

Abstract
MT1 melatonin receptors expressed in Chinese hamster ovary cells remain sensitive to a melatonin re-challenge even following chronic melatonin exposure when microtubules are depolymerized in the cell, an exposure that normally results in MT1 receptor desensitization. We extended our findings to MT2 melatonin receptors using both in vitro and in vivo approaches. Using CHO cells expressing human MT2 melatonin receptors, microtubule depolymerization prevents the loss in the number of high potency states of the receptor when compared to melatonin-treated cells. Additionally, microtubule depolymerization increases in melatonin-induced PKC activity but not PI hydrolysis via Gi proteins similar to that shown for MT1Rs. Furthermore, microtubule depolymerization in MT2-CHO cells enhances the exchange of GTP on Gi-proteins using a photoaffinity analog of GTP. To test whether microtubules are capable of modulating melatonin-induced phase-shifts, microtubules are depolymerized specifically within the suprachiasmatic nucleus of the hypothalamus of the Long Evans rat and the efficacy of melatonin to phase shift their circadian activity rhythms was assessed and compared to animals with intact SCN microtubules. We find that microtubule depolymerization in the SCN using either Colcemid or nocodazole enhances the efficacy of 10 pM melatonin to phase-shift the activity rhythms of the Long Evans rat. No enhancement occurs in the presence of β-lumicolchicine, the inactive analog of Colcemid. Taken together, these data suggest that microtubule dynamics can modulate melatonin-induced phase shifts of circadian activity rhythms which may explain, in part, why circadian disturbances occur in individuals afflicted with diseases associated with microtubule disturbances.
Keywords: MT2 melatonin receptors, microtubules, SCN, circadian rhythms, Alzherimer’s Disease, melatonin
Introduction
Microtubule disruption within the cell can lead to changes in cell architecture and alter G-protein coupled receptor (GPCR) signaling. In particular, depolymerization of microtubules into alpha-beta tubulin heterodimers or the heterodimers themselves in cells leads to enhanced G protein-mediated signaling as shown in numerous studies and for numerous GPCRs [1–5].
Recently, it has been shown that microtubule manipulation, either by reducing tubulin levels [6] or by depolymerization [7] enhances MT1 melatonin receptor (MT1R) function [6, 7], prevents melatonin-induced MT1R desensitization and MEK1/2 and ERK1/2 activation [8]. As melatonin receptors are involved in phase-shifting circadian activity rhythms, particularly, MT2 melatonin receptors (MT2Rs) [9–11], then changes in microtubule dynamics on these receptors may produce consequences on circadian activity patterns. This may lead to altered sleep/wake cycles that occurs in people suffering from Alzheimer’s Disease (AD), a disease characterized by microtubule derangements [12].
Sleep disturbances are reported in Alzheimer’s patients characterized by increases in both the frequency and duration of nocturnal awakenings and daytime naps [13–15]. These frequent nighttime awakenings are often the primary reason for institutional care [16–18]. Because circadian rhythm disturbances strongly contribute to cognitive deterioration and functional disabilities in AD patients [16, 19, 20], then a better knowledge of the major factors involved in the development of the disturbances may guide novel therapeutic strategies.
As reviewed [12], circadian rhythm disturbances during aging and AD are probably due to alterations in the circadian timing system. This may be due to (a) a diminished output and disrupted clock function in AD patients [21]; (b) alterations in the pineal gland and in melatonin secretion [22–24]; (c) a decreased input to the SCN [25]; and (d) alterations in the melatonin receptor [26, 27].
Although numerous studies have documented a role of melatonin and melatonin receptors in the etiology of AD [25–34], the focus of this study was to assess the impact of microtubule disruption on melatonin receptors involved in circadian entrainment using in vitro and in vivo approaches. Neurofibrillary tangles have been observed in the brains of AD patients [27] including the SCN [35], the master biological clock and the function of these microtubules are also impaired [36]. Reductions in both MT1R and MT2R receptors have also been observed [26, 27]; however, studies show that patients with AD remain sensitive to the phase-shifting effects of melatonin [12, 37–39]. The clinical findings argue in favor of disruption of the circadian timing system in AD since numerous rhythms are disturbed (e.g., body temperature, and concentrations of other hormones including glucocorticoids [12, 39]).
Even though some have suggested that damage to the SCN neurons may underlie these circadian rhythm disturbances [40, 41], another possibility could be an alteration in melatonin receptor sensitivity in the SCN of AD patients due to microtubule derangements. To test this, two approaches were taken. The first approach was to assess the effects of microtubule depolymerization on human MT2Rs expressed in CHO cells (MT2-CHO) similar to the approach taken for MT1Rs expressed in CHO cells (MT1-CHO) [7]. The second approach was to assess the effect of microtubule depolymerization in the SCN of the Long Evans rat on the efficacy of melatonin to phase-shift their circadian activity rhythms. The hypothesis of this study is that microtubule depolymerization will enhance the function of MT2Rs both in vitro and in vivo.
Experimental Procedures
Cell culture and treatments
Chinese hamster ovary (CHO) cells stably expressing the human MT2 melatonin receptor (MT2-CHO) were used in this study and are described elsewhere [42]. For the depolymerization studies, MT2-CHO cells were grown in F12 media (Life Technologies, Grand Island, NY) containing 10% fetal bovine serum and 1% penicillin G sodium (5000 units/ml)-streptomycin sulfate (5000 μg/ml) (Life Technologies, Grand Island, NY). After growing to approximately 75% confluence on a 10 cm dish, the media was removed, and the cells were re-fed with F12 media containing vehicle (ethanol 0.000001%), Colcemid (5 μM), melatonin (400 pM) (Sigma, St. Louis, MO) or combinations of each. The cells were incubated for 8 hr at 37°C in a 5% CO2 atmosphere. Following this exposure period, the media was then removed, and cells were re-fed with F12 media containing vehicle, Colcemid (5 μM), and incubated for an additional 16 hr at 37°C in a 5% CO2 atmosphere. This exposure paradigm was chosen to more closely mimic the natural rise and fall of melatonin levels throughout the 24 hr cycle as described [7].
Radioligand binding assays
MT2-CHO cell lysates were prepared following an 8 hr pretreatment and 16 hr withdrawal of melatonin. These lysates were subjected to saturation binding analysis using appropriate concentrations of 2-[125I]-iodomelatonin (1 to 1000 pM) in the absence or presence of melatonin (1 μM) as described [7]. The reaction was incubated for 1 hr at 25°C, filtered, washed and counted. All data were normalized against their vehicle controls.
Following exposure to vehicle, Colcemid, melatonin or combinations of melatonin and Colcemid, MT2-CHO cells were washed, lifted, and added to tubes containing 80–100 pM 2-[125I]-iodomelatonin in the absence (total binding) or presence of various concentrations of melatonin (1 fM to 1 μM) as already decribed [7]. All data were normalized against their totals.
Cyclic AMP accumulation assays
Cyclic AMP accumulation assays were carried out as described previously [7]. Briefly, MT2-CHO cells were plated equally in 24-well dishes and allowed to grow to confluence. The cells were exposed to F12 media containing vehicle, Colcemid, melatonin, or combinations of each for 8 hr. Afterwards, the media was removed and some cells were re-exposed to F12 media containing vehicle and some to Colcemid for an additional 16 hr. Following the 24 hr exposure period, 100 μM of forskolin and 30 μM of rolipram in F12 media (maximum response) or F12 media and rolipram (basal response) were added to the cells to stimulate the production of cAMP. In addition, some of the cells were exposed to increasing concentrations of melatonin (1 fM to 1 μM) for 10 min at 370C. Cyclic AMP formation was measured using the Correlate-EIA Direct cAMP kit (Assay Designs, Inc., Ann Arbor, MI) according to the manufacturer’s instructions. All data were normalized against their forskolin/rolipram (maximal) response and concentration-response curves were constructed. Data points were fit by 1-or 2-site nonlinear regression analysis based upon the lowest residual sum of squares (GraphPad Prism). The cAMP levels generated in the presence of forskolin for each treatment group were also normalized against their vehicle controls.
Immunoblot analysis
MT2-CHO cells were allowed to grow to confluence on 10 cmplates. The cells were exposed to F12 media containing vehicle or melatonin for 8 hr at 37°C. After 8 hr, the media was removed, and fresh F12 media was added. The cells were incubated an additional 16 hr. Afterwards, cell membranes were prepared whereby 10 μg were loaded onto gels and subjected to SDS-PAGE as described previously [7]. G-proteins (Giα2,3 or Gqα) were detected by enhanced chemiluminescence (ECL-Plus, Amersham, Arlingtion Heights, IL) using rabbit anti-Giα2, 3 or Gqα (1:1500) (Santa Cruz Biotechnology, CA) in a TBS/milk solution and an HRP-conjugated goat anti-rabbit IgG (1:5000) (Pierce Chemical Co., Rockford, IL). G-protein bands were quantified by densitometry (NIH-Image, Bethesda, MD) and normalized against vehicle controls.
Cell photography
MT2-CHO cells were grown to a sub-confluent density on 10 cm diameter dishes in F12 media and then subjected to the aforementioned treatment conditions as described [7]. After the drug exposures, the cells were fixed with 0.5% glutaraldehyde (Electron Microscopy Sciences, Fort Washington, PA) in PBS (minus Ca2+ and Mg2+) for 5 min and washed three times. Cells were observed using a Nikon TMS inverted microscope equipped with phase-contrast optics. Images were digitally captured using a Scion Series 7 image capture system (Scion Corporation, Frederick, MD), processed using Adobe Photoshop Software and printed with a Codonics NP-1660 Photographic printer.
[32P]-azidoanilido GTP exchange assays
Experiments were carried out exactly as described previously [7]. Briefly, MT2-CHO cells were grown to 80 to 85% confluence in a 6-well dish in F12 media under the conditions described previously. Next, cells were washed, permeabilized using saponin, washed and then labeled with 0.1 μM [32P] azidoanilido GTP ([32P]AAGTP) for 10 min at room temperature. Melatonin (1μM) was added to facilitate GTP exchange and then the reactions were cooled on ice, exposed to UV light (9W, 254 nm) and stopped. [32P]AAGTP incorporation into G-proteins was visualized by autoradiography following SDS-PAGE. The amount of [32P]AAGTP incorporated into G-proteins was quantified using NIH image software and then normalized against vehicle-treated cells. Data were analyzed by one-way ANOVA followed by a Newman-Keuls post-hoc t-test where significance was defined as p<0.05.
Phosphoinositide hydrolysis assays
Phosphoinositide hydrolysis assays were carried out as described previously [7]. Briefly, MT2-CHO cells were plated and grown to confluence. Next, the cells were exposed to F12 media containing vehicle, Colcemid, melatonin, or combinations of each for 8 hr. Afterwards, the media was removed and some cells were re-exposed to F12 media containing vehicle and some to Colcemid, for an additional 16 hr. Endogenous phospholipids within the MT2-CHO cell were labeled using myo-[2-3H]myoinositol (2 μM/mL; NEN/DuPont, Boston, MA) and then re-challenged with vehicle (basal) or melatonin (1pM to 30 μM). The formation of phosphoinositides was assessed using column chromatography and liquid scintillation counting.
PKC activity assays
To establish the concentration-response effects of melatonin on PKC activity in MT2-CHO cells, cells were grown to confluence in F12 media. Next, cells were lifted, lightly pelleted and resuspended in F12 media containing either vehicle (basal) or increasing concentrations of melatonin (1 pM-1 μM) for 20 min at room temp. To assess the effect of melatonin and Colcemid pretreatment on melatonin (10 nM)-induced PKC activation, cells were grown to confluence and then exposed to F12 media containing vehicle, Colcemid, melatonin, pertussis toxin or combinations of each for 8 hr. Afterwards, the media was removed and some cells were re-exposed to F12 media containing vehicle and some to Colcemid, or pertussis toxin for an additional 16 hr the various treatments as described [7]. Afterwards, the media was removed and then the cells were re-challenged with either vehicle (basal) or 10 nM melatonin. PKC activity was assessed in these cells using the SignaTECT™ Protein Kinase C (PKC) Assay System (Promega, Madison, WI) according to the manufacturer’s instructions. Radioactive spots were quantified by densitometric analysis and relative OD values obtained and PKC activities calculated. Each data p oint represents the mean ± SEM of 2–4 independent experiments. Data were analyzed by one-way ANOVA followed by a Newman-Keuls post-hoc where (*) was defined as p<0.05.
Long Evans rats and housing
In this study, 24 male Long Evans rats ranging in age between 1.5–2 mos and weighing approximately 280–320 g were used. Six animals were obtained from Charles River Laboratories at a time and housed singly in a temperature (22°C) and humidity-controlled room for up to 4 mos. Food and water were provided ad libitum. Animals were maintained for 2 wks in a 12/12 LD cycle and then were transferred to constant dim red light (DD) conditions following surgery to avoid any light mediated effects on circadian phase shifts. The animals were allowed to recover for 2 wks following the surgery to stabilize free-running activity rhythms. To avoid any non-photic entrainment of circadian activity rhythms [43], cages, food and water were changed at different times of the day. Each animal received approximately 4–5 drug treatments per study. All experiments followed guidelines of the NIH and Duquesne University policies for the care and use of laboratory animals.
Cannulations of the SCN
For the brain cannulations, animals were anesthetized with pentabarbital (i.p. 50 mg/kg) and placed into stereotaxic frame (Stulting stereotaxic instrument, Model number 51600) using a brain atlas (Paxinos and Watson) for specific coordinates as described with modification [44]. An incision was made exposing the dorsal aspect of the skull and a hole drilled over the SCN (MS; Bregma-0.92, 0.2 lateral). Lowering 9.2mm dorso-ventrally, a stainless steel cannula was permanently placed in the region.
Circadian activity measurements
The gross motor activity of the animals was measured via intra-abdominally transplanted VM-FH transmitters (Mini Mitter Co, Inc., Bend, OR) and a TR-3000 receiver (Mini Mitter Co., Inc, Bend, OR) placed under each animal’s cage. Animal activity rhythms were recorded continuously for up to 4 mos using Vital View and Actiview software (Mini Mitter Co., Inc, Bend, OR). For each drug infusion, a 7-day baseline recording was taken followed by a 7-day drug period followed by a 7-day washout period; a treatment paradigm already described [45]. On the day of a drug infusion, either DMSO/vehicle (0.001% of both) 1μl; melatonin (1 × 10−17 mole or 1 × 10−16 mole in 1μl; Colcemid (5 × 10−12 mole in 1μl), melatonin and Colcemid, (1 × 10−17 mole or 1 × 10−16 mole melatonin in 1μl and 5 × 10−12 mole Colcemid in 1μl), or melatonin and beta-lumicolchicine (1 × 10−17 mole melatonin in 1μl and 5 × 10−12 mole β-lumicolchicine in 1μl), or melatonin and nocodazole (1 × 10−17 mole melatonin in 1μl and 1 × 10−11 mole nocodazole in 1μl) were infused into the SCN through the guide cannula over 5 min at a rate of 0.2μl/min using a modified procedure already described [44] 2 hrs before their onset of activity. Average peak activity levels over a 7-day period for each animal before (baseline), during (drug treatment) and after (washout) each treatment was calculated using ActiView software (Mini Mitter Co., Inc, Bend, OR). The change in peak activity rhythms from baseline following each drug treatment was calculated for each animal and then averaged between animals where SEMs were calculated. All data were analyzed by 1-way ANOVA followed by a Newman-Keuls post-hoc t-test where significance (p) <0.05 using GraphPad Prism software (GraphPad, Inc., San Diego, CA).
Histology
At the completion of the study, some rats were anesthetized with pentobarbital (50 mg/kg) and were infused with 5 μM Colcemid 15 min prior to intracardial perfusion. Following the infusion, rats were perfused with 4% paraformaldehyde in 0.1M phosphate buffer, pH 7.4. After fixing, the brains were removed from the cranium and stored in 0.1M phosphate buffer containing 20% sucrose for 24 hr, frozen on powdered dry ice and stored at −80°C until further processing. Brains were then sectioned using a cryotome (25μm) sections. Sections containing the SCN region were processed for immunohistochemistry to confirm microtubule depolymerization following Colcemid infusion and also to show the probe placement over the cortex of the SCN using cresyl stain.
Immunohistochemical analysis of β-tubulin in the SCN
Briefly, sections were incubated in mouse β tubulin antibody (Sigma; 4 μg/ml) for 72 hrs at 4°C. Next, sections were rinsed thoroughly and incubated in biotinylated horse anti mouse secondary antibody. The sections were rinsed and incubated in Avidin-Biotin HRP complex 9 (1:500) using the Vector Elite kit (Vector Labs, Burlingame, CA) for 1h. Next, the tissues were incubated in tris acetate solution containing 3-3′ diamino benzidine (0.5mg/mL), H202 (0.01%) and NiCl2 (0.032%) for 10 min. Counter staining was performed using cresyl stain. Following the reaction, the sections were rinsed, mounted onto slides, dehydrated in graded ethanols, cleaned in xylene and coverslipped under DePex as referenced [46]. Sections were observed under a microscope and microphotographs taken.
Results
As shown in figure 1, Colcemid, when added during the 16 hr withdrawal period (/C) for either vehicle-pretreated (V/C) or melatonin-pretreated (M/C) produced the characteristic rounded morphology. No changes in cellular morphology occurred in vehicle-treated cells (V/V) or if Colcemid was added during the 8 hr pretreatment period (data not shown).
Fig. 1.

The effects of Colcemid on MT2-CHO cell morphology. Pretreatment of MT2-CHO cells with melatonin (400 pM for 8 hr) followed by a 16 hr withdrawal of melatonin (M/V) resulted in a morphology similar to vehicle-treated cells (V/V). The addition of Colcemid, during the 8 hr pretreatment with or without melatonin resulted in no change in cellular morphology when compared to vehicle-treated cells (data not shown). However, the addition of Colcemid, during the 16 hr withdrawal period with (M/C) or without (V/C) melatonin, resulted in a round morphology when compared to vehicle (V/V)-treated cells. Data shown are representative of one experiment repeated twice.
Pretreatment and withdrawal of melatonin (M/V) did not change the forskolin-induced cAMP accumulation when compared to vehicle-treated (V/V) cells (Fig. 2) nor did it change affinity (KD) [vehicle KD= 116 pM (66–204 pM) versus melatonin-treated KD= 115 pM (49–270 pM)] or Bmax values (melatonin-treated Bmax = 116 % ± 59% of vehicle-treated cells). Even though no increase in forskolin-induced cAMP accumulation occurred following melatonin exposure, the addition of Colcemid during the 16 hr withdrawal period (M/C) significantly attenuated this response compared to melatonin-treated cells. No effect of Colcemid on cAMP reduction was seen when added in combination with melatonin during the 8 hr pretreatment period in MT2-CHO cells when compared to either vehicle or melatonin exposed cells (data not shown).
Figure 2.
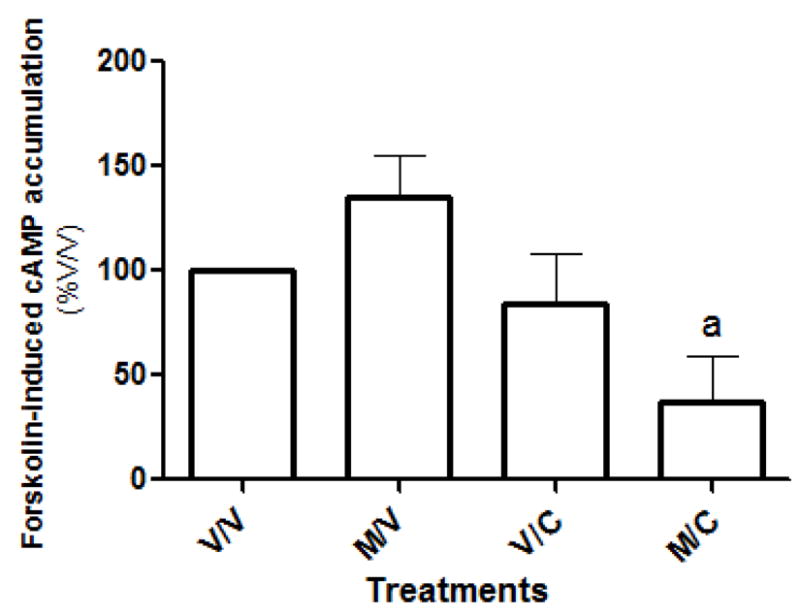
Pretreatment of MT2-CHO cells with melatonin (400 pM for 8 hr) followed by a 16 hr withdrawal of melatonin (M/V) resulted in no increase in forskolin-induced cAMP accumulation compared to vehicle (V/V)-treated cells. Even though no increase in forskolin-induced cAMP accumulation occurred in MT2-CHO cells following melatonin exposure, addition of Colcemid during the 16 hr withdrawal period (M/C), significantly attenuated forskolin-induced cAMP accumulation compared to melatonin (M/V)-treated cells. No effect of Colcemid was seen when added in combination with melatonin during the 8 hr pretreatment period (data not shown) or during the 16 hr withdrawal period (V/C). Each data point represents the mean ± SEM of 4–11 experiments performed in quadruplicate. Data were analyzed by one-way ANOVA followed by a Newman-Keuls post-hoc where (a) p<0.05 when compared to melatonin-treated cells.
The effects of microtubule depolymerization on the potency of melatonin to inhibit forskolin-induced cAMP accumulation was examined in cells exposed to vehicle, melatonin, Colcemid or combinations of each. Pretreatment of MT2-CHO cells with melatonin for 8h followed by withdrawal for 16 hr (M/V) resulted in a loss in the number of high potency sites of this receptor defined as a potency value of < 100 pM when compared to vehicle-treated cells (Fig. 3). The addition of Colcemid during the 16 hr melatonin withdrawal period (M/C) prevented the loss of high potency sites that occurs in melatonin-treated (M/V) cells. Colcemid alone was without effect whether it was added during the 8 hr pretreatment period (data not shown) or 16 hr withdrawal period compared to vehicle-treated cells (Fig. 3).
Fig. 3.
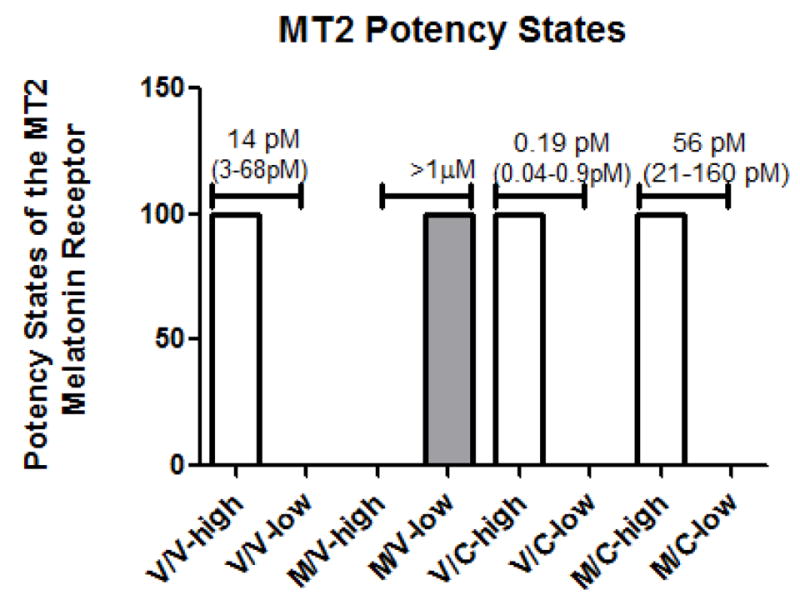
Pretreatment of MT2-CHO cells with melatonin (400 pM for 8 hr) followed by a 16 hr withdrawal of melatonin (M/V) resulted in a loss of high potency sites (-high) and a shift to low potency states (-low) when compared to vehicle (V/V)-exposed cells. However, Colcemid, added during the 16 hr withdrawal period (M/C), prevented the loss in the number of high-potency states when compared to melatonin-treated cells. The addition of Colcemid during the 8 hr pretreatment period (data not shown) or during the 16 hr withdrawal period (V/C) yielded effects similar to vehicle -treated cells. Data are representative of 3–7 experiments performed in triplicate or quadruplicate. Data points were fit by 1-site or 2-site fit non-linear regression analysis (GraphPad Prism, San Diego, CA) based on residual sum of squares. Potency (IC50) values shown above the graphs represent the mean and range of SEM of 3–5 experiments performed in duplicate.
The effects of microtubule depolymerization on the affinity of melatonin to compete for 2-[125I]-iodomelatonin binding was also examined. However, no changes in affinity (Ki) occurred in any of the treatment groups and when compared to melatonin-exposed cells [Ki values (range of SEM): V/V= 1.6 nM (1.2–2.2nM); M/V=1.3nM (0.98–1.9nM); V/C=1.7nM (0.91–3.2 nM); M/C=0.7nM (0.34–1.4 nM), n=3].
As shown in figure 4, melatonin increased the formation of PIs in a concentration-dependent increase in vehicle (V/V)-treated cells which was abolished if cells were pre-exposed with melatonin (M/V). Colcemid added in the 16 hr withdrawal period (M/C) was without effect on modulating melatonin-induced PI hydrolysis in MT2-CHO cells. Melatonin (1pM-1μM) increased PKC activity in MT2-CHO cells in a concentration-dependent manner up to 10 nM where the higher concentrations resulted in a decrease (Fig. 5). Exposure of MT2-CHO cells to melatonin (M/V) followed by a 1 nM melatonin rechallenge increased PKC activity similar to vehicle-treated (V/V) cells. However, microtubule depolymerization either following a vehicle-pretreatment (V/C) or melatonin pretreatment (M/C) enhanced melatonin-induced PKC activity compared to vehicle- or melatonin-treated cells. No effect of Colcemid was observed if it was added alone or in combination with melatonin during the 8 hr pretreatment period (data not shown). The addition of pertussis toxin (MP/CP) attenuated the enhancing effects of Colcemid on melatonin-induced PKC activity (M/C) while it was without effect if added alone (P/P; Fig. 6).
Fig. 4.
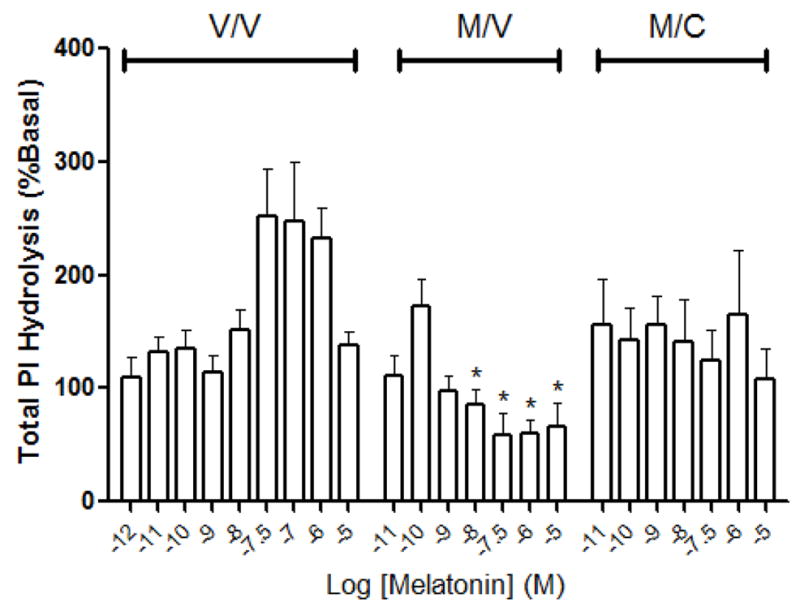
The effect of melatonin on PI hydrolysis in MT2-CHO cells following various treatments. As shown, melatonin (10 nM – 10 μM) increased the formation of PIs in cells exposed to vehicle (V/V) with peak levels occurring at 30 nM melatonin. No concentration-dependent increase in PIs occurred in cells exposed to melatonin (M/V) or melatonin and then Colcemid (M/C). Each data point represents the mean ± SEM of 5–13 independent experiments. Significance (*) is defined as p < 0.05 when compared to the levels of PI released in vehicle-treated cells stimulated with the same concentration of melatonin.
Fig. 5.
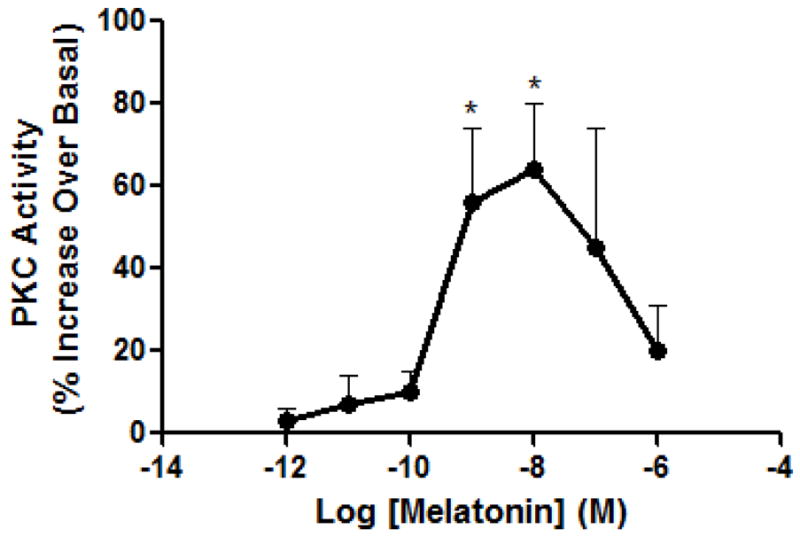
The effect of melatonin on PKC activation in MT2-CHO cells following various treatments. Melatonin (1 pM – 1 μM) enhanced PKC activity with a maximum occurring at 10 nM melatonin. Data are representative of 3–6 independent experiments. Significance (*) is defined as p < 0.05 when compared to basal levels of PKC activity.
Fig. 6.
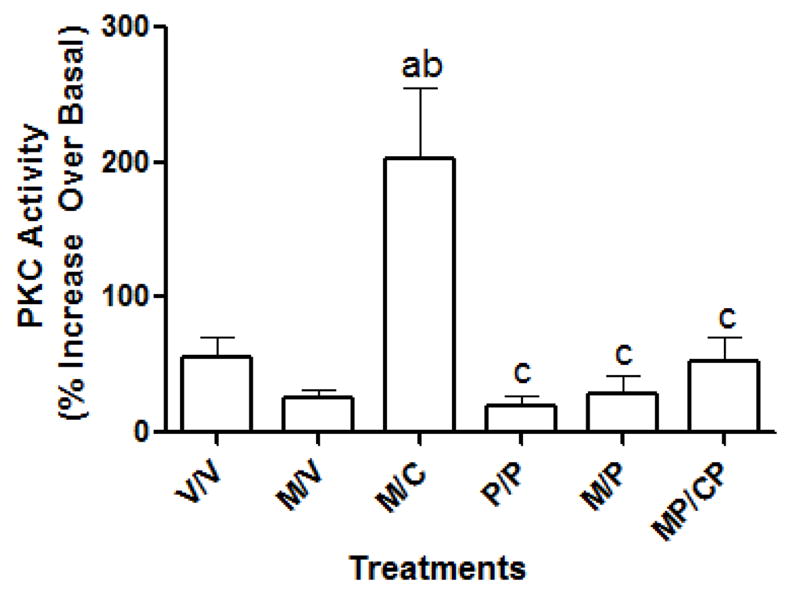
The effect of pertussis toxin on the enhancement of PKC induced following microtubule depolymerization. As shown, Colcemid, added during the 16 hr withdrawal period following either a vehicle pretreatment (V/C) or a melatonin pretreatment (M/C), enhanced PKC activity following a 10nM melatonin re-challenge when compared to vehicle-treated cells (V/V) or melatonin-treated cells (M/V), respectively. However, when pertussis toxin was added along with Colcemid during the 16 hr withdrawal period (MP/CP), this enhancement of PKC activity by 10 nM melatonin was blocked. The addition of pertussis toxin either after melatonin pretreatment (M/P) or alone (P/P) was without effect. All data were analyzed by 1-way ANOVA followed by a Newman-Keuls Post-hoc t-test where (a)= P<0.05 when compared to V/V; (b)=P<0.05 when compared to M/V; (c) =P<0.05 when compared to M/C.
As shown in figure 7A, an increase in the incorporation of [32P]AAGTP into G-proteins occurred when cells were exposed to Colcemid during the 16 hr withdrawal period (/C) and when compared to vehicle-pretreated (V/V) or melatonin-pretreated (M/V) cells. This incorporation of [32P]AAGTP was into Giα– and not Gqα– proteins as revealed through western blot analysis (Fig. 7B). To ensure that the increase in G protein function modulated by microtubules was not due to an up-regulation of Gi protein levels, western blot analysis was performed. No statistical difference in Gi protein levels occurred following melatonin exposure compared to vehicle-treated cells (data not shown).
Fig. 7.

The effect of microtubule depolymerization in MT2-CHO cells on the incorporation of [32P]-azidoanilido GTP into G-proteins. (A) Depicted is the amount of [32P]-azidoanilido GTP incorporated into G-proteins in MT2-CHO cells following exposure to vehicle, melatonin, Colcemid, or combinations of each. As shown, following depolymerization of microtubules during the 16 hr withdrawal period in vehicle (V/C)- or melatonin (M/C)-exposed cells, an increase in melatonin-mediated [32P]-azidoanilido GTP incorporation into G-proteins occurred when compared to vehicle-treated (V/V) or melatonin-treated (M/V) cells. No effect of Colcemid was seen when added during the 8 hr melatonin pretreatment period (M+C/V). The data shown in the graphs were expressed as a percentage of the vehicle-exposed cells. Data represent the mean ± SEM of 6 experiments. Data were analyzed by one-way ANOVA followed by a Newman-Keuls post-hoc t-test where (a) = p<0.05 when compared to vehicle (V/V)-treated cells; (b) = p<0.05 compared to melatonin (M/V)-treated cells; (c) = p<0.05 when compared to Colcemid (C/V)-treated cells; (d) = p<0.05 when compared to melatonin + Colcemid (M+C/V)-treated cells. [INSET: a representative autoradiogram of the [32P]-azidoanilido GTP incorporation into G-proteins in MT2-CHO cells. Note that the lane assignments are identical to the legends in the graphs]. (B) Depicted are immunoblots of Giα, Gqα (middle and right panels, respectively) of the autoradiograms of [32P]-azidoanilido GTP incorporation into G-proteins following activation by melatonin (1 μM) (left panel). The qualitative nature of the bands (doublets) as well as the size of the bands having incorporated the [32P]-azidoanilido GTP suggested that this labeling occurred predominantly into Giα and not Gqα. Samples were run in duplicate. Data are representative of one experiment repeated one other time.
In the studies involving the Long Evans rat, cresyl stains of the SCN showed that the guide cannulae were properly positioned in the SCN as revealed by the guide cannulae track marks (see arrow; Fig. 8). Immunohistochemical analysis of β-tubulin in the SCN showed that infusion of Colcemid through the guide cannula depolymerized microtubules within the SCN specifically reflected by the characteristic “rounding” of the SCN cells due to microtubule collapse (Fig. 9).
Fig. 8.

Cresyl stain of the SCN in the Long Evans rat. As shown, the track for the guide cannula can be distinguished in these brain slices (arrow, right side) when compared to an uncannulated SCN shown on the left. Magnifications are indicated in parentheses. The SCN region is outlined by a dotted circle.
Fig. 9.

Immunohistochemical analysis of β– tubulin within the SCN in the Long Evans rat. Colcemid was infused directly into the SCN of a Long Evans rat through a cannula 15 min prior to sacrifice. As shown, the cells within the SCN infused with Colcemid (left and middle panels) produced the characteristic “rounding” of cells when compared to the SCN infused with vehicle (right panel). These data demonstrate that the infusion of Colcemid through the guide cannula was able to selectively depolymerize the microtubules within the SCN. Magnifications are indicated in parentheses. The SCN region is outlined by a dotted circle.
Infusion of melatonin (100 pM) through the cannulae directly into the SCN produced a phase-shift in the average peak activity rhythms compared to 10 pM melatonin and vehicle (Fig. 10). Co-infusion of Colcemid (5μM) along with melatonin (10 pM) induced a significant phase-shift in peak activity rhythms compared to 10 pM melatonin alone. Co-infusion of β-lumicolchicine, an inactive analog of Colcemid, along with 10 pM melatonin was without effect on phase-shifting peak activity rhythms and was similar to both 10 pM- and vehicle-infusions. Colcemid (5μM) alone was without effect when compared to vehicle treatment. Nocodazole, added in combination with melatonin (10 pM) induced significant phase-shifts in peak activity rhythms compared to 10 pM melatonin (Fig. 11A) and vehicle infusions albeit to a lesser magnitude than the melatonin (10 pM)/Colcemid (5μM) infusions (Fig. 11B).
Fig. 10.

The effects of melatonin and Colcemid on the circadian activity rhythms of the Long Evans rat. As shown, infusion of melatonin (100 pM) through the cannula inserted into the SCN resulted in phase-shifts of circadian activity rhythms compared to vehicle or 10 pM melatonin infusions. Colcemid (5 μM), added in combination with melatonin (10 pM), enhanced the phase-shifts when compared to animals infused with melatonin (10 pM) alone. Beta-lumicolchicine, the inactive analog of Colcemid, when infused along with 10 pM melatonin, was without effect and was similar to vehicle and 10 pM melatonin infusions. Similarly, the infusion of Colcemid (5 μM) was without effect and similar to vehicle. Data are representative of 3–5 independent experiments. Data were analyzed by one-way ANOVA followed by a Newman-Keuls post-hoc t-test where (a) = p < 0.05 when compared to vehicle; (b) = p < 0.05 when compared to 10 pM melatonin; (c) = p < 0.05 when compared to 100 pM melatonin; (d) = p < 0.05 when compared to 10 pM melatonin + 5 μM Colcemid.
Fig. 11.
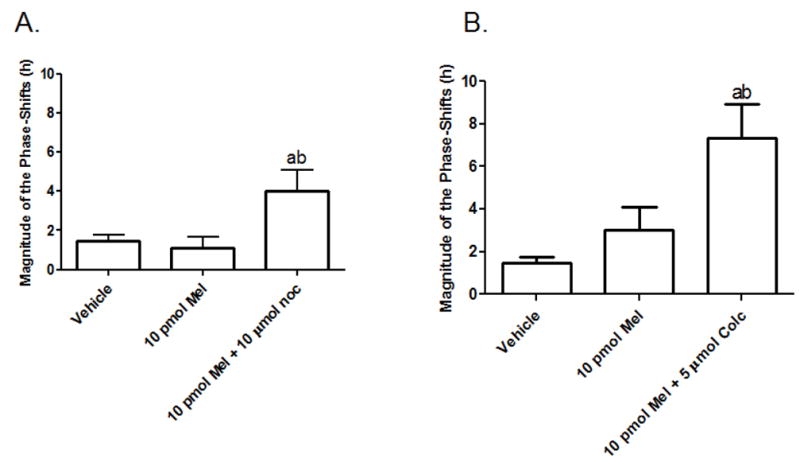
The effects of melatonin and nocodazole or Colcemid on phase-shifting the circadian activity rhythms of the Long Evans rat. (A) As shown, nocodazole (10 μM), co-infused with 10 pM melatonin, enhances phase-shifts of peak activity rhythms when compared to 10 pM melatonin and vehicle. However, the magnitude of the shifts are not as great as those induced with the 10 pM melatonin/5μM Colcemid infusions as shown in (B). Data are representative of 3 independent experiments for the nocodazole studies and 7 independent experiments for the Colcemid studies. Data were analyzed by one-way ANOVA followed by a Newman-Keuls post-hoc t-test where (a) = p < 0.05 when compared to vehicle; (b) = p < 0.05 when compared to 10 pM melatonin.
Discussion
MT1Rs and MT2Rs are responsible for the sleep promoting and circadian effects of melatonin [47]. The physiological roles of MT1Rs and MT2Rs in circadian entrainment is becoming clearer and shows that MT1Rs may be important for dampening the firing of SCN neurons and, as such, are involved in regulating the amplitude of circadian rhythmicity [48] and MT2Rs more involved in phase-shifting circadian activity rhythms [9–11]. Little is known regarding MT2R-mediated signaling and regulation. In our study and similar to other reports [42, 49, 50], melatonin inhibited forskolin-induced cAMP accumulation [42, 49, 50] and stimulation of PI hydrolysis [42]. Chronic exposure to melatonin in a manner that mimics the normal rise and fall of melatonin in vivo resulted in MT2R desensitization reflected by a complete loss in MT2R-mediated inhibition of forskolin-induced cAMP accumulation and an attenuation in MT2R-mediated increase in PI hydrolysis when compared to vehicle-exposed cells. Melatonin increased PKC activity in a concentration-dependent manner where peak activity was achieved at concentrations of melatonin between 1–10 nM. Similar effects (EC50 values in the 1–10 nM range) of melatonin to increase PKC activity are seen in SCN brain slices taken from the Long Evans rat [51]. Melatonin induces these phase-advances in the neuronal firing patterns of the SCN neurons through MT2Rs [11]. Concentrations of melatonin greater than 10 nM resulted in decreases in PKC activity, which is probably due to MT2R desensitization. Because MT2Rs are involved in phase-shifting circadian activity rhythms, then understanding the signaling mechanisms that underlie their function and regulation will aid in the understanding of their function in vivo.
Melatonin receptor sensitivity changes throughout the light/dark cycle in the SCN where the lowest receptor affinity and density occur during the hours of darkness when melatonin levels are highest [52–54]. Changes in melatonin receptor sensitivity may be essential to the role of melatonin receptors in vivo to modulate circadian rhythms. Disruptions in melatonin receptor sensitivity states may impact on melatonin regulated physiological processes like one’s sleep/wake cycles.
Microtubules have been shown to modulate the sensitivity states of both MT1Rs [7] and MT2Rs (present study). In particular, melatonin-induced MT1R desensitization [7] as well as MT2R desensitization (present study) is prevented when microtubules are depolymerized. The mechanism on how this occurs is most probably due to an increase in GTP exchange on the Gi-protein by alpha/beta tubulin heterodimers as depicted schematically in [7, 8]. This is supported in the [32P]AAGTP labeling studies which show that the incorporation of [32P]AAGTP into G-proteins was enhanced when microtubules were depolymerized by Colcemid. These data show that this enhancement in G-protein activation by Colcemid to increase Gα-GTP occurs on Gi-proteins and not Gq-proteins. This is further supported in the cAMP second messenger studies which show that the loss in high potency states of the MT2R (that normally occurs following melatonin exposure) is prevented in the presence of Colcemid. Melatonin-induced MT2R densitization is most probably being prevented because microtubule alpha/beta tubulin heterodimers are able to by-pass the MT2R and directly activate Gi-proteins in these cells. Direct binding of tubulin heterodimers to G-proteins has been demonstrated in other studies [55]. Furthermore, the attenuation in forskolin-induced cAMP accumulation in the presence of Colcemid supports microtubule-induced activation of Gi-protein as an increase in Giα-GTP would exert an inhibitory influence on adenylyl cyclase.
Surprisingly, though, were our findings on PI hydrolysis and PKC. As shown in our study, microtubule depolymerization enhanced melatonin-induced PKC activity but not melatonin-induced PI hydrolysis. Furthermore, these effects of Colcemid on enhancing melatonin-induced PKC activity were blocked in the presence of pertussis toxin. These data suggest that these alpha/beta tubulin heterodimers were modulating PKC activity in MT2-CHO cells through Gi. Similar effects of microtubules on both Gi-proteins and PKC activity is shown for MT1Rs expressed in CHO cells [7]. Also, the melatonin-induced phase-advances in the neuronal firing patterns in an SCN brain slice is shown to be mediated via MT2Rs [11], Gi-proteins and PKC [51]. Interestingly, manipulation of microtubules in MT2R-expressing cell line is able to modulate the same signaling mechanisms involved in clock rhythmicity. Perhaps, then, alterations in microtubule dynamics either through chemical manipulation (i.e., use of microtubule depolymerizing agents) or disease (i.e, AD) may impact on circadian rhythmicity in vivo. To test whether microtubule depolymerization can manipulate melatonin receptors involved in phase-shifting circadian activity rhythms, the SCN of the Long Evans rat was cannulated so that the microtubules within this region of the brain could be depolymerized specifically and in a controlled manner without impacting on other structures. Because the SCN controls circadian activity rhythms [47], it is hypothesized that microtubule depolymerization within the SCN will enhance the potency of melatonin to phase-shift their activity rhythms.
As shown, melatonin produced dose-dependent phase-shifts in the circadian activity rhythms of the Long Evans rat that were dose-dependent and similar in magnitude to the shifts induced by 3 pM melatonin in the neuronal firing patterns in the Long Evans rat SCN brain slice [11]. Dose-dependent effects of melatonin on activity rhythms have been shown [56]. In order to determine if microtubules could enhance melatonin-induced phase shifts, the dose of melatonin (i.e., 10 pM) that produced minimal, if any, shifts was chosen for the studies using the depolymerizing agents.
Depolymerization of microtubules in the SCN, using two different microtubule depolymerizing agents (i.e., Colcemid or nocodazole), enhanced the magnitude of the phase-shifting effects of 10 pM melatonin when compared to those shifts induced by 10 pM melatonin alone or vehicle. No enhancing effects of β-lumicolchicine, the inactive analog of Colcemid, on 10 pM melatonin-induced phase shifts was observed suggesting that these enhancing effects were due to Colcemid’s effects on microtubule and not due to its structural effects. This dose of Colcemid was chosen because it depolymerized microtubules in MT1-CHO cells [7] and in MT2-CHO cells (Fig. 1) as well as depolymerized the microtubules located within SCN cells (Fig. 9). Colcemid was also chosen because it acts rapidly and reversibly to prevent microtubule polymerization by acting at the alpha/beta tubulin interface.
Besides using chemical manipulation, microtubule derangements occur in disease states like AD; a disease characterized by neurofibrillary tangles. The neurofibrillary tangles are associated with hyperphosphorylation of tau protein; a microtubule associated protein that is associated with protein folding and microtubule assembly. A hyperphosphorylated tau protein leads to protein misfolding and inhibits tubulin assembly [57, 58]. Tau hyperphosphorylation occurs through an imbalance between tau protein kinases, glycogen synthase kinase-3 (GSK3β), cell division cycle 2 (cdk2), cyclin-dependent kinase-5 (cdk5), ERK 1 [59] and through its phosphatase, PP-2A [58].
Neurofibrillary tangles have been observed in the brains of people suffering from AD [27], including the SCN [35] and the neuronal microtubules are both morphologically and functionally abnormal in patients with advanced AD [36]. The disruption of microtubule dynamics in the brains of AD patients may be altering the function and sensitivity of melatonin receptors throughout the light/dark cycle that thus be one of the mechanisms underlying the frequent awakenings during the night and during naps [13–15].
Changes in MT1R and MT2R density in the brains of AD patients occurs, and the circadian timing system from the SCN of AD patients is also altered [12, 21]. Microtubule dynamics may be modifying the function of both melatonin receptors in the SCN; increasing MT1R activity during sleep to enhance arousal and increasing MT2R activity to phase-shift circadian activity rhythms. Microtubule depolymerization in CHO cells expressing either the MT1R or MT2R yielded similar effects on their coupling to adenylyl cyclase, on their activation of PKC and on their response to chronic melatonin exposure. These effects of MT1Rs and MT2Rs on sleep may be mediated via the PKC and MAPK pathways in the SCN. These signaling pathways have been shown to modulate clock rhythmicity. Mitogen activated protein kinase (MAPK) activity is shown to display circadian variations in the SCN and is induced by light [60]. Melatonin, acting through MT1Rs, is shown to increase MEK/ERK 1/2 activity in MT1-CHO cells [8] and microtubule depolymerization in MT1-CHO cells modulates MEK/ERK1/2 activity differentially, which is dependent upon the presence of agonist. For example, depolymerization of microtubules by Colcemid in the absence of melatonin increases MEK/ERK 1/2 activity in MT1-CHO cells when compared to vehicle-treated cells. However, microtubule depolymerization in the presence of melatonin decreases melatonin-induced MEK/ERK 1/2 activity compared to melatonin-treated cells [8]. Understanding the relationships between microtubules and signaling mechanisms involved in clock rhythmicity like MEK and ERK 1/2 within the SCN are important considering that the distribution of the active forms of both MEK and ERK 1/2 are found to correspond to the neurofibrillary changes in AD [58].
These data are intriguing and support a role for microtubules in modulating melatonin receptor function both in vitro and in vivo and should guide future studies examining the impact of microtubule depolymerization in the SCN on the phase-shifting effects as mediated through MT2Rs. Seeing that sleep disturbances in those suffering from AD are the primary reason for exaggerating cognitive impairment and the primary cause for their institutional care, then understanding the molecular mechanisms underlying these sleep disturbances to direct future therapies in the clinic for the treatment of AD is important [12]. At present, melatonin-directed therapies are being developed and show promise in vitro [61] and in vivo [33, 62–65].
Acknowledgments
This work was supported by grants NIH R15 DK 54070-01A1 and Duquesne University Faculty Development funds awarded to PAW-E and by an American Foundation for Pharmaceutical Education pre-doctoral fellowship awarded to MJJ. This work was also supported by grants MH 39595 and MH 57391 awarded to MMR. The authors would also like to thank Drs. Michael O. Fry and Mark Liberatore for their technical assistance on the SCN cannulations and to Dr. Robert Gibbs (University of Pittsburgh) for his technical assistance on the immunohistochemical and histological analysis.
References
- 1.YAN K, POPOVA JS, MOSS A, et al. Tubulin stimulates adenylyl cyclase activity in C6 glioma cells by bypassing the beta-adrenergic receptor: a potential mechanism of G protein activation. J Neurochem. 2001;76:182–90. doi: 10.1046/j.1471-4159.2001.00013.x. [DOI] [PubMed] [Google Scholar]
- 2.COTE M, PAYET MD, DUFOUR MN, et al. Association of the G protein alpha(q)/alpha11-subunit with cytoskeleton in adrenal glomerulosa cells: role in receptor-effector coupling. Endocrinology. 1997;138:3299–307. doi: 10.1210/endo.138.8.5319. [DOI] [PubMed] [Google Scholar]
- 3.JASPER JR, POST SR, DESAI KH, et al. Colchicine and cytochalasin B enhance cyclic AMP accumulation via postreceptor actions. J Pharmacol Exp Ther. 1995;274:937–42. [PubMed] [Google Scholar]
- 4.LEIBER D, JASPER JR, ALOUSI AA, et al. Alteration in Gs-mediated signal transduction in S49 lymphoma cells treated with inhibitors of microtubules. J Biol Chem. 1993;268:3833–7. [PubMed] [Google Scholar]
- 5.COTE M, PAYET MD, GALLO-PAYET N. Association of alpha S-subunit of the GS protein with microfilaments and microtubules: implication during adrenocorticotropin stimulation in rat adrenal glomerulosa cells. Endocrinology. 1997;138:69–78. doi: 10.1210/endo.138.1.4860. [DOI] [PubMed] [Google Scholar]
- 6.WITT-ENDERBY PA, JARZYNKA MJ, KRAWITT BJ, et al. Knock-down of RGS(4) and beta tubulin in CHO cells expressing the human MT(1) melatonin receptor prevents melatonin-induced receptor desensitization. Life Sci. 2004;75:2703–15. doi: 10.1016/j.lfs.2004.08.002. [DOI] [PubMed] [Google Scholar]
- 7.JARZYNKA MJ, PASSEY DK, IGNATIUS PF, et al. Modulation of melatonin receptors and G-protein function by microtubules. J Pineal Res. 2006;41:324–36. doi: 10.1111/j.1600-079X.2006.00371.x. [DOI] [PubMed] [Google Scholar]
- 8.BONDI CD, MCKEON RM, BENNETT JM, et al. MT1 melatonin receptor internalization underlies melatonin-induced morphologic changes in Chinese hamster ovary cells and these processes are dependent on Gi proteins, MEK 1/2 and microtubule modulation. J Pineal Res. 2008;44:288–98. doi: 10.1111/j.1600-079X.2007.00525.x. [DOI] [PubMed] [Google Scholar]
- 9.JIN X, VON GALL C, PIESCHL RL, et al. Targeted disruption of the mouse Mel(1b) melatonin receptor. Mol Cell Biol. 2003;23:1054–60. doi: 10.1128/MCB.23.3.1054-1060.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.DUBOCOVICH ML, YUN K, AL-GHOUL WM, et al. Selective MT2 melatonin receptor antagonists block melatonin-mediated phase advances of circadian rhythms. FASEB J. 1998;12:1211–20. doi: 10.1096/fasebj.12.12.1211. [DOI] [PubMed] [Google Scholar]
- 11.HUNT AE, AL-GHOUL WM, GILLETTE MU, et al. Activation of MT(2) melatonin receptors in rat suprachiasmatic nucleus phase advances the circadian clock. Am J Physiol Cell Physiol. 2001;280:C110–8. doi: 10.1152/ajpcell.2001.280.1.C110. [DOI] [PubMed] [Google Scholar]
- 12.WU YH, SWAAB DF. Disturbance and strategies for reactivation of the circadian rhythm system in aging and Alzheimer’s disease. Sleep Med. 2007;8:623–36. doi: 10.1016/j.sleep.2006.11.010. [DOI] [PubMed] [Google Scholar]
- 13.HARPER DG, STOPA EG, MCKEE AC, et al. Differential circadian rhythm disturbances in men with Alzheimer disease and frontotemporal degeneration. Arch Gen Psychiatry. 2001;58:353–60. doi: 10.1001/archpsyc.58.4.353. [DOI] [PubMed] [Google Scholar]
- 14.PRINZ PN, VITIELLO MV, RASKIND MA, et al. Geriatrics: sleep disorders and aging. N Engl J Med. 1990;323:520–6. doi: 10.1056/NEJM199008233230805. [DOI] [PubMed] [Google Scholar]
- 15.MCCURRY SM, LOGSDON RG, TERI L, et al. Characteristics of sleep disturbance in community-dwelling Alzheimer’s disease patients. J Geriatr Psychiatry Neurol. 1999;12:53–9. doi: 10.1177/089198879901200203. [DOI] [PubMed] [Google Scholar]
- 16.MOE KE, VITIELLO MV, LARSEN LH, et al. Symposium: Cognitive processes and sleep disturbances: Sleep/wake patterns in Alzheimer’s disease: relationships with cognition and function. J Sleep Res. 1995;4:15–20. doi: 10.1111/j.1365-2869.1995.tb00145.x. [DOI] [PubMed] [Google Scholar]
- 17.POLLAK CP, PERLICK D. Sleep problems and institutionalization of the elderly. J Geriatr Psychiatry Neurol. 1991;4:204–10. doi: 10.1177/089198879100400405. [DOI] [PubMed] [Google Scholar]
- 18.BIANCHETTI A, SCURATTI A, ZANETTI O, et al. Predictors of mortality and institutionalization in Alzheimer disease patients 1 year after discharge from an Alzheimer dementia unit. Dementia. 1995;6:108–12. doi: 10.1159/000106930. [DOI] [PubMed] [Google Scholar]
- 19.BONANNI E, MAESTRI M, TOGNONI G, et al. Daytime sleepiness in mild and moderate Alzheimer’s disease and its relationship with cognitive impairment. J Sleep Res. 2005;14:311–7. doi: 10.1111/j.1365-2869.2005.00462.x. [DOI] [PubMed] [Google Scholar]
- 20.MORAN M, LYNCH CA, WALSH C, et al. Sleep disturbance in mild to moderate Alzheimer’s disease. Sleep Med. 2005;6:347–52. doi: 10.1016/j.sleep.2004.12.005. [DOI] [PubMed] [Google Scholar]
- 21.WU YH, FISCHER DF, KALSBEEK A, et al. Pineal clock gene oscillation is disturbed in Alzheimer’s disease, due to functional disconnection from the “master clock”. FASEB J. 2006;20:1874–6. doi: 10.1096/fj.05-4446fje. [DOI] [PubMed] [Google Scholar]
- 22.WU YH, FEENSTRA MG, ZHOU JN, et al. Molecular changes underlying reduced pineal melatonin levels in Alzheimer disease: alterations in preclinical and clinical stages. J Clin Endocrinol Metab. 2003;88:5898–906. doi: 10.1210/jc.2003-030833. [DOI] [PubMed] [Google Scholar]
- 23.MISHIMA K, TOZAWA T, SATOH K, et al. Melatonin secretion rhythm disorders in patients with senile dementia of Alzheimer’s type with disturbed sleep-waking. Biol Psychiatry. 1999;45:417–21. doi: 10.1016/s0006-3223(97)00510-6. [DOI] [PubMed] [Google Scholar]
- 24.MAGRI F, SARRA S, CINCHETTI W, et al. Qualitative and quantitative changes of melatonin levels in physiological and pathological aging and in centenarians. J Pineal Res. 2004;36:256–61. doi: 10.1111/j.1600-079X.2004.00125.x. [DOI] [PubMed] [Google Scholar]
- 25.WU YH, ZHOU JN, VAN HEERIKHUIZE J, et al. Decreased MT1 melatonin receptor expression in the suprachiasmatic nucleus in aging and Alzheimer’s disease. Neurobiol Aging. 2007;28:1239–47. doi: 10.1016/j.neurobiolaging.2006.06.002. [DOI] [PubMed] [Google Scholar]
- 26.BRUNNER P, SOZER-TOPCULAR N, JOCKERS R, et al. Pineal and cortical melatonin receptors MT1 and MT2 are decreased in Alzheimer’s disease. Eur J Histochem. 2006;50:311–6. [PubMed] [Google Scholar]
- 27.SAVASKAN E, AYOUB MA, RAVID R, et al. Reduced hippocampal MT2 melatonin receptor expression in Alzheimer’s disease. J Pineal Res. 2005;38:10–6. doi: 10.1111/j.1600-079X.2004.00169.x. [DOI] [PubMed] [Google Scholar]
- 28.WU YH, ZHOU JN, BALESAR R, et al. Distribution of MT1 melatonin receptor immunoreactivity in the human hypothalamus and pituitary gland: colocalization of MT1 with vasopressin, oxytocin, and corticotropin-releasing hormone. J Comp Neurol. 2006;499:897–910. doi: 10.1002/cne.21152. [DOI] [PubMed] [Google Scholar]
- 29.REITER RJ, TAN DX, MANCHESTER LC, et al. Melatonin reduces oxidant damage and promotes mitochondrial respiration: implications for aging. Ann N Y Acad Sci. 2002;959:238–50. doi: 10.1111/j.1749-6632.2002.tb02096.x. [DOI] [PubMed] [Google Scholar]
- 30.PAPPOLLA MA, CHYAN YJ, POEGGELER B, et al. An assessment of the antioxidant and the antiamyloidogenic properties of melatonin: implications for Alzheimer’s disease. J Neural Transm. 2000;107:203–31. doi: 10.1007/s007020050018. [DOI] [PubMed] [Google Scholar]
- 31.FENG Z, ZHANG JT. Melatonin reduces amyloid beta-induced apoptosis in pheochromocytoma (PC12) cells. J Pineal Res. 2004;37:257–66. doi: 10.1111/j.1600-079X.2004.00164.x. [DOI] [PubMed] [Google Scholar]
- 32.MATSUBARA E, BRYANT-THOMAS T, PACHECO QUINTO J, et al. Melatonin increases survival and inhibits oxidative and amyloid pathology in a transgenic model of Alzheimer’s disease. J Neurochem. 2003;85:1101–8. doi: 10.1046/j.1471-4159.2003.01654.x. [DOI] [PubMed] [Google Scholar]
- 33.WANG JZ, WANG ZF. Role of melatonin in Alzheimer-like neurodegeneration. Acta Pharmacol Sin. 2006;27:41–9. doi: 10.1111/j.1745-7254.2006.00260.x. [DOI] [PubMed] [Google Scholar]
- 34.SCHMID HA, REQUINTINA PJ, OXENKRUG GF, et al. Calcium, calcification, and melatonin biosynthesis in the human pineal gland: a postmortem study into age-related factors. J Pineal Res. 1994;16:178–83. doi: 10.1111/j.1600-079x.1994.tb00098.x. [DOI] [PubMed] [Google Scholar]
- 35.STOPA EG, VOLICER L, KUO-LEBLANC V, et al. Pathologic evaluation of the human suprachiasmatic nucleus in severe dementia. J Neuropathol Exp Neurol. 1999;58:29–39. doi: 10.1097/00005072-199901000-00004. [DOI] [PubMed] [Google Scholar]
- 36.BOUTTE AM, NEELY MD, BIRD TD, et al. Diminished taxol/GTP-stimulated tubulin polymerization in diseased region of brain from patients with late-onset or inherited Alzheimer’s disease or frontotemporal dementia with parkinsonism linked to chromosome-17 but not individuals with mild cognitive impairment. J Alzheimers Dis. 2005;8:1–6. doi: 10.3233/jad-2005-8101. [DOI] [PubMed] [Google Scholar]
- 37.MISHIMA K, OKAWA M, HOZUMI S, et al. Supplementary administration of artificial bright light and melatonin as potent treatment for disorganized circadian rest-activity and dysfunctional autonomic and neuroendocrine systems in institutionalized demented elderly persons. Chronobiol Int. 2000;17:419–32. doi: 10.1081/cbi-100101055. [DOI] [PubMed] [Google Scholar]
- 38.MAHLBERG R, KUNZ D, SUTEJ I, et al. Melatonin treatment of day-night rhythm disturbances and sundowning in Alzheimer disease: an open-label pilot study using actigraphy. J Clin Psychopharmacol. 2004;24:456–9. doi: 10.1097/01.jcp.0000132443.12607.fd. [DOI] [PubMed] [Google Scholar]
- 39.CARDINALI DP, BRUSCO LI, LIBERCZUK C, et al. The use of melatonin in Alzheimer’s disease. Neuro Endocrinol Lett. 2002;23 (Suppl 1):20–3. [PubMed] [Google Scholar]
- 40.PANDI-PERUMAL SR, ZISAPEL N, SRINIVASAN V, et al. Melatonin and sleep in aging population. Exp Gerontol. 2005;40:911–25. doi: 10.1016/j.exger.2005.08.009. [DOI] [PubMed] [Google Scholar]
- 41.SKENE DJ, SWAAB DF. Melatonin rhythmicity: effect of age and Alzheimer’s disease. Exp Gerontol. 2003;38:199–206. doi: 10.1016/s0531-5565(02)00198-5. [DOI] [PubMed] [Google Scholar]
- 42.MACKENZIE RS, MELAN MA, PASSEY DK, et al. Dual coupling of MT(1) and MT(2) melatonin receptors to cyclic AMP and phosphoinositide signal transduction cascades and their regulation following melatonin exposure. Biochem Pharmacol. 2002;63:587–95. doi: 10.1016/s0006-2952(01)00881-4. [DOI] [PubMed] [Google Scholar]
- 43.SLOTTEN HA, KREKLING S, PEVET P. Photic and nonphotic effects on the circadian activity rhythm in the diurnal rodent Arvicanthis ansorgei. Behav Brain Res. 2005;165:91–7. doi: 10.1016/j.bbr.2005.06.046. [DOI] [PubMed] [Google Scholar]
- 44.JOHNSON DA, ZAMBON NJ, GIBBS RB. Selective lesion of cholinergic neurons in the medial septum by 192 IgG-saporin impairs learning in a delayed matching to position T-maze paradigm. Brain Res. 2002;943:132–41. doi: 10.1016/s0006-8993(02)02623-9. [DOI] [PubMed] [Google Scholar]
- 45.BENLOUCIF S, DUBOCOVICH ML. Melatonin and light induce phase shifts of circadian activity rhythms in the C3H/HeN mouse. J Biol Rhythms. 1996;11:113–25. doi: 10.1177/074873049601100204. [DOI] [PubMed] [Google Scholar]
- 46.GIBBS RB. Expression of estrogen receptor-like immunoreactivity by different subgroups of basal forebrain cholinergic neurons in gonadectomized male and female rats. Brain Res. 1996;720:61–8. doi: 10.1016/0006-8993(96)00106-0. [DOI] [PubMed] [Google Scholar]
- 47.PANDI-PERUMAL SR, SRINIVASAN V, SPENCE DW, et al. Role of the melatonin system in the control of sleep: therapeutic implications. CNS Drugs. 2007;21:995–1018. doi: 10.2165/00023210-200721120-00004. [DOI] [PubMed] [Google Scholar]
- 48.LIU C, WEAVER DR, JIN X, et al. Molecular dissection of two distinct actions of melatonin on the suprachiasmatic circadian clock. Neuron. 1997;19:91–102. doi: 10.1016/s0896-6273(00)80350-5. [DOI] [PubMed] [Google Scholar]
- 49.REPPERT SM, GODSON C, MAHLE CD, et al. Molecular characterization of a second melatonin receptor expressed in human retina and brain: the Mel1b melatonin receptor. Proc Natl Acad Sci U S A. 1995;92:8734–8. doi: 10.1073/pnas.92.19.8734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.GERDIN MJ, MASANA MI, REN D, et al. Short-term exposure to melatonin differentially affects the functional sensitivity and trafficking of the hMT1 and hMT2 melatonin receptors. J Pharmacol Exp Ther. 2003;304:931–9. doi: 10.1124/jpet.102.044990. [DOI] [PubMed] [Google Scholar]
- 51.MCARTHUR AJ, HUNT AE, GILLETTE MU. Melatonin action and signal transduction in the rat suprachiasmatic circadian clock: activation of protein kinase C at dusk and dawn. Endocrinology. 1997;138:627–34. doi: 10.1210/endo.138.2.4925. [DOI] [PubMed] [Google Scholar]
- 52.WITT-ENDERBY PA, BENNETT J, JARZYNKA MJ, et al. Melatonin receptors and their regulation: biochemical and structural mechanisms. Life Sci. 2003;72:2183–98. doi: 10.1016/s0024-3205(03)00098-5. [DOI] [PubMed] [Google Scholar]
- 53.SCHUSTER C, GAUER F, MALAN A, et al. The circadian clock, light/dark cycle and melatonin are differentially involved in the expression of daily and photoperiodic variations in mt(1) melatonin receptors in the Siberian and Syrian hamsters. Neuroendocrinology. 2001;74:55–68. doi: 10.1159/000054670. [DOI] [PubMed] [Google Scholar]
- 54.NEU JM, NILES LP. A marked diurnal rhythm of melatonin ML1A receptor mRNA expression in the suprachiasmatic nucleus. Brain Res Mol Brain Res. 1997;49:303–6. doi: 10.1016/s0169-328x(97)00218-0. [DOI] [PubMed] [Google Scholar]
- 55.ROYCHOWDHURY S, RASENICK MM. Tubulin-G protein association stabilizes GTP binding and activates GTPase: cytoskeletal participation in neuronal signal transduction. Biochemistry. 1994;33:9800–5. doi: 10.1021/bi00198a052. [DOI] [PubMed] [Google Scholar]
- 56.DEACON S, ARENDT J. Melatonin-induced temperature suppression and its acute phase-shifting effects correlate in a dose-dependent manner in humans. Brain Res. 1995;688:77–85. doi: 10.1016/0006-8993(95)96872-i. [DOI] [PubMed] [Google Scholar]
- 57.BENITEZ-KING G, TUNEZ I, BELLON A, et al. Melatonin prevents cytoskeletal alterations and oxidative stress induced by okadaic acid in N1E-115 cells. Exp Neurol. 2003;182:151–9. doi: 10.1016/s0014-4886(03)00085-2. [DOI] [PubMed] [Google Scholar]
- 58.PEI JJ, BRAAK H, AN WL, et al. Up-regulation of mitogen-activated protein kinases ERK1/2 and MEK1/2 is associated with the progression of neurofibrillary degeneration in Alzheimer’s disease. Brain Res Mol Brain Res. 2002;109:45–55. doi: 10.1016/s0169-328x(02)00488-6. [DOI] [PubMed] [Google Scholar]
- 59.LU Q, SORIA JP, WOOD JG. p44mpk MAP kinase induces Alzheimer type alterations in tau function and in primary hippocampal neurons. J Neurosci Res. 1993;35:439–44. doi: 10.1002/jnr.490350411. [DOI] [PubMed] [Google Scholar]
- 60.OBRIETAN K, IMPEY S, STORM DR. Light and circadian rhythmicity regulate MAP kinase activation in the suprachiasmatic nuclei. Nat Neurosci. 1998;1:693–700. doi: 10.1038/3695. [DOI] [PubMed] [Google Scholar]
- 61.RODRIGUEZ-FRANCO MI, FERNANDEZ-BACHILLER MI, PEREZ C, et al. Novel tacrine-melatonin hybrids as dual-acting drugs for Alzheimer disease, with improved acetylcholinesterase inhibitory and antioxidant properties. J Med Chem. 2006;49:459–62. doi: 10.1021/jm050746d. [DOI] [PubMed] [Google Scholar]
- 62.RIEMERSMA-VAN DER LEK RF, SWAAB DF, TWISK J, et al. Effect of bright light and melatonin on cognitive and noncognitive function in elderly residents of group care facilities: a randomized controlled trial. JAMA. 2008;299:2642–55. doi: 10.1001/jama.299.22.2642. [DOI] [PubMed] [Google Scholar]
- 63.DOWLING GA, BURR RL, VAN SOMEREN EJ, et al. Melatonin and bright-light treatment for rest-activity disruption in institutionalized patients with Alzheimer’s disease. J Am Geriatr Soc. 2008;56:239–46. doi: 10.1111/j.1532-5415.2007.01543.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.CHENG Y, FENG Z, ZHANG QZ, et al. Beneficial effects of melatonin in experimental models of Alzheimer disease. Acta Pharmacol Sin. 2006;27:129–39. doi: 10.1111/j.1745-7254.2006.00267.x. [DOI] [PubMed] [Google Scholar]
- 65.FENG Z, QIN C, CHANG Y, et al. Early melatonin supplementation alleviates oxidative stress in a transgenic mouse model of Alzheimer’s disease. Free Radic Biol Med. 2006;40:101–9. doi: 10.1016/j.freeradbiomed.2005.08.014. [DOI] [PubMed] [Google Scholar]