

1. Introduction to DNA topoisomerases
DNA topoisomerases I and II (Top1 and Top2) are established molecular targets of anticancer drugs.1–5 Mammalian somatic cells express six topoisomerase genes: two TOP1 (TOP1 and TOP1mt), two TOP2 (TOP2α and β), two topoisomerase III (TOP3α and β)6,7 (Figure 1A). The most recently discovered eukaryotic topoisomerase is mitochondrial Top1 (Top1mt), which we reported in 2001.8,9
Figure 1. Schematic architecture of the topoisomerase cleavage complexes.
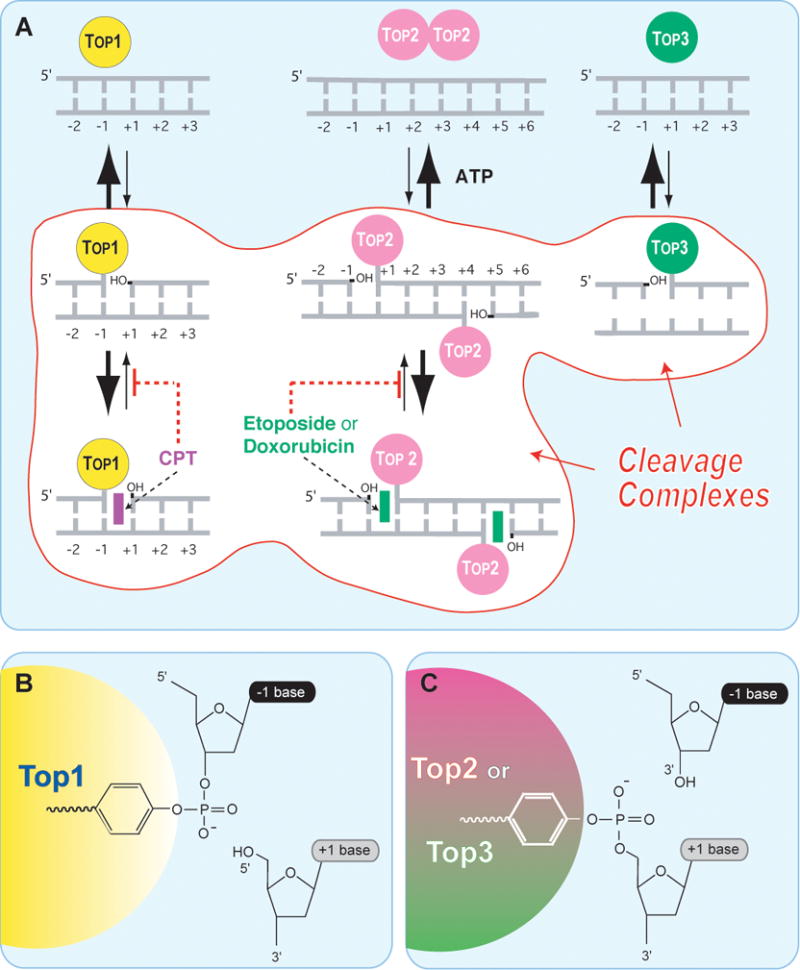
A. Topoisomerases I (Top1 nuclear and Top1mt) bind to double-stranded DNA and form covalent complexes at the 3′-end of the breaks. All other topoisomerases form covalent complexes at the 5′-end of the breaks. Top1 cleavage complexes are selectively stabilized by the natural alkaloid camptothecin (CPT). Topoisomerase II homodimers (Top2α and Top2β) bind to double-stranded DNA and form cleavage complexes with a canonical 4-base pair overhang. Top2 binds and hydrolyze ATP during catalysis. Top2 inhibitors stabilize the Top2 cleavage complexes and are potent anticancer drugs.1,3,4,134 Topoisomerases III (Top3α and Top3β) bind as monomers to non-canonical DNA structures (single-stranded DNA)14 in association with a RecQ helicase (BLM in humans, Sgs-1 in budding yeast, Rhq1 in fission yeast). Top3 has been proposed to resolve double-holiday junctions arising from stalled replication forks.18 Top3 inhibitors have not been reported. B. A topoisomerase catalytic tyrosine residue carries out the nucleophilic attack and breakage of a DNA phosphoester bond. The polarity depends on the topoisomerase type. Topoisomerases I (nuclear and mitochondrial Top1) form a covalent bond with the 3′-DNA end and generate a 5′-hydroxyl-end. This cleavage intermediate (Top1cc) allows controlled rotation of the 5′-end around the intact DNA strand 12. Under normal conditions, the reaction is reversible. Religation (back arrow from B -> A) is favored over cleavage and requires the alignment of the 5′-hydroxyl-end with the phosphoester tyrosyl-DNA bond for nucleophilic attack. All other human Topo enzymes (Top2 and Top3) have an opposite polarity compared to Top1. They form covalent bonds with the 5′-end of the break and generate 3′-hydroxyl ends.
A common feature of topoisomerases is their catalytic mechanism, which in all cases consists in a nucleophilic attack of a DNA phosphodiester bond by a catalytic tyrosyl residue from the topoisomerase. The resulting covalent attachment of the tyrosine to the DNA phosphate is either at the 3′-end of the broken DNA in the case of Top1 enzymes (Top1 and Top1mt) or at the 5′-end of the broken DNA for the other topoisomerases (Figure 1). Thus, Top1 enzymes are the only topoisomerases that form a covalent link with the 3′-end of the broken DNA while generating a 5′-hydroxyl end at the other end of the break. In that respect, the eukaryotic Top1 enzymes belong to the broader family of site-specific tyrosine recombinases of prokaryotes and yeast (e.g., XerCD of Escherichia coli, bacteriophage λ integrase and Cre recombinase, and Flp of Saccharomyces cerevisiae).
Another unique feature of the Top1 enzymes is their DNA relaxation mechanism by “controlled rotation” rather than by “strand passage”.10–12 In other words, Top1 enzymes relax DNA by letting the 5′-hydroxyl end swivel around the intact strand. This processive reaction does not require ATP or divalent metal binding, which is different from Top2 enzymes, which require both ATP hydrolysis and Mg2+.5,13 Top3 enzymes, which, like other type IA topoisomerases require Mg2+ (but no ATP) for catalysis14 are not very active in relaxing DNA supercoiling. They can relax DNA when it is very negatively supercoiled (single-stranded) one turn at a time.15 Moreover, both Top2 and Top3 enzymes change DNA topology by a strand passage distributive mechanism rather than by the processive controlled rotation of the Top1 enzymes. In the case of the Top2 enzymes, a full DNA duplex [referred to as the T (transported) strand] goes through the double-strand break made by an enzyme homodimer5,16,17 (Figure 1A). In the case of the Top3 enzymes, a single strand goes through the single-stranded break,14 typically at double-Holliday junction crossovers.18
The remarkable efficiency of the nicking-closing activity of Top1 enables the enzyme to relax both negatively and positively supercoiled DNA (even at 0°C)19 with similar efficiency.12 This is in contrast with Top2α, which relaxes more efficiently positive supercoiling.20 Of note, Top2β, like Top1 relaxes both positive and negative supercoils similarly.20 Removing positive supercoils is required for replication and transcription progression. Otherwise their accumulation in advance of replication and transcription complexes hinders the melting of the DNA duplex (by helicases) and consequently polymerase translocation along the DNA template.
The normal nicking-closing activity of Top1 can however be uncoupled when the 5′-hydroxyl end generated by the nicking reaction becomes misaligned; for instance at preexisting base lesions or DNA nicks.21,22 In such cases, the Top1 cleavage complex (Top1cc) remains without effective legitimate religation partner. Those Top1-DNA covalent complexes are commonly referred to as “suicide complexes”. Under such conditions, Top1 can nevertheless religate an illegitimate (“foreign”) 5-hydroxyl-DNA end and act as a recombinase.23 This property is routinely used for molecular cloning (TOPO® Cloning, Invitrogen) using vaccinia Top1.24
2. Camptothecins are uniquely targeted therapies
Camptothecin (CPT) is a plant alkaloid first identified from the Chinese tree, Camptotheca acuminata by Monroe Wall and coworkers.25,26 Soon after the discovery that CPT inhibited Top1 by trapping Top1cc,27,28 three lines of evidence demonstrated the selective poisoning of Top1 by CPT: 1/Only the natural CPT isomer was active against Top1;29,30 2/Genetically modified yeast deleted for Top1 (Top1Δ) was immune to CPT;31–33 3/Cells selected for CPT -resistance showed point mutations in the Top1 gene.34
One such mutation found in human leukemia cells,35 is Asn-722-Ser (see Figure 4F). Most remarkably the Asn-722-Ser mutation has recently been found in all the CPT-producing plants. That mutation probably enables plants to grow in the presence of CPT36 while being protected from predators.
Figure 4. Interfacial inhibition by Top1 inhibitor.

(A) Top1 is mostly associated non-covalently with chromatin. (B) Top1 relaxes DNA by making single-strand breaks that are generated by the covalent linkage of Top1 to the 3′-end of DNA (Top1cc; see Figure 1). (C) Camptothecins (see Figure 2) or non-camptothecin Top1 inhibitors (see Figure 3) bind reversibly to the Top1cc and slow down DNA religation. (D) Ternary complex including Top1 (yellow), DNA (dark blue ribbons), and an indenoisoquinoline or CPT (green and red in the middle).45,67,69,81 (E) Same structure except Top1 is in ribbon representation. (F) Hydrogen bond network between camptothecin and Top1 amino acid residues. (G). Hydrogen bond network between the indenoisoquinoline derivative MJ-238 and Top1. Note that mutation of asparagine 722 to serine (N722S), which confers resistance to camptothecin and only partially to indenoisoquinolines, is also present in camptothecin-producing plants.36
The discovery that CPT-producing plants bear a CPT-resistance Top1 mutation raises the question as to whether the Top1 mutation or the production of CPT came first during evolution. An interesting alternative might be that endophyte fungi that grow in the CPT-producing plants actually produce CPT.37–39 In which case, it is plausible that the production of CPT came first from the endophytes and the plants were selected for the Top1 mutations that rendered them immune to CPT.
3. Clinical overview of the camptothecins
Two water-soluble camptothecin derivatives are presently approved by the FDA for IV administration: topotecan and irinotecan (Figure 2). Topotecan (Hycamtin®) is used to treat ovarian cancers and small-cell lung cancers (SCLC). However, hematological toxicity is a common side effect due to the destruction of bone marrow progenitors. As a result, infections can occur due to loss of white blood cells, bruising or bleeding to the loss of platelets, and anemia with fatigue to loss of red blood cells. Within a day following infusion, patients generally feel sick with nausea and possibly vomiting, which can generally be controlled with anti-emetic drugs. Patients may also feel tired during the first weeks of treatment. Hair loss starts 3–4 weeks after the first dose. It is temporary. Hair re-grows once the treatment is finished. Because of potential teratogenic effects, it is recommended to use contraception during topotecan treatment and a few months afterwards.
Figure 2. Chemical structure of clinically relevant CPT derivatives and analogs.

The top scheme shows the equilibrium between the active lactone form and its inactive carboxylate derivative. The lactone is converted into the carboxylate within minutes in human serum at physiological pH 135. Topotecan (Hycamtin®) and irinotecan are routinely used for IV infusion in cancer treatment. Four CPT derivatives are in clinical trials. Gimatecan (Sigma-Tau®, Novartis®) is an oral derivative developed for the treatment of glioma. Belotecan (CKD602, Camtobell®; Chong Keun Dang Corp.) is a water-soluble derivative given IV. Limited information is available on the ongoing clinical status of lurtotecan and exatecan (Daiichi Pharmaceutical Co Ltd). E-ring modifications have been introduced to generate synthetic analogs with limited (but irreversible) E-ring opening (Diflometecan; Beaufour-Ipsen) 43,44,48,136 and no ring opening (S 39625; Servier) 50,51,137. Both E-ring-modified derivatives are given IV.
Irinotecan (CPT-11) is approved by the FDA for colorectal tumors. It is a prodrug and needs to be converted to its active metabolite SN-38 by carboxylesterase (Figure 2). The most severe side effect is diarrhea, which can be severe. Temporary liver dysfunction is generally asymptomatic. The other side effects are the same as those produced by topotecan.
Two newer camptothecin derivatives are in clinical trials: gimatecan and belotecan (Figure 2). Gimatecan is given orally and belotecan IV. Both have shown some activity in glioma.
4. Molecular pharmacology of novel CPT analogues with stabilized E-ring
One of the main limitations of all camptothecin derivatives is their spontaneous and rapid inactivation (within minutes) by E-ring opening (Figure 2). Although this reaction is potentially reversible, its equilibrium favors the carboxylate form at physiological neutral pH. Moreover, the active lactone derivatives is rapidly depleted in the blood stream due to the tight binding of the carboxylates to serum albumin.40
Two approaches have been taken to overcome the E-ring lactone instability. The first was to enlarge the E-ring by one carbon atom, which limits E-ring opening but also prohibits its reclosure 41–43. The corresponding compounds are synthetic and named homocamptothecins. Diflomotecan is the clinical derivative (Figure 2). This approach is however potentially problematic as irreversible E-ring opening inactivates homocamptothecin 44. From a chemical biology standpoint, one reason for studying the homocamptothecins was the presence of bound carboxylate in the crystal structure of topotecan,45 which is in contrast with the fact that the carboxylate form of CPT is clinically ineffective and inactive in trapping Top1cc.29,30 Since homocamptothecin are at least as potent as CPT in spite of limited E-ring opening,46 this suggests that E-ring opening was not necessary for trapping Top1cc.44 A second reason for studying homocamptothecins was to determine whether changing the E-ring could overcome the known drug efflux multidrug resistance mechanism to camptothecins.47 Interestingly, we found limited impact of ABCG2 drug efflux resistance for homocamptothecins, which gives them an advantage over the camptothecins.48,49
The second approach to stabilize the E-ring was to convert the E-ring from a 6- to a 5-membered ring. Complete stabilization of the E-ring has been successfully achieved with the synthesis of the α-keto derivatives50 exemplified by S39625.51 Removal of the lactone precludes E-ring opening.50 This novel series provided a further test for the relationship between lack of E-ring opening and trapping of Top1cc. The remarkable potency of this novel drug class against purified Top1 and in cells, with selective targeting of Top1, and persistent Top1cc indicate the tight binding of S39625 to Top1cc.51 These experiments provide further evidence that lactone E-ring opening is not necessary for the trapping of Top1cc by CPT derivatives. S39625 is under consideration for clinical trials.
5. The non-camptothecin Top1 inhibitors
Camptothecins are the only clinically approved Top1 inhibitors. In spite of their activity in colon, lung and ovarian cancers,52,53 camptothecins have limitations: 2,34,52,54–56 as discussed above, 1/camptothecins are chemically unstable and rapidly inactivated to carboxylate in blood; 2/Top1cc reverse within minutes after drug removal, which imposes long infusions; 3/Cells overexpressing the drug efflux membrane transporters ABCG2 and ABCB1 (Pgp)47–49 are cross-resistant to camptothecins; 4/The side effects of camptothecins are dose-limiting and potentially severe (diarrhea and neutropenia).
Because of those limitations, because camptothecins were the only known Top1 inhibitor chemotype, and because it is well-established that drugs with a common primary target exhibit different clinical activities (for instance doxorubicin, amsacrine and etoposide as Top2 inhibitors), we initiated the discovery of novel Top1 inhibitors using the Developmental Therapeutics Program (DTP-NCI) cell lines and drug databases.57,58 We reported the first indenoisoquinoline NSC 314622 in 1998.59 During the past 10 years, the indenoisoquinolines have been optimized for therapeutic development, and more than 400 derivatives have been synthesized and tested 59–87. Two derivatives (NSC 725776 and 724998; Figure 3) are under review for clinical trials at the NCI Bethesda using histone γ-H2AX as a pharmacodynamic biomarker.88
Figure 3. Chemical structure of clinically relevant non-CPT Top1 inhibitors.

Three chemical families are described (for further information see 52). Two indenoisoquinolines (NSC 725776 and NSC 724998) are in preclinical development at the NCI (Joint patent NCI-Purdue University). ARC-111 is a phenanthridine derivative licensed to Genzyme Co. The indolocarbazoles are further have been tested in clinical trials. Information is limited on their ongoing clinical development.
The indenoisoquinolines are one of the three classes of non-camptothecin Top1 inhibitors in clinical development (Figure 3).2,52 The indolocarbazoles were the first introduced. They underwent Phase I–II clinical trials but appear to hit other cellular targets besides Top1.52,89 Their current clinical development as anticancer drugs does not appear very active. More interesting are the phenanthridine derivatives (Figure 3), which have been licensed to Genzyme Inc. and should enter Phase I clinical trials in the near future.52,90,91 The phenanthridines analogs of ARC-111 exhibit chemical and biological similarities with the indenoisoquinolines,52 and it will be interesting to compare the clinical activities of the two classes of non-CPT Top1 inhibitors.
Two indenoisoquinolines have been selected for clinical development, and an IND has been filed. The Phase I trial is aimed at comparing the two drugs side-by-side and to use histoneγH2AX as a pharmacodynamic biomarker. The selected indenoisoquinolines have several favorable characteristics:61,63
1/They are chemically stable, which is not the case of camptothecins (see above); 2/They trap Top1cc at differential sites from camptothecins, which is indicative of potentially different gene targeting; 3/Their anti-proliferative activity is similar to or greater than camptothecins in the NCI60 cell lines; 4/They selectively target Top1 in cells,61,63,92 as demonstrated by high resistance of Top1-deficient P388 cells,63,93 and cross-resistance of cells with Top1-downregulation by shRNA;63,92 5/They are not substrates of ABC membrane transporters,63 which suggest an ability to overcome resistance to camptothecins; 6/Their antitumor activity in animal models61 is better correlated with effects on human bone marrow progenitors,94 suggesting that therapeutic doses in mice might be achievable in humans.95 The phenanthridine derivatives (ARC-111; Figure 3) share many of the same advantages as the indenoisoquinolines,90 which is not surprising considering the chemical similarities between the indenoisoquinoline and phenanthridine families (Figure 3).
6. The interfacial inhibition paradigm
With Kurt Kohn, we hypothesized in 1990 that topoisomerase inhibitors trap cleavage complexes by binding at the enzyme-DNA interface and proposed that the drugs stack between the base pairs flanking the cleavage site because of their planar aromatic structure (Figure 4C).96–98. Additional bonds also would link the drugs to the enzyme. The concept was proposed for both Top296,97 and Top198 inhibitors. The interfacial model has been validated after crystallization of topotecan, camptothecin, indenoisoquinolines and an indolocarbazole bound to Top1cc.67,69,81
Like other Top1 inhibitors (topotecan, CPT and an indolocarbazole),45,67,69,81 the indenoisoquinolines bind at the interface of the Top1cc-DNA complexes by intercalating at the cleavage site (π–π interactions) (Figure 4D–E) and by forming a network of H-bonds with critical Top1 residues involved in CPT resistance (Figure 4F–G).34
In fact, many natural products act as inter-facial inhibitors.99 They are characterized by their stereospecific and selective binding to a site involving two or more macromolecules (proteins and/or nucleic acids) within complexes undergoing conformational changes. Interfacial inhibitors trap (generally reversibly) an intermediate state of the complex, resulting in kinetic inactivation.
Potential inhibitors of nucleic acid-protein interfaces include inhibitors of Top2 (dexrazoxane, anthracyclines, epipodophyllotoxins), gyrase (ciprofloxacin), RNA polymerases (α-amanitin, actinomycin D), ribosomes (streptomycin, tetracycline, kirromycin, thiostrepton, and possibly cycloheximide), as well as HIV-1 integrase inhibitors.69,100 The interfacial inhibitor paradigm also applies to protein-protein interfaces such as GTP-binding protein complexes (brefeldin A), tubulin (taxol, colchicine, epothilone), 14-3-3-ATPase (fusicoccin), adenylcyclase hetero-dimers (forskolin), mTOR-related complexes (rapamycin).99 As for nucleic acid-protein complexes, these drugs take advantage of transient structural and energetic conditions created by the macromolecular complex giving rise to “hot spots” for drug binding.
From a drug discovery view point, the interfacial inhibition paradigm demonstrates the value of screening natural products, the importance of looking for non-competitive inhibitors and for setting up high throughput assays based on enhancement of macromolecular binding rather than only binding inhibition.
7. Conversion of Top1-DNA complexes into cellular DNA damage
The cytotoxicity of Top1 inhibitors is due to the trapping of Top1cc rather than to the inhibition of Top1 catalytic activity. This is because the Top1cc are converted into DNA damage by DNA replication and transcription (Figure 5B). That direct relationship between drug target level and cytotoxic activity is consistent with early studies showing that yeast strains without Top1 are immune to CPT,32 and with more recent studies showing that Top1 depletion in human cells confers resistance to CPT and indenoisoquinolines,92,101, and that the antitumor activity of camptothecins is positively correlated with cellular Top1 levels.53,102,103 Noticeably, high Top1 levels have also been correlated with oxaliplatin activity in patients.102
Figure 5. DNA damage and repair resulting from the trapping of Top1cc.
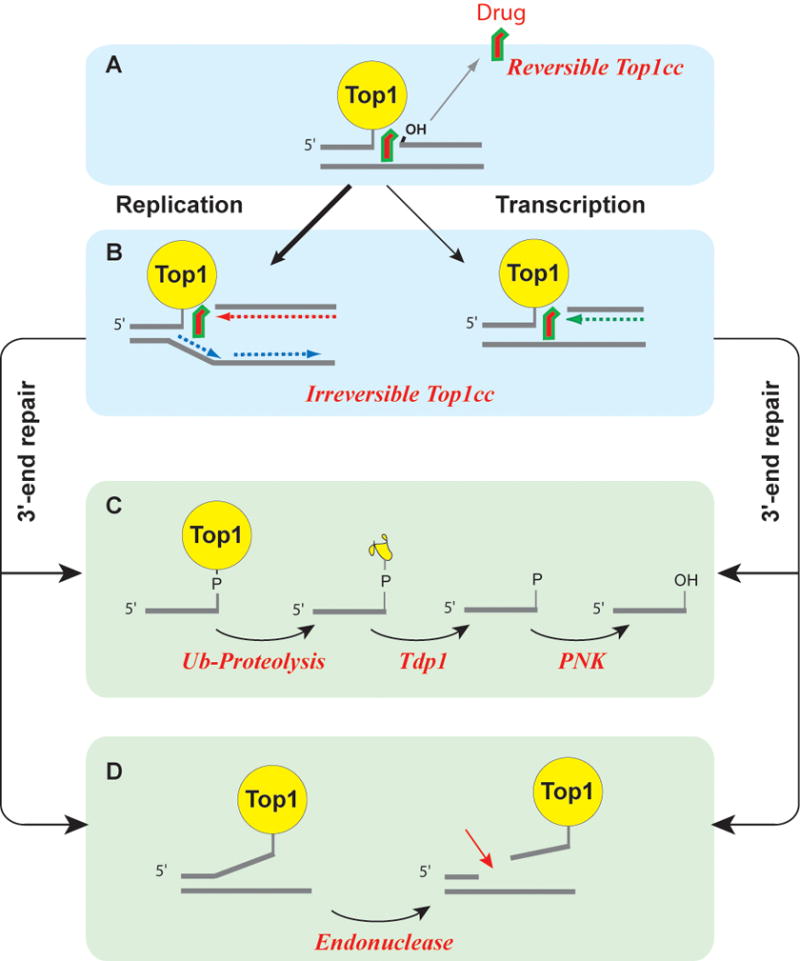
(A) Most Top1cc are reversible as Top1 inhibitors tend to rapidly dissociate from the Top1cc before DNA replication or transcription collision. (B) Collision between a replication fork and a stalled Top1cc (shown on the leading strand) produces a replication-induced double-strand end (“replication run-off”).138 Similarly, a stalled Top1cc can produce a transcription block and activate the DNA damage response.139,140 The repair of covalent Top1-DNA complexes is believed to involve two main pathways. (C) Tdp1 can hydrolyze the tyrosyl-phosphoester bond following the degradation of Top1 by ubiquitin-proteasomal degradation (for review see22,121). (D) Data obtained from yeast DNA repair mutant suggest an alternative endonuclease pathway, which is not yet validated in human cells. The candidate endonucleases are Mre11-CtIP, XPF-ERCC1 and Mus81-Eme1 (for review see2,22).
The trapping of Top1cc can occur irrespective of DNA supercoiling, as demonstrated by the fact that Top1cc can be efficiently produced in small oligonucleotides or linear DNA fragments.27,28,52,60,91,98 Nevertheless, recent studies show that the trapping of Top1cc by camptothecins can be influenced by DNA supercoiling. Indeed, Top1cc tend to be preferentially trapped by camptothecins in positively supercoiled vs. negatively supercoiled DNA, suggesting that the most lethal Top1cc tend to be in advance of replication and transcription complexes (Figure 5B).104–108
Although both nuclear and mitochondrial topoisomerases I (Top1 and Topmt) are sensitive to CPT,109,110 Top1 (nuclear Top1) rather than Top1mt (mitochondrial Top1) is relevant for the anticancer activity of camptothecins. Indeed, point mutations in Top1 at key interacting residues (see Figure 5) are sufficient to confer high resistance to CPT.34. We also sequenced the Top1mt of the CPT-resistant human leukemia CEM-C2 cells and found no Top1mt mutation in those cells (Zhang and Pommier, unpublished), indicating that Top1mt is probably not a relevant target of CPT at pharmacological concentrations. This might be due to the alkaline pH of mitochondria, which promotes lactone E-ring opening (see Figure 2), and to a lack of penetration of CPT into mitochondria because camptothecin lacks a positive charge, which is a common feature of mitochondria-targeted drugs. Of note, topotecan, which does possess a side chain with a potentially protonated nitrogen has been found to accumulate in mitochondria.111,112 The relevance of this accumulation in mitochondria remains to be determined.
8. Repair of Top1-associated DNA damage: relationship with anticancer activity
Of the two main DNA damaging pathways, replication and transcription (Fig. 5B), experimental evidence suggests that the replication pathway exerts a prominent role for cancer cells in culture.30,104 Nevertheless, it is also clear that the differential response of cancer cells compared to normal cells involves downstream cellular responses from the replication- and transcription-mediated DNA damage. Among those differences, it is likely that a variety of molecular defects in cell cycle and apoptotic checkpoints play critical roles in the antitumor activity of Top1 inhibitors.2,22
It is also likely that defects in DNA repair contribute to the cellular response to Top1 inhibitors. The repair is Top1-mediated DNA damage can be schematically divided in the repair/excision of the Top1 covalently linked to the DNA (3′-end processing) and the repair/religation of the 5′-end. The best characterized pathway for 3′-end processing utilizes tyrosyl-DNA-phosphodiesterase (Tdp1) (Figure 5C).113–115 However, an alternative pathway involving 3′-flap endonucleases (Mus81/Eme1; Mre11/Rad50; XPF/ERCC1) has been invoked (Figure 5D), primarily based on the hypersensitivity to CPT of yeast strains with mutations for those endonucleases.22,116–121
The conservation of Tdp1 from yeast to humans113,122 is consistent with the ubiquitous occurrence of Top1cc under physiological conditions. Tdp1 can also remove 3′-phospho-glycolates lesions.123,124 Tdp1 knockout mice are viable but hypersensitive to camptothecins and bleomycin.125,126 The hereditary Tdp1 mutation H493R leads to the rare neurodegenerative disease SCAN1 (spino-cerebellar ataxia with axonal neuropathy) 121,127. Experiments in SCAN1 lymphoblastoid cells treated with CPT demonstrated the importance of human Tdp1 for the repair of transcription-associated Top1cc 128,129.
Based on the redundancy of the DNA repair pathways for Top1cc, we proposed a rationale for combining Tdp1 and Top1 inhibitors, and for the discovery of Tdp1 inhibitors 121 (Figure 6). This rationale stems from the fact that genetic studies demonstrate that redundant pathways repair Top1cc in normal cells and that several of the corresponding genes are known to be inactivated in cancers [for instance the Mre11 or BRCA1] 22. Thus, Tdp1 inhibitors should selectively enhance the activity of Top1 inhibitors in cancer cells with such deficiencies [i.e. colon cancers 130,131] (Figure 6B) while sparing normal cells (Figure 6A). Because Tdp1 deficiency has only a mild phenotype in yeast113,118 and mice,125,126 it is anticipated that Tdp1 inhibitors will have limited side effects. Several chemical families have already been reported as leads for discovery of Tdp1 inhibitors.101,121,132,133
Figure 6. Rationale for Tdp1 inhibitors.

(A) In normal cells, Top1-DNA covalent complexes can be repaired by redundant mechanisms, which can be divided in two main pathways: i/the Tdp1 hydrolysis pathway (see Figure 5C), and ii/the 3′-endonuclease pathway (see Figure 5D). (B) Cancer cells might be more dependent on the Tdp1 pathway as a result of mutations and inactivation of DNA checkpoints (BRCA1, Chk2…). The expected effect of combining a Tdp1 inhibitor with a Top1 inhibitor would be an increase in the therapeutic index of the Top1 inhibitor as the Tdp1 inhibitor would sensitize preferentially the cancer cells.
Acknowledgments
Our studies are supported by the National Cancer Institute Intramural Program, Center for Cancer Research. We wish to thank Dr. Kurt W. Kohn for long term collaboration and many insights. We also wish to thank the past and current members of the Laboratory of Molecular Pharmacology for their dedication and enthusiasm over the years.
References
- 1.Li TK, Liu LF. Annu Rev Pharmacol Toxicol. 2001;41:53–77. doi: 10.1146/annurev.pharmtox.41.1.53. [DOI] [PubMed] [Google Scholar]
- 2.Pommier Y. Nat Rev Cancer. 2006;6:789–802. doi: 10.1038/nrc1977. [DOI] [PubMed] [Google Scholar]
- 3.McClendon AK, Osheroff N. Mutat Res. 2007;623:83–97. doi: 10.1016/j.mrfmmm.2007.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nitiss JL. Nat Rev Cancer. 2009;9:338–50. doi: 10.1038/nrc2607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nitiss JL. Nat Rev Cancer. 2009;9:327–37. doi: 10.1038/nrc2608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Champoux JJ. Annu Rev Biochem. 2001;70:369–413. doi: 10.1146/annurev.biochem.70.1.369. [DOI] [PubMed] [Google Scholar]
- 7.Wang JC. Nat Rev Mol Cell Biol. 2002;3:430–40. doi: 10.1038/nrm831. [DOI] [PubMed] [Google Scholar]
- 8.Zhang H, Barcelo JM, Lee B, Kohlhagen G, Zimonjic DB, Popescu NC, Pommier Y. Proc Natl Acad Sci U S A. 2001;98:10608–13. doi: 10.1073/pnas.191321998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhang H, Meng LH, Zimonjic DB, Popescu NC, Pommier Y. Nucleic Acids Res. 2004;32:2087–92. doi: 10.1093/nar/gkh525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Stewart L, Redinbo MR, Qiu X, Hol WGJ, Champoux JJ. Science. 1998;279:1534–1541. doi: 10.1126/science.279.5356.1534. [DOI] [PubMed] [Google Scholar]
- 11.Stivers JT, Harris TK, Mildvan AS. Biochemistry. 1997;36:5212–5222. doi: 10.1021/bi962880t. [DOI] [PubMed] [Google Scholar]
- 12.Koster DA, Croquette V, Dekker C, Shuman S, Dekker NH. Nature. 2005;434:671–4. doi: 10.1038/nature03395. [DOI] [PubMed] [Google Scholar]
- 13.Deweese JE, Burgin AB, Osheroff N. Nucleic Acids Res. 2008;36:4883–93. doi: 10.1093/nar/gkn466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Goulaouic H, Roulon T, Flamand O, Grondard L, Lavelle F, Riou JF. Nucleic Acids Res. 1999;27:2443–2450. doi: 10.1093/nar/27.12.2443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Champoux JJ. Proc Natl Acad Sci U S A. 2002;99:11998–2000. doi: 10.1073/pnas.202483499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schoeffler AJ, Berger JM. Biochem Soc Trans. 2005;33:1465–70. doi: 10.1042/BST0331465. [DOI] [PubMed] [Google Scholar]
- 17.Corbett KD, Benedetti P, Berger JM. Nat Struct Mol Biol. 2007;14:611–9. doi: 10.1038/nsmb1264. [DOI] [PubMed] [Google Scholar]
- 18.Wu L, Hickson ID. Nature. 2003;426:870–4. doi: 10.1038/nature02253. [DOI] [PubMed] [Google Scholar]
- 19.Covey JM, Jaxel C, Kohn KW, Pommier Y. Cancer Res. 1989;49:5016–5022. [PubMed] [Google Scholar]
- 20.McClendon AK, Rodriguez AC, Osheroff N. J Biol Chem. 2005;280:39337–45. doi: 10.1074/jbc.M503320200. [DOI] [PubMed] [Google Scholar]
- 21.Pourquier P, Pommier Y. Adv Cancer Res. 2001;80:189–216. doi: 10.1016/s0065-230x(01)80016-6. [DOI] [PubMed] [Google Scholar]
- 22.Pommier Y, Barcelo JM, Rao VA, Sordet O, Jobson AG, Thibaut L, Miao ZH, Seiler JA, Zhang H, Marchand C, Agama K, Nitiss JL, Redon C. Prog Nucleic Acid Res Mol Biol. 2006;81:179–229. doi: 10.1016/S0079-6603(06)81005-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pommier Y, Jenkins J, Kohlhagen G, Leteurtre F. Mutat Res. 1995;337:135–45. doi: 10.1016/0921-8777(95)00019-g. [DOI] [PubMed] [Google Scholar]
- 24.Shuman S. J Biol Chem. 1992;267:8620–8627. [PubMed] [Google Scholar]
- 25.Wall ME, Wani MC, Cooke CE, Palmer KH, McPhail AT, Slim GA. J Am Chem Soc. 1966;88:3888–3890. [Google Scholar]
- 26.Wall ME, Wani MC. Cancer Res. 1995;55:753–760. [PubMed] [Google Scholar]
- 27.Hsiang YH, Hertzberg R, Hecht S, Liu LF. J Biol Chem. 1985;260:14873–8. [PubMed] [Google Scholar]
- 28.Dexheimer TS, Pommier Y. Nat Protoc. 2008;3:1736–50. doi: 10.1038/nprot.2008.174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jaxel C, Kohn KW, Wani MC, Wall ME, Pommier Y. Cancer Res. 1989;49:1465–1469. [PubMed] [Google Scholar]
- 30.Hsiang YH, Liu LF, Wall ME, Wani MC, Nicholas AW, Manikumar G, Kirschenbaum S, Silber R, Potmesil M. Cancer Res. 1989;49:4385–4389. [PubMed] [Google Scholar]
- 31.Eng WK, Faucette L, Johnson RK, Sternglanz R. Mol Pharmacol. 1988;34:755–760. [PubMed] [Google Scholar]
- 32.Nitiss J, Wang JC. Proc Natl Acad Sci U S A. 1988;85:7501–7505. doi: 10.1073/pnas.85.20.7501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bjornsti MA, Benedetti P, Viglianti GA, Wang JC. Cancer Res. 1989;49:6318–6323. [PubMed] [Google Scholar]
- 34.Pommier Y, Pourquier P, Urasaki Y, Wu J, Laco G. Drug Resist Updat. 1999;2:307–318. doi: 10.1054/drup.1999.0102. [DOI] [PubMed] [Google Scholar]
- 35.Fujimori A, Harker WG, Kohlhagen G, Hoki Y, Pommier Y. Cancer Res. 1995;55:1339–1346. [PubMed] [Google Scholar]
- 36.Sirikantaramas S, Yamazaki M, Saito K. Proc Natl Acad Sci U S A. 2008;105:6782–6. doi: 10.1073/pnas.0801038105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rehman S, Shawl AS, Kour A, Andrabi R, Sudan P, Sultan P, Verma V, Qazi GN. Appl Biochem Microbiol. 2008;44:203–209. [Google Scholar]
- 38.Kusari S, Zuhlke S, Spiteller M. J Nat Prod. 2009;72:2–7. doi: 10.1021/np800455b. [DOI] [PubMed] [Google Scholar]
- 39.Amna T, Puri SC, Verma V, Sharma JP, Khajuria RK, Musarrat J, Spiteller M, Qazi GN. Can J Microbiol. 2006;52:189–96. doi: 10.1139/w05-122. [DOI] [PubMed] [Google Scholar]
- 40.Burke TG, Mi ZM. J Med Chem. 1994;37:40–46. doi: 10.1021/jm00027a005. [DOI] [PubMed] [Google Scholar]
- 41.Lavergne O, Lesueur-Ginot L, Pla Rodas F, Kasprzyk PG, Pommier J, Demarquay D, Prevost G, Ulibarri G, Rolland A, Schiano-Liberatore AM, Harnett J, Pons D, Camara J, Bigg DCH. J Med Chem. 1998;41:5410–9. doi: 10.1021/jm980400l. [DOI] [PubMed] [Google Scholar]
- 42.Chen AY, Shih SJ, Garriques LN, Rothenberg ML, Hsiao M, Curran DP. Mol Cancer Ther. 2005;4:317–24. [PubMed] [Google Scholar]
- 43.Tangirala RS, Antony S, Agama K, Pommier Y, Anderson BD, Bevins R, Curran DP. Bioorg Med Chem. 2006;14:6202–12. doi: 10.1016/j.bmc.2006.05.073. [DOI] [PubMed] [Google Scholar]
- 44.Urasaki Y, Takebayashi Y, Pommier Y. Cancer Res. 2000;60:6577–80. [PubMed] [Google Scholar]
- 45.Staker BL, Hjerrild K, Feese MD, Behnke CA, Burgin AB, Jr, Stewart L. Proc Natl Acad Sci U S A. 2002;99:15387–92. doi: 10.1073/pnas.242259599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bailly C, Lansiaux A, Dassonneville L, Demarquay D, Coulomb H, Huchet M, Lavergne O, Bigg DC. Biochemistry. 1999;38:15556–63. doi: 10.1021/bi990947h. [DOI] [PubMed] [Google Scholar]
- 47.Brangi M, Litman T, Ciotti M, Nishiyama K, Kohlhagen G, Takimoto C, Robey R, Pommier Y, Fojo T, Bates SE. Cancer Res. 1999;59:5938–46. [PubMed] [Google Scholar]
- 48.Bates SE, Medina-Perez WY, Kohlhagen G, Antony S, Nadjem T, Robey RW, Pommier Y. J Pharmacol Exp Ther. 2004;310:836–842. doi: 10.1124/jpet.103.063149. [DOI] [PubMed] [Google Scholar]
- 49.Liao Z, Robey RW, Guirouilh-Barbat J, To KK, Polgar O, Bates SE, Pommier Y. Mol Pharmacol. 2008;73:490–7. doi: 10.1124/mol.107.041178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hautefaye P, Cimetiere B, Pierre A, Leonce S, Hickman J, Laine W, Bailly C, Lavielle G. Bioorg Med Chem Lett. 2003;13:2731–5. doi: 10.1016/s0960-894x(03)00534-1. [DOI] [PubMed] [Google Scholar]
- 51.Takagi K, Dexheimer TS, Redon C, Sordet O, Agama K, Lavielle G, Pierre A, Bates SE, Pommier Y. Mol Cancer Ther. 2007;6:3229–38. doi: 10.1158/1535-7163.MCT-07-0441. [DOI] [PubMed] [Google Scholar]
- 52.Teicher BA. Biochem Pharmacol. 2008;75:1262–1271. doi: 10.1016/j.bcp.2007.10.016. [DOI] [PubMed] [Google Scholar]
- 53.Burgess DJ, Doles J, Zender L, Xue W, Ma B, McCombie WR, Hannon GJ, Lowe SW, Hemann MT. Proc Natl Acad Sci U S A. 2008;105:9053–8. doi: 10.1073/pnas.0803513105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Meng LH, Liao ZY, Pommier Y. Curr Topics Med Chem. 2003;3:305–320. doi: 10.2174/1568026033452546. [DOI] [PubMed] [Google Scholar]
- 55.Pommier Y. Curr Med Chem Anti-Canc Agents. 2004;4:429–34. doi: 10.2174/1568011043352777. [DOI] [PubMed] [Google Scholar]
- 56.Pommier Y, Cushman M. Mol Cancer Ther. 2008 doi: 10.1158/1535-7163.MCT-08-0706. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Paull KD, Shoemaker RH, Hodes L, Monks A, Scudiero DA, Rubinstein L, Plowman J, Boyd M. J Natl Cancer Inst. 1989;81:1088–1092. doi: 10.1093/jnci/81.14.1088. [DOI] [PubMed] [Google Scholar]
- 58.Shoemaker RH. Nat Rev Cancer. 2006;6:813–23. doi: 10.1038/nrc1951. [DOI] [PubMed] [Google Scholar]
- 59.Kohlhagen G, Paull K, Cushman M, Nagafufuji P, Pommier Y. Mol Pharmacol. 1998;54:50–58. doi: 10.1124/mol.54.1.50. [DOI] [PubMed] [Google Scholar]
- 60.Antony S, Jayaraman M, Laco G, Kohlhagen G, Kohn KW, Cushman M, Pommier Y. Cancer Res. 2003;63:7428–35. [PubMed] [Google Scholar]
- 61.Antony S, Kohlhagen G, Agama K, Jayaraman M, Cao S, Durrani FA, Rustum YM, Cushman M, Pommier Y. Mol Pharmacol. 2005;67:523–530. doi: 10.1124/mol.104.003889. [DOI] [PubMed] [Google Scholar]
- 62.Antony S, Agama KK, Miao ZH, Hollingshead M, Holbeck SL, Wright MH, Varticovski L, Nagarajan M, Morrell A, Cushman M, Pommier Y. Mol Pharmacol. 2006;70:1109–20. doi: 10.1124/mol.106.024372. [DOI] [PubMed] [Google Scholar]
- 63.Antony S, Agama KK, Miao ZH, Takagi K, Wright MH, Robles AI, Varticovski L, Nagarajan M, Morrell A, Cushman M, Pommier Y. Cancer Res. 2007;67:10397–405. doi: 10.1158/0008-5472.CAN-07-0938. [DOI] [PubMed] [Google Scholar]
- 64.Cinelli MA, Morrell A, Dexheimer TS, Scher ES, Pommier Y, Cushman M. J Med Chem. 2008;51:4609–19. doi: 10.1021/jm800259e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Cushman M, Jayaraman M, Vroman JA, Fukunaga AK, Fox BM, Kohlhagen G, Strumberg D, Pommier Y. J Med Chem. 2000;43:3688–98. doi: 10.1021/jm000029d. [DOI] [PubMed] [Google Scholar]
- 66.Fox BM, Xiao X, Antony S, Kohlhagen G, Pommier Y, Staker BL, Stewart L, Cushman M. J Med Chem. 2003;46:3275–3282. doi: 10.1021/jm0300476. [DOI] [PubMed] [Google Scholar]
- 67.Ioanoviciu A, Antony S, Pommier Y, Staker BL, Stewart L, Cushman M. J Med Chem. 2005;48:4803–4814. doi: 10.1021/jm050076b. [DOI] [PubMed] [Google Scholar]
- 68.Jayaraman M, Fox BM, Hollingshead M, Kohlhagen G, Pommier Y, Cushman M. J Med Chem. 2002;45:242–9. doi: 10.1021/jm000498f. [DOI] [PubMed] [Google Scholar]
- 69.Marchand C, Antony S, Kohn KW, Cushman M, Ioanoviciu A, Staker BL, Burgin AB, Stewart L, Pommier Y. Mol Cancer Ther. 2006;5:287–95. doi: 10.1158/1535-7163.MCT-05-0456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Morrell A, Antony S, Kohlhagen G, Pommier Y, Cushman M. Bioorg Med Chem Lett. 2004;14:3659–63. doi: 10.1016/j.bmcl.2004.05.022. [DOI] [PubMed] [Google Scholar]
- 71.Morrell A, Antony S, Kohlhagen G, Pommier Y, Cushman M. Bioorg Med Chem Lett. 2006;16:1846–9. doi: 10.1016/j.bmcl.2006.01.008. [DOI] [PubMed] [Google Scholar]
- 72.Morrell A, Jayaraman M, Nagarajan M, Fox BM, Meckley MR, Ioanoviciu A, Pommier Y, Antony S, Hollingshead M, Cushman M. Bioorg Med Chem Lett. 2006;16:4395–9. doi: 10.1016/j.bmcl.2006.05.048. [DOI] [PubMed] [Google Scholar]
- 73.Morrell A, Antony S, Kohlhagen G, Pommier Y, Cushman M. J Med Chem. 2006;49:7740–53. doi: 10.1021/jm060974n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Morrell A, Placzek MS, Steffen JD, Antony S, Agama K, Pommier Y, Cushman M. J Med Chem. 2007;50:2040–2048. doi: 10.1021/jm0613119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Morrell A, Placzek M, Parmley S, Antony S, Dexheimer TS, Pommier Y, Cushman M. J Med Chem. 2007;50:4419–4430. doi: 10.1021/jm070361q. [DOI] [PubMed] [Google Scholar]
- 76.Morrell A, Placzek M, Parmley S, Grella B, Antony S, Pommier Y, Cushman M. J Med Chem. 2007;50:4388–4404. doi: 10.1021/jm070307+. [DOI] [PubMed] [Google Scholar]
- 77.Nagarajan M, Xiao X, Antony S, Kohlhagen G, Pommier Y, Cushman M. J Med Chem. 2003;46:5712–24. doi: 10.1021/jm030313f. [DOI] [PubMed] [Google Scholar]
- 78.Nagarajan M, Morrell A, Fort BC, Meckley MR, Antony S, Kohlhagen G, Pommier Y, Cushman M. J Med Chem. 2004;47:5651–5661. doi: 10.1021/jm040025z. [DOI] [PubMed] [Google Scholar]
- 79.Nagarajan M, Morrell A, Antony S, Kohlhagen G, Agama K, Pommier Y, Ragazzon PA, Garbett NC, Chaires JB, Hollingshead M, Cushman M. J Med Chem. 2006;49:5129–5140. doi: 10.1021/jm060046o. [DOI] [PubMed] [Google Scholar]
- 80.Nagarajan M, Morrell A, Ioanoviciu A, Antony S, Kohlhagen G, Agama K, Hollingshead M, Pommier Y, Cushman M. J Med Chem. 2006;49:6283–9. doi: 10.1021/jm060564z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Staker BL, Feese MD, Cushman M, Pommier Y, Zembower D, Stewart L, Burgin AB. J Med Chem. 2005;48:2336–45. doi: 10.1021/jm049146p. [DOI] [PubMed] [Google Scholar]
- 82.Strumberg D, Pommier Y, Paull K, Jayaraman M, Nagafuji P, Cushman M. J Med Chem. 1999;42:446–457. doi: 10.1021/jm9803323. [DOI] [PubMed] [Google Scholar]
- 83.Xiao X, Antony S, Kohlhagen G, Pommier Y, Cushman M. Bioorg Med Chem. 2004;12:5147–60. doi: 10.1016/j.bmc.2004.07.027. [DOI] [PubMed] [Google Scholar]
- 84.Xiao X, Antony S, Kohlhagen G, Pommier Y, Cushman M. J Org Chem. 2004;69:7495–7501. doi: 10.1021/jo048808f. [DOI] [PubMed] [Google Scholar]
- 85.Xiao X, Antony S, Pommier Y, Cushman M. J Med Chem. 2005;48:3231–8. doi: 10.1021/jm050017y. [DOI] [PubMed] [Google Scholar]
- 86.Xiao X, Miao ZH, Antony S, Pommier Y, Cushman M. Bioorg Med Chem Lett. 2005;15:2795–8. doi: 10.1016/j.bmcl.2005.03.101. [DOI] [PubMed] [Google Scholar]
- 87.Xiao X, Antony S, Pommier Y, Cushman M. J Med Chem. 2006;49:1408–12. doi: 10.1021/jm051116e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Bonner WM, Redon CE, Dickey JS, Nakamura AJ, Sedelnikova OA, Solier S, Pommier Y. Nat Rev Cancer. 2008;8:957–967. doi: 10.1038/nrc2523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Urasaki Y, Laco G, Takebayashi Y, Bailly C, Kohlhagen G, Pommier Y. Cancer Res. 2001;61:504–508. [PubMed] [Google Scholar]
- 90.Kurtzberg LS, Battle T, Rouleau C, Bagley RG, Agata N, Yao M, Schmid S, Roth S, Crawford J, Krumbholz R, Ewesuedo R, Yu XJ, Wang F, Lavoie EJ, Teicher BA. Mol Cancer Ther. 2008;7:3212–22. doi: 10.1158/1535-7163.MCT-08-0568. [DOI] [PubMed] [Google Scholar]
- 91.Li TK, Houghton PJ, Desai SD, Daroui P, Liu AA, Hars ES, Ruchelman AL, LaVoie EJ, Liu LF. Cancer Res. 2003;63:8400–7. [PubMed] [Google Scholar]
- 92.Miao ZH, Player A, Shankavaram U, Wang YH, Zimonjic DB, Lorenzi PL, Liao ZY, Liu H, Shimura T, Zhang HL, Meng LH, Zhang YW, Kawasaki ES, Popescu NC, Aladjem MI, Goldstein DJ, Weinstein JN, Pommier Y. Cancer Res. 2007;67:8752–8761. doi: 10.1158/0008-5472.CAN-06-4554. [DOI] [PubMed] [Google Scholar]
- 93.Mattern MR, Hofman GA, Polsky RM, Funk LR, McCabe FL, Johnson RK. Oncol Res. 1993;5:467–474. [PubMed] [Google Scholar]
- 94.Erickson-Miller CL, May RD, Tomaszewski J, Osborn B, Murphy MJ, Page JG, Parchment RE. Cancer Chemother Pharmacol. 1997;39:467–72. doi: 10.1007/s002800050600. [DOI] [PubMed] [Google Scholar]
- 95.Giovanella BC, Stehlin JS, Wall ME, Wani MC, Nicholas AW, Liu LF, Silber R, Potmesil M. Science. 1989;246:1046–1048. doi: 10.1126/science.2555920. [DOI] [PubMed] [Google Scholar]
- 96.Capranico G, Kohn KW, Pommier Y. Nucleic Acids Res. 1990;18:6611–6619. doi: 10.1093/nar/18.22.6611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Pommier Y, Capranico G, Orr A, Kohn KW. Nucleic Acids Res. 1991;19:5973–5980. doi: 10.1093/nar/19.21.5973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Jaxel C, Capranico G, Kerrigan D, Kohn KW, Pommier Y. J Biol Chem. 1991;266:20418–20423. [PubMed] [Google Scholar]
- 99.Pommier Y, Cherfils J. Trends Pharmacol Sci. 2005;28:136–145. doi: 10.1016/j.tips.2005.01.008. [DOI] [PubMed] [Google Scholar]
- 100.Pommier Y, Marchand C. Curr Med Chem Anti-Canc Agents. 2005;5:421–429. doi: 10.2174/1568011054222337. [DOI] [PubMed] [Google Scholar]
- 101.Antony S, Marchand C, Stephen AG, Thibaut L, Agama KK, Fisher RJ, Pommier Y. Nucleic Acids Res. 2007;35:4474–4484. doi: 10.1093/nar/gkm463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Braun MS, Richman SD, Quirke P, Daly C, Adlard JW, Elliott F, Barrett JH, Selby P, Meade AM, Stephens RJ, Parmar MK, Seymour MT. J Clin Oncol. 2008;26:2690–8. doi: 10.1200/JCO.2007.15.5580. [DOI] [PubMed] [Google Scholar]
- 103.Pfister TD, Reinhold WC, Agama K, Gupta S, Khin SA, Kinders R, Parchment RE, Tomaszewski JE, Doroshow JH, Pommier Y. Mol Cancer Ther. 2009 doi: 10.1158/1535-7163.MCT-09-0016. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Holm C, Covey JM, Kerrigan D, Pommier Y. Cancer Res. 1989;49:6365–6368. [PubMed] [Google Scholar]
- 105.Hsiang YH, Lihou MG, Liu LF. Cancer Res. 1989;49:5077–5082. [PubMed] [Google Scholar]
- 106.Wu J, Liu LF. Nucleic Acids Res. 1997;25:4181–4186. doi: 10.1093/nar/25.21.4181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.McClendon AK, Osheroff N. Biochemistry. 2006;45:3040–50. doi: 10.1021/bi051987q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Koster DA, Palle K, Bot ES, Bjornsti MA, Dekker NH. Nature. 2007;448:213–7. doi: 10.1038/nature05938. [DOI] [PubMed] [Google Scholar]
- 109.Zhang H, Barcelo JM, Lee B, Kohlhagen G, Zimonjic DB, Popescu NC, Pommier Y. Proc Natl Acad Sci U S A. 2001;98:10608–13. doi: 10.1073/pnas.191321998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Zhang H, Pommier Y. Biochemistry. 2008;47:11196–203. doi: 10.1021/bi800774b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Croce AC, Bottiroli G, Supino R, Favini E, Zuco V, Zunino F. Biochem Pharmacol. 2004;67:1035–45. doi: 10.1016/j.bcp.2003.10.034. [DOI] [PubMed] [Google Scholar]
- 112.Diaz de la Loza MC, Wellinger RE. Nucleic Acids Res. 2009;37:e26. doi: 10.1093/nar/gkn1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Pouliot JJ, Yao KC, Robertson CA, Nash HA. Science. 1999;286:552–555. doi: 10.1126/science.286.5439.552. [DOI] [PubMed] [Google Scholar]
- 114.Debethune L, Kohlhagen G, Grandas A, Pommier Y. Nucleic Acids Res. 2002;30:1198–204. doi: 10.1093/nar/30.5.1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Interthal H, Pouliot JJ, Champoux JJ. Proc Natl Acad Sci U S A. 2001;98:12009–14. doi: 10.1073/pnas.211429198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Vance JR, Wilson TE. Mol Cell Biol. 2001;21:7191–8. doi: 10.1128/MCB.21.21.7191-7198.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Vance JR, Wilson TE. Proc Natl Acad Sci U S A. 2002;99:13669–74. doi: 10.1073/pnas.202242599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Pouliot JJ, Robertson CA, Nash HA. Genes Cells. 2001;6:677–87. doi: 10.1046/j.1365-2443.2001.00452.x. [DOI] [PubMed] [Google Scholar]
- 119.Liu C, Pouliot JJ, Nash HA. Proc Natl Acad Sci U S A. 2002;99:14970–5. doi: 10.1073/pnas.182557199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Deng C, Brown JA, You D, Brown JM. Genetics. 2005;170:591–600. doi: 10.1534/genetics.104.028795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Dexheimer TS, Antony S, Marchand C, Pommier Y. Anticancer Agents Med Chem. 2008;8:381–9. doi: 10.2174/187152008784220357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Yang SW, Burgin AB, Huizenga BN, Robertson CA, Yao KC, Nash HA. Proc Natl Acad Sci USA. 1996;93:11534–11539. doi: 10.1073/pnas.93.21.11534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Inamdar KV, Pouliot JJ, Zhou T, Lees-Miller SP, Rasouli-Nia A, Povirk LF. J Biol Chem. 2002;277:27162–27168. doi: 10.1074/jbc.M204688200. [DOI] [PubMed] [Google Scholar]
- 124.Zhou T, Lee JW, Tatavarthi H, Lupski JR, Valerie K, Povirk LF. Nucleic Acids Res. 2005;33:289–297. doi: 10.1093/nar/gki170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Hirano R, Interthal H, Huang C, Nakamura T, Deguchi K, Choi K, Bhattacharjee MB, Arimura K, Umehara F, Izumo S, Northrop JL, Salih MA, Inoue K, Armstrong DL, Champoux JJ, Takashima H, Boerkoel CF. EMBO J. 2007;26:4732–43. doi: 10.1038/sj.emboj.7601885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Katyal S, el-Khamisy SF, Russell HR, Li Y, Ju L, Caldecott KW, McKinnon PJ. EMBO J. 2007;26:4720–31. doi: 10.1038/sj.emboj.7601869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Takashima H, Boerkoel CF, John J, Saifi GM, Salih MA, Armstrong D, Mao Y, Quiocho FA, Roa BB, Nakagawa M, Stockton DW, Lupski JR. Nat Genet. 2002;32:267–72. doi: 10.1038/ng987. [DOI] [PubMed] [Google Scholar]
- 128.El-Khamisy SF, Saifi GM, Weinfeld M, Johansson F, Helleday T, Lupski JR, Caldecott KW. Nature. 2005;434:108–13. doi: 10.1038/nature03314. [DOI] [PubMed] [Google Scholar]
- 129.Miao ZH, Agama K, Sordet O, Povirk L, Kohn KW, Pommier Y. DNA Repair (Amst) 2006;5:1489–94. doi: 10.1016/j.dnarep.2006.07.004. [DOI] [PubMed] [Google Scholar]
- 130.Takemura H, Rao VA, Sordet O, Furuta T, Miao ZH, Meng L, Zhang H, Pommier Y. J Biol Chem. 2006;281:30814–23. doi: 10.1074/jbc.M603747200. [DOI] [PubMed] [Google Scholar]
- 131.Giannini G, Rinaldi C, Ristori E, Ambrosini MI, Cerignoli F, Viel A, Bidoli E, Berni S, D’Amati G, Scambia G, Frati L, Screpanti I, Gulino A. Oncogene. 2004;23:2640–7. doi: 10.1038/sj.onc.1207409. [DOI] [PubMed] [Google Scholar]
- 132.Liao Z, Thibaut L, Jobson A, Pommier Y. Mol Pharmacol. 2006;70:366–372. doi: 10.1124/mol.105.021865. [DOI] [PubMed] [Google Scholar]
- 133.Marchand C, Lea WA, Jadhav A, Dexheimer TS, Austin CP, Inglese J, Pommier Y, Simeonov A. Mol Cancer Ther. 2009;8:240–8. doi: 10.1158/1535-7163.MCT-08-0878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Pommier Y, Goldwasser F. In: Cancer Chemotherapy and Biotherapy: Principles and Practice. 4. Chabner BA, Longo DL, editors. Lippincott Williams & Wilkins; Philadelphia: 2006. [Google Scholar]
- 135.Mi Z, Burke TG. Biochemistry. 1994;33:10325–10336. doi: 10.1021/bi00200a013. [DOI] [PubMed] [Google Scholar]
- 136.Lesueur-Ginot L, Demarquay D, Kiss R, Kasprzyk PG, Dassonneville L, Bailly C, Camara J, Lavergne O, Bigg DC. Cancer Res. 1999;59:2939–43. [PubMed] [Google Scholar]
- 137.Lansiaux A, Leonce S, Kraus-Berthier L, Bal-Mahieu C, Mazinghien R, Didier S, David-Cordonnier MH, Hautefaye P, Lavielle G, Bailly C, Hickman JA, Pierre A. Mol Pharmacol. 2007;72:311–9. doi: 10.1124/mol.107.034637. [DOI] [PubMed] [Google Scholar]
- 138.Strumberg D, Pilon AA, Smith M, Hickey R, Malkas L, Pommier Y. Mol Cell Biol. 2000;20:3977–87. doi: 10.1128/mcb.20.11.3977-3987.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Lin CP, Ban Y, Lyu YL, Desai SD, Liu LF. J Biol Chem. 2008;283:21074–83. doi: 10.1074/jbc.M803493200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Sordet O, Larochelle S, Nicolas E, Stevens EV, Zhang C, Shokat KM, Fisher RP, Pommier Y. J Mol Biol. 2008;381:540–9. doi: 10.1016/j.jmb.2008.06.028. [DOI] [PMC free article] [PubMed] [Google Scholar]