

Abstract
Background
Chronic pancreatitis is a significant cause of morbidity and a known risk factor for pancreatic adenocarcinoma. Interleukin-1β is a proinflammatory cytokine involved in pancreatic inflammation. We sought to determine whether targeted overexpression of interleukin-1β in the pancreas could elicit localized inflammatory responses and chronic pancreatitis.
Methods
We created a transgenic mouse model (elastase sshIL-1β) in which the rat elastase promoter drives the expression of human interleukin-1β. Mice were followed up for up to 2 years. Pancreata of elastase sshIL-1β mice were analyzed for chronic pancreatitis-associated histologic and molecular changes. In order to study the potential effect of p53 mutation in chronic pancreatitis, elastase sshIL-1β mice were crossed with p53R172H mice.
Results
Three transgenic lines were generated, and in each line the pancreas was atrophic and occasionally showed dilation of pancreatic and biliary ducts secondary to proximal fibrotic stenosis. Pancreatic histology showed typical features of chronic pancreatitis. There was evidence for increased acinar proliferation and apoptosis, along with prominent expression of TNF-α, CXCL1, SDF1, TGF-β1, MMP2-7-9, TIMP1 and COX2. The severity of the lesions correlated well with the level of human interleukin-1β expression. Older mice displayed acinar-ductal metaplasia but did not develop mPanIN or tumors. Elastase sshIL-1β*p53R172H/+ mice had increased frequency of tubular complexes, some of which were acinar-ductal metaplasia.
Conclusion
Overexpression of interleukin-1β in the murine pancreas induces chronic pancreatitis. Elastase sshIL-1β mice consistently develop severe chronic pancreatitis and constitute a promising model for studying chronic pancreatitis and its relationship with pancreatic adenocarcinoma.
Keywords: Interleukin-1, Pancreas, Pancreatitis, Pancreatic Cancer, p53
Introduction
Chronic pancreatitis leads to a progressive and irreversible destruction of the pancreatic gland.1 In advanced cases, this may result in pancreatic exocrine insufficiency and / or diabetes. Significant morbidity is also associated with occasional biliary or pancreatic ductal obstruction secondary to fibrotic stenosis or ductal obliteration. Furthermore, chronic pancreatitis is a well described risk factor for pancreatic adenocarcinoma,2, 3 especially in cases of hereditary chronic pancreatitis.4
While the pathogenesis of the initial pancreatic lesion depends on the etiology of the disease, it is believed subsequent progression to chronic pancreatitis implies a common mechanism. Two main hypotheses have been proposed. The first one is called the necrosis-fibrosis sequence hypothesis, and is based on clinical and experimental data which suggest that chronic pancreatitis could result from repetitive episodes of acute pancreatitis. However, chronic pancreatitis is sometimes observed without evidence of associated necrosis, suggesting a different pathogenic mechanism, also referred to as the sentinel acute pancreatitis episode (SAPE) hypothesis. According to the latter, an initial episode of acute pancreatitis elicits chronic inflammation by recruiting immune cells and activating pancreatic stellate cells. Finally, it is possible that both mechanisms occur simultaneously, and their respective contributions vary depending on the etiology of the disease and the presence of additional host or environmental factors.
Several mouse models of chronic pancreatitis have been reported to date, supporting the different aforementioned hypotheses. The most commonly employed model consists of repeated injections of cerulein, an analogue of cholecystokinin, which induces chronic pancreatitis if injected repeatedly over a sufficient period of time, in agreement with the necrosis-fibrosis sequence hypothesis.5 Pancreatic duct ligation is another model that can induce inflammation and pancreatic atrophy.6 Recently, transgenic mouse models have been developed, some of which are based on genetic mutations associated with hereditary pancreatitis. Archer et al reported a model relying on the acinar expression of a mutated version of cationic trypsinogen PRSS1,7 while Durie et al. reported CFTR knock out mice.8 Both mouse models showed somewhat mild pancreatic lesions. Other models have also been described with a variable degree of similarity with human chronic pancreatitis. These are based on defective ciliary function,9 overexpression of keratin 8,10 disruption of genes encoding factors associated with the TGF-β signaling pathway,11 and knock-out of PARK,12 or EIF1 and 2.13
An interesting observation in human patients is the variability in individual susceptibility to chronic pancreatitis.14 This could be related to variability in the immune response, which has been demonstrated to influence the penetrance and severity of chronic inflammatory diseases. Interleukin-1β is an inflammatory cytokine15, 16 and polymorphisms in this gene have been shown to affect the immune response.17 This cytokine has also been shown to have an important role in the pathogenesis of pancreatitis.18
We sought to determine whether local activation of the immune system within the pancreas could elicit chronic pancreatitis. We hypothesized that expression of a single proinflammatory cytokine in the pancreas could induce chronic pancreatitis and consequently we created a transgenic mouse, elastase sshIL-1β, in which overexpression of interleukin-1β is targeted to the pancreas using the elastase promoter.19 Elastase sshIL-1β mice consistently developed severe chronic pancreatitis that exhibited an early onset and phenocopied well the human disease.
Materials and Methods
Mice
All mice procedures were performed according to the protocol (AC-AAAA5113) approved by the Institutional Animal Use and Care Committee of Columbia University.
The sshIL-1β gene 15, 20 was amplified by PCR, adding EcoR I restriction sites at 5′ and 3′ ends. After EcoR I digestion, sshIL-1β was inserted downstream the rat elastase promoter (kind gift from Doris Stoffer 19) and the rabbit β-globin enhancer, and upstream a rabbit β-globin polyA sequence (Fig. 1A). Orientation was checked by Hind III digestion and the plasmid was sequenced. Elastase sshIL-1β transgene was then extracted from pBluescript by Xho I – Xba I digestion and microinjected in the pronucleus of fertilized CBA × C57BL/6J oocytes. Pups were genotyped using three different pairs of primers spanning the entire transgene (see supplementary table 1, genotyping primers). Positive founders were mated with C57BL/6J mice, and F1 or F2 generations were sacrificed for analysis. Two hours before sacrifice, mice were injected with 50mg/kg of BrdU (Sigma-Aldrich, St. Louis, MO, USA) intra-peritoneally.
Figure 1.
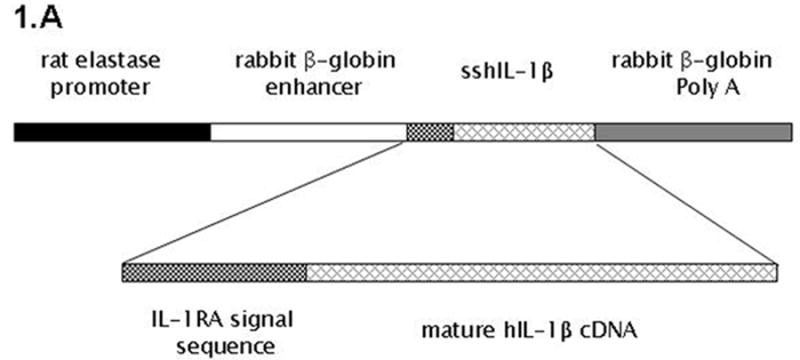
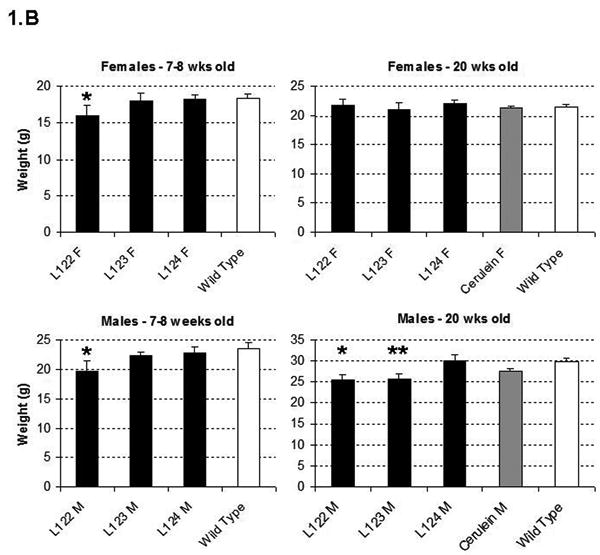

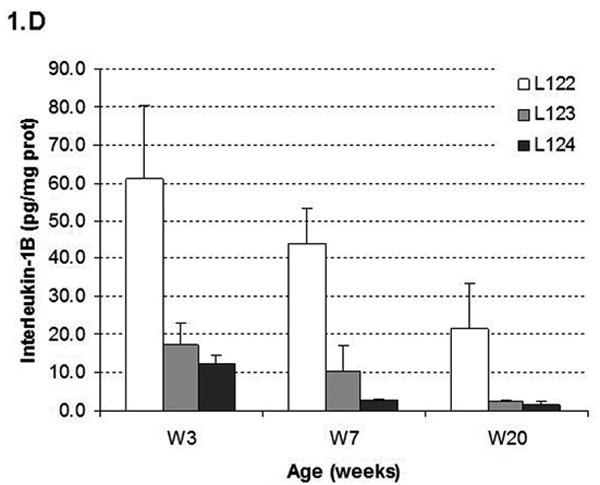
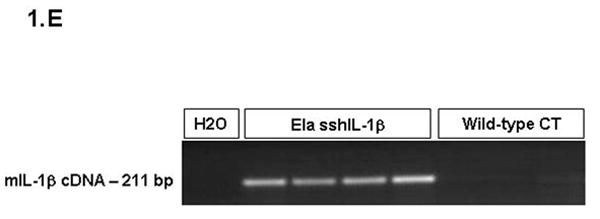
(A) Map of the elastase sshIL-1β construct: the mature human interleukin-1β cDNA was fused with the signal sequence of human interleukin-1 receptor antagonist, allowing secretion without caspase 1 cleavage. Expression of sshIL-1β transgene was driven by the rat elastase promoter. (B) Average weight (error bar is S.E.M.) of elastase sshIL-1β mice, wild-type littermate controls, and cerulein treated mice (20 weeks treatment) (n=6 to 22): upper left panel: 7-8 week-old females, * P=0.01 (/wild-type, t-test); upper right panel: 7-8 week-old males, * P=0.06 (/wild-type); lower left panel: 20 week-old females; lower right panel: 20 week-old males, * P=0.00001 (/wild-type), ** P=0.00005 (/wild-type). (C) Detection of sshIL-1β mRNA in pancreatic tissues of transgenic mice from the three different lines. PCR was performed on cDNAs isolated from 7-8 week-old mice using primers specific for sshIL-1β (forward primer on the sshIL1RA part and reverse primer on the hIL1β part of the transgene). Controls were wild-type littermate (WT) and samples in which reverse transcriptase enzyme had been omitted (RT- line) to control for possible genomic DNA contamination. (D) ELISA quantification of human interleukin-1β in pancreatic protein extracts from 7-8 week-old mice from three different founders (n=3-4). Average concentrations of sshIL-1β (error bar is S.E.M.) are reported in pg/mg of pancreatic protein at 3 different ages (3, 7, and 20 weeks old). (E) Murine IL-1β mRNA expression in pancreas of 7 week-old mice detected by RT-PCR. The expected size of the amplified product from tissue cDNA is 211 bp. Lane 1 (left): negative control. Lanes 2 to 5, pancreata of elastase sshIL-1β mice (age 7 weeks): 2-3 L122, 4-5 L124. Lanes 6 to 8, pancreata of wild-type control mice.
In order to generate elastase sshIL-1β mice heterozygous for the p53R172H germline mutation (elastase sshIL-1β*p53R172H/+), elastase sshIL-1β mice (L124) were crossed with mice homozygous for p53R172H mutation (kind gift from Tyler Jacks, Massachusetts Institute of Technology, Cambridge, MA, USA).21
For the cerulein based model of chronic pancreatitis C57BL/6 mice were injected intra-peritoneally with 50 μg/kg of cerulein (American Peptide Company, Inc, Sunnyvale, CA, USA - dissolved in sterile water) on seven consecutive hours, twice a week for ten weeks, and once a week for 10 additional weeks. Sacrifice was performed one week after the last cerulein injection.
Tissue Preparation
Histology, Immunohistochemistry, Immunofluorescence, and Microscopy
Histologic Analysis and Quantification of Fibrosis
H&E sections were analyzed and lesions were graded using a modification of the scoring system previously described by Demols et al,22 taking into account the extent of inflammation, glandular atrophy and tubular complexes, and the percentage of area involved (supplementary table 2). For fibrosis quantification, six non overlapping 100X magnification fields of Sirius red stained slides were captured. Morphometric analysis was performed using the IPLab 3.6 software (BD Biosciences, Franklin Lakes, NJ, USA). Preneoplastic lesions were defined according to the criteria of Hruban et al.23
Proliferation and Apoptosis Quantification
cDNA Synthesis and Polymerase Chain Reaction
Interleukins Quantification
Flow Cytometry
Magnetic Resonance Imaging
Glucose Tolerance Test
Research of Fat Malabsorption
Statistical Analysis
Statistical analysis was performed using t-test or Wilcoxon signed-rank test when appropriate and p values <0.05 were considered significant. Data are expressed as means +/- S.E.M.
Results
Generation of Elastase sshIL-1β Mice
Three founder mice were generated and subsequently bred to generate three separate lines (L122, L123, L124). The litter size was normal for all three lines. For lines L122 and L123, approximately 15% of the pups showed significant growth retardation and needed to be euthanized. The remainder of the elastase sshIL-1β mice displayed normal behavior and growth although the average weight was significantly lower in L122 mice at 7 weeks and in L122 and L123 male mice at 20 weeks (Fig. 1B). For line 124, mice lived up to 2 years, while the oldest mice from line 123 are currently 22 months old and appear healthy. While a detailed survival analysis was not carried out, the elastase sshIL-1β mice do not appear to show any significant decreased longevity compared with wild-type control mice. We confirmed the expression of the sshIL-1β transgene in the pancreas by RT-PCR and by ELISA measurement of human interleukin-1β peptide in pancreatic protein extracts (Fig. 1C-D). Expression of sshIL-1β could not be detected in any of the other tissues we analyzed, including liver, sub-maxillary glands, kidney, lung and spleen (data not shown). The level of human interleukin-1β expression was different in the three transgenic lines, and these were classified as high (L122), medium (L123) and low (L124) expressers. No human interleukin-1β was detectable in control mice, which were age matched wild-type mice or mice with cerulein induced chronic pancreatitis. It is noteworthy that the level of human interleukin-1β expression decreased with time in all three lines, probably related to progressive acinar atrophy as also described in other acinar promoter based transgenic mice.7 IL-1β itself is a known downstream target of the NF-kB pathway. Consistent with the notion that sshIL-1β activates the NF-kB pathway, we could detect strong expression of endogenous murine IL-1β in the pancreata of elastase sshIL-1β mice at the age of 7 weeks while there was only very weak expression in the majority of age-matched wild-type control mice (Fig. 1E). In addition, in order to demonstrate that human IL-1β can directly activate the murine IL-1 receptor, we isolated myeloid cells from the bone marrow of mice and stimulated these cells with human IL-1β. Treatment of marrow-derived myeloid cells with standard doses of human IL-1β resulted in a >3-fold increase in IL-6 mRNA and a >7 fold increase in IL-6 protein secretion (not shown). Since IL-6 is the most commonly used downstream surrogate for IL-1 receptor signaling and NF-kB pathway activation, these findings indicate that human IL-1β can directly stimulate the murine IL-1 receptor.
Elastase sshIL-1β Mice Display Chronic Pancreatitis
Pancreata of elastase sshIL-1β mice were atrophic (Fig. 2A). In addition, as early as 7 weeks, L122 and L123 mice displayed dilatation of the main pancreatic duct alone, or, in some cases, associated with dilatation of the biliary duct (Fig. 2B) due to fibrotic stenosis at the level of the head of the pancreas, mimicking human chronic pancreatitis. Histologic analysis showed other features typical of chronic pancreatitis, including acinar atrophy, tubular complexes, mixed inflammatory infiltrate, and fibrosis, the latter confirmed by trichrome staining (Fig. 3). These lesions appeared as early as one week after birth, starting with a mixed inflammatory infiltrate and initially mild fibrosis. Interestingly, we did not observe pancreatic necrosis at any time point. Complete autopsy did not reveal abnormality in any other organ.
Figure 2.
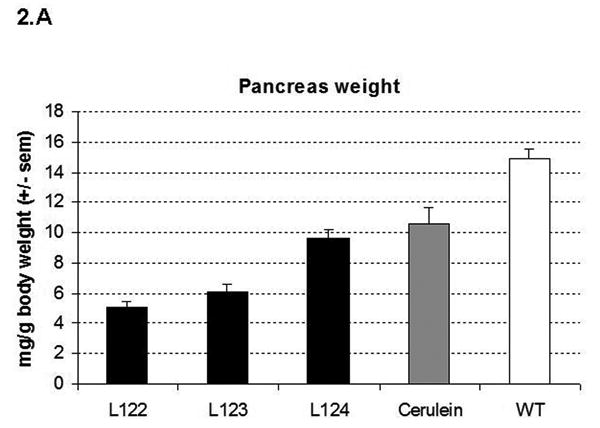
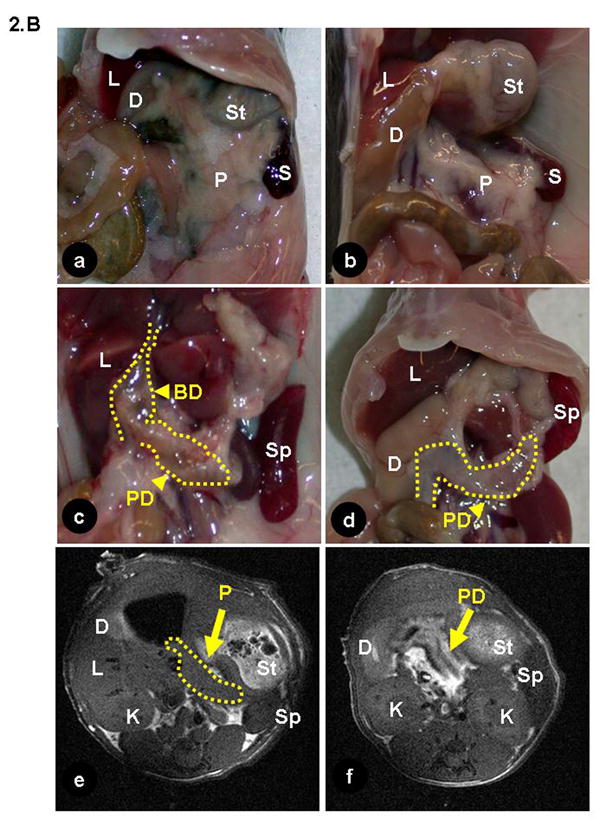
(A) Pancreas to body weight ratio of 7-8 week-old elastase sshIL-1β mice, cerulein treated mice (20 weeks treatment), and wild-type littermate (average +/- S.E.M., n=4). (B) (a-d) Necropsy of elastase sshIL-1β mice: (a) wild-type littermate; (b) Cerulein treated mice (20-week treatment) (c) elastase sshIL-1β mice: biliary and pancreatic duct dilatation due to fibrosis of the pancreatic head (L123, 11 weeks old); (d) dilatation of the pancreatic duct and atrophic pancreas (L122, 8 weeks old); (e, f): T1 weighted magnetic resonance imaging picture (e) wild-type mouse; the pancreatic duct is not visible; (f) 25 week-old elastase sshIL-1β mouse (L122), displaying a dilated pancreatic duct (arrow). BD: biliary duct, D: duodenum, K: kidney, L: liver, P: pancreas, PD: pancreatic duct, Sp: spleen, St: stomach.
Figure 3.
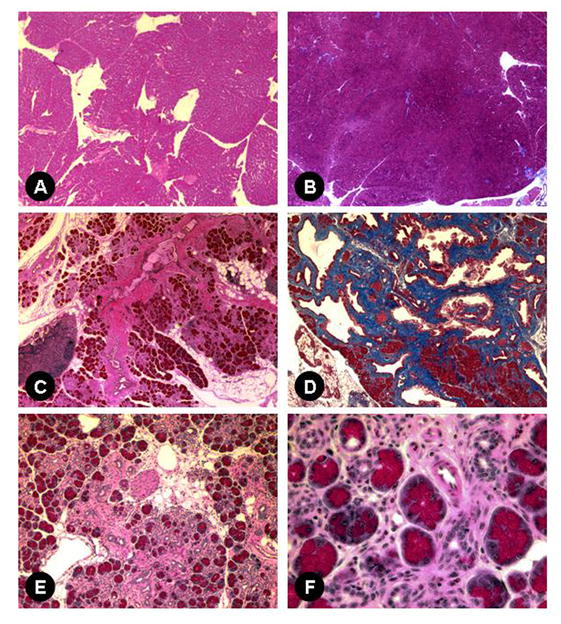
H&E (A, C, E, and F) and Masson's trichrome staining (B, D). Wild-type pancreas, 40X (A, B) and pancreas of 7-8 week-old transgenic mice: (C) elastase sshIL-1β L124, acinar atrophy, inflammatory infiltrate and fibrosis surrounding a major duct (40X); (D) L122, extensive fibrosis evidenced by Masson's trichrome staining (40X); (E) L124, loss of acinar cells, extensive fibrosis, tubular complexes, mixed cellular infiltrate, and increased fat (100X); (F) higher magnification (400X): acini and tubular complexes amid mixed inflammatory infiltrate composed of lymphocytes, macrophages and granulocytes (400X).
The inflammatory infiltrate consisted mainly of lymphocytes, but variable numbers of macrophages and granulocytes were seen admixed. Characterization of the infiltrate by flow cytometry analysis (Fig. 4A) demonstrated a predominance of T-cells (CD3+), and confirmed the presence of macrophages (CD11b+), granulocytes (gr1+) and B-cells (CD19+). The predominance of T lymphocytes was in agreement with the chronic nature of the inflammation.
Figure 4.
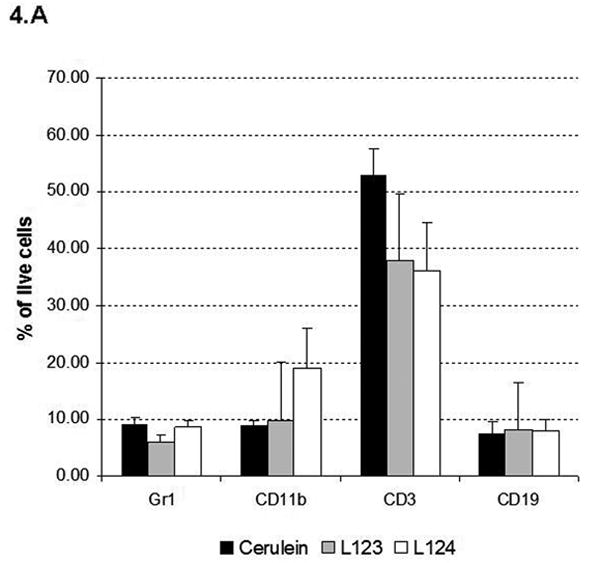
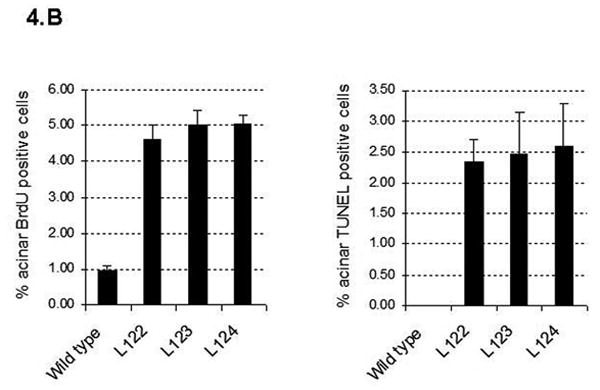
(A) Flow cytometry analysis of pancreatic inflammatory infiltrate in 20 week-old elastase sshIL-1β (L123, L124) and cerulein treated mice using cell specific markers: CD3 (T lymphocyte), CD19 (B lymphocyte), Gr1 (granulocyte), and CD11b (macrophage). Results are expressed as average percentage of live cells (error bar is S.E.M.) obtained after digestion of pancreatic samples (n=4). (B) Acinar proliferation (BrdU immunohistochemical staining – left panel) and apoptosis (TUNEL assay– right panel) indexes in 7-8 week-old elastase sshIL-1β mice (n=4). Controls are wild-type littermates. Results are expressed as means, error bars are S.E.M.
Increased proliferation and apoptosis in the exocrine pancreas are hallmarks of chronic pancreatitis in humans. We quantified proliferation using immunohistochemical staining for BrdU, and apoptosis by the TUNEL assay (Fig. 4B). Both proliferation and apoptosis were significantly elevated compared to age matched wild-type controls.
Chronic pancreatitis is associated with a stromal reaction involving inflammatory cell infiltration as well as proliferation and activation of a specific type of mesenchymal cells called pancreatic stellate cells.24 These cells can be detected by staining for desmin. Figure 5A shows increased number of pancreatic stellate cells in elastase sshIL-1β mice, interspersed between acini, located around ducts or regenerative ductules, or scattered amongst the inflammatory infiltrate. Some of these cells, located in the periacinar or periductal area were activated pancreatic stellate cells, as demonstrated by expression of α-smooth muscle actin.
Figure 5.
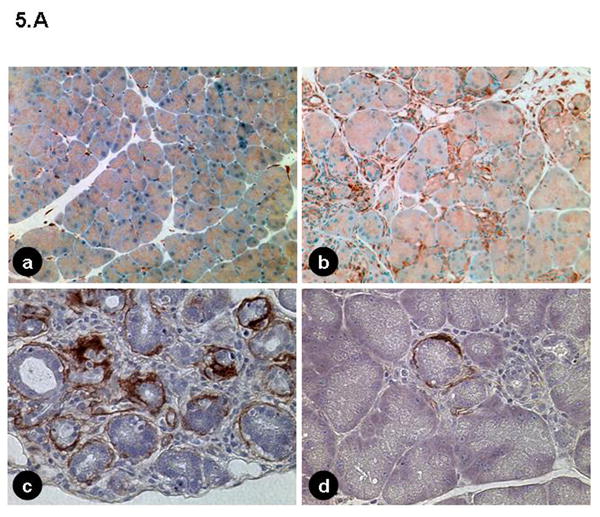
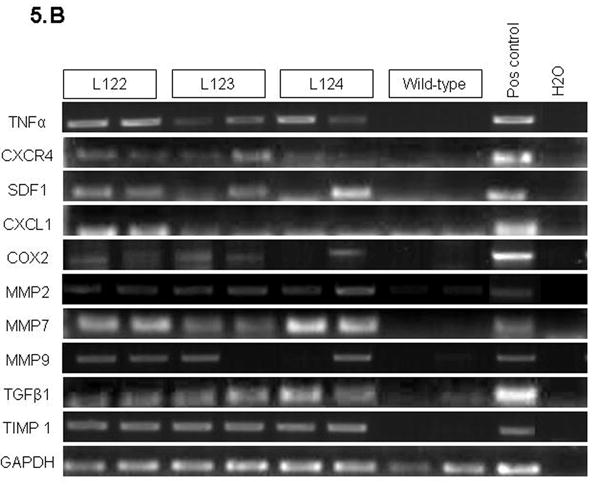
(A) (a, b) Immunohistochemical staining for desmin in 7-8 week-old mice: (a) wild-type (40X); (b) elastase sshIL-1β mouse (100X), pancreatic stellate cells are interspersed between acinar and ductal structures or amid the inflammatory infiltrate. (c, d) Immunohistochemical staining for α-smooth muscle actin in elastase sshIL-1β mouse: Activated pancreatic stellate cells surrounding acini and tubular complexes (400X). (B) Reverse transcription PCR performed on cDNAs isolated from 7-8 week-old elastase sshIL-1β mice. Controls are wild-type littermates. Transgenic mice show increased mRNA expression of cytokines (TNF-α), chemokines (CXCL1, SDF1 and its receptor CXCR4), COX2, metalloproteases (MMP2-7-9, TIMP1), and TGF-β1.
By reverse transcription PCR (Fig. 5B), we detected the expression of genes associated with chronic pancreatitis, including TNF-α, which is also a downstream target of interleukin-1β; chemokines, like SDF1 and CXCL1; TGF-β1, a growth factor that is involved in pancreatic fibrosis and the activation of pancreatic stellate cells; metalloproteases involved in the remodeling of the extracellular matrix (MMP 2-7-9, TIMP 1); and cyclooxygenase 2. These genes all showed minimal expression in the pancreas of wild-type control mice.
Severity of Chronic Pancreatitis is Correlated with the Expression Level of Human Interleukin-1β
In order to correlate the extent and severity of the lesions with the level of human interleukin-1β expression, we graded the lesions and quantified fibrosis by morphometric analysis after Sirius red staining (Fig. 6A, B). Mice from lines 122 and 123 (with high and moderate level of sshIL-1β expression, respectively) had more severe lesions, and higher levels of fibrosis than mice from the low expressing line (line 124). We also compared the histologic findings in 20 week-old transgenic mice with those of mice treated for 20 weeks with repeated injections of cerulein, since the latter has been the most commonly employed model of murine chronic pancreatitis. Despite the protracted protocol, mice which received cerulein had milder lesions and less fibrosis than elastase sshIL-1β mice from any of the three lines.
Figure 6.
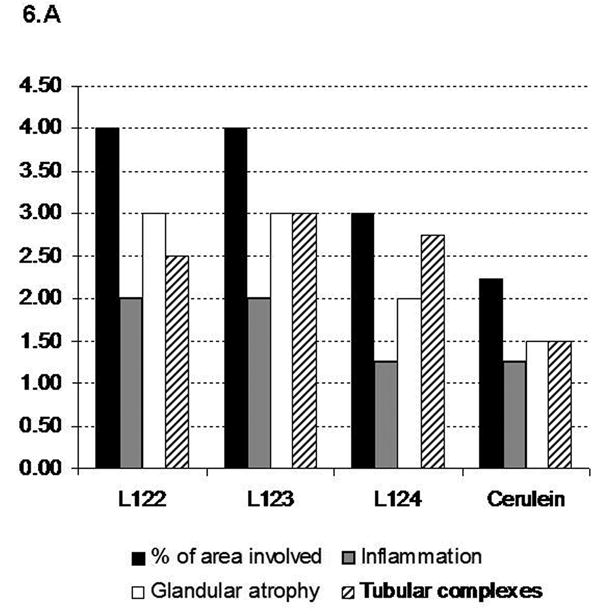
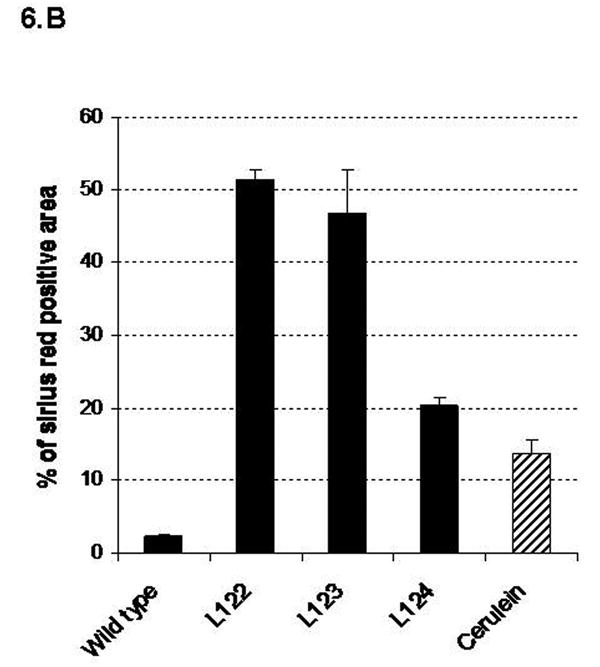
(A) Histologic scoring of pancreatic sections based on H&E stained sections from 20 week-old elastase sshIL-1β mice, and wild-type mice treated for 20 weeks with cerulein. Results are expressed as means (n=4). (B) Quantification of collagen area (mean percentage +/- S.E.M.) by morphometric analysis after Sirius red staining in the same groups of mice.
Elastase sshIL-1β Mice Do Not Develop Significant Pancreatic Insufficiency
Severe cases of pancreatitis can be associated with pancreatic endocrine and/or exocrine insufficiency. To assess glucose tolerance in these mice, we performed a glucose tolerance test, which did not demonstrate significant abnormalities of glucose tolerance in transgenic mice compared to wild-type controls (Supplementary fig. 1). We also evaluated pancreatic exocrine function by assessing for fat malabsorption in mice fed a high fat diet. We performed Oil Red O staining on stool smears (Supplementary fig. 2) but did not detect any fat in the stools of elastase sshIL-1β mice. It is noteworthy these tests were performed in mice 8-10 months old in order to study mice with advanced disease.
Elastase sshIL-1β Mice Develop Acinar-Ductal Metaplasia
Chronic pancreatitis is a well described risk factor for pancreatic adenocarcinoma. We followed a cohort of transgenic elastase sshIL-1β up to 2 years to determine whether they might develop spontaneous pancreatic neoplasia. Acinar atrophy and fibrosis was progressive, and the majority of the pancreatic gland was replaced over time by adipose tissue. None of the mice developed pancreatic tumors or mPanIN. Eosinophilic metaplasia was seen in the large dilated ducts in a few mice, but this lesion is typically considered a reactive change (Fig. 7A). We also observed acinar-ductal metaplasia starting between 20 (L122-123) and 40 weeks of age (L124) (Fig. 7B). This lesion was confirmed by performing immunofluorescence staining for amylase and cytokeratin 19 (Fig. 7C): in some tubular complexes, cells expressing both acinar and ductal markers were observed. On staining for BrdU (fig 7D) these lesions had a low proliferation index suggesting acinar trans-differentiation25 rather than expansion of centroacinar cells as suggested by some studies.26
Figure 7.
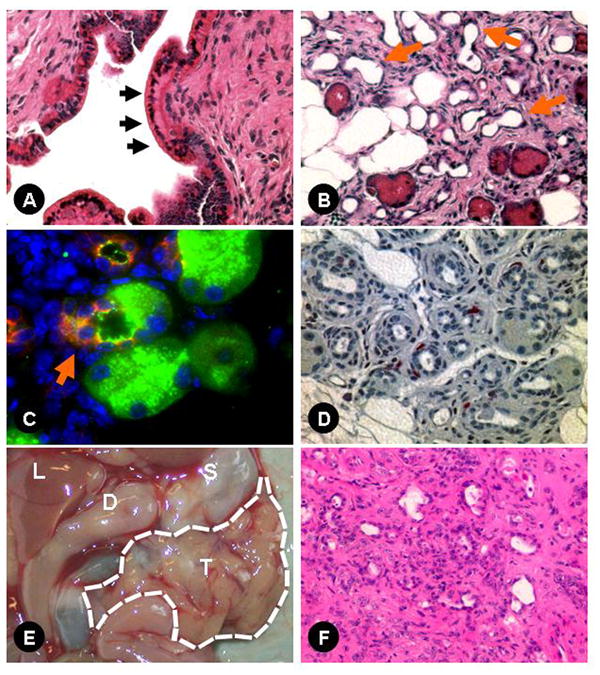
(A) Eosinophilic metaplasia in the main pancreatic duct of an 8 week-old elastase sshIL-1β mouse (L122) (400X); (B) acinar-ductal metaplasia (arrows) in a 20 week-old elastase sshIL-1β mouse (200X); (C) acinar-ductal metaplasia: immunofluorescence for amylase (FITC-green) and cytokeratin 19 (Texas-Red) showing a cell (arrow) co-expressing both acinar and ductal markers (900X); (D) representative picture of BrdU immunohistochemical staining in a 20 week-old elastase sshIL-1β mouse showing low epithelial proliferation in tubular complexes (400X); (E) pancreatic tumor in a 43 week-old elastase sshIL-1β*p53R172H/+ mouse: T-large nodular tumor replacing the entire pancreas, S-stomach, D-duodenum, L-liver; (F) H&E section of this pancreatic tumor showing sarcomatoid and giant cell differentiation (200X).
In patients with hereditary chronic pancreatitis, exposure to tobacco, a known mutagen, significantly increases the risk of pancreatic adenocarcinoma.27 We thus hypothesized that, in addition to chronic inflammation, an additional genetic event might be required to initiate carcinogenesis. Consequently, we crossed elastase sshIL-1β (L124) with mice harboring a R172H mutation in p53,21 and generated elastase sshIL-1β mice heterozygous for p53R172H mutation (elastase sshIL-1β*p53R172H/+). These mice had normal growth and development compared to elastase sshIL-1β mice. On comparing the severity of lesions in elastase sshIL-1β mice with or without p53R172H/+ mutation, no significant difference in the extent of inflammation or acinar atrophy was observed, but a significantly increased number of tubular complexes, including some showing evidence of acinar-ductal metaplasia, was noted in 40 week-old elastase sshIL-1β*p53R172H/+ mice (Fig. 8A). No difference was observed at 20 weeks old (data not shown). As p53 is involved in apoptosis regulation,21 we compared acinar apoptotic indexes according to p53 status and found that elastase sshIL-1β*p53R172H/+ mice had a lower apoptotic index, although the difference was not statistically significant (Fig. 8B). We did not observe any mPanIN lesion in elastase sshIL-1β*p53R172H/+ mice, even in mice up to 15 months of age. Follow up of this cohort was shorter than that of elastase sshIL-1β mice as, starting at 12 months, mice began to die from p53 germline mutation associated tumors (lymphomas and sarcomas essentially). These tumors occurred with the same frequency as in the single transgenic p53R172H/+ control group. Finally, we observed one single case of pancreatic tumor (out of 15 mice), an adenocarcinoma with sarcomatoid and giant cell features in a 43 week-old mouse (Fig. 7E-F). The pancreas was enlarged and nodular and the tumor showed extensive invasion with peritoneal carcinomatosis. No liver or lung metastases were identified. In areas of non-tumoral pancreas, chronic pancreatitis was evident with acinar-ductal metaplasia, but no mPanIN was observed.
Figure 8.
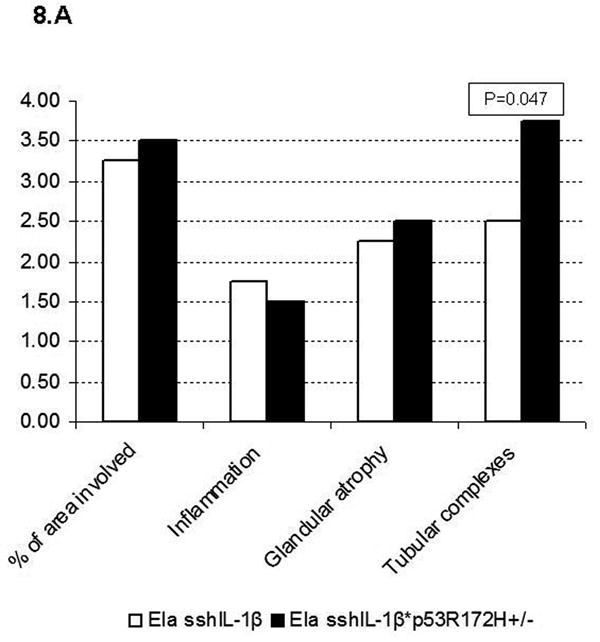
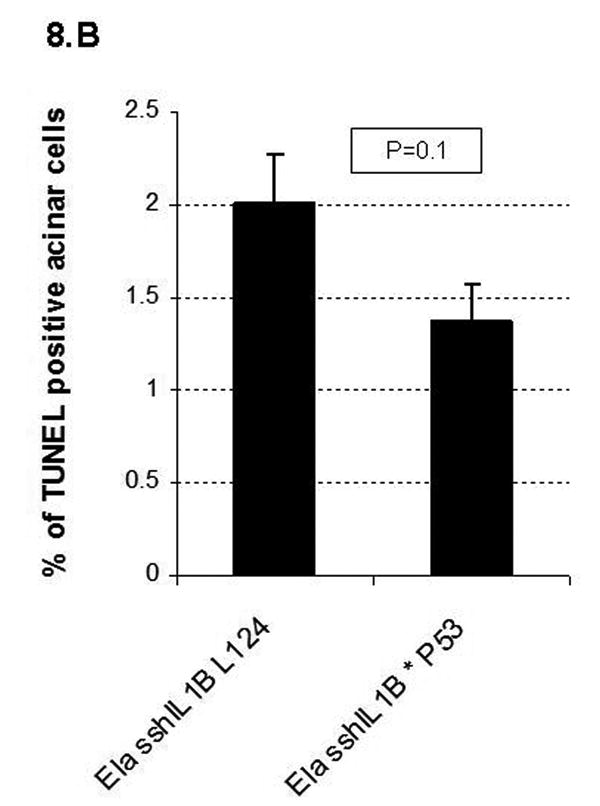
(A) Histologic scoring of pancreatic sections from 40 week-old elastase sshIL-1β*p53R172H/+ mice compared to single transgenic elastase sshIL-1β mice from the same founder line (L124), n=4. Results are shown as means. Extent of tubular complexes is significantly higher in elastase sshIL-1β*p53R172H/+ mice (Wilcoxon test). (B) Acinar apoptotic index determined by the TUNEL assay in 40 week-old elastase sshIL-1β mice with or without p53R172H/+ mutation. p=0.1 (t-test). Results are shown as means (n=4), error bars are S.E.M.
Discussion
Elastase sshIL-1β mice have a phenotype of early onset chronic pancreatitis, with chronic inflammatory infiltrate consisting predominantly of T lymphocytes, progressive glandular atrophy with acinar cell loss, and severe fibrosis, particularly in murine lines with moderate to high levels of human interleukin-1β expression, leading to biliary and pancreatic duct stenosis. The phenotype was strikingly similar to that of chronic pancreatitis in humans. It was associated with accelerated cellular turnover in the exocrine pancreas, and with prominent expression of cytokines (TNFα), chemokines (CXCL1, SDF1), growth factors (TGF-β1), metalloproteases (MMP2-7-9, TIMP1) and cyclooxygenase 2. Importantly, these mice did not consistently develop pancreatic ductal neoplasm, even in the setting of p53 mutation, but exhibited acinar-ductal metaplasia.
Our results indicate that interleukin-1β can induce chronic inflammation in the murine pancreas, as it has previously been reported in the lung.16 This conclusion is supported by the fact that all three transgenic lines displayed a similar phenotype, which severity correlated with the level of expression of human interleukin-1β, and by in vitro results showing activation of the IL-1 receptor pathway by human IL-1β. Although sources of IL-1β in human chronic pancreatitis are believed to be leukocytes and stromal cells, we found that targeting hIL-1β expression to acinar cell could consistently induce the disease. As in many transgenic mouse models, overexpression of this proinflammatory cytokine in pancreatic epithelial cells proved to be effective in replicating human disease but of course does not in any way implicate pancreatic acinar cells as a source of IL-1β production in patients with pancreatitis. Another consequence of the acinar expression of IL-1β could be the induction of an immune response secondary to the expression of the human peptide. Although this appears unlikely, we can not completely exclude such hypothesis.
Interestingly, some mice with higher levels of human interleukin-1β expression had a lower body weight, despite the absence of any evidence of fat malabsorption or diabetes. Although decreased body weight has not been reported in other animal models of interleukin-1β overexpression,15, 16 it is possible that the lower bodyweight was due to the systemic effect of interleukin-1β as reported by Matsuki et al.28 Another explanation could possibly be alimentary restriction secondary to the pain associated with food intake.
We did not observe acinar necrosis in the transgenic mice, supporting the concept that the activation of the immune system and of pancreatic stellate cells - in our case resulting from interleukin-1β activity - is sufficient to induce chronic pancreatitis. This suggests that interleukin-1β might have a major role in the persistent activation of the immune system leading to chronic pancreatitis. These findings could have important consequences for the understanding and treatment of chronic pancreatitis in humans. For example, polyphormisms in interleukin-1β gene could influence the penetrance and severity of the disease, and therapy aiming at blocking the action of interleukin-1β could be considered.
The precise mechanisms of action and direct downstream targets of interleukin-1β remain unclear. In our model, it appears that overexpression of sshIL-1β led to recruitment and activation of numerous inflammatory cells that further propagated inflammation via secretion of cytokines and chemokines, possibly entailing paracrine and autocrine events. Such view is supported by previous reports of interleukin-1β function: It has been observed that interleukin-1β can recruit leukocytes, indirectly, through up regulation of chemokines16 such as CXCL1 or SDF1, as observed in our mice, or directly, attracting T lymphocytes.29 This latter cell population is particularly interesting since T lymphocytes can be directly activated by interleukin-1β 15 and it has been suggested that T cells could have a direct cytotoxic role in chronic pancreatitis.30, 31 Pancreatic stellate cells are another cell type that could be involved in the response to interleukin-1β. These cells are known to be involved in the pathogenesis of chronic pancreatitis by synthesizing components and modifiers of the extracellular matrix, contributing to fibrosis, and acting upon the microenvironment through paracrine and autocrine mechanisms.24 Interleukin-1β is a well described activator of pancreatic stellate cells, and in agreement, we observed an increase in the number of activated pancreatic stellate cells.
Other murine models of chronic pancreatitis have previously been described. They are based on different mechanisms or modes of cellular injury: cerulein administration,5 genetic engineering,7-9 chemical injury,32 or duct ligation.6 The most widely employed model is the cerulein injection model. However, the model is cumbersome, requiring repeated injections over long periods of time, and results in pancreatic lesions milder than in our model. Chemical and duct ligation models also have limitations in that the lesions tend to decrease over time after the initial injury. More recently, several transgenic models have been described, some of them based on the genetic alterations of hereditary pancreatitis 7, 8, but these models are limited by the low penetrance and significant latency of the phenotype. In this regard, the severity and high penetrance of the phenotype make the elastase sshIL-1β mice a convenient and promising model of chronic pancreatitis.
Chronic pancreatitis is a known risk factor for pancreatic adenocarcinoma.2, 3 Unfortunately, information regarding the mechanistic basis of pancreatic carcinogenesis in the setting of chronic inflammation is scarce, particularly with respect to early genetic events. Recently, it has been reported that cerulein can contribute to pancreatic carcinogenesis in the context of activating KRAS mutation.33, 34 We followed a cohort of transgenic mice for up to two years but could not detect any tumors or mPanIN lesions. p53 mutation is frequent in pancreatic adenocarcinoma,35 and recent animal models have demonstrated its critical role in neoplastic transformation in the pancreas.33 Based on these observations we generated elastase sshIL-1β mice heterozygous for p53R172H mutation.21 Only one mouse developed pancreatic adenocarcinoma at 43 weeks of age. Pancreatic cancer was not observed in our p53R172H controls, but this type of tumor has been reported to occur rarely in mice harboring p53R172H mutation.21
We did observe eosinophilic metaplasia and acinar-ductal metaplasia 23 in our double transgenic mice. Eosinophilic metaplasia, which was also seen in our single transgenic mice, has generally been considered a reactive change. The association of acinar-ductal metaplasia with preneoplasia remains the subject of debate, but it is noteworthy that this type of lesion was more extensive in elastase sshIL-1β*p53R172H/+ mice. Since p53 mutations have been reported to confer resistance to apoptotic stimuli,23 it is tempting to speculate that the higher frequency of acinar-ductal metaplasia in elastase sshIL-1β*p53R172H/+ mice be the result, as our results suggest it, of decreased apoptosis of acinar cells, which subsequently differentiate into ductal cells.
Overall, our results suggest that p53 mutation is not critical for the initiation of carcinogenesis in the setting of chronic pancreatitis, and other molecular pathways may be more relevant. Guerra et al recently reported the effect of cerulein in the setting of activating KRAS mutation.34 SMAD4 is also a critical antioncogene in pancreatic carcinogenesis,35-39 and is involved in the signal transduction of TGF-β, which is up regulated in chronic pancreatitis.32 Future studies are required to investigate the relevance of these pathways for pancreatitis-associated neoplasia.
In conclusion, our results demonstrate that overexpression of interleukin-1β in the murine pancreas induces chronic pancreatitis, highlighting the potential importance of interleukin-1β in immune activation in the pancreas. Elastase sshIL-1β mice do not develop spontaneous pancreatic ductal neoplasms but appear to constitute a promising model for studies of chronic pancreatitis and of the role of chronic inflammation in pancreatic carcinogenesis.
Supplementary Material
Acknowledgments
the authors thank Doris Stoffers for providing the rat elastase promoter, Tyler Jacks for providing p53R172H mice, Chyuan-Sheng Lin for his help in generating elastase sshIL-1β mice, James Masciotti for performing magnetic resonance imaging, and Vivian Ng and Jacqueline Baumgartner for their help with animal procedures.
Financial support: Frederic Marrache was supported by a postdoctoral fellowship from the Association pour la Recherche contre le Cancer, Villejuif, France
Footnotes
Conflict of interest disclosure: The authors do no have any conflict of interest to disclose
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Witt H, Apte MV, Keim V, et al. Chronic pancreatitis: challenges and advances in pathogenesis, genetics, diagnosis, and therapy. Gastroenterology. 2007;132:1557–73. doi: 10.1053/j.gastro.2007.03.001. [DOI] [PubMed] [Google Scholar]
- 2.Malka D, Hammel P, Maire F, et al. Risk of pancreatic adenocarcinoma in chronic pancreatitis. Gut. 2002;51:849–52. doi: 10.1136/gut.51.6.849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lowenfels AB, Maisonneuve P, Cavallini G, et al. Pancreatitis and the risk of pancreatic cancer. International Pancreatitis Study Group. N Engl J Med. 1993;328:1433–7. doi: 10.1056/NEJM199305203282001. [DOI] [PubMed] [Google Scholar]
- 4.Lowenfels AB, Maisonneuve P, DiMagno EP, et al. Hereditary pancreatitis and the risk of pancreatic cancer. International Hereditary Pancreatitis Study Group. J Natl Cancer Inst. 1997;89:442–6. doi: 10.1093/jnci/89.6.442. [DOI] [PubMed] [Google Scholar]
- 5.Neuschwander-Tetri BA, Burton FR, Presti ME, et al. Repetitive self-limited acute pancreatitis induces pancreatic fibrogenesis in the mouse. Dig Dis Sci. 2000;45:665–74. doi: 10.1023/a:1005423122127. [DOI] [PubMed] [Google Scholar]
- 6.Watanabe S, Abe K, Anbo Y, et al. Changes in the mouse exocrine pancreas after pancreatic duct ligation: a qualitative and quantitative histological study. Arch Histol Cytol. 1995;58:365–74. doi: 10.1679/aohc.58.365. [DOI] [PubMed] [Google Scholar]
- 7.Archer H, Jura N, Keller J, et al. A mouse model of hereditary pancreatitis generated by transgenic expression of R122H trypsinogen. Gastroenterology. 2006;131:1844–55. doi: 10.1053/j.gastro.2006.09.049. [DOI] [PubMed] [Google Scholar]
- 8.Durie PR, Kent G, Phillips MJ, et al. Characteristic multiorgan pathology of cystic fibrosis in a long-living cystic fibrosis transmembrane regulator knockout murine model. Am J Pathol. 2004;164:1481–93. doi: 10.1016/S0002-9440(10)63234-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cano DA, Sekine S, Hebrok M. Primary cilia deletion in pancreatic epithelial cells results in cyst formation and pancreatitis. Gastroenterology. 2006;131:1856–69. doi: 10.1053/j.gastro.2006.10.050. [DOI] [PubMed] [Google Scholar]
- 10.Casanova ML, Bravo A, Ramirez A, et al. Exocrine pancreatic disorders in transsgenic mice expressing human keratin 8. J Clin Invest. 1999;103:1587–95. doi: 10.1172/JCI5343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bottinger EP, Jakubczak JL, Roberts IS, et al. Expression of a dominant-negative mutant TGF-beta type II receptor in transgenic mice reveals essential roles for TGF-beta in regulation of growth and differentiation in the exocrine pancreas. Embo J. 1997;16:2621–33. doi: 10.1093/emboj/16.10.2621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Harding HP, Zeng H, Zhang Y, et al. Diabetes mellitus and exocrine pancreatic dysfunction in perk-/- mice reveals a role for translational control in secretory cell survival. Mol Cell. 2001;7:1153–63. doi: 10.1016/s1097-2765(01)00264-7. [DOI] [PubMed] [Google Scholar]
- 13.Iglesias A, Murga M, Laresgoiti U, et al. Diabetes and exocrine pancreatic insufficiency in E2F1/E2F2 double-mutant mice. J Clin Invest. 2004;113:1398–407. doi: 10.1172/JCI18879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Uomo G, Manes G. Risk factors of chronic pancreatitis. Dig Dis. 2007;25:282–4. doi: 10.1159/000103903. [DOI] [PubMed] [Google Scholar]
- 15.Bjorkdahl O, Akerblad P, Gjorloff-Wingren A, et al. Lymphoid hyperplasia in transgenic mice over-expressing a secreted form of the human interleukin-1beta gene product. Immunology. 1999;96:128–37. doi: 10.1046/j.1365-2567.1999.00655.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lappalainen U, Whitsett JA, Wert SE, et al. Interleukin-1beta causes pulmonary inflammation, emphysema, and airway remodeling in the adult murine lung. Am J Respir Cell Mol Biol. 2005;32:311–8. doi: 10.1165/rcmb.2004-0309OC. [DOI] [PubMed] [Google Scholar]
- 17.Macarthur M, Hold GL, El-Omar EM. Inflammation and Cancer II. Role of chronic inflammation and cytokine gene polymorphisms in the pathogenesis of gastrointestinal malignancy. Am J Physiol Gastrointest Liver Physiol. 2004;286:G515–20. doi: 10.1152/ajpgi.00475.2003. [DOI] [PubMed] [Google Scholar]
- 18.Norman JG, Fink GW, Sexton C, et al. Transgenic animals demonstrate a role for the IL-1 receptor in regulating IL-1beta gene expression at steady-state and during the systemic stress induced by acute pancreatitis. J Surg Res. 1996;63:231–6. doi: 10.1006/jsre.1996.0253. [DOI] [PubMed] [Google Scholar]
- 19.Heller RS, Stoffers DA, Bock T, et al. Improved glucose tolerance and acinar dysmorphogenesis by targeted expression of transcription factor PDX-1 to the exocrine pancreas. Diabetes. 2001;50:1553–61. doi: 10.2337/diabetes.50.7.1553. [DOI] [PubMed] [Google Scholar]
- 20.Wingren AG, Bjorkdahl O, Labuda T, et al. Fusion of a signal sequence to the interleukin-1 beta gene directs the protein from cytoplasmic accumulation to extracellular release. Cell Immunol. 1996;169:226–37. doi: 10.1006/cimm.1996.0113. [DOI] [PubMed] [Google Scholar]
- 21.Olive KP, Tuveson DA, Ruhe ZC, et al. Mutant p53 gain of function in two mouse models of Li-Fraumeni syndrome. Cell. 2004;119:847–60. doi: 10.1016/j.cell.2004.11.004. [DOI] [PubMed] [Google Scholar]
- 22.Demols A, Van Laethem JL, Quertinmont E, et al. Endogenous interleukin-10 modulates fibrosis and regeneration in experimental chronic pancreatitis. Am J Physiol Gastrointest Liver Physiol. 2002;282:G1105–12. doi: 10.1152/ajpgi.00431.2001. [DOI] [PubMed] [Google Scholar]
- 23.Hruban RH, Adsay NV, Albores-Saavedra J, et al. Pathology of genetically engineered mouse models of pancreatic exocrine cancer: consensus report and recommendations. Cancer Res. 2006;66:95–106. doi: 10.1158/0008-5472.CAN-05-2168. [DOI] [PubMed] [Google Scholar]
- 24.Omary MB, Lugea A, Lowe AW, et al. The pancreatic stellate cell: a star on the rise in pancreatic diseases. J Clin Invest. 2007;117:50–9. doi: 10.1172/JCI30082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Means AL, Meszoely IM, Suzuki K, et al. Pancreatic epithelial plasticity mediated by acinar cell transdifferentiation and generation of nestin-positive intermediates. Development. 2005;132:3767–76. doi: 10.1242/dev.01925. [DOI] [PubMed] [Google Scholar]
- 26.Stanger BZ, Stiles B, Lauwers GY, et al. Pten constrains centroacinar cell expansion and malignant transformation in the pancreas. Cancer Cell. 2005;8:185–95. doi: 10.1016/j.ccr.2005.07.015. [DOI] [PubMed] [Google Scholar]
- 27.Lowenfels AB, Maisonneuve P, Whitcomb DC, et al. Cigarette smoking as a risk factor for pancreatic cancer in patients with hereditary pancreatitis. Jama. 2001;286:169–70. doi: 10.1001/jama.286.2.169. [DOI] [PubMed] [Google Scholar]
- 28.Matsuki T, Horai R, Sudo K, et al. IL-1 plays an important role in lipid metabolism by regulating insulin levels under physiological conditions. J Exp Med. 2003;198:877–88. doi: 10.1084/jem.20030299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hunninghake GW, Glazier AJ, Monick MM, et al. Interleukin-1 is a chemotactic factor for human T-lymphocytes. Am Rev Respir Dis. 1987;135:66–71. doi: 10.1164/arrd.1987.135.1.66. [DOI] [PubMed] [Google Scholar]
- 30.Demols A, Le Moine O, Desalle F, et al. CD4(+)T cells play an important role in acute experimental pancreatitis in mice. Gastroenterology. 2000;118:582–90. doi: 10.1016/s0016-5085(00)70265-4. [DOI] [PubMed] [Google Scholar]
- 31.Hunger RE, Mueller C, Z'Graggen K, et al. Cytotoxic cells are activated in cellular infiltrates of alcoholic chronic pancreatitis. Gastroenterology. 1997;112:1656–63. doi: 10.1016/s0016-5085(97)70048-9. [DOI] [PubMed] [Google Scholar]
- 32.Sparmann G, Merkord J, Jaschke A, et al. Pancreatic fibrosis in experimental pancreatitis induced by dibutyltin dichloride. Gastroenterology. 1997;112:1664–72. doi: 10.1016/s0016-5085(97)70049-0. [DOI] [PubMed] [Google Scholar]
- 33.Hingorani SR, Wang L, Multani AS, et al. Trp53R172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer Cell. 2005;7:469–83. doi: 10.1016/j.ccr.2005.04.023. [DOI] [PubMed] [Google Scholar]
- 34.Guerra C, Schuhmacher AJ, Canamero M, et al. Chronic pancreatitis is essential for induction of pancreatic ductal adenocarcinoma by K-Ras oncogenes in adult mice. Cancer Cell. 2007;11:291–302. doi: 10.1016/j.ccr.2007.01.012. [DOI] [PubMed] [Google Scholar]
- 35.Tuveson DA, Hingorani SR. Ductal pancreatic cancer in humans and mice. Cold Spring Harb Symp Quant Biol. 2005;70:65–72. doi: 10.1101/sqb.2005.70.040. [DOI] [PubMed] [Google Scholar]
- 36.Bardeesy N, DePinho RA. Pancreatic cancer biology and genetics. Nat Rev Cancer. 2002;2:897–909. doi: 10.1038/nrc949. [DOI] [PubMed] [Google Scholar]
- 37.Izeradjene K, Combs C, Best M, et al. Kras(G12D) and Smad4/Dpc4 haploinsufficiency cooperate to induce mucinous cystic neoplasms and invasive adenocarcinoma of the pancreas. Cancer Cell. 2007;11:229–43. doi: 10.1016/j.ccr.2007.01.017. [DOI] [PubMed] [Google Scholar]
- 38.Ijichi H, Chytil A, Gorska AE, et al. Aggressive pancreatic ductal adenocarcinoma in mice caused by pancreas-specific blockade of transforming growth factor-beta signaling in cooperation with active Kras expression. Genes Dev. 2006;20:3147–60. doi: 10.1101/gad.1475506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bardeesy N, Cheng KH, Berger JH, et al. Smad4 is dispensable for normal pancreas development yet critical in progression and tumor biology of pancreas cancer. Genes Dev. 2006;20:3130–46. doi: 10.1101/gad.1478706. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.