

Abstract
Epigenetic gene silencing, and associated promoter CpG island DNA hypermethylation, is an alternative mechanism to mutations by which tumor suppressor genes may be inactivated within a cancer cell 1-4,5-7. These epigenetic changes are prevalent in all types of cancer, and their appearance may precede genetic changes in pre-malignant cells and foster the accumulation of additional genetic and epigenetic hits8. These epigenetically modified genes constitute important categories of tumor suppressor genes including cell cycle regulators, pro-differentiation factors, and anti-apoptotic genes3, and many of these genes are known to play a role in normal development 9-11. While the silencing of these genes may play an essential role in tumor initiation or progression, the mechanisms underlying the specific targeting of these genes for DNA hypermethylation remains to be determined. The large numbers of epigenetically silenced genes that may be present in any given tumor, and the clustering of silenced genes within single cell pathways (ref), begs the question of whether gene silencing is a series of random events resulting in an enhanced survival of a pre-malignant clone, or whether silencing is the result of a directed, instructive program for silencing initiation reflective of the cells of origin for tumors. In this regard, the current review stresses the latter hypothesis and the important possibility that the program is linked, at least for silencing of some cancer genes, to the epigenetic control of stem/precursor cell gene expression patterns.
Many groups have suggested that stem cells or early progenitor cells may be the precursors from which cancer cells are derived12-15. All of these studies cite the fact that tumors harbor stem/precursor cell population markers, have heterogeneous cell populations among which only cell subsets have tumor regeneration capacity, and are driven by the up-regulation of many pathways important to maintenance of stem/precursor cells12-15. Our studies have focused on cancer genes with abnormal gene silencing associated with aberrant DNA CpG island hypermethylation, changes which often manifest early in tumor progression and are present in pre-malignant lesions. We have hypothesized that epigenetic silencing of tumor suppressor genes may facilitate abnormal survival of cells under chronic stress conditions, such as chronic wound healing and inflammation8,16. In turn, three groups, including our own17-19, have recently compiled evidence that transcriptionally repressive chromatin modifications associated with the promoters of these silenced genes in cancer cells resemble the chromatin modifications inherent to those same genes in normal embryonic stem cells which may normally help prevent stem/precursor cells from conversion to committed cells until programmed to do so. The three groups have hypothesized that the above chromatin patterns may render groups of genes vulnerable to errors which result in recruitment of aberrant promoter DNA methylation during early progression of adult cancers which abnormally locks in the silencing of the genes and helps foster selective clonal expansion of cells harboring these epigenetic abnormalities. This, in turn, helps set the stage for tumor progression driven by the additional epigenetic and genetic hits observed in cancer cells. The evidence for this hypothesis potentially linking cancer epigenetics to the epigenetics of normal and neoplastic stem/precursor cells cells is the focus of the following discussions.
In a recent study17, we compiled a panel of 29 tumor suppressor, and candidate tumor suppressor, genes identified by our group and others to be frequently DNA hypermethylated and silenced in adult cancers. Genes were identified from a mix of cancer cell lines and primary tumor samples, and they encompassed a wide range of tissue types including brain, breast, colon, esophageal, gastric, head and neck, hematopoetic, kidney/renal, liver, lung, ovarian, pancreatic, prostate, and uterine cancer. All of the genes were found to be hypermethylated in multiple tumor types, and hypermethylation of several of these genes (CDH1, CDKN2A, and RASSF1) has been observed in all of these tumor types. An immediate initial observation upon compilation of this list was the high frequency of targeting of genes involved in normal development, including genes such as p16 which can regulate stem cell number and cell cycle functions20, GATA transcription factors which regulate differentiation21, morphogenesis control genes such as CDH1, and anti-apoptotic genes such as DAP-kinase3. The nature of some of the genes raised for us the question of whether a stem or precursor cell origin for cancer, and specifically the underlying epigenetic controls responsible for controlling expression of these genes in normal stem and progenitor cells, provide an explanation for the targeting of at least some genes for DNA hypermethylation and gene silencing during tumor initiation and progression.
Other recent work in our lab, focused on understanding the chromatin modifications present when genes are DNA hypermethylated in adult cancers, has also suggested links to stem cells. Studies in colon and breast cancer cells indicated that enrichment of multiple components of transcriptionally repressive chromatin is characteristic of cancer gene promoters silenced in association with aberrant DNA methylation8. Perhaps, most interesting, have been findings that these silenced cancer gene promoters, when reactivated by DNA demethylation do not return to a fully euchromatic chromatin state8. Rather, while active marks are restored, most repressive histone modification marks remain, including H3K27me3. This histone modification is catalyzed by EZH2, a key component of the Polycomb group (PcG) complexes which maintain long-term gene silencing from lower organisms to man and are essential for the normal state of stem/progenitor cells and their commitment to various tissue types 22-25. Recent studies have linked EZH2 to promoter recruitment of the enzymes that catalyze DNA methylation, the DNA methyltransferases (DNMT's), and have suggested a role for this protein during the induction and targeting of DNA methylation 26. High steady state levels of PcG complexes are especially present in stem and progenitor cells, as well as in tumor cells that have similar properties24,27,28. Furthermore, a critical link between our genes under study, and ES/precursor cells has emerged from several recently published seminal studies of the epigenetic profiles of genes in ES and embryonic fibroblasts (EF). These genome tiling array approaches have identified that promoter regions for a group of genes important to development, morphogenesis, organogenesis, etc, comprising ~ 1,000 genes and between ~8% (ES cells) 29 and ~14% (EF cells) 30 of the annotated genome to be PcG regulated in these cell types as marked by PcG components and/or the H3K27methylation mark.
Given the above information, we matched our list of genes frequently DNA hypermethylated in adult cancers to the full list of genes associated with PcG targeting in the above studies of ES and EF. Remarkably, we found 68% of our genes were associated with PcG in either cell type 17. Similar results were seen using 23 genes newly identified as hypermethylated in HCT-116 cells (manuscript submitted) for which 56.5% were identified as PcG targets in either ES or EF cells. These data were striking, particularly considering, as noted previously above, that for each of the candidate genes we have studied in depth in adult cancer cells, DNA hypermethylation is accompanied by an H3K27 repressive mark8. This is in agreement with data published simultaneously by two additional groups. Widschwendter et.al. show that PRC targets are up to 12-fold more likely to have cancer-specific DNA hypermethylation than non-targets when comparing 177 genes identified as hypermethylated in primary human colorectal tumors 19. Additionally, Schlesinger et.al. demonstrate that of 24 genes identified as specifically DNA hypermethylated in the colon cancer line Caco-2, all also demonstrate concurrent enrichment of the H3K27me3 modification, which is generally lacking in unmethylated controls, and >60% of these genes are PRC regulated in the above studies of ES and EF cells 18.
All of the preceding associations led us to compare the epigenetic status for the group of our genes under study in normal embryonic stem (ES) cells versus embryonal carcinomas (EC) 17. EC are malignant counterparts of normal embryonic stem (ES) cells, but in contrast to adult cancers, retain a marked ability to manifest spontaneous differentiation and multi-lineage commitment in vitro and in vivo 31-34, though not to the degree of their normal ES cell counterparts. Our studies reveal that genes frequently DNA hypermethylated and deeply transcriptionally silenced in adult cancers usually lack such DNA methylation in normal (ES) and neoplastic (EC) embryonic cells. However, in both cell types, there is localization of key PcG proteins including SUZ12, EZH2, and SirT1, plus the H3K27me3 mark to their promoters 17. Interestingly, concurrent with enrichment of this repressive chromatin modification at the promoters of these genes, the active mark, H3K4me2, was simultaneously enriched. This dual marking bears another key relationship to findings for genes in ES cells. A “bivalent” combination of active (H3K4me2) and repressive (H3K27me3) histone modifications has recently been described in normal murine ES cells for a subset of developmental genes which are maintained in a low expression state 35,36. This bivalent state is resolved to a primarily active or repressive chromatin conformation with differentiation depending on which direction the transcription of the involved genes changes with differentiation cues 35.
As noted in the above study by Bernstein et. al. defining the concept of bivalent chromatin35, among embryonic cells, the degree of lineage commitment may determine the balance of this chromatin modification at gene promoters and the basal expression levels of involved genes. Similarly, this may be true for compartments of precursor cells in adult renewing cell systems from which adult cancers derive. Interestingly, for the genes from our original list that are identified as PcG targets, approximately 50% were listed as PcG targets in both ES and EF cells. However, the remaining 50% were listed as unique PcG targets in either ES or EF cells. Those that were PcG targets in ES cells were generally expressed at low levels in EC and up-regulated with differentiation, and those for EF cells were generally expressed at higher levels in EC cells and down-regulated with differentiation 17. This could explain how different patterns of hypermethylated genes in adult cancers might, then, be reflective of their chromatin status in a cell of origin. While the H3K27me3 mark appears to be ubiquitous at the small subset of DNA hypermethylated cancer gene promoters studied to date, direct global comparisons of PRC regulation between cancer cells and their proposed stem or progenitor cell of origin should help clarify and measure the full extent of the link between PcG regulation and DNA hypermethylation.
Our work in EC cells provides us with at least one additional clue necessary in order to understand the transition of a gene promoter from a transient “transcription ready” state to one of heritable, permanent gene silencing via DNA hypermethylation in a pre-malignant cell. While EC cells retain the bivalent marks found in ES cells, they show additional enrichment of two key repressive marks: H3K9me3, which is characteristic of silenced transcription in pericentromeric regions 37, and to a lower and more variable extent, H3K9me2 17. Both of these H3K9 marks are characteristic of DNA hypermethylated genes in adult cancers 8,38-43. In both Neurospora and Arabidopsis, mutations in histone methyltransferases which catalyze H3K9 methylation cause significant loss of genomic DNA methylation 15,44-47. Interestingly, in the EC cells, global levels of both of the H3K9 repressive marks are increased considerably as compared to ES cells17, suggesting a permissive background for the promoter changes in the neoplastic cells.
In adult cancers, the repressive chromatin present for DNA hypermethylated genes is initially more enriched for H3K9me2 8,40 than is seen in the EC cells, and this mark is the only repressive mark uniformly reduced when DNA hypermethylated genes are de-methylated in adult cancer cells8. In EC cells the fully methylated RASSF1a gene and the minimally methylated sFRP5 gene both demonstrate an increased presence of the H3K9me2 mark at their promoters as compared to unmethylated genes, and overexpression of the PcG protein Bmi1 can cause progressive promoter DNA methylation of the SFRP5 gene in EC cells and an increase in H3K9me2 17. Additional studies will be required to gain an understanding of the exact relationships between H3K9me2 and DNA hypermethylation and the factors responsible for the recruitment of each.
One additional finding in our recent study is key to understanding the pivotal role of the abnormal promoter DNA methylation seen in adult cancers for stable loss of key gene function. Even with the addition of the H3K9 silencing marks to the genes studied in the EC, the genes remain responsive to differentiation cues for availability to increase transcription. Thus, with retinoic acid induced neural differentiation of the cells, or with increased epithelial differentiation seen when EC cells are grown as explants in immune deficient mice, many of the genes had distinctly increased transcription17. This plasticity of expression in EC cells is not seen for these same genes when they are DNA methylated and deeply, heritably repressed in adult cancers and difficult to reactivate unless the DNA methylation is removed8.
In terms of human cancer biology, our findings and those of others, suggest that a stem cell like promoter “ground state” for these genes may be indicative of the contribution of stem cell and/or progenitor cells to the derivation of adult cancers. We and others have suggested that stem/progenitor cells are especially at risk for cancer initiation due to their continued expansion during states such as chronic wound healing and inflammation8,16. The stem cell chromatin state that we now show may provide a program for making key tumor suppressor genes vulnerable for imposition of promoter CpG island DNA methylation. This renders a transient “transcription ready” state to one of heritable, permanent gene silencing and may foster the abnormal cell expansion (Fig. 1.), and enhance the likelihood, in such cell clones, of subsequent tumor initiation and progression.
Figure 1. Model for how abnormal DNA methylation may be recruited to most tumor suppressor genes in adult cancer cells, reflecting a stem/progenitor cell of origin.
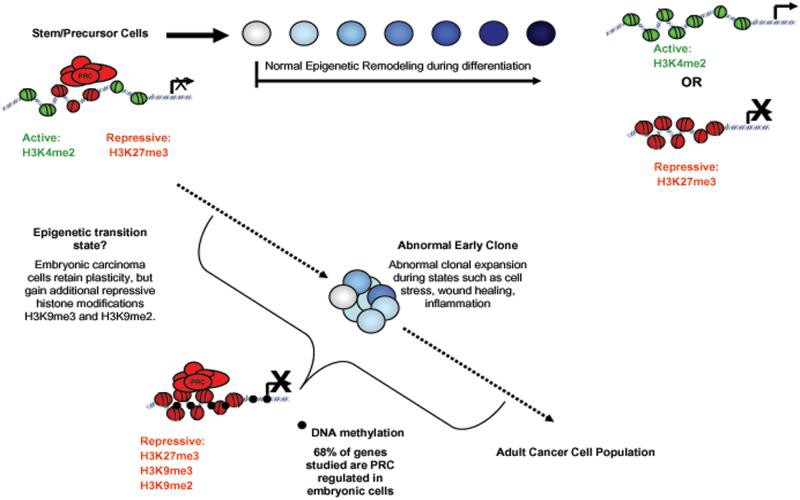
The promoter chromatin state for many genes that are frequently DNA hypermethylated in cancer is virtually identical between embryonic and adult stem and precursor cells and adult cancer cells. This “bivalent” chromatin state is characterized by simultaneous presence of both key active (green) and repressive (red) histone modifications and gene silencing polycomb group (PcG) proteins, and is also similar to that of CpG island promoters of multiple, developmentally related, genes which have low expression in ES cells35,36 and is resolved be either primarily active or repressive chromatin via normal epigenetic remodeling during differentiation. During states of abnormal cell renewal, such as cell stress, chronic injury repair, and inflammation, there is expansion of adult stem or precursor cells with the above bivalent chromatin states at the genes we have studied. The presence of PcG proteins and H3K27me3, plus addition of H3K9me2, may be responsible for recruitment of abnormal DNA methylation (black circles) to CpG islands at the gene promoters and this leads to cells with heritable, deep silencing of these genes. The loss of function of these genes, in turn, locks stem/precursor cells into abnormal clonal expansion which begins a process of neoplastic initiation and progression.
References
- 1.Jones PA, Laird PW. Cancer epigenetics comes of age. Nat.Genet. 1999;21:163–167. doi: 10.1038/5947. [DOI] [PubMed] [Google Scholar]
- 2.Feinberg AP, Tycko B. The history of cancer epigenetics. Nat Rev Cancer. 2004;4:143–53. doi: 10.1038/nrc1279. [DOI] [PubMed] [Google Scholar]
- 3.Herman JG, Baylin SB. Gene silencing in cancer in association with promoter hypermethylation. N Engl J Med. 2003;349:2042–54. doi: 10.1056/NEJMra023075. [DOI] [PubMed] [Google Scholar]
- 4.Jones PA, Baylin SB. The fundamental role of epigenetic events in cancer. Nat Rev Genet. 2002;3:415–28. doi: 10.1038/nrg816. [DOI] [PubMed] [Google Scholar]
- 5.Hahn WC, et al. Creation of human tumour cells with defined genetic elements. Nature. 1999;400:464–8. doi: 10.1038/22780. [DOI] [PubMed] [Google Scholar]
- 6.Aaltonen LA, et al. Clues to the pathogenesis of familial colorectal cancer. Science. 1993;260:812–6. doi: 10.1126/science.8484121. [DOI] [PubMed] [Google Scholar]
- 7.Kinzler KW, Vogelstein B. Cancer-susceptibility genes. Gatekeepers and caretakers. Nature. 1997;386:761–763. doi: 10.1038/386761a0. [DOI] [PubMed] [Google Scholar]
- 8.McGarvey KM, et al. Silenced Tumor Suppressor Genes Reactivated by DNA Demethylation Do Not Return to a Fully Euchromatic Chromatin State. Cancer Res. 2006;66:3541–3549. doi: 10.1158/0008-5472.CAN-05-2481. [DOI] [PubMed] [Google Scholar]
- 9.Gregorieff A, Clevers H. Wnt signaling in the intestinal epithelium: from endoderm to cancer. Genes Dev. 2005;19:877–90. doi: 10.1101/gad.1295405. [DOI] [PubMed] [Google Scholar]
- 10.Furukawa Y. Cell cycle control genes and hematopoietic cell differentiation. Leuk Lymphoma. 2002;43:225–31. doi: 10.1080/10428190290005973. [DOI] [PubMed] [Google Scholar]
- 11.Burch JB. Regulation of GATA gene expression during vertebrate development. Semin Cell Dev Biol. 2005;16:71–81. doi: 10.1016/j.semcdb.2004.10.002. [DOI] [PubMed] [Google Scholar]
- 12.Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci U S A. 2003;100:3983–8. doi: 10.1073/pnas.0530291100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Clarke MF, Fuller M. Stem cells and cancer: two faces of eve. Cell. 2006;124:1111–5. doi: 10.1016/j.cell.2006.03.011. [DOI] [PubMed] [Google Scholar]
- 14.Meydan N, et al. Inhibition of acute lymphoblastic leukaemia by a Jak-2 inhibitor. Nature. 1996;379:645–8. doi: 10.1038/379645a0. [DOI] [PubMed] [Google Scholar]
- 15.Jackson JP, et al. Dimethylation of histone H3 lysine 9 is a critical mark for DNA methylation and gene silencing in Arabidopsis thaliana. Chromosoma. 2004;112:308–15. doi: 10.1007/s00412-004-0275-7. [DOI] [PubMed] [Google Scholar]
- 16.Beachy PA, Karhadkar SS, Berman DM. Tissue repair and stem cell renewal in carcinogenesis. Nature. 2004;432:324–31. doi: 10.1038/nature03100. [DOI] [PubMed] [Google Scholar]
- 17.Ohm JE, et al. A stem cell-like chromatin pattern may predispose tumor suppressor genes to DNA hypermethylation and heritable silencing. Nat Genet. 2007;39:237–42. doi: 10.1038/ng1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schlesinger Y, et al. Polycomb-mediated methylation on Lys27 of histone H3 pre-marks genes for de novo methylation in cancer. Nat Genet. 2007;39:232–6. doi: 10.1038/ng1950. [DOI] [PubMed] [Google Scholar]
- 19.Widschwendter M, et al. Epigenetic stem cell signature in cancer. Nat Genet. 2007;39:157–8. doi: 10.1038/ng1941. [DOI] [PubMed] [Google Scholar]
- 20.Park IK, et al. Bmi-1 is required for maintenance of adult self-renewing haematopoietic stem cells. Nature. 2003;423:302–5. doi: 10.1038/nature01587. [DOI] [PubMed] [Google Scholar]
- 21.Laverriere AC, et al. GATA-4/5/6, a subfamily of three transcription factors transcribed in developing heart and gut. J Biol Chem. 1994;269:23177–84. [PubMed] [Google Scholar]
- 22.Lund A. a. v. L. Polycomb complexes and silencing mechanisms. Current Opinion in Genetics and Development. 2004;16:1–8. doi: 10.1016/j.ceb.2004.03.010. M. [DOI] [PubMed] [Google Scholar]
- 23.Valk-Lingbeek ME, Bruggeman SW, van Lohuizen M. Stem cells and cancer; the polycomb connection. Cell. 2004;118:409–18. doi: 10.1016/j.cell.2004.08.005. [DOI] [PubMed] [Google Scholar]
- 24.Kuzmichev A, et al. Composition and histone substrates of polycomb repressive group complexes change during cellular differentiation. PNAS. 2005;102:1859–1864. doi: 10.1073/pnas.0409875102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Otte AP, Kwaks TH. Gene repression by Polycomb group protein complexes: a distinct complex for every occasion? Curr Opin Genet Dev. 2003;13:448–54. doi: 10.1016/s0959-437x(03)00108-4. [DOI] [PubMed] [Google Scholar]
- 26.Vire E, et al. The Polycomb group protein EZH2 directly controls DNA methylation. Nature. 2006;439:871–4. doi: 10.1038/nature04431. [DOI] [PubMed] [Google Scholar]
- 27.Kleer CG, et al. EZH2 is a marker of aggressive breast cancer and promotes neoplastic transformation of breast epithelial cells. Proc Natl Acad Sci U S A. 2003;100:11606–11. doi: 10.1073/pnas.1933744100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kirmizis A, Bartley SM, Farnham PJ. Identification of the polycomb group protein SU(Z)12 as a potential molecular target for human cancer therapy. Mol Cancer Ther. 2003;2:113–21. [PubMed] [Google Scholar]
- 29.Lee TI, et al. Control of developmental regulators by polycomb in human embryonic stem cells. Cell. 2006;125:301–13. doi: 10.1016/j.cell.2006.02.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bracken AP, Dietrich N, Pasini D, Hansen KH, Helin K. Genome-wide mapping of Polycomb target genes unravels their roles in cell fate transitions. Genes Dev. 2006;20:1123–1136. doi: 10.1101/gad.381706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Andrews PW. Human teratocarcinomas. Biochim Biophys Acta. 1988;948:17–36. doi: 10.1016/0304-419x(88)90003-0. [DOI] [PubMed] [Google Scholar]
- 32.Andrews PW. Retinoic acid induces neuronal differentiation of a cloned human embryonal carcinoma cell line in vitro. Dev Biol. 1984;103:285–93. doi: 10.1016/0012-1606(84)90316-6. [DOI] [PubMed] [Google Scholar]
- 33.Mintz B, Illmensee K. Normal genetically mosaic mice produced from malignant teratocarcinoma cells. Proc Natl Acad Sci U S A. 1975;72:3585–9. doi: 10.1073/pnas.72.9.3585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Palmiter RD, Chen HY, Brinster RL. Differential regulation of metallothionein-thymidine kinase fusion genes in transgenic mice and their offspring. Cell. 1982;29:701–10. doi: 10.1016/0092-8674(82)90186-6. [DOI] [PubMed] [Google Scholar]
- 35.Bernstein BE, et al. A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell. 2006;125:315–226. doi: 10.1016/j.cell.2006.02.041. [DOI] [PubMed] [Google Scholar]
- 36.Azuara V, et al. Chromatin signatures of pluripotent cell lines. Nat Cell Biol. 2006 doi: 10.1038/ncb1403. [DOI] [PubMed] [Google Scholar]
- 37.Schotta G, et al. A silencing pathway to induce H3-K9 and H4-K20 trimethylation at constitutive heterochromatin. Genes Dev. 2004;18:1251–62. doi: 10.1101/gad.300704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lachner M, O'Sullivan RJ, Jenuwein T. An epigenetic road map for histone lysine methylation. J Cell Sci. 2003;116:2117–24. doi: 10.1242/jcs.00493. [DOI] [PubMed] [Google Scholar]
- 39.Nguyen CT, Gonzales FA, Jones PA. Altered chromatin structure associated with methylation-induced gene silencing in cancer cells: correlation of accessibility, methylation, MeCP2 binding and acetylation. Nucleic Acids Res. 2001;29:4598–606. doi: 10.1093/nar/29.22.4598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fahrner JA, Eguchi S, Herman JG, Baylin SB. Dependence of histone modifications and gene expression on DNA hypermethylation in cancer. Cancer Res. 2002;62:7213–8. [PubMed] [Google Scholar]
- 41.Kouzarides T. Histone methylation in transcriptional control. Curr Opin Genet Dev. 2002;12:198–209. doi: 10.1016/s0959-437x(02)00287-3. [DOI] [PubMed] [Google Scholar]
- 42.Briggs SD, et al. Gene silencing: trans-histone regulatory pathway in chromatin. Nature. 2002;418:498. doi: 10.1038/nature00970. [DOI] [PubMed] [Google Scholar]
- 43.Fischle W, Wang Y, Allis CD. Histone and chromatin cross-talk. Curr Opin Cell Biol. 2003;15:172–83. doi: 10.1016/s0955-0674(03)00013-9. [DOI] [PubMed] [Google Scholar]
- 44.Tamaru H, Selker EU. A histone H3 methyltransferase controls DNA methylation in Neurospora crassa. Nature. 2001;414:277–83. doi: 10.1038/35104508. [DOI] [PubMed] [Google Scholar]
- 45.Tamaru H, et al. Trimethylated lysine 9 of histone H3 is a mark for DNA methylation in Neurospora crassa. Nat Genet. 2003;34:75–9. doi: 10.1038/ng1143. [DOI] [PubMed] [Google Scholar]
- 46.Johnson L, Cao X, Jacobsen S. Interplay between two epigenetic marks. DNA methylation and histone H3 lysine 9 methylation. Curr Biol. 2002;12:1360–7. doi: 10.1016/s0960-9822(02)00976-4. [DOI] [PubMed] [Google Scholar]
- 47.Malagnac F, Bartee L, Bender J. An Arabidopsis SET domain protein required for maintenance but not establishment of DNA methylation. Embo J. 2002;21:6842–52. doi: 10.1093/emboj/cdf687. [DOI] [PMC free article] [PubMed] [Google Scholar]