

Abstract
Background:
Six candidate gene studies report a genetic association of DNA variants within the paraoxonase locus with sporadic amyotrophic lateral sclerosis (ALS). However, several other large studies, including five genome-wide association studies, have not duplicated this finding.
Methods:
We conducted a meta-analysis of 10 published studies and one unpublished study of the paraoxonase locus, encompassing 4,037 ALS cases and 4,609 controls, including genome-wide association data from 2,018 ALS cases and 2,425 controls.
Results:
The combined fixed effects odds ratio (OR) for rs662 (PON1 Q192R) was 1.09 (95% confidence interval [CI], 1.02–1.16, p = 0.01); the genotypic OR for RR homozygotes at Q192R was 1.25 (95% CI, 1.07–1.45, p = 0.0004); the combined OR for rs854560 (PON1 L55M) was 0.97 (95% CI, 0.86–1.10, p = 0.62); the OR for rs10487132 (PON2) was 1.08 (95% CI, 0.92–1.27, p = 0.35). Although the rs662 polymorphism reached a nominal level of significance, no polymorphism was significant after multiple testing correction. In the subanalysis of samples with genome-wide data from which population outliers were removed, rs662 had an OR of 1.06 (95% CI, 0.97–1.16, p = 0.22).
Conclusions:
In contrast to previous positive smaller studies, our genetic meta-analysis showed no significant association of amyotrophic lateral sclerosis (ALS) with the PON locus. This is the largest meta-analysis of a candidate gene in ALS to date and the first ALS meta-analysis to include data from whole genome association studies. The findings reinforce the need for much larger and more collaborative investigations of the genetic determinants of ALS.
GLOSSARY
- ALS
= amyotrophic lateral sclerosis;
- CI
= confidence interval;
- GWAS
= genome-wide association studies;
- OR
= odds ratio;
- SALS
= sporadic ALS;
- SNP
= single nucleotide polymorphism.
Amyotrophic lateral sclerosis (ALS) is a rapidly progressive and devastating neurodegenerative disorder that usually leads to death in only 2 to 5 years. Over 90% of ALS cases occur sporadically and are hypothesized to result from a combination of risk from genes and environment. Among candidate genes proposed as ALS susceptibility factors,1,2 recent attention has focused on the paraoxonase gene family. Six recent case-control association studies document an association of polymorphisms in the paraoxonase locus, which includes the PON1, PON3, and PON2 genes on chromosome 7 q 21.3-q22.1.3–8 However, one large candidate gene study and five genome-wide association studies (GWAS) have not supported this association.6,9–13 These conflicting results may reflect the relatively small sample sizes and the heterogeneity of single nucleotide polymorphisms chosen for analysis in each report. We hypothesized that a meta-analysis combining all available studies of the PON locus would increase the power to detect a true association and that analyzing a small subset of PON polymorphisms from GWAS data would reduce the multiple testing burden and increase the sensitivity for smaller effect sizes.
Paraoxonase 1 (PON1) is an esterase that metabolizes oxidized lipids and organophosphate insecticides such as chlorpyrifos, diazinon, and to a lesser extent parathion.14,15 Epidemiologic studies report an increased risk for sporadic ALS (SALS) after exposure to insecticides and pesticides.16–19 Genetic variation across the paraoxonase gene locus is believed to affect individual susceptibility to these exogenous compounds, including the PON1 coding variants L55M and Q192R and the regulatory region polymorphisms C-108T and A-162G.20,21 PON2 and PON3 share with PON1 activity metabolizing oxidized lipids,22 but do not metabolize organophosphates.
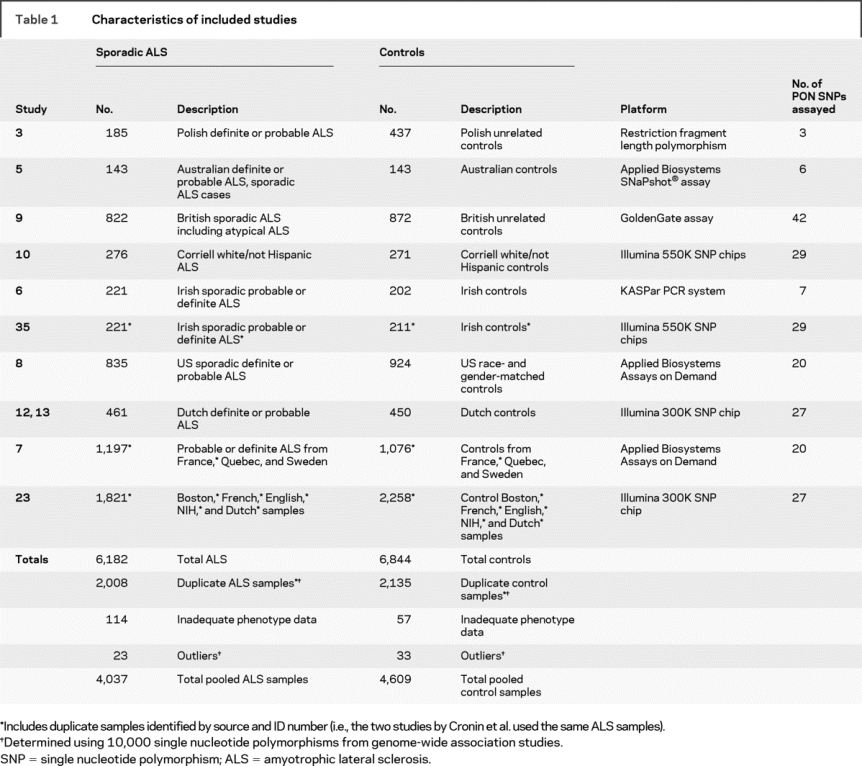
To clarify the evidence linking the PON locus to ALS, we performed a meta-analysis of 10 published association studies and one unpublished study of ALS that included paraoxonase gene polymorphisms (table 1). To reduce publication bias, we also actively sought genotyping results from unpublished data and from GWA studies. By combining individual genotyping results, we were able to eliminate duplicate samples between studies as well as population outliers for a total of 4,037 ALS cases and 4,609 controls. This is the largest genetic meta-analysis to date in ALS and the first to include data from recent genome-wide association studies.
Table 1 Characteristics of included studies
METHODS
This meta-analysis complies with the recommendations of the Meta-analysis Of Observational Studies in Epidemiology Group.
Included studies.
Our analysis included all SNP association studies of the PON locus in sporadic ALS conducted before August 1, 2008. To accomplish this task, the MEDLINE and PubMed databases were searched using keywords relating to the paraoxonase genes (e.g., “paraoxonase,” “PON,” “PON1,” “PON2,” “PON3”) or genome-wide analysis (“genome-wide,” “whole genome”) in combination with ALS (e.g., “amyotrophic lateral sclerosis,” “ALS,” “motor neuron disease”). To reduce the likelihood of publication bias, unpublished data, negative data from candidate gene studies, and GWAS data were also actively sought. We identified seven candidate gene studies, of which three assayed functional polymorphisms3,5,6 and four used tagging single nucleotide polymorphisms (SNPs).4,7–9 All of the authors of these studies were contacted to request individual genotyping results; genotypic and phenotypic information was available for inclusion from six of these candidate gene studies, totaling 3,403 ALS cases and 3,654 controls (table 1).3,5–9 One published candidate gene study,4 which reported an association based on an affected patient-parent trio cohort using paraoxonase tagging SNPs, was excluded because raw data were not made available for this analysis.4 However, the case-control arm of that same study found no association with ALS and therefore exclusion of these data are unlikely to have affected our results. To reduce the likelihood of publication bias, we also searched both unpublished data and negative data in the form of GWA studies using the methods above. Five published10–13,35 and one unpublished23 genome-wide studies were identified. The authors of five of these studies10,12,13,23,35 provided individually genotyped Illumina chip data for pooling in our analysis. One study was excluded because the PON region was only assayed using pooled DNA in the discovery phase11 and thus individual genotypes could not be utilized. After elimination of identified duplicate samples, a total of 2,041 cases and 2,468 controls were included in the overall analysis from the GWA studies. These samples were used in the subanalysis of GWAS data from which population outliers were also removed.
Data abstraction.
The following information was abstracted from each study: study design, geographic location, ethnicity of participants, inclusion criteria, numbers of cases and controls, DNA extraction and genotyping methods, SNPs tested, frequency of genotypes, consistency of genotype frequencies with Hardy-Weinberg equilibrium, and proportion of men. Confirmation of genotype frequencies and genotyping procedures was achieved by requesting individual genotyping data. In the few instances in which genotype frequencies provided by the investigators in tabular data differed slightly from published figures, the tabular data were used. The diagnosis of ALS was limited to definite or probable by El Escorial criteria and individuals with a diagnosis of possible or atypical ALS were excluded from the combined raw genotyping analysis.24,25 Ethnic origin was limited in almost all of the studies to white Caucasians of European ancestry; in the combined analysis, other ethnicities were excluded because genotype frequencies vary substantially between Caucasian and African and Asian populations.
Statistical methods for meta-analysis.
Owing to the heterogeneity of studied polymorphisms in each report, we used three methods to analyze risk for ALS attributable to each individual PON locus polymorphism. First, we performed a meta-analysis for each candidate SNP using the summary data from all studied populations. Results were combined using either a fixed effects (Mantel-Haenszel) model or random effects model depending on the heterogeneity between studies. Heterogeneity was calculated using Cochran's Q statistic and the I2 statistic, where I2 greater than 50% is considered significantly heterogeneous.26,27 Funnel plots were also generated to look for evidence of study bias.28 Forest and funnel plots were generated using Review Manager 4.2 (The Nordic Cochrane Center, Copenhagen, Denmark). We did not impute missing genotypes because of the strong possibility of spurious findings.29
As a second method, we formally combined all available raw genotyping results to calculate an association statistic for each SNP. This allowed us to exclude individuals with atypical phenotypes or non-Caucasian ethnicity who had been included in the original reports. Duplicates were eliminated in this analysis using sample ID (as in the case of NIH Coriell samples used in several studies)10 and by excluding samples with identity by descent >0.75 based on an analysis of 10,000 SNPs from the GWA studies (see below). This method also allowed us to examine risk using a variety of genetic association models. Logistic regression and allelic and genotypic association tests were performed using PLINK software.30,31
Finally, we performed a third analysis restricted to data from the whole genome association studies.10,12,13,23,35 Combining only GWAS data allowed us to eliminate duplicate samples using identity by descent >0.75, and to perform detailed population stratification bias analysis and haplotype analyses. Population structure refers to differences in allele frequencies due to variations in ancestry within a study population. Stratification bias refers to a false genetic association due to differences in the population structures of the case and control populations, and can be corrected by the exclusion of population outliers. Our GWA dataset was subjected to stratification analysis using 10,000 unlinked SNPs from other regions of the genome using PLINK, which is greater than the minimum 200 unlinked biallelic markers required to test for differences in population stratification.32 Based on the distribution of pairwise genome-wide identity-by-state distances, we applied complete linkage hierarchical cluster analysis. As a result, 66 population outliers, defined as 3 SDs from the group mean, were eliminated, leaving 2,018 cases and 2,425 controls.
All genotypes were tested for deviation from Hardy-Weinberg equilibrium using PLINK software. Genetic association tests were performed in PLINK including subsidiary analysis of dominant, recessive, genotypic, and Cochrane-Armitage trend tests. The per-allele OR (relative risk) of the rare allele (i.e., R192) was calculated using the logistic regression function of PLINK. Results were adjusted for multiple testing using the Bonferroni correction using either PLINK or SAS software (SAS Institute, Cary, NC) and visualized using Haploview 4.0.33,34 Bonferroni correction deflates the reported p value to take into account the number of tests performed, using the formula 1 − (1 − α)1/n (often approximated by α/n, where α equals 0.05).
Findings of data abstraction.
A total of 13 relevant studies were identified, of which 11 were included (table 1). Studies were conducted primarily in the United States and Europe, and all included samples were white/Caucasian from Poland, Australia, the United States, England, Ireland, Holland, Canada, France, or Sweden. All 11 included studies were case-control retrospective studies with controls drawn at random from approximately general populations, except for two studies which also included spousal controls.5,6 Only the French controls used in one study were not age- and gender-matched.23 Two studies3,5 used restriction fragment length polymorphism with standard restriction enzymes (AlwI or DpnII for PON1 Q192R and NlaIII for PON1 L55M and DdeI for PON 2 C311S). One candidate gene study also used the Applied BiosystemsSNaPshot® assay (Foster City, CA),5 one candidate study used the KASPar PCR system (KBiosciences, UK),6 one used the GoldenGate assay on an Illumina BeadArray station (San Diego, CA),9 and the remaining candidate gene studies used TaqMan SNP Assay probes (Applied Biosystems).7,8 All of the genome-wide association studies included used Illumina Infinium II SNP Chip assays, either the Human Hap550K6,10 or the HumanHap 300K SNP chips12,13,23 (Illumina), which facilitated the pooling of data across all sites.
Combining the individual genotyping data, after subtracting duplicate samples and population outliers, there were 8,646 individuals with confirmed phenotypic information, for a total of 4,037 cases and 4,609 controls. Across the GWA studies, there were 25 SNPs genotyped in common in 2,018 ALS subjects and 2,425 controls (after exclusion of overlapping samples and outliers). This included case-control populations from the United States, Ireland, the United Kingdom, France, and The Netherlands.
RESULTS
Meta-analysis and pooled genotype data for PON1 coding variants and promoter polymorphisms.
Among the coding and regulatory region PON1 polymorphisms with known putative functional effects, the Q192R polymorphism (rs662) was the most studied; it was included in 11 studies (9 distinct study populations) for a total of 4,151 cases of ALS (65.4% men) and 4,727 controls (66.7% men).3,5–10,12,23 The candidate SNP study6 and GWAS study35 of Cronin et al. used the same study population, as did the two articles by van Es et al.,12,13 and therefore their populations were included only once. We used a fixed effects model to analyze the published results for PON1 Q192R (rs662) because the amount of heterogeneity was not significant among the nine included studies (p = 0.15). The fixed-effects odds ratio (OR) for the R allele was 1.09 (95% confidence interval [CI], 1.02–1.16, p = 0.01) (figure 1A, left). The recessive model (RR genotype) had an OR of 1.25 (95% CI, 1.07–1.45, p = 0.004) (figure 1A, right). Eliminating the discovery study by Slowik et al.3 reduced the fixed effects OR for the R allele to 1.07 (95% CI, 1.0–1.15, p = 0.05). The funnel plot in figure 1B demonstrates no significant asymmetry to suggest publication bias, due in part to the inclusion of large published and unpublished negative studies, including the GWAS. We calculate that the total sample size of 4,151 ALS cases and 4,727 controls included in the rs662 analysis provided 80% power to detect an association with a relative risk of 1.1 at a significance level of 0.01 and greater than 90% power to detect a difference at a significance level of 0.05.36

Figure 1 Forest plots of nine studies of PON1 rs662 and six studies of rs854560
Study numbers refer to reference numbers. Published numbers include some duplicate samples, not included in the combined raw genotyping analysis. Areas of squares are proportional to the effective sample size, horizontal lines indicate confidence intervals (CI), and shaded diamonds denote grand totals. The horizontal axis is plotted on a log10 scale. (A) Forest plot of paraoxonase PON1 Q192R/rs662 polymorphism in amyotrophic lateral sclerosis. The left side of the figure shows a forest plot based on the odds ratios (ORs) for the arginine allele in each study. The right side of the graph shows a forest plot based on the frequency of RR homozygotes from the same studies. The overall ORs for both were calculated using a fixed effects model due to low heterogeneity. Cochran's Q statistic (a test of heterogeneity which follows a χ2 distribution) for the allelic analysis was not significant (12.12 with 8 degrees of freedom, p = 0.15) and the I2 statistic equaled 35% (less than 50%). For the analysis of RR homozygotes, Cochran's Q statistic equaled 5.71 with 8 degrees of freedom (p = 0.68) and the I2 statistic equaled 0%. (B) Funnel plot of the nine studies included in the rs662 analysis. The vertical axis shows the standard error of the log OR for each study, while the horizontal axis, the OR, is again plotted in a log10 scale. (C) Forest plot of the PON1 L55M/rs854560 polymorphism in amyotrophic lateral sclerosis. The overall OR was calculated using a random effects model due to significant heterogeneity (Cochran's Q statistic = 12.26 with 5 degrees of freedom, p = 0.03, and I2 = 59.2%).
Using the raw genotyping data, we excluded duplicate samples and samples with atypical phenotypes, resulting in 3,978 confirmed cases and 4,560 controls. The overall OR for the R allele remained 1.09 (95% CI, 1.02–1.16, uncorrected p = 0.013); for the homozygous recessive model, the OR was 1.21 (95% CI, 1.048–1.39, uncorrected p = 0.0029). Similarly, logistic regression calculated that the per-allele relative risk of the R192 variant for ALS was 1.08 (95% CI, 1.02–1.16, p = 0.013). Table 2 includes allelic, genotypic, dominant, and recessive models for the seven SNPs that were tested in at least 50% of the samples, shown graphically in figure 2, three of which were in high linkage disequilibrium with each other (D'). The R192 association was not significant after correction for 35 genotypic tests for the seven most commonly tested polymorphisms (Bonferroni-corrected p value = 0.103, table 2).
Table 2 Association testing of the seven most frequently assayed PON polymorphisms in 4,035 cases of sporadic amyotrophic lateral sclerosis and 4,603 controls
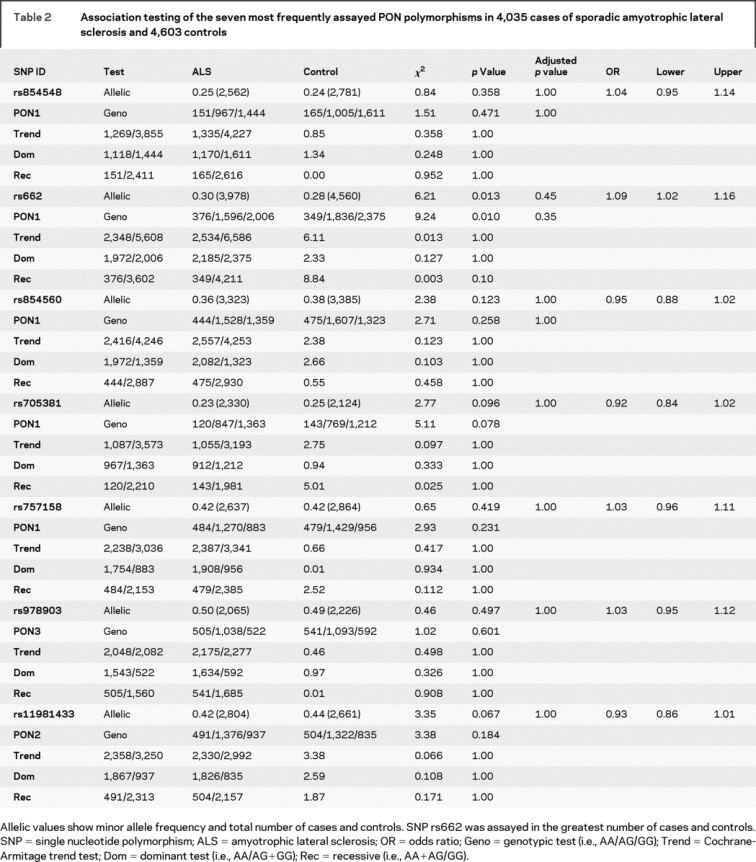

Figure 2 Association testing of seven single nucleotide polymorphisms within the PON cluster in 4,035 cases of sporadic amyotrophic lateral sclerosis and 4,603 controls
(A) Negative log10 of the unadjusted p value for each test in table 2 shown on y axis. (B) Exon mapping of the three paraoxonase genes, including the chromosomal location. (C) Pairwise linkage disequilibrium (D') and log of the odds ratio (lod) values were calculated using Haploview 4.0 for the seven tested single nucleotide polymorphisms. The color code on the Haploview plot follows the standard color scheme for Haploview: white (D' <1, lod <2); shades of pink/red (D' <1, lod >2); blue (D' = 1, lod <2).
The rs854560 (PON1 L55M) coding variant was assayed in six studies totaling 3,323 ALS cases and 3,385 controls.3,5–9 There was a significant amount of heterogeneity (I2 = 59%); therefore, we used a random-effects model which takes additional account of study variation. The combined OR was 0.97 (95% CI, 0.86–1.10, p = 0.62) (figure 1C, table 2). The combined OR for rs854560 using validated individual genotypes was 0.95 (95% CI, 0.88–1.015, p = 0.12). The promoter SNP rs705381 (PON1 G-162A) had a pooled OR of 0.92 (95% CI, 0.84–1.01, p = 0.1) in 2,330 ALS cases and 2,124 controls from four studies5–8 (table 2). Regulatory SNP rs705379 (PON1 T-108C) was only tested in a total of 364 ALS cases and 345 controls and therefore was not included in this analysis.
Meta-analysis of PON2 and PON3 polymorphisms.
PON2 rs11981433 was included in three studies of 2,804 ALS cases and 2,661 controls7–9 for which the combined OR was 0.93 (95% CI, 0.86–1.01, p = 0.07) (table 2). Combining 2,176 ALS cases and 1,934 controls from three studies6–8 for rs10487132 (PON2) produced an OR of 1.05 (95% CI, 0.96–1.14, p = 0.31). PON2 C311S/rs7493/rs6954345 had an OR of 1.16 (95% CI, 0.99–1.36, p = 0.06) in the pooled analysis of four studies.5–7,9 For a complete list of SNPs tested in at least 25% of samples, see table e-1 on the Neurology® Web site at www.neurology.org). None of these 34 SNPs reached significance after multiple testing correction.
Combined genome-wide association data.
Restricting the analysis to GWAS data permitted the elimination of duplicate samples and testing for population stratification using 10,000 unlinked SNPs from other regions of the genome using PLINK. After excluding 1,460 duplicates and 66 outliers, there were 2,018 unique ALS subjects and 2,425 unique controls with GWAS data (figure e-1 shows a multidimensional scaling analysis of the remaining samples based on identity-by-state distances). A total of 25 PON SNPs were tested in common, none of which was significant after Bonferroni correction (table e-2 and figure e-2). PON1 Q192R/rs662 was not significant in the pooled stratification-corrected genome-wide dataset (OR = 1.06, 95% CI, 0.97–1.16, p = 0.22); this suggests that stratification bias could account for the significant association noted in the combined analysis above for this SNP. Sliding-window haplotype analysis of the genome-wide data using PLINK software did not identify a single haplotype that was significantly associated with ALS after multiple test correction (table e-3).30,31
DISCUSSION
This meta-analysis of 11 genetic association studies, involving 4,037 ALS cases and 4,609 controls, provides the most comprehensive assessment so far of the relevance to ALS of polymorphisms within the paraoxonase locus. In addition, it is the largest meta-analysis of ALS to date, and the first to include genotyping data from recent genome-wide association studies in ALS. By including genome-wide studies, we were able to reduce publication bias. Our meta-analysis of all 11 studies found a nominally significant overall OR for the G allele (R192) of rs662 of 1.09 (95% CI, 1.02–1.16, p = 0.01). However, of the seven most commonly tested SNPs, including several that had previously been reported to be significant, no single variant, including rs662, showed robust association with risk for SALS after Bonferroni correction.
When our analysis was restricted to GWAS data from which population outliers had been removed, we found a lower nonsignificant OR for rs662 (1.06, 95% CI, 0.97–1.16, p = 0.22) than in our larger analysis. Although the sample size and statistical power of the genome-wide data were significantly smaller (with only 65% power to detect a relative risk of 1.1 at an alpha of 0.05), GWAS data allowed us to explore the influence of population stratification on our meta-analysis findings. The result suggests that the greater effect size observed in the candidate studies may have been falsely positive due to stratification. In our analysis of the genome-wide data, we found no PON polymorphism or haplotype with a Bonferroni-corrected p value below 0.05.
Our study has several limitations that must be considered when interpreting its findings. First, it was not possible to implement stratification control for the candidate gene studies; however, this is unlikely to result in a false negative finding unless there is a reversal of association in different subpopulations (Simpson's paradox), which was not observed in the forest plot of rs662. In addition, given that our analysis was limited to white Caucasians of European descent, population-specific differences in PON SNP frequencies, as described in African American populations with AD, would not be expected.37 Second, some of the regulatory promoter polymorphisms for PON1 were not tested in the majority of the samples. Finally, because data on exposure to exogenous toxins were only available in one study,5 we could not control for the heterogeneity of environmental toxin substrates across included populations.
Our meta-analysis suggests that common variants across the PON locus do not alter risk for ALS, although the rs662 polymorphism reached an uncorrected level of significance. The results also imply that candidate gene studies in ALS are likely to discover false positive associations, possibly due to population stratification. This supports the inclusion of unlinked genetic markers to test for stratification in future candidate gene studies. It also supports the use of unbiased association study data such as that generated in genome-wide analyses.
AUTHORS' AFFILIATIONS
From the Cecil B. Day Neuromuscular Research Laboratory (A.-M.W., J.E.L., R.H.B.), Massachusetts General Hospital, Charlestown; Department of Neurology (S.C., O.H.), Beaumont Hospital, Dublin, Ireland; Department of Neurology (A.S.), Jagiellonian University, Krakow, Poland; Department of Neurodegenerative Disease (D.K., E.M.C.F.), UCL Institute of Neurology, London, UK; Netherlands ALS Center Department of Neurology (M.A.V.E., L.H.V.d.B.), Rudolf Magnus Institute of Neuroscience, University Medical Center, Utrecht, The Netherlands; The Stacey MND Laboratory (J.M.M., R.P.), Department of Pathology, The University of Sydney, Australia; Department of Molecular and Clinical Genetics (J.M.M.), Royal Prince Alfred Hospital, Australia; Center of Excellence in Neuromics (P.N.V., G.A.R.), University of Montreal, CHUM Research Center, and the Department of Medicine, University of Montreal, Canada; Centre Référent SLA (V.M.), Hôpital Salpêtrière, Paris, France; Department of Human Genetics (J.M.), Hadassah University Hospital, Jerusalem, Israel; MRC Centre for Neurodegeneration Research (C.E.S., A.A.-C.), King's College London, Department of Clinical Neuroscience, Institute of Psychiatry, London, UK; Academic Neurology Unit (P.J.S,), Medical School, University of Sheffield, UK; Department of Clinical Neurosciences (K.E.M.), Institute of Biomedical Research, The Medical School, University of Birmingham, UK; University of Michigan (D.A.F.), Ann Arbor, MI and the ALS Society of Canada; Department of Neurology (P.M.A.), Umeå University Hospital, Sweden; Trinity College Institute of Neuroscience (O.H.), Trinity College, Dublin, Ireland; and Center for Human Genetics Research, Richard B. Simches Research Building (S.P.), Massachusetts General Hospital, Boston, MA.
SUPPORT INFORMATION
Support for the Boston-based investigations was provided by the ALS Therapy Alliance, Project ALS, the Angel Fund, the Pierre L. de Bourgknecht ALS Research Foundation, the Al-Athel ALS Research Foundation (R.H.B., J.E.L.), the ALS Family Charitable Foundation, the American Academy of Neurology Foundation/ALS Association (A.-M.W.), and the National Institute of Neurological Disorders and Stroke (R.H.B.). The Boston team also received funding from the Harvard Business School Class of 2007, the Yamner family, Avichai Kremer, Guy Yamen, Nate Boaz, and Professor Robert S. Kaplan. Support was also received from the Muscular Dystrophy Association, USA, and the NIH-NIEHS R01 ES013482-01 (D.A.F.). In Ireland, funding was received from the Muscular Dystrophy Association (United States) (S.C., O.H.) and the Irish Institute of Clinical Neuroscience Travel Award (S.C.). In England, support was provided by the MND Association of Great Britain and Northern Ireland (A.A.C., K.E.M., P.J.S.), the UK Medical Research Council (A.A.C., E.F., D.K., K.E.M., P.J.S.), the Wellcome Trust (P.N.L., C.E.S., P.J.S.), and Psychiatry Research Trust (Tim Perkins Fund) (P.N.L., A.A.C., C.E.S.). Support for the investigations in France was provided by Assistance Publique, Hopitaux de Paris, INSERM, ARS (Association pour la Recherche sur la SLA) and the Ministere de l'Enseignement Superieur et de la Recherche. Also supported by the DNA banks maintained by the Coriell Institute (with support from the National Institute of Neurological Disorders and Stroke) and Genethon. This study used data from the SNP Database at the National Institute of Neurological Disorders and Stroke Human Genetics Resource Center DNA and Cell Line Repository (http://ccr.coriell.org/ninds/). In The Netherlands, the project was supported by The Netherlands Organisation for Scientific Research (NWO) and the “Prinses Beatrix Fonds” (L.H.V.d.B.). In Australia, the project was supported by the Australian Rotary Health Fund, the Aimee Stacey Memorial Bequest, the Motor Neuron Disease Research Institute, and the National Health and Medical Research Council.
AUTHOR CONTRIBUTIONS
A.-M.W. takes full responsibility for the data, the analyses and interpretation, and the conduct of the research and has had full access to all of the data. A.-M.W., S.C., and J.E.L. drafted and initiated the report. All the investigators were involved in the design, interpretation, and redrafting. Statistical analyses were conducted by A.-M.W., Instructor in Neurology, Harvard Medical School, and Department of Neurology, Massachusetts General Hospital.
ACKNOWLEDGMENT
The authors thank all the clinicians who assisted in recruiting cases and controls, and particularly clients with ALS and their families.
Supplementary Material
Address correspondence and reprint requests to Dr. Wills, CNY 114-3125, 114 16th St, Charlestown, MA 02129 awills@partners.org.
Supplemental data at www.neurology.org
Editorial, page 11.
e-Pub ahead of print on March 25, 2009, at www.neurology.org.
Support information is provided at the end of the article.
Disclosure: The authors report no disclosures.
Devices: GoldenGate assay, Illumina BeadArray station, Infinium II SNP Chip assays, Human Hap550K SNP chips, HumanHap 300K SNP chips (Illumina, San Diego, CA); KASPar PCR system (KBiosciences, Hertfordshire, UK); SNaPshot® assay (Applied Biosystems, Foster City, CA).
Received September 12, 2008. Accepted in final form January 5, 2009.
REFERENCES
- 1.Schymick JC, Talbot K, Traynor BJ. Genetics of sporadic amyotrophic lateral sclerosis. Hum Mol Genet 2007;16:R233–242. [DOI] [PubMed] [Google Scholar]
- 2.Simpson CL, Al-Chalabi A. Amyotrophic lateral sclerosis as a complex genetic disease. Biochim Biophys Acta 2006;1762:973–985. [DOI] [PubMed] [Google Scholar]
- 3.Slowik A, Tomik B, Wolkow PP, et al. Paraoxonase gene polymorphisms and sporadic ALS. Neurology 2006;67:766–770. [DOI] [PubMed] [Google Scholar]
- 4.Saeed M, Siddique N, Hung WY, et al. Paraoxonase cluster polymorphisms are associated with sporadic ALS. Neurology 2006;67:771–776. [DOI] [PubMed] [Google Scholar]
- 5.Morahan JM, Yu B, Trent RJ, Pamphlett R. A gene-environment study of the paraoxonase 1 gene and pesticides in amyotrophic lateral sclerosis. Neurotoxicology 2007;28:532–540. [DOI] [PubMed] [Google Scholar]
- 6.Cronin S, Greenway MJ, Prehn JH, Hardiman O. Paraoxonase promoter and intronic variants modify risk of sporadic amyotrophic lateral sclerosis. J Neurol Neurosurg Psychiatry 2007;78:984–986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Valdmanis P, Kabashi E, Dyck A, et al. Association of paraoxonase gene cluster polymorphisms with ALS in France, Quebec and Sweden. Neurology 2008;71:514–520. [DOI] [PubMed] [Google Scholar]
- 8.Landers JE, Shi L, Cho TJ, et al. A common haplotype within the PON1 promoter region is associated with sporadic ALS. Amyotroph Lateral Scler 2008:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kasperaviciute D, Weale ME, Shianna KV, et al. Large-scale pathways-based association study in amyotrophic lateral sclerosis. Brain 2007;130:2292–2301. [DOI] [PubMed] [Google Scholar]
- 10.Schymick JC, Scholz SW, Fung HC, et al. Genome-wide genotyping in amyotrophic lateral sclerosis and neurologically normal controls: first stage analysis and public release of data. Lancet Neurol 2007;6:322–328. [DOI] [PubMed] [Google Scholar]
- 11.Dunckley T, Huentelman MJ, Craig DW, et al. Whole-genome analysis of sporadic amyotrophic lateral sclerosis. N Engl J Med 2007;357:775–788. [DOI] [PubMed] [Google Scholar]
- 12.van Es MA, Van Vught PW, Blauw HM, et al. ITPR2 as a susceptibility gene in sporadic amyotrophic lateral sclerosis: a genome-wide association study. Lancet Neurol 2007;6:869–877. [DOI] [PubMed] [Google Scholar]
- 13.van Es MA, van Vught PW, Blauw HM, et al. Genetic variation in DPP6 is associated with susceptibility to amyotrophic lateral sclerosis. Nat Genet 2008;40:29–31. [DOI] [PubMed] [Google Scholar]
- 14.Aldridge WN. Serum esterases: II: An enzyme hydrolysing diethyl p-nitrophenyl phosphate (E600) and its identity with the A-esterase of mammalian sera. Biochem J 1953;53:117–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Furlong CE, Richter RJ, Seidel SL, Costa LG, Motulsky AG. Spectrophotometric assays for the enzymatic hydrolysis of the active metabolites of chlorpyrifos and parathion by plasma paraoxonase/arylesterase. Anal Biochem 1989;180:242–247. [DOI] [PubMed] [Google Scholar]
- 16.Muddasir Qureshi M, Hayden D, Urbinelli L, et al. Analysis of factors that modify susceptibility and rate of progression in amyotrophic lateral sclerosis (ALS). Amyotroph Lateral Scler 2006;7:173–182. [DOI] [PubMed] [Google Scholar]
- 17.Rosati G, Pinna L, Granieri E, et al. Studies on epidemiological, clinical, and etiological aspects of ALS disease in Sardinia, Southern Italy. Acta Neurol Scand 1977;55:231–244. [DOI] [PubMed] [Google Scholar]
- 18.Holloway SM, Emery AE. The epidemiology of motor neuron disease in Scotland. Muscle Nerve 1982;5:131–133. [DOI] [PubMed] [Google Scholar]
- 19.McGuire V, Longstreth WT Jr, Nelson LM, et al. Occupational exposures and amyotrophic lateral sclerosis: a population-based case-control study. Am J Epidemiol 1997;145:1076–1088. [DOI] [PubMed] [Google Scholar]
- 20.Brophy VH, Jampsa RL, Clendenning JB, McKinstry LA, Jarvik GP, Furlong CE. Effects of 5′ regulatory-region polymorphisms on paraoxonase-gene (PON1) expression. Am J Hum Genet 2001;68:1428–1436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Davies HG, Richter RJ, Keifer M, Broomfield CA, Sowalla J, Furlong CE. The effect of the human serum paraoxonase polymorphism is reversed with diazoxon, soman and sarin. Nat Genet 1996;14:334–336. [DOI] [PubMed] [Google Scholar]
- 22.Mackness B, Durrington PN, Mackness MI. The paraoxonase gene family and coronary heart disease. Curr Opin Lipidol 2002;13:357–362. [DOI] [PubMed] [Google Scholar]
- 23.Landers J, Melki J, Meininger V, et al. Genetic variants in kinesin-associated protein gene KIFAP3 modify survival in sporadic amyotrophic lateral sclerosis. Submitted 2009. [DOI] [PMC free article] [PubMed]
- 24.Brooks BR. El Escorial World Federation of Neurology criteria for the diagnosis of amyotrophic lateral sclerosis: Subcommittee on Motor Neuron Diseases/Amyotrophic Lateral Sclerosis of the World Federation of Neurology Research Group on Neuromuscular Diseases and the El Escorial “Clinical limits of amyotrophic lateral sclerosis” workshop contributors. J Neurol Sci 1994;124(suppl):96–107. [DOI] [PubMed] [Google Scholar]
- 25.Brooks BR, Miller RG, Swash M, Munsat TL. El Escorial revisited: revised criteria for the diagnosis of amyotrophic lateral sclerosis. Amyotroph Lateral Scler Other Motor Neuron Disord 2000;1:293–299. [DOI] [PubMed] [Google Scholar]
- 26.Lau J, Ioannidis JP, Schmid CH. Quantitative synthesis in systematic reviews. Ann Intern Med 1997;127:820–826. [DOI] [PubMed] [Google Scholar]
- 27.Higgins JP, Thompson SG, Deeks JJ, Altman DG. Measuring inconsistency in meta-analyses. BMJ 2003;327:557–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Egger M, Davey Smith G, Schneider M, Minder C. Bias in meta-analysis detected by a simple, graphical test. BMJ 1997;315:629–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Xing C, Schumacher FR, Conti DV, Witte JS. Comparison of missing data approaches in linkage analysis. BMC Genet 2003;4(suppl 1):S44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Purcell S. Whole genome association analysis toolset. In: PLINK v1.00. Available at: http://pngu.mgh.harvard.edu/purcell/plink/. Accessed May 2008.
- 31.Purcell S, Neale B, Todd-Brown K, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet 2007;81:559–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Price AL, Patterson NJ, Plenge RM, Weinblatt ME, Shadick NA, Reich D. Principal components analysis corrects for stratification in genome-wide association studies. Nat Genet 2006;38:904–909. [DOI] [PubMed] [Google Scholar]
- 33.Barrett JC, Fry B, Maller J, Daly MJ. Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics 2005;21:263–265. [DOI] [PubMed] [Google Scholar]
- 34.Barrett J, Fry B, Maller J, Daly MJ. Haploview 4.0. Available at: http://www.broad.mit.edu/mpg/haploview/. Accessed May 2008.
- 35.Cronin S, Berger S, Ding J, et al. A genome-wide association study of sporadic ALS in a homogenous Irish population. Hum Mol Genet 2008;17:768–774. [DOI] [PubMed] [Google Scholar]
- 36.Purcell S, Cherny SS, Sham PC. Genetic Power Calculator: design of linkage and association genetic mapping studies of complex traits. Bioinformatics 2003;19:149–150. [DOI] [PubMed] [Google Scholar]
- 37.Erlich PM, Lunetta KL, Cupples LA, et al. Polymorphisms in the PON gene cluster are associated with Alzheimer disease. Hum Mol Genet 2006;15:77–85. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.