

Abstract
Excessive Ca2+ loading is central to most hypotheses of excitotoxic neuronal damage. We examined dendritic Ca2+ signals in single CA1 neurons, injected with fluorescent indicators, after extended exposures to a low concentration of NMDA (5 μm). As shown previously, NMDA produces an initial transient Ca2+ elevation of several micromolar, followed by recovery to submicromolar levels. Then after a delay of ∼20–40 min, a large Ca2+ elevation appears in apical dendrites and propagates to the soma. We show here that this large delayed Ca2+ increase is required for ultimate loss of membrane integrity. However, transient removal of extracellular Ca2+ for varying epochs before and after NMDA exposure does not delay the propagation of these events. In contrast to compound Ca2+ elevations, intracellular Na+ elevations are monophasic and were promptly reversed by the NMDA receptor antagonist MK-801 [(+)-5-methyl-10,11-dihydro-5H-dibenzo [a,d] cyclohepten-5,10-imine maleate]. MK-801 applied after the transient Ca2+ elevations blocked the delayed propagating Ca2+ increase. Even if applied after the propagating response was visualized, MK-801 restored resting Ca2+ levels. Propagating Ca2+ increases in dendrites were delayed or prevented by (1) reducing extracellular Na+, (2) injecting ATP together with the Ca2+ indicator, or (3) provision of exogenous pyruvate. These results show that extended NMDA exposure initiates degenerative signaling generally in apical dendrites. Although very high Ca2+ levels can report the progression of these responses, Ca2+ itself may not be required for the propagation of degenerative signaling along dendrites. In contrast, metabolic consequences of sustained Na+ elevations may lead to failure of ionic homeostasis in dendrites and precede Ca2+-dependent cellular compromise.
Keywords: excitotoxicity, CA1, hippocampus, sodium ion, ATP, MK-801, brain slice
Introduction
Overstimulation of glutamate receptors occurs during pathological conditions as a consequence of inappropriate transmitter release and/or compromised glutamate uptake (Olney, 1978; Choi, 1988a). As a consequence, multiple types of glutamate receptor are strongly activated, but, among these, the NMDA subtype has received particular attention in excitotoxicity (Simon et al., 1984; Choi, 1988b; Wahlestedt et al., 1993). Partial depolarization and relief of Mg2+ block after ischemic injuries may result in persistent low-level NMDA receptor activation (Novelli et al., 1988; Hori and Carpenter, 1994) and could contribute to increases in infarct size that occur after stroke injury in vivo. From studies of cultured neurons, excessive Ca2+ loading has been demonstrated after sustained NMDA exposures, and this Ca2+ overload has become central to most hypotheses of excitotoxic neuronal injury (Choi, 1988b; Arundine and Tymianski, 2003). NMDA receptor activation also results in substantial intracellular Na+ loading, and it has long been suggested that consequences of Na+ accumulation could be a significant contributor to neuronal injury (Rothman, 1985; Olney et al., 1986). Na+ extrusion by Na+/K+/ATPase is a major consumer of neuronal energy (Attwell and Laughlin, 2001), and ATP depletion that results from excessive Na+ entry could contribute to neuronal injury (Novelli et al., 1988; Fried et al., 1995).
ATP depletion may underlie Ca2+ overload after exposure of neuronal cultures to glutamate receptor agonists in neuronal cultures. As reviewed by Nicholls et al. (2007), intracellular Ca2+ accumulation and mitochondrial reactive oxygen species production has been suggested to cause unregulated Ca2+ increases and rapid neuronal death after NMDA receptor activation. However, studies of cerebellar granule cells in culture suggest that oxidative damage can be a consequence (rather than a cause) of unregulated Ca2+ overload that follows collapse of mitochondrial membrane potential (Vesce et al., 2005; Johnson-Cadwell et al., 2007). Demands for mitochondrial ATP synthesis to handle Na+ extrusion was suggested to be major contributor to subsequent deregulation of Ca2+ homeostasis in somata of these cultured neurons (Nicholls et al., 2007). It is not clear whether the relationships between energy depletion and Ca2+ homeostasis that have been described in neurons maintained in tissue culture also apply to neuronal preparations acutely prepared from the adult brain.
When CA1 neurons acutely isolated from adult hippocampus were briefly exposed to glutamate receptor agonists, Ca2+ deregulation could be localized to restricted regions of apical dendrites (Connor et al., 1988; Wadman and Connor, 1992). This was demonstrated in acutely prepared hippocampal slices, in which the full dendritic arbor remains essentially intact (Connor and Cormier, 2000), and dendritic Ca2+ overload has been implicated as an important determinant of excitotoxic vulnerability (Shuttleworth and Connor, 2001). We recently described propagating dendritic Ca2+ responses in CA1 neurons after extended exposures to NMDA and found that reversal of an Na+/Ca2+ exchanger did not contribute to Ca2+ overload (Dietz et al., 2007). The studies described here use the same NMDA model to examine whether Ca2+ itself is a critical causative agent for feedforward progression of degenerative signaling and/or whether other consequences of Na+ loading may contribute to initiation of dendritic injury.
Materials and Methods
Slice preparation.
Experiments were performed in accordance with the National Institute of Health guidelines for the humane treatment of laboratory animals, and the protocol for these procedures was reviewed annually by the Institutional Animal Care and Use Committee at the University of New Mexico School of Medicine. Male FVB/N mice were obtained from Harlan (Bar Harbor, ME) at 4 weeks of age and housed under standard conditions (12 h light/dark cycle) for a maximum of 2 weeks before they were killed. Mice were deeply anesthetized with a mixture of ketamine and xylazine (85 and 15 mg/ml, respectively; 150 μl, s.c.) and decapitated. Brains were quickly removed and placed in ice-cold cutting solution containing the following (in mm): 2 KCl, 1.25 NaH2PO4, 6 MgSO4, 26 NaHCO3, 0.2 CaCl2, 10 glucose, 220 sucrose, and 0.43 ketamine. Coronal sections (350 μm) were cut using a vibratome, and slices were transferred into room-temperature artificial CSF (ACSF) [in mm: 126 NaCl, 2 KCl, 1.25 NaH2PO4, 1 MgSO4 26 NaHCO3, 2 CaCl2, and 10 glucose (equilibrated with 95%O2/5%CO2)]. Cutting and recording solutions were both 315–320 mOsm. After warming to 35°C and holding for 1 h, ACSF was exchanged, and slices were held at room temperature until used for recording. Individual slices were transferred to the recording chamber on a fixed-stage microscope and superfused with warmed (32°C), oxygenated ACSF at a flow rate of 2–2.5 ml/min.
Indicator loading.
CA1 pyramidal neurons were visualized by using a 40× water-immersion objective on an Olympus (Tokyo, Japan) microscope (BX51WI) equipped with differential interference contrast optics. Whole-cell recordings were made by using pipettes fabricated from borosilicate glass using a horizontal pipette puller (Sutter Instruments, Novato, CA). The pipette solution contained the following (in mm): 135 K-gluconate, 8 NaCl, 1 MgCl2, 10 HEPES, 2 Mg2+-ATP, and 1 indicator [fura-6F, fura-2, or sodium-binding benzofuran isophthalate (SBFI)], pH 7.25 adjusted with KOH. Voltage-clamp recordings were made using a Multiclamp 700 A amplifier (Molecular Devices, Sunnyvale, CA), and cells were discarded (1) if the current required to maintain a holding voltage of −70 mV exceeded ± 100 pA or (2) if the membrane resistance was below 100 MΩ or exceeded 250 MΩ. After obtaining whole-cell access, dialysis (indicator loading) was strictly limited to 3 min. Pipettes were then slowly removed (∼0.5 μm/s) while continuously monitoring membrane resistance and soma Ca2+ or Na+ levels. Cells were discarded if soma Ca2+ or Na+ elevations were detected. The patch pipette removal process took ∼1 min to complete. Cells were then allowed to recover for 25 min after pipette removal before NMDA challenge, to allow diffusion of indicator into dendritic processes. This time was judged to be sufficient to allow equilibrium in the distal processes based on measurements of indicator concentration quite remote from somata. Reliable measurements could be made 200–250 μm from the somata and showed an initial rise in fluorescence at 0–15 min and a plateau until ∼30 min. To see whether this plateau represented actual equilibration of the dendritic and not simply equal influx from the soma and efflux to more distal parts of the dendritic tree, we monitored the decrease in soma fluorescence after electrode removal. This quantity declined initially but reached a near-asymptotic value at 20–25 min, indicating that there was no (or little) outflow from the soma and that the dendritic measurement reflected equilibration or near equilibration. To further address this point, we also tested the effects of doubling the incubation time (to 40 min) and found that the characteristics of propagating dendritic Ca2+ signals (described below) were unchanged, still occurring with mean delay of 35–40 min and propagating from apical dendrites. We have made estimates of indicator concentration at the soma (based on fluorescence intensity comparisons with neurons extensively dialyzed with known indicator concentrations) and conclude a concentration ∼200 μm, which is consistent with previous reports (Petrozzino and Connor, 1994).
Ca2+ and Na+ measurements.
Fluorescence measurements were made using a CCD-based system (Polychrome IV monochromator, IMAGO camera; T.I.L.L. Photonics, Pleasanton, CA). Indicators were excited at 350/380 nm, and emission was collected at 510 ± 40 nm. Image pairs were collected at 0.2 Hz. For figure presentation, images were first background subtracted, and then ratio images were generated and filtered using 3 × 3 pixel averaging. Final images were masked using an image generated from raw 380 nm fluorescence images. Conversion to Ca2+ concentrations was done using unfiltered pixel values, using an equation described previously (Grynkiewicz et al., 1985): [Ca2+] = KD(R − Rmin/Rmax − R)(Sf2/Sb2), where Rmin and Rmax are ratios at zero and saturating Ca2+ levels, and Sf2/Sb2 is the ratio of calcium-free to calcium-bound fluorescence at 380 nm excitation. Rmin, Rmax, and Sf2/Sb2 values were determined from calibration solutions (containing 1 mm Mg2+) that were loaded into thin-walled capillary tubes to prevent quenching effects and imaged within the recording chamber with the same water-immersion objectives as used for neuronal recordings. A KD value of 224 nm was assumed for fura-2 studies [calculated for 1 mm Mg2+ solutions (Grynkiewicz et al., 1985)] and 5.3 μm for fura-6F (Invitrogen, Carlsbad, CA). Because of the assumptions made in using these values, the calculated values should only be considered estimates of intracellular Ca2+ concentrations.
Calibration of SBFI signals was attempted but not found to be reliable in these preparations. Comparison of in situ ratios with in vitro standards (containing 135 mm K+) showed that in vitro comparisons were not appropriate, because Rmax values in vitro were less than maximal responses in neurons. Attempts at in situ calibrations (using monesin and gramicidin) were greatly complicated by substantial swelling of the slice and significant and rapid loss of 380 nm indicator signal. For these reasons, SBFI results are presented as ratio changes throughout the manuscript.
Reagents and solutions.
NMDA was applied in modified ACSF lacking added Mg2+. This solution was applied without preexposure to Mg2+-free ACSF, so there is expected to be some delay in NMDA receptor activation attributable to the time taken to reduce extracellular Mg2+ concentrations from the slice. The L-type Ca2+ channel blocker nimodipine (10 μm) was routinely included in all solutions after indicator loading. This is similar to the use of nifedipine (10 μm) in studies of protracted NMDA exposures in cultured cerebellar granule neurons (Rego et al., 2001). In preliminary experiments, nimodipine was found to reduce somewhat the incidence of nonrecoverable initial Ca2+ loading in CA1 somata soon after onset of NMDA exposures. However, nimodipine did not modify the kinetics of initial or sustained Ca2+ overload described below (n = 3; data not shown). In experiments to test the effects of elevated [ATP] in the pipette solution, Mg2+ATP was increased from 2 to 10 mm, which was calculated to increase free [ATP] from 0.18 to 1.03 mm [WEBMAXCLITE version 1.15 software, http://www.stanford.edu/∼cpatton/maxc.html (Patton et al., 2004)].
Except when noted, all reagents were from Sigma (St. Louis, MO). Stocks of nimodipine and ifenprodil were prepared in ethanol. Stocks of NMDA and TTX (in citrate buffer) (Calbiochem, San Diego, CA) were prepared in water. RO 256981 [R-(R,S)-α-(4-hydroxyphenyl)-β-methyl-4-(phenylmethyl)-1-piperidine propranol] (Fischer et al., 1997) and MK-801 [(+)-5-methyl-10,11-dihydro-5H-dibenzo [a,d] cyclohepten-5,10-imine maleate] stocks were in DMSO. Nominally Ca2+-free ACSF was prepared without added CaCl2 (without added chelators) and resulted in free Ca2+ concentrations of <10 μm (Chinopoulos et al., 2007). Reduced Na+ ACSF was prepared by supplementing 50% of NaCl with LiCl to produce a 41% reduction in total Na+.
Results
Ca2+ responses in CA1 pyramidal neurons produced by NMDA
Persistent NMDA exposure (5 μm, Mg2+-free ACSF; see Materials and Methods) produced a complex pattern of Ca2+ increases in CA1 neurons in slice (Fig. 1). An initial transient increase in cytosolic Ca2+ concentration (“Ca2+ spike”) in the soma and dendrites was followed, after a long delay, by a large sustained Ca2+ increase (“Ca2+ overload”) that originated in distal apical dendrites that propagated toward the soma, ultimately involving the entire neuron (Dietz et al., 2007, their Fig. 3). Because a goal of the present study was to determine the contribution of Ca2+ itself to the initiation and/or propagation of Ca2+ overload responses, the characteristics of Ca2+ transients were first considered here in more detail.
Figure 1.

Propagating secondary Ca2+ increases, initiated in apical dendrites. A, Ca2+ responses in a CA1 neuron during sustained exposure to NMDA. Left shows raw fura-6F fluorescence. Panels 1–6 are masked images pseudocolored to map Ca2+ concentration before NMDA (1) and during 40 min NMDA exposure (2–6). After an initial Ca2+ transient (2), the cell recovered to near-resting Ca2+ levels (3). After a long delay, sustained Ca2+ increases appeared first in a dendrite branch (4) and propagated through the entire neuron (5, 6). B, Data extracted from regions of interest in the soma and dendrite (location indicated by arrow) from the neuron shown in A. C, Population data, showing Ca2+ responses recorded from somata of 18 neurons, from slices prepared from 12 animals.
Figure 3.

A, Lack of effect of transient Ca2+ removal on the progression of subsequent Ca2+ overload. Soma Ca2+ levels (measured with fura-6F) from neurons exposed to NMDA. NMDA was present throughout all recordings, and initial Ca2+ transients have been aligned for all neurons at the 0 min time point (arrow). In seven experiments (dashed lines), the superfusate was switched to Ca2+-free ACSF for 10 min (horizontal bar) after the initial Ca2+ transient. Ca2+ was then restored to 2 mm, and sustained Ca2+ increases were monitored. These experiments were interleaved with seven experiments in which 2 mm Ca2+ was maintained throughout NMDA exposures (solid lines), and the two datasets showed overlapping ranges of responses. B, Preventing initial Ca2+ transients did not delay subsequent Ca2+ overload. Slices were superfused with Ca2+-free ACSF for 20 min, beginning 10 min before onset of NMDA exposure. Initial Ca2+ transients were not observed, but, after restoration of extracellular Ca2+, sustained Ca2+ overloads still reliably propagated from apical dendrites to somata. The overall delay in overload responses was similar to responses in Figure 3A, but note that intervals are measured here from the onset of NMDA exposure to the time of arrival of Ca2+ overload at somata (rather than between initial Ca2+ transients and Ca2+ overload in Fig. 3A). Six neurons from five animals; fura-6F was used throughout.
When measured using the low-affinity indicator fura-6F, initial Ca2+ spikes occurred after a delay of 6.8 ± 0.5 min after onset of NMDA exposures. The peak of these events were estimated at 12.6 ± 1.0 μm and were 16.5 ± 1.8 s in duration (n = 18) (Fig. 1C). Initial Ca2+ spikes were followed by substantial, but not complete, recovery of Ca2+ concentrations (to 0.83 ± 0.07 μm measured 5 min after each peak; n = 18). Some experiments were repeated with the high-affinity indicator fura-2 to more accurately assess the delay before initial Ca2+ spikes. Fura-2 revealed smaller Ca2+ oscillations (<500 nm) (Fig. 2) first detected 3.5 ± 1.0 min after NMDA exposure. These events occurred at a mean frequency of 4.0 ± 0.2/min and summated to ∼1 μm before the larger transient increase (which saturated fura-2) was observed. A 10 min preexposure to Mg2+-free ACSF before NMDA/Mg2+-free solution greatly reduced the delay before onset of oscillations, implying that Mg2+ washout from the slice contributed significantly to the delay (data not shown). After recovery from the initial Ca2+ spike, somatic Ca2+ levels were estimated with fura-2 to be 264 ± 43 nm (n = 4). The mechanisms underlying initiation and recovery from Ca2+ spikes were not investigated further because these events were not required for triggering propagating sustained Ca2+ elevations in dendrites (see below). However, it is possible that mitochondrial Ca2+ accumulation and/or forward operation sodium-calcium exchanger (NCX) activity could contribute to the initial recovery of cytosolic Ca2+ levels.
Figure 2.

A, The higher-affinity Ca2+ indicator fura-2 revealed initial Ca2+ oscillations. In this representative example from a fura-2-loaded neuron, small Ca2+ oscillations were observed soon after the onset of NMDA exposure. Individual small events summed together before a single much larger transient was triggered. The initial Ca2+ oscillations seen here were too small to be reliably detected in the other figures in which the lower-affinity indicator fura-6F was used. B, Rapid indicator loss after establishment of sustained Ca2+ increases in somata. Sustained Ca2+ increases are shown for regions in the proximal apical dendrite (50–75 μm from somata; triangles) and within somata (circles), during persistent NMDA exposures. Records have been aligned to the onset of sustained somatic Ca2+ elevations in each neuron. Indicator levels were monitored in somata at the isobestic point for fura-6F (360 nm; squares, relative to levels after initial loading) and showed prompt decreases after the arrival of sustained Ca2+ increases. All data represent mean ± SEM from five neurons.
Subsequent Ca2+ overload responses originated in distal apical dendrite segments. In most cases (>90%), the initiation sites were outside the field of view (>150–200 μm from somata). In the remaining neurons, there was no delay before the onset of Ca2+ overload, and these neurons were excluded from additional analysis. Dendritic Ca2+ overload responses were observed as a steep Ca2+ front that propagated slowly along the primary apical dendrites toward somata. These events usually were visualized first in a single branch of the apical dendritic tree, before they spread to involve all processes (Fig. 1A). An average propagation rate of 1.1 ± 0.2 μm/s was calculated from neurons in which an unbranched dendrite segment was fully within the plane of focus for a region of 30–80 μm from the soma (7 of 18 neurons). On average, sustained Ca2+ increases reached the soma 28.5 ± 1.5 min after the initial Ca2+ spike in each neuron (n = 18). The average total time taken for Ca2+ overload responses to arrive at the soma (after onset of NMDA exposure) was 35.3 ± 1.5 min (range, 27.7–48.1 min; n = 18) (Fig. 1C). The range may be contributed to, in part, by non-uniform propagation rates along branched dendrites and/or differences in the location of initiation sites of dendritic Ca2+ deregulation.
Sustained Ca2+ increases were persistent and ultimately were sufficient to saturate the indicator in most cases (estimated at >30 μm; see Materials and Methods). After invasion of somata, sustained Ca2+ increases were accompanied by prompt indicator loss (Fig. 2B), suggesting significant compromise of membrane integrity (Randall and Thayer, 1992; Avignone et al., 2005).
In the current study, we concentrated our analysis on signals in apical dendrites, because, in every case using extended NMDA exposure, neuronal injury (loss of somatic Ca2+ homeostasis and loss of indicator) did not occur until the arrival of sustained Ca2+ increases from the apical dendrite. However, there were a small number of cases (5 of 25 in a sample of control neurons) in which a segment of basal dendrite close to the soma did demonstrate Ca2+ elevations during NMDA exposure. These basal dendrite responses sometimes remained elevated for 20–30 min before sustained Ca2+ elevations were noted in apical dendrites but never themselves progressed into somata and triggered degeneration. A similar small proportion of basal dendrite signals was also noted in a previous study with NMDA stimulation (2 of 22 neurons tested). This contrasted with responses to inhibition of Na+/K+/ATPase activity with ouabain, in which responses invariably initiated in basal dendrites and invaded somata (Dietz et al., 2007). As discussed previously, differences in NMDA receptor distribution between apical and basal dendrites could contribute to these differences between responses to NMDA and ouabain (Dietz et al., 2007). In addition, ATP depletion is not expected during ouabain exposure, in contrast to the effects of NMDA stimulation (see below).
Effects of transient Ca2+ removal on arrival of propagating dendritic Ca2+ increases at somata
We tested the hypothesis that Ca2+ accumulation was required for feedforward propagation of Ca2+ overload along dendrites under this stimulus protocol. In the first set of experiments, the initial Ca2+ spike was allowed to develop, and then extracellular Ca2+ was transiently washed out. If Ca2+ influx is required for the propagation of Ca2+ overload responses along dendritic process, we reasoned that transient removal (10 or 20 min) of extracellular Ca2+ (see Materials and Methods) should result in a corresponding delay in arrival of overload Ca2+ responses at somata, after restoration of extracellular Ca2+ concentration to 2 mm.
Figure 3A shows that, when Ca2+ was removed for 10 min immediately after initial Ca2+ transients, there was no significant delay in the arrival of the Ca2+ overload (interval between initial Ca2+ spike and arrival of response at somata; 27.8 ± 1.9 min control and 27.2 ± 3.8 with Ca2+ removal; p = 0.87). In separate interleaved experiments, longer periods of transient Ca2+ washout also did not result in a significant delay in the interval between initial and sustained Ca2+ (10 min washout 31.3 ± 2.9 min vs 20 min washout 29.6 ± 1.95; p = 0.64; n = 7 each).
Figure 3B shows results from a second set of experiments, from which we found that the initial Ca2+ spikes were not required for initiation of the Ca2+ overload responses in dendrites. Extracellular Ca2+ was removed for 10 min before NMDA application and Ca2+-free media was continued for an additional 10 min (n = 6) (as illustrated in Fig. 3B) or 20 min (n = 6) after initiation of NMDA exposure. In each case, there was no Ca2+ spike, implying that its generation was dependent on Ca2+ influx. In addition, there was not a detectable increase in somatic or dendritic Ca2+ after reintroduction of 2 mm Ca2+ ACSF. In every case, the prevention of initial Ca2+ spikes did not prevent the subsequent initiation and propagation of sustained Ca2+ increases along apical dendrites. Furthermore, the duration of Ca2+ removal did not significantly affect the mean arrival time of sustained Ca2+ increases at somata (measured from onset of NMDA exposure), consistent with the results described above (33.3 ± 1.93 vs 37.9 ± 2.2 min; 20 and 30 min total Ca2+ washout time, respectively; p = 0.16; n = 6 each). These values are very close to those described above for control responses in which 2 mm Ca2+ was maintained throughout the experiment (35.3 ± 1.5 min) (described above for Fig. 1C).
These results suggested that sustained Ca2+ increases in dendrites may simply report the location of a propagating degenerative response rather than being a required agent for feedforward of ionic deregulation along CA1 dendrites.
Effects of protracted Ca2+ removal
Much longer periods of Ca2+ removal supported the idea that Ca2+ was not required for progression of degeneration along dendrites but also suggested that Ca2+ overload was required for ultimate loss of membrane integrity. Extracellular Ca2+ was washed out for 10 min period before NMDA onset and was withheld for an additional 70 min of NMDA exposure. In nominally Ca2+-free ACSF, progressive dendritic Ca2+ increases were never observed. However, after reintroduction of 2 mm Ca2+, all six neurons showed prompt and rapid dendritic Ca2+ increases that appeared in multiple dendritic processes almost immediately after reestablishment of Ca2+ levels in the recording chamber. The propagation rate of sustained Ca2+ elevations along the proximal apical dendrite was approximately threefold faster than in control conditions (3.1 ± 0.9 μm/s; n = 4) and had fully involved the somata within 3.5 ± 0.9 min after Ca2+ had reached the recording chamber (Fig. 4A).
Figure 4.

Effects of protracted Ca2+ removal. A, Each trace shows the somatic Ca2+ levels in a fura-6F-loaded neuron. Extracellular Ca2+ was washed out 10 min before the start of NMDA and then withheld for an additional 70 min of NMDA exposure (5 μm). After reintroduction of 2 mm Ca2+, all six neurons showed prompt and rapid dendritic Ca2+ increases that progressed almost immediately into somata. B, Somatic fura-6F levels (measured at 360 nm) were well maintained throughout the protracted NMDA exposure in Ca2+-free ACSF but decreased dramatically after reintroduction of 2 mm Ca2+. This representative example shows the coincident decrease in 360 nm fluorescence (solid line) as somatic Ca2+ levels rise to very high levels (dashed line).
Throughout this time course, there was no evidence of membrane compromise in neuronal somata, because indicator fluorescence (monitored at 360 nm) in single loaded neurons remained stable during the entire 70 min 0-Ca2+ NMDA period (Fig. 4B). However, indicator levels promptly declined after Ca2+ was reintroduced and somatic Ca2+ elevations were elevated to sustained high levels. These results suggest that Ca2+-independent mechanisms underlie progression of degenerative signaling along dendrites but that Ca2+ elevations are required for ultimate neuronal demise.
Intracellular Na+ increases resulting from persistent NMDA exposure
In contrast to the compound Ca2+ responses, NMDA produced monotonic increases in intracellular Na+ levels (Fig. 5). The characteristics of intracellular Na+ increases were investigated using the ratiometric indicator SBFI. Because attempts to calibrate these signals in slices were not successful, the results are presented as SBFI ratio changes (see Materials and Methods). NMDA produced mean SBFI ratios increases of 0.99 ± 0.06 (n = 11) that reached half-maximal levels 5.4 ± 0.7 min after NMDA onset (90% peak at 7.3 ± 0.7 min). As shown in Figure 5C, SBFI ratios did not show any significant recovery toward baseline levels throughout the NMDA exposures, in contrast to the prompt recovery of initial Ca2+ transients (Fig. 1).
Figure 5.

Intracellular Na+ increases. A, Responses of the Na+-sensitive indicator SBFI in a CA1 neuron during sustained exposure to NMDA. Left shows raw SBFI fluorescence. Panels 1–5 are masked images pseudocolored to map SBFI fluorescence ratio changes (350/380 nm excitation) before NMDA (1) and during 40 min NMDA exposure (2–5). B, Data extracted from two regions of interest (soma and dendrite, the latter indicated by the arrow in the left image). C, Population data showing somatic responses from 11 neurons, from slices prepared from five animals.
Effects of NMDA receptor block on Ca2+ and Na+ elevations
We examined whether continuous NMDA receptor activation was required for the events described above. An alternative possibility is that transient NMDA receptor stimulation might be sufficient to set in motion a chain of events that ultimately resulted in delayed Ca2+ overload.
In the first set of studies, the open-channel NMDA receptor blocker MK-801 (50 μm) was applied immediately after initial Ca2+ transients. In no case was a sustained Ca2+ increase observed in apical dendrites or somata (monitored for 60 min) (Fig. 6A). When applied 10 min after onset of NMDA exposure, MK-801 caused SBFI ratios to promptly return toward basal levels (5.6 ± 1.1 min to 90% recovery to prestimulus levels; n = 6) (Fig. 6B). These results suggest that persistent NMDA receptor activation was required to maintain sustained Na+ elevations and delayed sustained Ca2+ elevations.
Figure 6.

Effects of MK-801, applied before the development of sustained Ca2+ responses. A, Fura-6F experiments; NMDA was present throughout, and the noncompetitive receptor antagonist MK-801 (50 μm) was timed to arrive immediately after initial Ca2+ increases. Initial Ca2+ increases for all preparations have been aligned in the figure to the 0 min time point. MK-801 prevented the development of secondary Ca2+ responses in all neurons tested (n = 6). B, SBFI experiments, in which MK-801 was applied 10 min after the onset of exposure to NMDA. MK-801 promptly restored intracellular SBFI ratios to prestimulus levels in each neuron tested (n = 6).
MK-801 also reversed sustained Ca2+ increases that had already been established in proximal locations of dendrites. MK-801 was applied rapidly to the slice after a sustained dendritic Ca2+ response was visualized. In four of five neurons tested, Ca2+ returned promptly to near-basal intracellular Ca2+ concentrations (Fig. 7). In one of the neurons, the response was halted temporarily before the response progressed very slowly into the soma.
Figure 7.

MK-801 was still effective when applied after the establishment of Ca2+ overload in an apical dendrite. This example is representative of four trials in which a sustained Ca2+ response was allowed to propagate along the primary apical dendrite before MK-801 (50 μm) was applied. NMDA was present throughout. A, Raw fluorescence image of the neuron (left) and pseudocolor images (1–6) showing progression of a sustained Ca2+ response along the apical dendrite. MK-801 was applied as the sustained Ca2+ response advanced almost to the soma (4) and resulted in complete retraction of the response from all dendritic processes. B, Extracted Ca2+ concentrations from a somatic region (black) and a region of dendrite (red) at the site indicated by the arrow in A. Numbers correspond to panels illustrated in A.
Figure 8 shows that NR2B-containing NMDA receptors do not appear to be a major factor in the responses studied here with sustained NMDA receptor activation. NR2B-containing receptors are sensitive to inhibition by ifenprodil (Williams, 1993) and RO 256981 (Fischer et al., 1997) and have been suggested to contribute preferentially to excitotoxic injury (Liu et al., 2007). Preexposure to ifenprodil (10 μm) for 10 min before NMDA exposure did not prevent initiation and/or propagation of Ca2+ elevations. The total time taken (after NMDA onset) for arrival of Ca2+ overload responses at somata in the presence of 10 μm ifenprodil were 31.9 ± 1.6 min (n = 6), a value similar to controls (Fig. 1C). Na+ elevations were not obviously modified by either RO 256981 or ifenprodil. Figure 8B shows that (in contrast to the actions of MK-801) ifenprodil or RO 256981 did not reverse SBFI ratios, once they had been established by NMDA exposures. In addition, preexposure to both antagonists did not modify initial SBFI ratio increases (data not shown).
Figure 8.

Lack of effect of antagonists with selectivity for NMDA receptors containing NR2B subunits. A, Slices were preexposed to ifenprodil (10 μm) for 10 min before onset of NMDA exposure and remained present throughout. Ca2+ dynamics (somatic fura-6F measurements shown here) were similar to control responses described above (see Fig. 1), with initial Ca2+ transients followed by sustained Ca2+ overloads that initiated in apical dendrites before propagating to produce sustained somatic Ca2+ elevations. B, Representative examples showing that exposure to either ifenprodil (10 μm; left) or RO 256981 (10 μm; right) did not reverse Na+ elevations that had been established by NMDA exposures. SBFI ratios were measured in somatic compartments.
Although bath application of NMDA might indirectly activate neurons throughout the slice, indirect excitation does not appear to be a significant contributor to Ca2+ and Na+ dynamics in CA1 neurons recorded here. Preexposure of slices to TTX (1 μm) had little influence on Ca2+ responses. Initial Ca2+ spikes were still observed 5.9 ± 0.5 min after onset of NMDA exposure; Ca2+ overload responses originated in apical dendrites and arrived at somata 35.8 ± 3.6 min (n = 6). These characteristics are similar to those described for NMDA in ACSF (Fig. 1). Likewise, TTX did not alter the Na+ dynamics when compared with controls. The delay to half-maximal Na+ increase for TTX was 5.9 ± 0.7 min (n = 6), and the peak ratio change was 0.98 ± 0.03 (compared with delay of 5.4 ± 0.7 min and ratio change of 0.99 ± 0.06 in control conditions; see above). The noncompetitive AMPA channel blocker GYKI 53655 [1-(4-aminophenyl)-3-methylcarbamyl-4-methyl7,8-methylenedioxy-3,4-dihydro-5H-2,3-benzodiazepine] (50 μm) (Wilding and Huettner, 1995) did not significantly modify the amplitude of initial transient Ca2+ increases (21.8 ± 1.7 vs 16.4 ± 1.7 μm for control and GYKI 53655, respectively; n = 4 each; p = 0.07) or the delay in arrival of Ca2+ overload at somata (40.0 ± 13.4 vs 40.1 ± 2.5 min for control and interleaved GYKI 53655 experiments, respectively; n = 4; p = 0.9). These results suggest that persistent, direct excitation of NMDA receptors on CA1 neurons is the most likely cause of the persistent Na+ influx and Ca2+ deregulation described above.
Reducing extracellular Na+ delayed progression of dendritic Ca2+ elevations
We tested whether Na+ influx contributed to progressive Ca2+ elevation in dendrites. Responses in control ACSF with interleaved slices exposed to modified ACSF in which extracellular Na+ was reduced to 59% of control concentrations (using LiCl as a surrogate; see Materials and Methods). Li+ permeates NMDA receptors less well than Na+ (Yamaguchi and Ohmori, 1990) and is also a poorer substrate of Na+/K+/ATPase activity than is Na+ (Kang et al., 2004). In reduced Na+ conditions, the initial Ca2+ spike was smaller (3.0 ± 0.5 μm) than in controls (10.5 ± 3.3; p < 0.05; n = 6 each). In the control set, Ca2+ overload was observed in somata 43.2 ± 3.2 min (n = 6) after onset of NMDA exposure. In reduced Na+ ACSF, one neuron showed an unusually early sustained Ca2+ response (at 27.6 min), but the remaining five of six neurons showed no sign of dendritic Ca2+ overload, even when monitored for 60 min of NMDA exposure.
Effects of increased metabolic capacity on dendritic Ca2+ increase response
Extrusion of Na+ by Na+/K+/ATPase activity is a major consumer of neuronal ATP (Attwell and Laughlin, 2001). We tested the possibility that delayed Ca2+ overload is a consequence of ATP depletion, which follows from the metabolic demand of sustained Na+ extrusion. This was done by increasing ATP levels in the loading pipette or supplementing the superfusate with pyruvate.
Simply supplementing the pipette intracellular solution with additional ATP (10 mm rather than 2 mm added; see Materials and Methods) was sufficient to significantly delay the onset of Ca2+ overload responses in dendrites. As shown in Figure 9A, elevated intracellular ATP did not delay the initial Ca2+ spike but significantly decreased the amplitude (25.0 ± 3.6 vs 9.8 ± 2.8 μm for control and elevated ATP, respectively; n = 6 each; p < 0.008). The delay in Ca2+ overload was increased compared with control (36.5 ± 1.5 vs 57.9 ± 1.5 min for control and elevated ATP, respectively; n = 6 each; p < 0.01). Once initiated, there was no significant decrease in the rate of propagation of Ca2+ overload along apical dendrites (0.82 ± 0.16 vs 0.93 ± 0.25 μm/s for control and elevated ATP, respectively; n = 6 and 5; p = 0.6).
Figure 9.
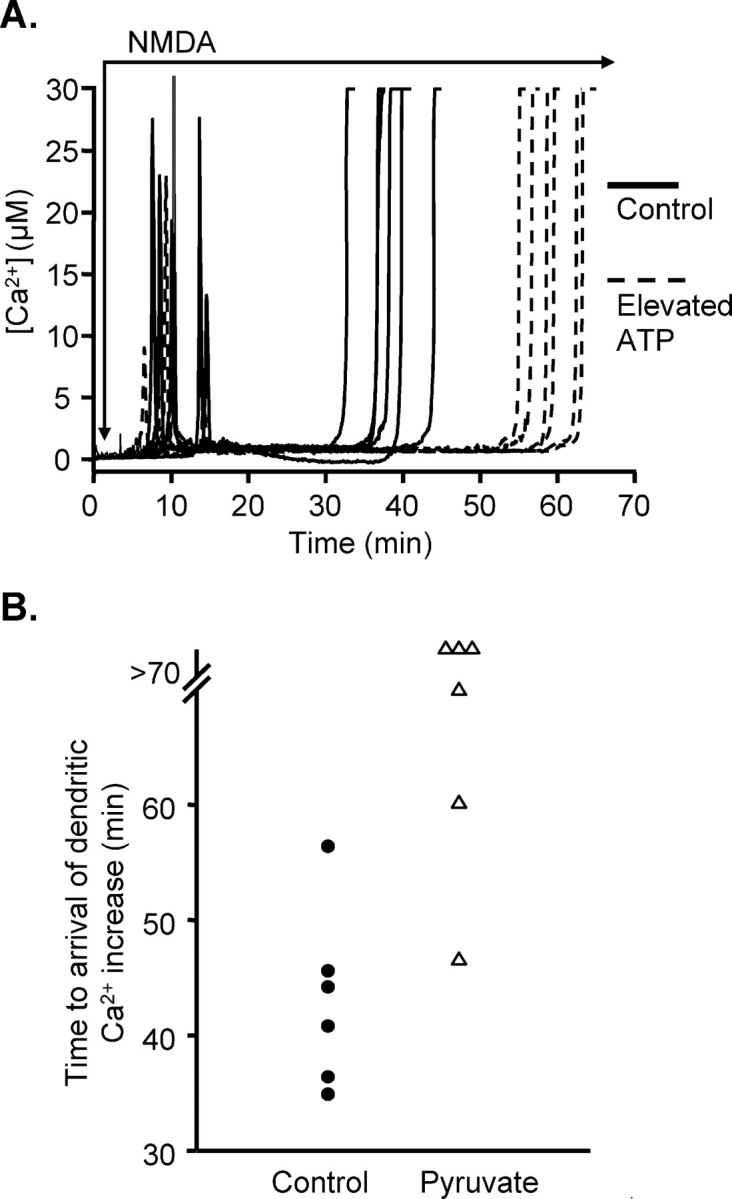
A, Increasing ATP concentration in the loading pipette delayed propagation of dendritic Ca2+ overload to somata. Solid lines show responses to NMDA exposure in neurons loaded for 3 min with fura-6F, in standard intracellular solution (2 mm added ATP; solid lines) (see Results). These experiments were interleaved with experiments in which 10 mm ATP was added to the intracellular loading solution (dashed lines). All other experimental conditions were unchanged. This manipulation did not change the average time to onset of the initial Ca2+ increases but significantly increased the time taken for sustained Ca2+ overload responses to arrive from apical dendrites to somata. B, Supplementation of ACSF with pyruvate prevented or delayed propagation of dendritic Ca2+ overload to somata. Each symbol represents the time taken for a Ca2+ overload response to arrive at the soma in a single preparation, after onset of exposure to NMDA. Filled circles show responses when slices were challenged in standard ACSF (10 mm glucose), as shown above (see Fig. 1). These experiments were interleaved with slices superfused with modified ACSF containing 10 mm pyruvate plus 10 mm glucose (20 min preexposure and maintained throughout NMDA challenge, indicated by triangles). Three of the pyruvate-supplemented preparations did not show any dendritic Ca2+ overload within the 70 min observation period. Ca2+ overload was observed in the other three pyruvate-supplemented preparations but occurred later than was seen in five of six of the matched control preparations.
In the second set of studies (Fig. 9B), slices were preexposed to pyruvate (10 mm in otherwise normal ACSF) for 15 min before the onset of NMDA exposure and maintained throughout the NMDA exposure. Pyruvate reduced the rate NMDA-stimulated SBFI ratio increases (0.32 ± 0.03 vs 0.17 ± 0.04 units/s; n = 5; p < 0.0017) and caused a small reduction in the peak ratio change (ΔRatio, 1.05 ± 0.04 vs 0.93 ± 0.02 for control and pyruvate, respectively; n = 5 each; p < 0.04). Pyruvate preexposure greatly decreased the mean amplitude of initial Ca2+ transients (from 16.1 ± 2.9 to 1.7 ± 0.4 μm; n = 6; p < 0.001). In addition, in three of six neurons tested, no evidence of subsequent dendritic Ca2+ overload was observed with observation times up to 70 min. In three of six neurons tested, dendritic Ca2+ overload was observed but was delayed compared with five of six responses in untreated slices (mean delay of 58.3 ± 6.8 min for three responders in pyruvate vs 43.1 ± 3.2 min for interleaved controls; n = 6).
Discussion
This study examines mechanisms by which protracted exposure to NMDA initiates severe Ca2+ increases in apical dendrites of CA1 neurons, which propagate slowly throughout the neuron. We conclude that Ca2+ accumulation itself is not required for the initiation or progression of disrupted ionic homeostasis during this type of agonist exposure. The initial Ca2+ spike was not necessary for the initiation of subsequent Ca2+ overload (Fig. 3), and, when the effects of transient Ca2+ removal were tested, it was found that the final, catastrophic somatic Ca2+ increase was not delayed to any significant extent (Fig. 3). In contrast to Ca2+ elevations, Na+ accumulation was persistent in somatic and dendritic compartments, and extracellular Na+ reductions delayed or prevented the propagation of dendritic Ca2+ increases. Furthermore, results obtained with ATP or pyruvate supplementation are consistent with the hypothesis that metabolic consequences of Na+ loading may underlie progressive regional metabolic dysfunction and consequent Ca2+ overload.
Na+ loading
In contrast to Ca2+ elevations, Na+ accumulation was monotonic and nearly contemporaneous in somatic and dendritic compartments. Previous work examining glutamate- or NMDA-induced Na+ increases in cultured neurons have reported stable Na+ increases ranging from 45 to 80 mm (Kiedrowski et al., 1994; Pinelis et al., 1994). The ratiometric Na+ measurements made in the present study could not be reliably calibrated (see Materials and Methods), but it is likely that similar values are achieved here.
The major route of Na+ entry was via NMDA receptors on CA1 neurons. SBFI ratio increases were promptly reversed by MK-801, and, from the lack of effect of TTX and GYKI 53655, it appears that other ionotropic glutamate receptors and voltage-gated Na+ channels made little direct contribution. NR2B-containing NMDA receptors have been implicated in other studies of excitotoxic injury (Reyes et al., 1998; Liu et al., 2007), but antagonists selective for NR2B NMDA receptors were without effect on Na+ or Ca2+ dynamics (Fig. 8). It is suggested that the contribution of NR2B receptor mechanisms to NMDA-mediated toxicity is quite low in slices from adult animals [when compared with neonatal tissue (Zhou and Baudry, 2006)], and this developmental change may explain the lack of NR2B contribution here.
Mechanisms coupling sustained Na+ loads to dendritic Ca2+ increases
Among the several possible outcomes of extended intracellular Na+ elevation, demands of increased Na+/K+/ATPase activity is a critical consequence (Chinopoulos et al., 2000; Greenwood et al., 2007), and our results appear consistent with the idea that ATP depletion is a major mechanism underlying the ultimate loss of Ca2+ homeostasis. Increasing the concentration of ATP in the dialysis pipette significantly delayed the arrival of dendritic Ca2+ increases at the soma (Fig. 9A), and supplying exogenous pyruvate provided sufficient additional metabolic competence to delay or even prevent the loss of Ca2+ regulation (Fig. 9B). Importantly, Na+ loading was not greatly decreased during pyruvate exposures, suggesting that ATP depletion is downstream of Na+ loading rather than the bulk of the Na+ increase resulting from a progressive loss of ATP-dependent extrusion. Intracellular Ca2+ levels are expected to rise dramatically if ATP-dependent Ca2+ extrusion and sequestration mechanisms become compromised in the face of heightened Ca2+ influx through NMDA receptors. Progressive failure of Na+/K+/ATPase activity would also be expected, and we have shown previously that direct inhibition of Na+/K+/ATPase with ouabain can produce dendritic Ca2+ deregulation (Dietz et al., 2007). The reason why supplementation with either ATP or pyruvate is effective here, despite the continual superfusion of slices with 10 mm glucose, is currently unknown. It is possible that neuronal glucose transport becomes limiting, and/or glial-neuronal metabolic coupling is insufficient under these stimulation conditions.
Cytosolic Na+ loading can also drive cytosolic Ca2+ elevations more directly, as a consequence of Na+/Ca2+ exchange at the plasma membrane (Kiedrowski, 2007) and mitochondrial inner membrane (Hoyt et al., 1998b). Although it is possible that exchange at both sites contributes to Ca2+ overload measured here, we showed recently that an inhibitor selective for the reverse-operation mode of plasma membrane NCX1–NCX3 isoforms did not influence dendritic Ca2+ overload responses to NMDA under very similar circumstances, suggesting little contribution from plasma membrane Na+-dependent NCX reversal with this particular stimulus (Dietz et al., 2007). Na+ loading and activation of mitochondrial 2Na+/Ca2+ exchange contributes to somatic Ca2+ elevations during oxygen and glucose removal from hippocampal slices (Zhang and Lipton, 1999) (see also Scanlon et al., 2000). A similar exchange may contribute to initial Ca2+ transients observed here, but it might be expected that mitochondrial Ca2+ capacity would be progressively drained by this pathway during sustained Na+ elevations (Hoyt et al., 1998a) and thus not be able to contribute significantly directly to the delayed Ca2+ overload. The relative contribution of these pathways, together with metabolic depletion, remains to be determined.
Relationship to previous studies of progressive calcium deregulation in dendrites
It is noted that the slowly propagating Ca2+ overload described here is quite similar in appearance to a previously reported phenomenon in dendrites of adult hippocampal neurons, termed a “secondary Ca2+ response” (Connor et al., 1988; Wadman and Connor, 1992; Connor and Cormier, 2000; Shuttleworth and Connor, 2001). Both responses first appear in distal dendrites before slowly invading the rest of the neuron, but, despite the similarities, key differences suggest that different underlying mechanisms are involved. First, initiation of the “secondary response” was accomplished by multiple, relatively brief exposures to an excitatory amino acid, and, after establishment of the secondary response, no additional exposure to the agonist was required and glutamate receptor antagonists were ineffective in reversing it (Connor et al., 1988; Wadman and Connor, 1992). In the present example, persistent NMDA exposure was required, and MK-801 was immediately effective in reversing the response (Fig. 7). Second, secondary Ca2+ responses were triggered by repeated Ca2+ overloads, which then set in motion a feedforward progression of Ca2+ deregulation along the dendrite. In this study, there was no requirement at all for an initial Ca2+ spike to initiate a delayed dendritic Ca2+ overload (Fig. 3). The model used here depends on continuous receptor activation, which presumably causes a progressive metabolic crisis attributable to Na+ influx, occurring first in the smallest apical dendritic compartments and ending in the mitochondria-rich somata. It seems likely that a rich NMDA receptor distribution, together with large surface area/volume ratios of small apical dendrites combine to cause large local demands on ATP production. However, it is also possible that events initiate in remote dendritic processes attributable to a relative lack of ATP-generating machinery at these sites.
Similar mechanistic differences may contribute to the diversity of responses seen after glutamate receptor stimulation in neuronal cultures. For example, in many studies of cultured hippocampal or cortical neurons, brief exposures to glutamate receptor agonists triggers a large initial cytosolic Ca2+ increase required for delayed toxicity (Randall and Thayer, 1992). A cascade of deleterious effects can be triggered by a brief period of mitochondrial Ca2+ loading (Stout et al., 1998) and then proceeds without receptor activation. Brief exposures of cultured rat hippocampal neurons to high concentrations of glutamate lead to very large sustained inward current and Ca2+ influx that persist after glutamate removal (Deshpande et al., 2007) (see also Chen et al., 1997). This current appears to be mediated by a novel Ca2+-permeable Gd3+-sensitive current (Deshpande et al., 2007). It seems possible that such mechanisms could contribute to the secondary Ca2+ responses seen in dendrites after brief intense receptor stimulation but not the responses seen here (because MK-801 effectively blocked Ca2+ elevations).
Something more akin to the present work may be seen in some studies of cultured cerebellar granule neurons. Glutamate exposures resulted in collapse of the ATP/ADP ratio (Budd and Nicholls, 1996) and ATP deficiencies were suggested to be important initiators for subsequent uncontrolled Ca2+ increases (Brocard et al., 2001; Jekabsons and Nicholls, 2004). Interestingly, if NMDA receptor-mediated influx was terminated with MK-801, somatic Ca2+ deregulation was prevented in some neurons, but other neurons within the same cultures still underwent deregulation (Castilho et al., 1998). It was proposed that MK-801 was not protective if somata of neurons were about to deregulate or had already done so (Castilho et al., 1998). It would be interesting to know whether the window for protection is related to the propagation of events from dendritic initiation sites to somata. Although localized Ca2+ overload has been identified in dendrites in culture after glutamate receptor stimulation (Randall and Thayer, 1992; Bindokas and Miller, 1995), the idea that dendritic Ca2+ deregulation always precedes and determines somatic deregulation in neuronal cultures has not yet been directly established.
The above is not to deny the critical importance of Ca2+ overload to ultimate neuronal injury. Limiting Ca2+ influx during glutamate exposure has been shown by numerous studies to be neuroprotective against cell death (Choi, 1988b; Arundine and Tymianski, 2003), and, indeed, this study clearly shows that Ca2+ elevations were required ultimately for cellular injury (Fig. 4B). Once established, persistently high Ca2+ increases may trigger numerous Ca2+-dependent processes that are critical for the full expression of neuronal injury. However, the present work suggests that triggering these processes can be dependent on dendritic mechanisms upstream of Ca2+ overload.
Footnotes
This work was supported by National Institutes of Health Grants NS 43458 and 51288. We thank Drs F. Valenzuela and W. Müller for helpful suggestions throughout the course of these experiments.
References
- Arundine M, Tymianski M. Molecular mechanisms of calcium-dependent neurodegeneration in excitotoxicity. Cell Calcium. 2003;34:325–337. doi: 10.1016/s0143-4160(03)00141-6. [DOI] [PubMed] [Google Scholar]
- Attwell D, Laughlin SB. An energy budget for signaling in the grey matter of the brain. J Cereb Blood Flow Metab. 2001;21:1133–1145. doi: 10.1097/00004647-200110000-00001. [DOI] [PubMed] [Google Scholar]
- Avignone E, Frenguelli BG, Irving AJ. Differential responses to NMDA receptor activation in rat hippocampal interneurons and pyramidal cells may underlie enhanced pyramidal cell vulnerability. Eur J Neurosci. 2005;22:3077–3090. doi: 10.1111/j.1460-9568.2005.04497.x. [DOI] [PubMed] [Google Scholar]
- Bindokas VP, Miller RJ. Excitotoxic degeneration is initiated at non-random sites in cultured rat cerebellar neurons. J Neurosci. 1995;15:6999–7011. doi: 10.1523/JNEUROSCI.15-11-06999.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brocard JB, Tassetto M, Reynolds IJ. Quantitative evaluation of mitochondrial calcium content in rat cortical neurones following a glutamate stimulus. J Physiol (Lond) 2001;531:793–805. doi: 10.1111/j.1469-7793.2001.0793h.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budd SL, Nicholls DG. Mitochondria, calcium regulation, and acute glutamate excitotoxicity in cultured cerebellar granule cells. J Neurochem. 1996;67:2282–2291. doi: 10.1046/j.1471-4159.1996.67062282.x. [DOI] [PubMed] [Google Scholar]
- Castilho RF, Hansson O, Ward MW, Budd SL, Nicholls DG. Mitochondrial control of acute glutamate excitotoxicity in cultured cerebellar granule cells. J Neurosci. 1998;18:10277–10286. doi: 10.1523/JNEUROSCI.18-24-10277.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen QX, Perkins KL, Choi DW, Wong RK. Secondary activation of a cation conductance is responsible for NMDA toxicity in acutely isolated hippocampal neurons. J Neurosci. 1997;17:4032–4036. doi: 10.1523/JNEUROSCI.17-11-04032.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chinopoulos C, Tretter L, Rozsa A, Adam-Vizi V. Exacerbated responses to oxidative stress by an Na+ load in isolated nerve terminals: the role of ATP depletion and rise of [Ca2+]i. J Neurosci. 2000;20:2094–2103. doi: 10.1523/JNEUROSCI.20-06-02094.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chinopoulos C, Connor JA, Shuttleworth CW. Emergence of a spermine-sensitive, non-inactivating conductance in mature hippocampal CA1 pyramidal neurons upon reduction of extracellular Ca2+: dependence on intracellular Mg2+ and ATP. Neurochem Int. 2007;50:148–158. doi: 10.1016/j.neuint.2006.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi DW. Glutamate neurotoxicity and diseases of the nervous system. Neuron. 1988a;1:623–634. doi: 10.1016/0896-6273(88)90162-6. [DOI] [PubMed] [Google Scholar]
- Choi DW. Calcium-mediated neurotoxicity: relationship to specific channel types and role in ischemic damage. Trends Neurosci. 1988b;11:465–469. doi: 10.1016/0166-2236(88)90200-7. [DOI] [PubMed] [Google Scholar]
- Connor JA, Cormier RJ. Cumulative effects of glutamate microstimulation on Ca2+ responses of CA1 hippocampal pyramidal neurons in slice. J Neurophysiol. 2000;83:90–98. doi: 10.1152/jn.2000.83.1.90. [DOI] [PubMed] [Google Scholar]
- Connor JA, Wadman WJ, Hockberger PE, Wong RK. Sustained dendritic gradients of Ca2+ induced by excitatory amino acids in CA1 hippocampal neurons. Science. 1988;240:649–653. doi: 10.1126/science.2452481. [DOI] [PubMed] [Google Scholar]
- Deshpande LS, Limbrick DD, Sombati S, Delorenzo RJ. Activation of a novel injury-induced calcium permeable channel that plays a key role in causing extended neuronal depolarization and initiating neuronal death in excitotoxic neuronal injury. J Pharmacol Exp Ther. 2007;322:443–452. doi: 10.1124/jpet.107.123182. [DOI] [PubMed] [Google Scholar]
- Dietz RM, Kiedrowski L, Shuttleworth CW. Contribution of Na+/Ca2+ exchange to excessive Ca2+ loading in dendrites and somata of CA1 neurons in acute slice. Hippocampus. 2007;17:1049–1059. doi: 10.1002/hipo.20336. [DOI] [PubMed] [Google Scholar]
- Fischer G, Mutel V, Trube G, Malherbe P, Kew JN, Mohacsi E, Heitz MP, Kemp JA. Ro 25–6981, a highly potent and selective blocker of N-methyl-d-aspartate receptors containing the NR2B subunit. Characterization in vitro. J Pharmacol Exp Ther. 1997;283:1285–1292. [PubMed] [Google Scholar]
- Fried E, Amorim P, Chambers G, Cottrell JE, Kass IS. The importance of sodium for anoxic transmission damage in rat hippocampal slices: mechanisms of protection by lidocaine. J Physiol (Lond) 1995;489:557–565. doi: 10.1113/jphysiol.1995.sp021072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenwood SM, Mizielinska SM, Frenguelli BG, Harvey J, Connolly CN. Mitochondrial dysfunction and dendritic beading during neuronal toxicity. J Biol Chem. 2007;282:26235–26244. doi: 10.1074/jbc.M704488200. [DOI] [PubMed] [Google Scholar]
- Grynkiewicz G, Poenie M, Tsien RY. A new generation of Ca2+ indicators with greatly improved fluorescence properties. J Biol Chem. 1985;260:3440–3450. [PubMed] [Google Scholar]
- Hori N, Carpenter DO. Transient ischemia causes a reduction of Mg2+ blockade of NMDA receptors. Neurosci Lett. 1994;173:75–78. doi: 10.1016/0304-3940(94)90153-8. [DOI] [PubMed] [Google Scholar]
- Hoyt KR, Arden SR, Aizenman E, Reynolds IJ. Reverse Na+/Ca2+ exchange contributes to glutamate-induced intracellular Ca2+ concentration increases in cultured rat forebrain neurons. Mol Pharmacol. 1998a;53:742–749. [PubMed] [Google Scholar]
- Hoyt KR, Stout AK, Cardman JM, Reynolds IJ. The role of intracellular Na+ and mitochondria in buffering of kainate-induced intracellular free Ca2+ changes in rat forebrain neurones. J Physiol (Lond) 1998b;509:103–116. doi: 10.1111/j.1469-7793.1998.103bo.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jekabsons MB, Nicholls DG. In situ respiration and bioenergetic status of mitochondria in primary cerebellar granule neuronal cultures exposed continuously to glutamate. J Biol Chem. 2004;279:32989–33000. doi: 10.1074/jbc.M401540200. [DOI] [PubMed] [Google Scholar]
- Johnson-Cadwell LI, Jekabsons MB, Wang A, Polster BM, Nicholls DG. “Mild Uncoupling” does not decrease mitochondrial superoxide levels in cultured cerebellar granule neurons but decreases spare respiratory capacity and increases toxicity to glutamate and oxidative stress. J Neurochem. 2007;101:1619–1631. doi: 10.1111/j.1471-4159.2007.04516.x. [DOI] [PubMed] [Google Scholar]
- Kang Y, Notomi T, Saito M, Zhang W, Shigemoto R. Bidirectional interactions between h-channels and Na+-K+ pumps in mesencephalic trigeminal neurons. J Neurosci. 2004;24:3694–3702. doi: 10.1523/JNEUROSCI.5641-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiedrowski L. Critical role of sodium in cytosolic [Ca2+] elevations in cultured hippocampal CA1 neurons during anoxic depolarization. J Neurochem. 2007;100:915–923. doi: 10.1111/j.1471-4159.2006.04308.x. [DOI] [PubMed] [Google Scholar]
- Kiedrowski L, Wroblewski JT, Costa E. Intracellular sodium concentration in cultured cerebellar granule cells challenged with glutamate. Mol Pharmacol. 1994;45:1050–1054. [PubMed] [Google Scholar]
- Liu Y, Wong TP, Aarts M, Rooyakkers A, Liu L, Lai TW, Wu DC, Lu J, Tymianski M, Craig AM, Wang YT. NMDA receptor subunits have differential roles in mediating excitotoxic neuronal death both in vitro and in vivo. J Neurosci. 2007;27:2846–2857. doi: 10.1523/JNEUROSCI.0116-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholls DG, Johnson-Cadwell L, Vesce S, Jekabsons M, Yadava N. Bioenergetics of mitochondria in cultured neurons and their role in glutamate excitotoxicity. J Neurosci Res. 2007;85:3206–3212. doi: 10.1002/jnr.21290. [DOI] [PubMed] [Google Scholar]
- Novelli A, Reilly JA, Lysko PG, Henneberry RC. Glutamate becomes neurotoxic via the N-methyl-d-aspartate receptor when intracellular energy levels are reduced. Brain Res. 1988;451:205–212. doi: 10.1016/0006-8993(88)90765-2. [DOI] [PubMed] [Google Scholar]
- Olney JW. Neurotoxicity of excitatory amino acids. In: McGreer EG, Olney JW, McGreer PL, editors. Kainic acid as a tool in neurobiology. New York: Raven; 1978. pp. 95–121. [Google Scholar]
- Olney JW, Price MT, Samson L, Labruyere J. The role of specific ions in glutamate neurotoxicity. Neurosci Lett. 1986;65:65–71. doi: 10.1016/0304-3940(86)90121-7. [DOI] [PubMed] [Google Scholar]
- Patton C, Thompson S, Epel D. Some precautions in using chelators to buffer metals in biological solutions. Cell Calcium. 2004;35:427–431. doi: 10.1016/j.ceca.2003.10.006. [DOI] [PubMed] [Google Scholar]
- Petrozzino JJ, Connor JA. Dendritic Ca2+ accumulations and metabotropic glutamate receptor activation associated with an N-methyl-d-aspartate receptor-independent long-term potentiation in hippocampal CA1 neurons. Hippocampus. 1994;4:546–558. doi: 10.1002/hipo.450040504. [DOI] [PubMed] [Google Scholar]
- Pinelis VG, Segal M, Greenberger V, Khodorov BI. Changes in cytosolic sodium caused by a toxic glutamate treatment of cultured hippocampal neurons. Biochem Mol Biol Int. 1994;32:475–482. [PubMed] [Google Scholar]
- Randall RD, Thayer SA. Glutamate-induced calcium transient triggers delayed calcium overload and neurotoxicity in rat hippocampal neurons. J Neurosci. 1992;12:1882–1895. doi: 10.1523/JNEUROSCI.12-05-01882.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rego AC, Ward MW, Nicholls DG. Mitochondria control AMPA/kainate receptor-induced cytoplasmic calcium deregulation in rat cerebellar granule cells. J Neurosci. 2001;21:1893–1901. doi: 10.1523/JNEUROSCI.21-06-01893.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reyes M, Reyes A, Opitz T, Kapin MA, Stanton PK. Eliprodil, a non-competitive, NR2B-selective NMDA antagonist, protects pyramidal neurons in hippocampal slices from hypoxic/ischemic damage. Brain Res. 1998;782:212–218. doi: 10.1016/s0006-8993(97)01280-8. [DOI] [PubMed] [Google Scholar]
- Rothman SM. The neurotoxicity of excitatory amino acids is produced by passive chloride influx. J Neurosci. 1985;5:1483–1489. doi: 10.1523/JNEUROSCI.05-06-01483.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scanlon JM, Brocard JB, Stout AK, Reynolds IJ. Pharmacological investigation of mitochondrial Ca2+ transport in central neurons: studies with CGP-37157, an inhibitor of the mitochondrial Na+-Ca2+ exchanger. Cell Calcium. 2000;28:317–327. doi: 10.1054/ceca.2000.0171. [DOI] [PubMed] [Google Scholar]
- Shuttleworth CW, Connor JA. Strain-dependent differences in calcium signaling predict excitotoxicity in murine hippocampal neurons. J Neurosci. 2001;21:4225–4236. doi: 10.1523/JNEUROSCI.21-12-04225.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon RP, Swan JH, Griffiths T, Meldrum BS. Blockade of N-methyl-d-aspartate receptors may protect against ischemic damage in the brain. Science. 1984;226:850–852. doi: 10.1126/science.6093256. [DOI] [PubMed] [Google Scholar]
- Stout AK, Raphael HM, Kanterewicz BI, Klann E, Reynolds IJ. Glutamate-induced neuron death requires mitochondrial calcium uptake. Nat Neurosci. 1998;1:366–373. doi: 10.1038/1577. [DOI] [PubMed] [Google Scholar]
- Vesce S, Jekabsons MB, Johnson-Cadwell LI, Nicholls DG. Acute glutathione depletion restricts mitochondrial ATP export in cerebellar granule neurons. J Biol Chem. 2005;280:38720–38728. doi: 10.1074/jbc.M506575200. [DOI] [PubMed] [Google Scholar]
- Wadman WJ, Connor JA. Persisting modification of dendritic calcium influx by excitatory amino acid stimulation in isolated Ca1 neurons. Neuroscience. 1992;48:293–305. doi: 10.1016/0306-4522(92)90491-j. [DOI] [PubMed] [Google Scholar]
- Wahlestedt C, Golanov E, Yamamoto S, Yee F, Ericson H, Yoo H, Inturrisi CE, Reis DJ. Antisense oligodeoxynucleotides to NMDA-R1 receptor channel protect cortical neurons from excitotoxicity and reduce focal ischaemic infarctions. Nature. 1993;363:260–263. doi: 10.1038/363260a0. [DOI] [PubMed] [Google Scholar]
- Wilding TJ, Huettner JE. Differential antagonism of alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid-preferring and kainate-preferring receptors by 2,3-benzodiazepines. Mol Pharmacol. 1995;47:582–587. [PubMed] [Google Scholar]
- Williams K. Ifenprodil discriminates subtypes of the N-methyl-d-aspartate receptor: selectivity and mechanisms at recombinant heteromeric receptors. Mol Pharmacol. 1993;44:851–859. [PubMed] [Google Scholar]
- Yamaguchi K, Ohmori H. Voltage-gated and chemically gated ionic channels in the cultured cochlear ganglion neurone of the chick. J Physiol (Lond) 1990;420:185–206. doi: 10.1113/jphysiol.1990.sp017907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Lipton P. Cytosolic Ca2+ changes during in vitro ischemia in rat hippocampal slices: major roles for glutamate and Na+-dependent Ca2+ release from mitochondria. J Neurosci. 1999;19:3307–3315. doi: 10.1523/JNEUROSCI.19-09-03307.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou M, Baudry M. Developmental changes in NMDA neurotoxicity reflect developmental changes in subunit composition of NMDA receptors. J Neurosci. 2006;26:2956–2963. doi: 10.1523/JNEUROSCI.4299-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]