting
Forty-eight hours following behavioral assessment on test day the saline-paired, cocaine-paired, and cocaine-unpaired mice were reintroduced to the saline-, cocaine-, or saline-paired chambers, respectively and remained there for 20 min. Immediately following, all mice were rapidly decapitated and NAc and DHC tissue was used for western blot analysis.
Nucleus accumbens
Examination of total protein levels in the NAc of mice from saline-paired, cocaine-paired, and cocaine-unpaired groups revealed no difference in CREB, DARPP-32, ERK2, or GluR1 between the three groups (Fig. 2b). Analysis of phosphorylated forms of the above proteins (Fig. 3) revealed a main effect of treatment condition on levels of Ser133 P-CREB (F2,21 = 3.545; p < 0.05), Thr34 P-DARPP-32 (F2,14 = 3.796; p < 0.05), and Thr75 P-DARPP-32 (F2,20 = 4.636; p < 0.05). Ser133 P-CREB and Thr34 P-DARPP-32 levels were significantly higher and Thr75 P-DARPP-32 was significantly lower in the NAc of cocaine-paired mice compared with saline-paired and cocaine-unpaired mice. No significant difference in phosphorylated protein levels of P-ERK2 or Ser845 phospho-GluR1 was observed between the three groups (Fig. 3).
Fig. 3.

Quantitative analysis of phosphorylated protein levels in the nucleus accumbens (NAc) of saline-paired, cocaine-paired, and cocaine-unpaired mice. Phosphorylation levels of Ser133 P-CREB, Thr34 and Thr75 P-DARPP-32, Tyr 185 P-ERK2 and Ser845 P-GluR1 in saline-paired (SAL), cocaine-paired (COC) and cocaine-unpaired (UNP) mice were detected by western blot analysis. Protein phosphorylation levels were normalized to their respective total protein levels and values are shown as fold-induction (mean optical density ± SEM) compared with saline-paired controls. *p < 0.05 versus saline-paired and cocaine-unpaired groups; n = 6–9 mice per group for each protein examined.
Hippocampus
Examination of total protein levels in the DHC of mice from saline-paired, cocaine-paired, and cocaine-opposite treatment groups revealed no difference in CREB, ERK2, or GluR1 between the three groups (Fig. 4b). Analysis of phosphorylated forms of the above proteins (Fig. 5) revealed a main effect of treatment condition on Ser133 P-CREB (F2,21 = 3.478; p < 0.05), Tyr185 P-ERK2 (F2,21 = 4.907; p < 0.05), and Ser845 phospho-GluR1 (F2,21 = 5.635; p < 0.05) levels. Ser133 P-CREB, Tyr185 P-ERK2, and Ser845 phospho-GluR1 levels were significantly higher in the DHC of cocaine-paired mice exposed to the chamber in which they received cocaine compared with saline-paired and cocaine-unpaired mice exposed to the chamber in which they received saline (Fig. 5).
Fig. 4.

Quantitative analysis of protein levels in the dorsal hippocampus (DHC) of saline-paired, cocaine-paired, and cocaine-unpaired mice. (a) Punches of DHC tissue +2.7 mm relative to bregma (Paxinos and Franklin 2003), were obtained from mice from saline-paired (SAL), cocaine-paired (COC), and cocaine-unpaired (UNP) treatment groups. (b) Analysis of protein levels of CREB, ERK2, and GluR1 in the DHC of mice using western blot analysis from the above three treatment groups. Protein levels were normalized to actin and values are shown as fold-induction (mean optical density ± SEM) compared with saline-paired controls. No differences were observed between any of the treatment conditions; n = 6–9 mice per treatment group for each protein examined.
Discussion
The present study examined changes in total protein levels and phosphorylated forms of the molecular substrates CREB, DARPP-32, ERK2, and GluR1 in the NAc and DHC in mice that had undergone cocaine CPP followed by re-exposure to the environment in which cocaine or saline was given. Upon re-exposure to the chamber in which the mice were given cocaine higher P-CREB levels were found in both the NAc and DHC compared with saline or cocaine-treated mice re-exposed to the saline-paired environment. In the NAc there were correspondingly higher levels of Thr34 P-DARPP-32 and lower levels of Thr75 P-DARPP-32. Examination of P-ERK2 and phospho-GluR1 levels revealed no difference. In the DHC higher levels of P-ERK2 and phospho-GluR1 were observed. These data suggest that association of environmental context with drug reward involves recruitment of the DHC–NAc circuit by activation of the DARPP-32/CREB pathway in the NAc and the ERK/CREB pathway in the DHC.
Nucleus accumbens pathways and cocaine-conditioned place preference
Dopamine in the NAc is the key neurotransmitter that mediates drug reward (Wise and Rompre 1989; Wise and Hoffman 1992) primarily via D1 dopamine receptors. Systemic D1 but not D2 dopamine receptor antagonists block the development of place preference to cocaine (Cervo and Samanin 1995; Graham et al. 2007). Similarly microinjection of the D1 antagonist SCH23390, but not the D2 antagonist sulpiride into the NAc blocks cocaine place preference (Baker et al. 1996, 1998).
The precise signaling pathway that is involved in mediating cocaine preference is not yet fully understood. We found that mice exhibiting cocaine preference have higher levels of CREB phosphorylated at Ser133. One route to CREB phosphorylation in the NAc is via D1 receptor activated PKA, which leads to phosphorylation of DARPP-32 at Thr34 thus inhibiting protein phosphatase 1, a potent inhibitor of Ser133 CREB phosphorylation (Hemmings et al. 1984). Additionally, cocaine-mediated decreases in phosphorylation of DARPP-32 at Thr75 has been shown to release the inhibition of PKA (Bibb et al. 1999). Both of these events lead to increases in Ser133 P-CREB. Based on the above observations, our findings of increased levels of Thr34 P-DARPP-32 and P-CREB with a corresponding decrease in Thr75 P-DARPP-32 suggests that the D1/PKA pathway is recruited in the NAc upon re-exposure of cocaine-treated mice to the environment in which they were given cocaine. This agrees with previously published work suggesting recruitment of the D1 dopamine pathway following exposure to drug cues (Crombag et al. 2002) and D1 receptors specifically in the NAc following cocaine-primed reinstatement (Schmidt et al. 2006) (Fig. 6).
Fig. 6.
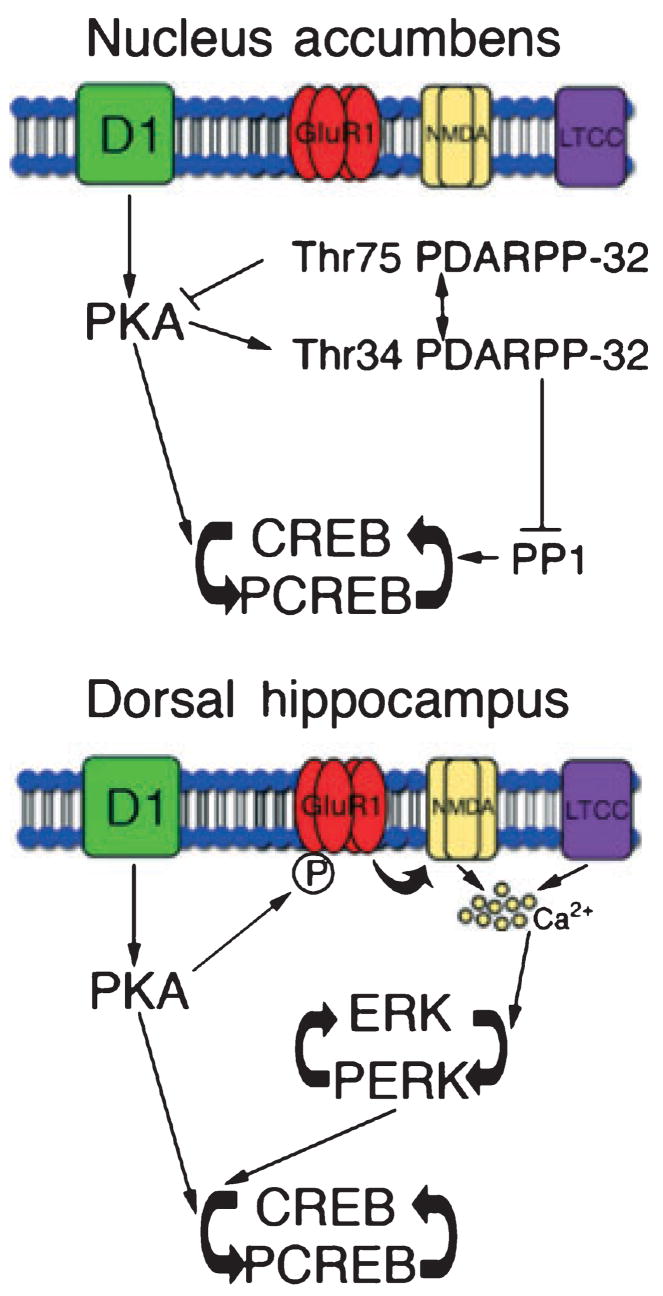
Schematic representation of signal transduction pathways activated in the nucleus accumbens (NAc) and dorsal hippocampus (DHC) in cocaine-place preference behavior. We propose that in the NAc activation of the D1/PKA/DARPP-32 pathway results in higher levels of P-CREB following re-exposure to a previously rewarding environment. Higher levels of Thr34 P-DARPP-32 inhibits protein phosphatase 1 (PP1), a CREB phosphatase and lower levels of Thr75 P-DARPP-32, a PKA inhibitor would lead to increased PKA activity. Both of these events would lead to higher levels of P-CREB. In the DHC, we propose that in addition to the D1/PKA pathway the GluR1/NMDA/L-type Ca2+ channel/Ca2+ pathway via ERK results in higher levels of P-CREB. Lines with arrows indicate activation while lines with bars indicate inhibition.
In our model, the finding of increased Thr34 P-DARPP-32 in the NAc is in agreement with the important role of phosphorylation of DARPP-32 at Thr34 in cocaine CPP; and is consistent with the fact that Thr34 mutants do not develop a place preference to cocaine (Zachariou et al. 2006). However, our finding of a lack of an increase in P-ERK2 following re-exposure differs from previous work showing an important role for the ERK pathway in cocaine CPP (Valjent et al. 2000, 2004; Miller and Marshall 2005). For example, ERK1 knockout mice that have increased ERK2 signaling show an increased ability to develop a place preference (Ferguson et al. 2006). Furthermore, Miller and Marshall (2005) show that during the test period place preference can be blocked by mitogen-activated protein kinase kinase (MEK) inhibitor microinjection into the NAc core thus blocking ERK phosphorylation. The difference between our results and those of Miller and Marshall may be attributable to (i) the differences between a chamber preference test performed by Miller and Marshall versus drug-paired chamber re-exposure, (ii) the difference in time points that were examined (on test day by Miller and Marshal vs. 48 h following test day) or (iii) the difference in anatomical regions examined. Our data reflect ERK phosphorylation levels examined in the mouse NAc core and shell whereas Miller and Marshall found increased levels of ERK phosphorylation specifically in the rat NAc core and not the NAc shell.
In our study, we did not observe a change in total GluR1 levels in the NAc when examined 2 days following cocaine treatment or a change in phospho-GluR1 levels after re-exposure to the cocaine-paired environment, which is consistent with others who have found that GluR1 does not play a role in cocaine cue-induced drug seeking (by using either over-expression of GluR1 in the NAc or GluR1 genetic knockout mice) (Mead et al. 2007; Bachtell et al. 2008). However intra-NAc administration of α-amino-3-hydroxy-5-methylisoxazole-4-propionate (AMPA) receptor antagonists have been shown to block cue-induced cocaine-seeking behavior (Di Ciano and Everitt 2001; Backstrom and Hyytia 2007) and NAc GluR1 has been shown to mediate incubation of cue-induced cocaine seeking by enhancing drug seeking 45 days following drug self-administration (Conrad et al. 2008). In addition, an important role of AMPA receptor regulation in cocaine-induced drug-seeking (Cornish and Kalivas 2000; Park et al. 2002; Suto et al. 2004) and sensitizing behaviors (Pierce et al. 1996; Boudreau and Wolf 2005; Boudreau et al. 2007; Kourrich et al. 2007) has been reported. The different time points examined in this study compared with Conrad et al. (2008) that also examined GluR1 protein levels may explain the divergent results. We examined protein changes 2 days following cocaine treatment, a time point at which Conrad et al. (2008) demonstrate that GluR1 contributes to a lesser extent to cue-induced drug seeking than at later time points. Examination of GluR1 and Ser845 phospho-GluR1 levels at later time points in our CPP protocol could possibly uncover a role for GluR1. Furthermore, the differences observed between our study and drug-seeking behavior could be attributed to the different molecular pathways involved in non-contingent drug re-exposure and drug-seeking behavior following self-administration versus drug context re-exposure and place preference in the CPP model. Further studies would be necessary to delineate the specific molecular pathways underlying the discrete aspects of CPP versus self-administration behavior.
Hippocampal pathways and cocaine-conditioned place preference
Dorsal hippocampus function is essential to cocaine CPP as ablation of the DHC has been shown to block development of cocaine CPP (Meyers et al. 2003). The molecular changes we report here in the DHC in cocaine CPP is consistent with previous work that has shown an essential role of the DHC in self-administration reinstatement by contextual cues but not explicit conditioned stimuli or cocaine-priming (Fuchs et al. 2005), nor recall of contextual memories (Hall et al. 2001).
Dopaminergic neurons originating from the VTA project to the CA1 and dentate gyrus of the hippocampus (Gasbarri et al. 1994) and are known to modulate the formation of long-term memory (Lisman and Grace 2005). The DHC dopaminergic systems’ involvement in cocaine reward has not been well studied. However, previous work has shown that stimulation of dopamine D1 or D2 receptors in the DHC facilitates the development of morphine-induced CPP (Rezayof et al. 2003). Given that both morphine and cocaine activate VTA-NAc dopaminergic neurons (Beitner-Johnson and Nestler 1991), it is possible that morphine and cocaine have similar effects on VTA-hippocampal projections.
Dopaminergic D1 receptor activation of DHC neurons can lead to increased synaptic plasticity via increased AMPA and NMDA function (Yang 2000; Gao et al. 2006). In the hippocampus PKA activation directly phosphorylates GluR1 at Ser845 (Esteban et al. 2003). In our model following re-exposure to the chamber in which cocaine was administered we find higher levels of Ser845 phospho-GluR1, which may result from dopamine activation of the D1-PKA pathway. This heightened phosphorylation of GluR1 suggests that mechanisms of drug reward result in changes in synaptic plasticity of the DHC (Fig. 6).
In the DHC, in addition to dopamine D1 receptors, activation of glutamatergic, L-type Ca2+ channel, or neurotrophin signaling could result in activation of the CREB and ERK pathway (Ghosh and Greenberg 1995; Wu et al. 2001; Minichiello et al. 2002; Deisseroth et al. 2003; Pokorska et al. 2003) (Fig. 6). This is in agreement with cocaine’s ability to increase ERK phosphorylation in the hippocampus independent of D1 receptors (Valjent et al. 2004). In our model, we find higher levels of ERK phosphorylation in the DHC in mice re-exposed to the environment in which cocaine was given that could result via either pathway. A recent study has reported increased expression of the neurotrophin receptor tyrosine receptor kinase B (TrkB) in the hippocampus following amphetamine CPP (Shen et al. 2006) and a role for hippocampal Cav1.2 L-type Ca2+ channel-activated CREB and ERK has been identified in spatial memory (Moosmang et al. 2005). An essential role for the CREB/ERK pathway in contextual memory has been shown in the contextual fear model (Ahi et al. 2004; Thomas and Huganir 2004; Chen et al. 2005; Sindreu et al. 2007). Higher levels of CREB and ERK phosphorylation in the CA1 region of the hippocampus (sampled in our study) were observed following contextual fear conditioning and inhibitors of ERK phosphorylation blocked the development of context- and tone-dependent fear conditioning (Ahi et al. 2004). Additionally, in a contextual fear memory recall task higher levels of ERK phosphorylation were observed in the DHC (Chen et al. 2005). This suggests that activation of ERK in the DHC is important for recall of contextual memories related to drugs and fear.
CREB and cocaine-conditioned place preference
The link between CREB function and drug reward has been characterized in numerous reviews (Blendy and Maldonado 1998; Carlezon et al. 1998; Chao and Nestler 2004; Nestler 2004). Despite extensive research studying this link the evidence for CREB’s role in drug reward remains difficult to interpret. Upon acute cocaine exposure CREB phosphorylation transiently increases in the NAc (Walters et al. 2003), which leads to increased excitability of NAc neurons (Dong et al. 2006), a potential mechanism of drug reward. However, studies using transgenic mice with reduced CREB function and viral vector delivery of inhibitors of CREB imply that decreases in CREB, not increases in CREB, mediate the rewarding properties of cocaine, amphetamine, and morphine (Carlezon et al. 1998; Walters and Blendy 2001; Barrot et al. 2002). It seems possible that the induction of CREB phosphorylation following acute cocaine initiates a negative feedback mechanism causing reduced basal dopamine activity (Cole et al. 1995). According to this theory, exposure to cocaine would lead to increased dopamine activity leading to euphoria during periods of drug use, and dysphoria during drug-free periods because of the reduced basal dopamine activity (Carlezon et al. 1998). It is clear from the present results that exposure to a drug-associated context remains a potent activator of signaling pathways leading to CREB phosphorylation in the reward pathways of the brain indicating a sustained susceptibility to plasticity-related protein activation during drug-related cue exposure.
The CREB function in the DHC of mice given morphine has shown that CREB phosphorylation increases during the conditioning phase, as well as during morphine-reinstatement of place preference behavior (Zhou and Zhu 2006), which may suggest a functional role for CREB in the development and recall of morphine-induced place preference. Furthermore, a reduction in CREB function in the DHC of mice impairs delayed spatial-task memory suggesting that CREB in the DHC is essential for long-term memory formation (Bourtchuladze et al. 1994). CREB has also been shown to be responsible for the stability of retrieved or reactivated fear conditioned contextual memories (Kida et al. 2002). Taken together it seems CREB activation in the DHC plays an important role in the development and stability of long-term contextual memories.
In conclusion, an in-depth analysis of changes in the molecular pathways of drug reward and aberrant learning resulting from drug exposure is essential to understanding the progression to compulsive drug use and abuse relapse. Evidence from our study shows that discrete molecular signaling pathways play an important role in drug abuse, providing insights regarding molecular substrates in the NAc and DHC that are associated with context-induced adaptations.
Acknowledgments
This work has been supported by the Department of Pediatrics, Division of Pediatric Neurology, Weill Cornell Medical College funds (AMR) and National Institute of Drug Abuse (NIDA) Grant, KO2DA00354 (BEK). We thank Gagandeep Kaur for her technical assistance with the western blot analysis.
Abbreviations
- AMPA
α-amino-3-hydroxy-5-methylisoxazole-4-propionate
- CPP
cocaine-conditioned place preference
- DARPP-32
dopamine- and cyclic AMP-regulated phosphoprotein 32
- DHC
dorsal hippocampus
- ERK
extracellular signal-regulated kinase
- NAc
nucleus accumbens
- P-CREB
phospho-cAMP-response element binding protein
- P-DARPP-32
phospho-DARPP-32
- P-ERK
phospho-ERK
- phospho-GluR1
GluR1 phosphorylation
- PKA
protein kinase A
- VTA
ventral tegmental area
References
- Ahi J, Radulovic J, Spiess J. The role of hippocampal signaling cascades in consolidation of fear memory. Behav Brain Res. 2004;149:17–31. doi: 10.1016/s0166-4328(03)00207-9. [DOI] [PubMed] [Google Scholar]
- Bachtell RK, Choi KH, Simmons DL, Falcon E, Monteggia LM, Neve RL, Self DW. Role of GluR1 expression in nucleus accumbens neurons in cocaine sensitization and cocaine-seeking behavior. Eur J Neurosci. 2008;27:2229–2240. doi: 10.1111/j.1460-9568.2008.06199.x. [DOI] [PubMed] [Google Scholar]
- Backstrom P, Hyytia P. Involvement of AMPA/kainate, NMDA, and mGlu5 receptors in the nucleus accumbens core in cue-induced reinstatement of cocaine seeking in rats. Psychopharmacology (Berl) 2007;192:571–580. doi: 10.1007/s00213-007-0753-8. [DOI] [PubMed] [Google Scholar]
- Baker DA, Khroyan TV, O’Dell LE, Fuchs RA, Neisewander JL. Differential effects of intraaccumbens sulpiride on cocaine-induced locomotion and conditioned place preference. J Pharmacol Exp Ther. 1996;279:392–401. [PubMed] [Google Scholar]
- Baker DA, Fuchs RA, Specio SE, Khroyan TV, Neisewander JL. Effects of intraaccumbens administration of SCH-23390 on cocaine-induced locomotion and conditioned place preference. Synapse. 1998;30:181–193. doi: 10.1002/(SICI)1098-2396(199810)30:2<181::AID-SYN8>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- Bardo MT. Neuropharmacological mechanisms of drug reward: beyond dopamine in the nucleus accumbens. Crit Rev Neurobiol. 1998;12:37–67. doi: 10.1615/critrevneurobiol.v12.i1-2.30. [DOI] [PubMed] [Google Scholar]
- Bardo MT, Bevins RA. Conditioned place preference: what does it add to our preclinical understanding of drug reward? Psychopharmacology (Berl) 2000;153:31–43. doi: 10.1007/s002130000569. [DOI] [PubMed] [Google Scholar]
- Barrot M, Olivier JD, Perrotti LI, et al. CREB activity in the nucleus accumbens shell controls gating of behavioral responses to emotional stimuli. Proc Natl Acad Sci USA. 2002;99:11435–11440. doi: 10.1073/pnas.172091899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beitner-Johnson D, Nestler EJ. Morphine and cocaine exert common chronic actions on tyrosine hydroxylase in dopaminergic brain reward regions. J Neurochem. 1991;57:344–347. doi: 10.1111/j.1471-4159.1991.tb02133.x. [DOI] [PubMed] [Google Scholar]
- Berke JD, Hyman SE. Addiction, dopamine, and the molecular mechanisms of memory. Neuron. 2000;25:515–532. doi: 10.1016/s0896-6273(00)81056-9. [DOI] [PubMed] [Google Scholar]
- Bibb JA, Snyder GL, Nishi A, et al. Phosphorylation of DARPP-32 by Cdk5 modulates dopamine signalling in neurons. Nature. 1999;402:669–671. doi: 10.1038/45251. [DOI] [PubMed] [Google Scholar]
- Blendy JA, Maldonado R. Genetic analysis of drug addiction: the role of cAMP response element binding protein. J Mol Med. 1998;76:104–110. doi: 10.1007/s001090050197. [DOI] [PubMed] [Google Scholar]
- Boudreau AC, Wolf ME. Behavioral sensitization to cocaine is associated with increased AMPA receptor surface expression in the nucleus accumbens. J Neurosci. 2005;25:9144–9151. doi: 10.1523/JNEUROSCI.2252-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boudreau AC, Reimers JM, Milovanovic M, Wolf ME. Cell surface AMPA receptors in the rat nucleus accumbens increase during cocaine withdrawal but internalize after cocaine challenge in association with altered activation of mitogen-activated protein kinases. J Neurosci. 2007;27:10621–10635. doi: 10.1523/JNEUROSCI.2163-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourtchuladze R, Frenguelli B, Blendy J, Cioffi D, Schutz G, Silva AJ. Deficient long-term memory in mice with a targeted mutation of the cAMP-responsive element-binding protein. Cell. 1994;79:59–68. doi: 10.1016/0092-8674(94)90400-6. [DOI] [PubMed] [Google Scholar]
- Brami-Cherrier K, Valjent E, Herve D, Darragh J, Corvol JC, Pages C, Arthur SJ, Girault JA, Caboche J. Parsing molecular and behavioral effects of cocaine in mitogen- and stress-activated protein kinase-1-deficient mice. J Neurosci. 2005;25:11444–11454. doi: 10.1523/JNEUROSCI.1711-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brightwell JJ, Smith CA, Neve RL, Colombo PJ. Long-term memory for place learning is facilitated by expression of cAMP response element-binding protein in the dorsal hippocampus. Learn Mem. 2007;14:195–199. doi: 10.1101/lm.395407. [DOI] [PubMed] [Google Scholar]
- Carlezon WA, Jr, Thome J, Olson VG, Lane-Ladd SB, Brodkin ES, Hiroi N, Duman RS, Neve RL, Nestler EJ. Regulation of cocaine reward by CREB. Science. 1998;282:2272–2275. doi: 10.1126/science.282.5397.2272. [DOI] [PubMed] [Google Scholar]
- Cervo L, Samanin R. Effects of dopaminergic and glutamatergic receptor antagonists on the acquisition and expression of cocaine conditioning place preference. Brain Res. 1995;673:242–250. doi: 10.1016/0006-8993(94)01420-m. [DOI] [PubMed] [Google Scholar]
- Chao J, Nestler EJ. Molecular neurobiology of drug addiction. Annu Rev Med. 2004;55:113–132. doi: 10.1146/annurev.med.55.091902.103730. [DOI] [PubMed] [Google Scholar]
- Chen X, Garelick MG, Wang H, Lil V, Athos J, Storm DR. PI3 kinase signaling is required for retrieval and extinction of contextual memory. Nat Neurosci. 2005;8:925–931. doi: 10.1038/nn1482. [DOI] [PubMed] [Google Scholar]
- Cole RL, Konradi C, Douglass J, Hyman SE. Neuronal adaptation to amphetamine and dopamine: molecular mechanisms of prodynorphin gene regulation in rat striatum. Neuron. 1995;14:813–823. doi: 10.1016/0896-6273(95)90225-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conrad KL, Tseng KY, Uejima JL, Reimers JM, Heng LJ, Shaham Y, Marinelli M, Wolf ME. Formation of accumbens GluR2-lacking AMPA receptors mediates incubation of cocaine craving. Nature. 2008 doi: 10.1038/nature06995. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cornish JL, Kalivas PW. Glutamate transmission in the nucleus accumbens mediates relapse in cocaine addiction. J Neurosci. 2000;20:RC89. doi: 10.1523/JNEUROSCI.20-15-j0006.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crombag HS, Grimm JW, Shaham Y. Effect of dopamine receptor antagonists on renewal of cocaine seeking by reexposure to drug-associated contextual cues. Neuropsychopharmacology. 2002;27:1006–1015. doi: 10.1016/S0893-133X(02)00356-1. [DOI] [PubMed] [Google Scholar]
- Deak M, Clifton AD, Lucocq LM, Alessi DR. Mitogen-and stress-activated protein kinase-1 (MSK1) is directly activated by MAPK and SAPK2/p38, and may mediate activation of CREB. EMBO J. 1998;17:4426–4441. doi: 10.1093/emboj/17.15.4426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deisseroth K, Mermelstein PG, Xia H, Tsien RW. Signaling from synapse to nucleus: the logic behind the mechanisms. Curr Opin Neurobiol. 2003;13:354–365. doi: 10.1016/s0959-4388(03)00076-x. [DOI] [PubMed] [Google Scholar]
- Di Ciano P, Everitt BJ. Dissociable effects of antagonism of NMDA and AMPA/KA receptors in the nucleus accumbens core and shell on cocaine-seeking behavior. Neuropsychopharmacology. 2001;25:341–360. doi: 10.1016/S0893-133X(01)00235-4. [DOI] [PubMed] [Google Scholar]
- Dong Y, Green T, Saal D, Marie H, Neve R, Nestler EJ, Malenka RC. CREB modulates excitability of nucleus accumbens neurons. Nat Neurosci. 2006;9:475–477. doi: 10.1038/nn1661. [DOI] [PubMed] [Google Scholar]
- Esteban JA, Shi SH, Wilson C, Nuriya M, Huganir RL, Malinow R. PKA phosphorylation of AMPA receptor subunits controls synaptic trafficking underlying plasticity. Nat Neurosci. 2003;6:136–143. doi: 10.1038/nn997. [DOI] [PubMed] [Google Scholar]
- Ferguson SM, Fasano S, Yang P, Brambilla R, Robinson TE. Knockout of ERK1 enhances cocaine-evoked immediate early gene expression and behavioral plasticity. Neuropsychopharmacology. 2006;31:2660–2668. doi: 10.1038/sj.npp.1301014. [DOI] [PubMed] [Google Scholar]
- Fuchs RA, Evans KA, Ledford CC, Parker MP, Case JM, Mehta RH, See RE. The role of the dorsomedial prefrontal cortex, basolateral amygdala, and dorsal hippocampus in contextual reinstatement of cocaine seeking in rats. Neuropsychopharmacology. 2005;30:296–309. doi: 10.1038/sj.npp.1300579. [DOI] [PubMed] [Google Scholar]
- Gao C, Sun X, Wolf ME. Activation of D1 dopamine receptors increases surface expression of AMPA receptors and facilitates their synaptic incorporation in cultured hippocampal neurons. J Neurochem. 2006;98:1664–1677. doi: 10.1111/j.1471-4159.2006.03999.x. [DOI] [PubMed] [Google Scholar]
- Gasbarri A, Verney C, Innocenzi R, Campana E, Pacitti C. Mesolimbic dopaminergic neurons innervating the hippocampal formation in the rat: a combined retrograde tracing and immunohistochemical study. Brain Res. 1994;668:71–79. doi: 10.1016/0006-8993(94)90512-6. [DOI] [PubMed] [Google Scholar]
- Ghosh A, Greenberg ME. Calcium signaling in neurons: molecular mechanisms and cellular consequences. Science. 1995;268:239–247. doi: 10.1126/science.7716515. [DOI] [PubMed] [Google Scholar]
- Gonzalez GA, Montminy MR. Cyclic AMP stimulates somatostatin gene transcription by phosphorylation of CREB at serine 133. Cell. 1989;59:675–680. doi: 10.1016/0092-8674(89)90013-5. [DOI] [PubMed] [Google Scholar]
- Graham DL, Hoppenot R, Hendryx A, Self DW. Differential ability of D1 and D2 dopamine receptor agonists to induce and modulate expression and reinstatement of cocaine place preference in rats. Psychopharmacology (Berl) 2007;191:719–730. doi: 10.1007/s00213-006-0473-5. [DOI] [PubMed] [Google Scholar]
- Hall J, Thomas KL, Everitt BJ. Fear memory retrieval induces CREB phosphorylation and Fos expression within the amygdala. Eur J Neurosci. 2001;13:1453–1458. doi: 10.1046/j.0953-816x.2001.01531.x. [DOI] [PubMed] [Google Scholar]
- Halpain S, Girault JA, Greengard P. Activation of NMDA receptors induces dephosphorylation of DARPP-32 in rat striatal slices. Nature. 1990;343:369–372. doi: 10.1038/343369a0. [DOI] [PubMed] [Google Scholar]
- Hemmings HC, Jr, Nairn AC, Aswad DW, Greengard P. DARPP-32, a dopamine- and adenosine 3′:5′-monophosphate-regulated phosphoprotein enriched in dopamine-innervated brain regions. II Purification and characterization of the phosphoprotein from bovine caudate nucleus. J Neurosci. 1984;4:99–110. doi: 10.1523/JNEUROSCI.04-01-00099.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyman SE. Addiction: a disease of learning and memory. Am J Psychiatry. 2005;162:1414–1422. doi: 10.1176/appi.ajp.162.8.1414. [DOI] [PubMed] [Google Scholar]
- Hyman SE, Malenka RC. Addiction and the brain: the neurobiology of compulsion and its persistence. Nat Rev Neurosci. 2001;2:695–703. doi: 10.1038/35094560. [DOI] [PubMed] [Google Scholar]
- Hyman SE, Malenka RC, Nestler EJ. Neural mechanisms of addiction: the role of reward-related learning and memory. Annu Rev Neurosci. 2006;29:565–598. doi: 10.1146/annurev.neuro.29.051605.113009. [DOI] [PubMed] [Google Scholar]
- Kelley AE. Memory and addiction: shared neural circuitry and molecular mechanisms. Neuron. 2004;44:161–179. doi: 10.1016/j.neuron.2004.09.016. [DOI] [PubMed] [Google Scholar]
- Kida S, Josselyn SA, de Ortiz SP, Kogan JH, Chevere I, Masushige S, Silva AJ. CREB required for the stability of new and reactivated fear memories. Nat Neurosci. 2002;5:348–355. doi: 10.1038/nn819. [DOI] [PubMed] [Google Scholar]
- Kourrich S, Rothwell PE, Klug JR, Thomas MJ. Cocaine experience controls bidirectional synaptic plasticity in the nucleus accumbens. J Neurosci. 2007;27:7921–7928. doi: 10.1523/JNEUROSCI.1859-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuo YM, Liang KC, Chen HH, Cherng CG, Lee HT, Lin Y, Huang AM, Liao RM, Yu L. Cocaine-but not methamphetamine-associated memory requires de novo protein synthesis. Neurobiol Learn Mem. 2007;87:93–100. doi: 10.1016/j.nlm.2006.06.004. [DOI] [PubMed] [Google Scholar]
- Lisman JE, Grace AA. The hippocampal-VTA loop: controlling the entry of information into long-term memory. Neuron. 2005;46:703–713. doi: 10.1016/j.neuron.2005.05.002. [DOI] [PubMed] [Google Scholar]
- Mayr B, Montminy M. Transcriptional regulation by the phosphorylation-dependent factor CREB. Nat Rev Mol Cell Biol. 2001;2:599–609. doi: 10.1038/35085068. [DOI] [PubMed] [Google Scholar]
- Mead AN, Zamanillo D, Becker N, Stephens DN. AMPA-receptor GluR1 subunits are involved in the control over behavior by cocaine-paired cues. Neuropsychopharmacology. 2007;32:343–353. doi: 10.1038/sj.npp.1301045. [DOI] [PubMed] [Google Scholar]
- Meyers RA, Zavala AR, Neisewander JL. Dorsal, but not ventral, hippocampal lesions disrupt cocaine place conditioning. Neuroreport. 2003;14:2127–2131. doi: 10.1097/00001756-200311140-00023. [DOI] [PubMed] [Google Scholar]
- Miller CA, Marshall JF. Molecular substrates for retrieval and reconsolidation of cocaine-associated contextual memory. Neuron. 2005;47:873–884. doi: 10.1016/j.neuron.2005.08.006. [DOI] [PubMed] [Google Scholar]
- Minichiello L, Calella AM, Medina DL, Bonhoeffer T, Klein R, Korte M. Mechanism of TrkB-mediated hippocampal long-term potentiation. Neuron. 2002;36:121–137. doi: 10.1016/s0896-6273(02)00942-x. [DOI] [PubMed] [Google Scholar]
- Moosmang S, Haider N, Klugbauer N, et al. Role of hippocampal Cav1.2 Ca2+ channels in NMDA receptor-independent synaptic plasticity and spatial memory. J Neurosci. 2005;25:9883–9892. doi: 10.1523/JNEUROSCI.1531-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nairn AC, Svenningsson P, Nishi A, Fisone G, Girault JA, Greengard P. The role of DARPP-32 in the actions of drugs of abuse. Neuropharmacology. 2004;47 (Suppl 1):14–23. doi: 10.1016/j.neuropharm.2004.05.010. [DOI] [PubMed] [Google Scholar]
- Nestler EJ. Molecular basis of long-term plasticity underlying addiction. Nat Rev Neurosci. 2001;2:119–128. doi: 10.1038/35053570. [DOI] [PubMed] [Google Scholar]
- Nestler EJ. Molecular mechanisms of drug addiction. Neuropharmacology. 2004;47 (Suppl 1):24–32. doi: 10.1016/j.neuropharm.2004.06.031. [DOI] [PubMed] [Google Scholar]
- Park WK, Bari AA, Jey AR, Anderson SM, Spealman RD, Rowlett JK, Pierce RC. Cocaine administered into the medial prefrontal cortex reinstates cocaine-seeking behavior by increasing AMPA receptor-mediated glutamate transmission in the nucleus accumbens. J Neurosci. 2002;22:2916–2925. doi: 10.1523/JNEUROSCI.22-07-02916.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paxinos G, Franklin K. The Mouse Brain in Stereotaxic Coordinates. 2. Academic Press; New York: 2003. [Google Scholar]
- Pierce RC, Bell K, Duffy P, Kalivas PW. Repeated cocaine augments excitatory amino acid transmission in the nucleus accumbens only in rats having developed behavioral sensitization. J Neurosci. 1996;16:1550–1560. doi: 10.1523/JNEUROSCI.16-04-01550.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pokorska A, Vanhoutte P, Arnold FJ, Silvagno F, Hardingham GE, Bading H. Synaptic activity induces signalling to CREB without increasing global levels of cAMP in hippocampal neurons. J Neurochem. 2003;84:447–452. doi: 10.1046/j.1471-4159.2003.01504.x. [DOI] [PubMed] [Google Scholar]
- Rezayof A, Zarrindast MR, Sahraei H, Haeri-Rohani A. Involvement of dopamine receptors of the dorsal hippocampus on the acquisition and expression of morphine-induced place preference in rats. J Psychopharmacol. 2003;17:415–423. doi: 10.1177/0269881103174005. [DOI] [PubMed] [Google Scholar]
- Roma PG, Riley AL. Apparatus bias and the use of light and texture in place conditioning. Pharmacol Biochem Behav. 2005;82:163–169. doi: 10.1016/j.pbb.2005.08.004. [DOI] [PubMed] [Google Scholar]
- Schmidt HD, Anderson SM, Pierce RC. Stimulation of D1-like or D2 dopamine receptors in the shell, but not the core, of the nucleus accumbens reinstates cocaine-seeking behaviour in the rat. Eur J Neurosci. 2006;23:219–228. doi: 10.1111/j.1460-9568.2005.04524.x. [DOI] [PubMed] [Google Scholar]
- Self DW. Regulation of drug-taking and -seeking behaviors by neuroadaptations in the mesolimbic dopamine system. Neuropharmacology. 2004;47 (Suppl 1):242–255. doi: 10.1016/j.neuropharm.2004.07.005. [DOI] [PubMed] [Google Scholar]
- Shen F, Meredith GE, Napier TC. Amphetamine-induced place preference and conditioned motor sensitization requires activation of tyrosine kinase receptors in the hippocampus. J Neurosci. 2006;26:11041–11051. doi: 10.1523/JNEUROSCI.2898-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sindreu CB, Scheiner ZS, Storm DR. Ca2+-stimulated adenylyl cyclases regulate ERK-dependent activation of MSK1 during fear conditioning. Neuron. 2007;53:79–89. doi: 10.1016/j.neuron.2006.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suto N, Tanabe LM, Austin JD, Creekmore E, Pham CT, Vezina P. Previous exposure to psychostimulants enhances the reinstatement of cocaine seeking by nucleus accumbens AMPA. Neuropsychopharmacology. 2004;29:2149–2159. doi: 10.1038/sj.npp.1300533. [DOI] [PubMed] [Google Scholar]
- Svenningsson P, Nishi A, Fisone G, Girault JA, Nairn AC, Greengard P. DARPP-32: an integrator of neurotransmission. Annu Rev Pharmacol Toxicol. 2004;44:269–296. doi: 10.1146/annurev.pharmtox.44.101802.121415. [DOI] [PubMed] [Google Scholar]
- Taubenfeld SM, Wiig KA, Bear MF, Alberini CM. A molecular correlate of memory and amnesia in the hippocampus. Nat Neurosci. 1999;2:309–310. doi: 10.1038/7217. [DOI] [PubMed] [Google Scholar]
- Thomas GM, Huganir RL. MAPK cascade signalling and synaptic plasticity. Nat Rev Neurosci. 2004;5:173–183. doi: 10.1038/nrn1346. [DOI] [PubMed] [Google Scholar]
- Valjent E, Corvol JC, Pages C, Besson MJ, Maldonado R, Caboche J. Involvement of the extracellular signal-regulated kinase cascade for cocaine-rewarding properties. J Neurosci. 2000;20:8701–8709. doi: 10.1523/JNEUROSCI.20-23-08701.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valjent E, Pages C, Herve D, Girault JA, Caboche J. Addictive and non-addictive drugs induce distinct and specific patterns of ERK activation in mouse brain. Eur J Neurosci. 2004;19:1826–1836. doi: 10.1111/j.1460-9568.2004.03278.x. [DOI] [PubMed] [Google Scholar]
- Walters CL, Blendy JA. Different requirements for cAMP response element binding protein in positive and negative reinforcing properties of drugs of abuse. J Neurosci. 2001;21:9438–9444. doi: 10.1523/JNEUROSCI.21-23-09438.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walters CL, Kuo YC, Blendy JA. Differential distribution of CREB in the mesolimbic dopamine reward pathway. J Neurochem. 2003;87:1237–1244. doi: 10.1046/j.1471-4159.2003.02090.x. [DOI] [PubMed] [Google Scholar]
- Wang JQ, Arora A, Yang L, Parelkar NK, Zhang G, Liu X, Choe ES, Mao L. Phosphorylation of AMPA receptors: mechanisms and synaptic plasticity. Mol Neurobiol. 2005;32:237–249. doi: 10.1385/MN:32:3:237. [DOI] [PubMed] [Google Scholar]
- Wise RA, Hoffman DC. Localization of drug reward mechanisms by intracranial injections. Synapse. 1992;10:247–263. doi: 10.1002/syn.890100307. [DOI] [PubMed] [Google Scholar]
- Wise RA, Rompre PP. Brain dopamine and reward. Annu Rev Psychol. 1989;40:191–225. doi: 10.1146/annurev.ps.40.020189.001203. [DOI] [PubMed] [Google Scholar]
- Wolf ME, Sun X, Mangiavacchi S, Chao SZ. Psychomotor stimulants and neuronal plasticity. Neuropharmacology. 2004;47 (Suppl 1):61–79. doi: 10.1016/j.neuropharm.2004.07.006. [DOI] [PubMed] [Google Scholar]
- Wu GY, Deisseroth K, Tsien RW. Spaced stimuli stabilize MAPK pathway activation and its effects on dendritic morphology. Nat Neurosci. 2001;4:151–158. doi: 10.1038/83976. [DOI] [PubMed] [Google Scholar]
- Xing J, Ginty DD, Greenberg ME. Coupling of the RAS–MAPK pathway to gene activation by RSK2, a growth factor-regulated CREB kinase. Science. 1996;273:959–963. doi: 10.1126/science.273.5277.959. [DOI] [PubMed] [Google Scholar]
- Yang SN. Sustained enhancement of AMPA receptor- and NMDA receptor-mediated currents induced by dopamine D1/D5 receptor activation in the hippocampus: an essential role of post-synaptic Ca2+ Hippocampus. 2000;10:57–63. doi: 10.1002/(SICI)1098-1063(2000)10:1<57::AID-HIPO6>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- Zachariou V, Sgambato-Faure V, Sasaki T, Svenningsson P, Berton O, Fienberg AA, Nairn AC, Greengard P, Nestler EJ. Phosphorylation of DARPP-32 at threonine-34 is required for cocaine action. Neuropsychopharmacology. 2006;31:555–562. doi: 10.1038/sj.npp.1300832. [DOI] [PubMed] [Google Scholar]
- Zhou LF, Zhu YP. Changes of CREB in rat hippocampus, prefrontal cortex and nucleus accumbens during three phases of morphine induced conditioned place preference in rats. J Zhejiang Univ Sci B. 2006;7:107–113. doi: 10.1631/jzus.2006.B0107. [DOI] [PMC free article] [PubMed] [Google Scholar]