

Abstract
Dysregulation of the ubiquitin-proteasome pathway plays an essential role in tumor growth and development. Shikonin, a natural naphthoquinone isolated from the traditional Chinese medicine Zi Cao (gromwell), has been reported to possess tumor cell-killing activity, and results from a clinical study using a shikonin-containing mixture demonstrated its safety and efficacy for the treatment of late-stage lung cancer. In the present study, we reported that shikonin is an inhibitor of tumor proteasome activity in vitro and in vivo. Our computational modeling predicts that the carbonyl carbons C1 and C4 of shikonin potentially interact with the catalytic site of β5 chymotryptic subunit of the proteasome. Indeed, shikonin potently inhibits the chymotrypsin-like activity of purified 20S proteasome (IC50 12.5 μmol/L) and tumor cellular 26S proteasome (IC50 between 2-16 μmol/L). Inhibition of the proteasome by shikonin in murine hepatoma H22, leukemia P388 and human prostate cancer PC-3 cultures resulted in accumulation of ubiquitinated proteins and several proteasome target proapoptotic proteins (IκB-α, Bax and p27), followed by induction of cell death. Shikonin treatment resulted in tumor growth inhibition in both H22 allografts and PC-3 xenografts, associated with suppression of the proteasomal activity and induction of cell death in vivo. Finally, shikonin treatment significantly prolonged the survival period of mice bearing P388 leukemia. Our results indicate that the tumor proteasome is one of the cellular targets of shikonin, and inhibition of the proteasome activity by shikonin contributes to its anti-tumor property.
Keywords: Shikonin, medicinal compounds, proteasome inhibitor, hepatoma, prostate cancer
Shikonin is an active naphthoquinone that is mainly isolated from Chinese medicine Zi Cao (gromwell), the dried root of Lithospermum erythrorhizon Sieb. et Zucc, Arnebia euchroma (Royle) Johnst, or Arnebia guttata Bunge.1 Dating back to 5th century, Zi Cao had been used for the treatment of throat sore, burns, cut and skin diseases such as macular eruption, measles and carbuncles in China.2
Shikonin exerted anti-inflammatory effect by inhibiting tumor necrosis factor α (TNF-α) that is important for initiation and persistence of inflammation.3,4 Shikonin inhibited transcription factor IID binding to TNF-α promoter3 and blocked TNF-α pre-mRNA splicing,4 resulting in TNF-α suppression. Shikonin also possessed anti-bacteria, anti-fungi, and anti-HIV properties.1,5,6
Sankawa et al. reported that shikonin inhibited the tumor growth in murine Sarcoma-180.7 Subsequent in vitro studies revealed that shikonin induced apoptotic cell death in a variety of cancers, which involved multiple cellular targets.8-16 Shikonin induced apoptosis by activation of caspase-3 in leukemia, bladder and cervical cancer cells.8-10 In caspase-3-negative MCF-7 cells, it triggered necroptosis that contributed to overcome Bcl-2- and Bcl-XL-mediated apoptotic resistance.11 Shikonin inhibited the activity of topoisomerase II and NF-κB, both of which are potential targets for chemotherapy.12,13 Treatment with shikonin led to cell cycle arrest through up-regulation of p53 and down-regulation of cyclin-dependent protein kinase 4 in malignant melanoma.14 Shikonin reacted with endogenous thiols including glutathione, which in turn induced apoptosis in HL-60 cells.15 Shikonin-induced apoptosis could be protected by N-acetylcysteine (NAC) in SK-Hep-1 hepatoma,16 suggesting that it targeted an oxidative stress-mediated pathway.
A clinical trial using shikonin in 19 cases of late-stage lung cancer revealed that use of a shikonin-containing mixture reduced lung cancer growth with the effective rate of 63.3%, remission rate of 36.9%, and 1-year survival rate of 47.3%.17 Furthermore, administration of the shikonin-containing mixture increased body weight and appetite of the patients. No harmful effects on peripheral system, heart, kidney and liver were observed after shikonin treatment.17
Although several cellular proteins or activities could be affected by shikonin treatment as stated above, its specific molecular target has not been discovered. The chemical structure of shikonin suggested to us that it might target the proteasome. Shikonin contains at least two carbonyl carbons which are electrophilic in nature since the oxygen pulls electron density away from the carbon. The eukaryotic proteasome (26S proteasome) is a multicatalytic protease complex with a 20S preteolytic core and 19S regulatory caps.18-20 The proteasomal subunits β5, β2 and β1 in 20S catalytic core are responsible for three main proteolytic activities of the proteasome, chymotrypsin (CT)-like, trypsin-like, and peptidyl-glutamyl peptide-hydrolyzing (PGPH)-like or caspase-like activities, respectively. A threonine residue at the N terminus (Thr 1) of these β subunits imparts the catalytic activity of the proteasome.21 The atom Oγ of Thr 1 (Thr 1 Oγ) is activated to be nucleophilic by proton shuttling from Thr 1 Oγ to the proton acceptor Thr 1 N.22 Compounds with electrophilic functional groups are able to react with the nucleophilic Thr 1 Oγ,22 causing interference of the proteasomal activity.
Consistently, in a computational modeling study, shikonin was docked to the proteasomal β5 subunit in an orientation and conformation that was suitable for nucleophilic attack by Thr 1 of the β5 subunit. Shikonin directly inhibited the chymotrypsin-like activity of purified 20S proteasome in vitro. Since our laboratories have established animal models using murine hepatoma H22, leukemia P388 or human PC-3 prostate cancer23, these cell lines were selected to test whether shikonin could inhibit 26S proteasome in both cultured tumor cells and tumor tissues and, if yes, whether the proteasome inhibition was associated with cell death induction and tumor growth inhibition.
Materials and Methods
Materials
Purified shikonin (>98%) was purchased from National Institute for the Control Pharmaceutical and Biological Products (Beijing, China) and BIOMOL International LP (Plymouth Meeting, PA) for the studies in murine hepatoma H22 and human prostate cancer PC-3 cell line, respectively. Shikonin was dissolved in DMSO (Sigma; St. Louis, MO) at a stock concentration of 50 mM, aliquoted and stored at -80 °C. Cremophor EL, ethanol and propidium iodide (PI) were purchased from Sigma (St. Louis, MO). Fetal bovine serum (FBS) was from Tissue Culture Biologicals (Tulare, CA). RPMI 1640, penicillin and streptomycin were purchased from Invitrogen Co. (Carlsbad, CA). Purified rabbit 20S proteasome and fluorogenic peptide substrates Suc-LLVY-AMC (for the proteasomal chymotrypsin-like activity) were from Calbiochem Inc. (Gibbstown, NJ). Mouse monoclonal antibody against human poly(ADP-ribose) polymerase (PARP) was purchased from BIOMOL International LP (Plymouth Meeting, PA). Mouse monoclonal antibodies against Bax (B-9), p27 (F-8), Ubiquitin (P4D1) and rabbit polyclonal antibody against inhibitor of nuclear factor κB-α (IκB-α) (C-15), GAPDH (FL-335) and goat polyclonal antibody against actin (C-11) were from Santa Cruz Biotechnology Inc. (Santa Cruz, CA). Rabbit polyclonal antibody Hsp70 (SPA-812) was purchased from Stressgen Bioreagents (Ann Arbor, MI). Mouse monoclonal antibody NCL-p27 was purchased from Novocastra Laboratories Ltd (Benton Lane, Newcasle upon Tyne, UK). Enhanced Chemiluminescence Reagent was from Amersham Biosciences (Piscataway, NJ). Apoptag Peroxidase In Situ Apoptosis Detection Kit was from Chemicon International, Inc. (Temecula, CA). The terminal deoxynucleotidyl transferase-mediated dUTP nick end-labeling (TUNEL) in situ apoptosis detection kit was from Roche (Mannheim, Germany). Annexin V-FITC Apoptosis Detection Kit was from Bipec Biopharma Corporation (Cambridge, MA).
Nucleophilic susceptibility analysis
Analysis of electron density surface colored by nucleophilic susceptibility was generated using Quantum CAChe (Fujitsu; Fairfield, NJ). Highly susceptible atoms for nucleophilic attack were showed by two-colored “bull's-eyes”.
Computational modeling
The crystal structure of the eukaryotic yeast 20S proteasome used for all the docking studies was obtained from the Protein Database.24,25 The yeast 20S proteasome is structurally very similar to the mammalian 20S proteasome, and the chymotrypsin proteolytic site between the two species is highly conserved. The AutoDock 3.0 suite of programs and docking parameters were set up as described.24,25 The Autodock software was run on an i386 architecture computer operating with Redhat Linux 6.0 ™ operating system. The selected dockings for shikonin were the two clusters with more members and lower binding free energies. Structural output from Autodock was visualized using PyMOL software.
Inhibition of purified 20S proteasome activity by shikonin
Purified rabbit 20S proteasome (35 ng) was incubated with 40 μmol/L of fluorogenic peptide substrate Suc-LLVY-AMC (for the proteasomal chymotrypsin-like activities) in 100 μl assay buffer (20 mM Tris-HCl, pH 7.5) in the presence of shikonin at different concentrations or the solvent DMSO for 2 h at 37°C, followed by measurement of hydrolysis of the fluorogenic substrates using a Wallac Victor3™ multilabel counter with 355-nm excitation and 460-nm emission wavelengths.
Cell cultures
Murine hepatoma H22, human prostate cancer PC-3 and murine leukemia P388 cells were purchased from American Type Culture Collection (Manassas, VA) and grown in RPMI 1640 supplemented with 10% FBS, 100 units/ml of penicillin, and 100 μg/ml of streptomycin or cultured as recommended by the supplier. Cell cultures were maintained at 37 °C and 5% CO2.
Whole cell extract preparation and Western blotting analysis
A whole cell extract was prepared as described previously.26 Western blotting assay using Enhanced Chemiluminescence Reagent was performed as described previously.27
Inhibition of the cellular proteasome activity by shikonin
Prostate cancer PC-3 or murine leukemia P388 cells were treated with different concentrations of shikonin for indicated times. The prepared whole cell extracts (7.5 μg) were incubated with the substrate Suc-LLVY-AMC for 2 h, followed by fluorescent measurement as described above.
Annexin V-FITC binding assay
H22 or P388 cells were treated with shikonin as indicated. Harvested cells were washed with cold PBS and re-suspended with 1×binding buffer, followed by Annexin V- FITC incubation for 15 min and PI staining for another 15 min at 4 °C in dark. The apoptosis rates were detected by flow cytometry.
Flow cytometry analysis by propidium iodide (PI)
H22 cells were treated with 4 μmol/L shikonin for different hours, followed by fixation with 70% ethanol and staining with PI. Cell viability was quantified as previously described.28
Establishment and treatment of H22 allografts
Male KMF mice aged 5 weeks were purchased from Guangdong Animal Center and housed in accordance with protocols approved by the Guangdong Animal Center. Murine hepatoma H22 cells (10×106) suspended in 0.2 mL of serum-free RPMI 1640 were inoculated s.c. in the left armpit of each mouse. After 24 h of inoculation, mice were randomly divided into three groups (10 mice per group) and treated with either vehicle (10% DMSO, 15% ethanol, and 75% PBS) or shikonin (4.0 or 8.0 mg/kg) for consecutive 7 days. Two days later, the mice were sacrificed, and the tumor tissues were weighed and stored at -70 °C.
Establishment and treatment of PC-3 xenografts
Male athymic nude mice aged 5 weeks were purchased from Taconic Research Animal Services (Hudson, NY) and housed in accordance with protocols approved by the Institutional Laboratory Animal Care and Use Committee of Wayne State University. PC-3 xenografts were generated by s.c. inoculation as described before23 and treated with either vehicle [10% DMSO, 20% Cremophor:ethanol (3:1) and 70% PBS] or shikonin (5 mg/kg) twice a week. Tumors were measured by caliper and tumor volume was calculated using standard formula: Width2×Length/2. When the tumor reached to 1200 mm3 in the vehicle group, the experiment was terminated and tumor tissues were removed for biological analysis.
Proteasome inhibition and apoptosis assays using tumor tissue samples
Western blotting using animal tumor tissue samples was performed similarly as described above using cultured cancer cells. TUNEL assay and p27 immunostaining in tumor tissues were performed according to manufactory protocols.
Effect of shikonin on cumulative survival in KMF mice bearing mouse leukemia P388 cells
Male KMF aged 5 weeks were i.p. inoculated with murine leukemia P388 cells (1×107) suspended in 0.2 mL of serum-free RPMI 1640. After 24 h, mice were randomly divided into two groups (10 mice per group) and treated with i.p. bolus injections of either the drug vehicle (10% DMSO, 15% ethanol, and 75% PBS) or shikonin (4 mg/kg) for 7 consecutive days. The mice were then kept for additional 60 days and mouse survival was monitored every day.
Statistical analysis
The difference with respect to tumor growth inhibition multiple groups was analyzed by ANOVA, followed by Dunnett Test. P<0.01 was considered as significant difference.
Results
Computational modeling studies predict that shikonin interacts with the proteasomal β5 chymotryptic subunit and inhibits the chymotrypsin-like activity
Toward the goal of searching for a putative specific target of shikonin in tumor cells, we analyzed the chemical structure of shikonin and noticed that it contained two carbonyl carbons, C1 and C4 (Fig 1A). Some compounds containing eletrophilic carbonyl carbons were susceptible towards attack by nucleophilic catalytic site of the proteasome.29 To investigate whether shikonin was susceptible to be attacked by the proteasome, we performed computational electron density analysis for shikonin molecule. The result suggests that carbonyl carbons C1 and C4 of shikonin molecule have high susceptibility, as denoted by “bull's eyes” with red center (Fig. 1B), towards a nucleophilic attack. Results from computational docking indicated that shikonin could bind to the β5 chymotrypsin site in an orientation and conformation that was suitable for a nucleophilic attack by the OH group of Thr 1 of β5 subunit (Fig. 1C). There were two major docking modes out of total seven obtained. One mode with the lowest docked free energy (-10.12 kcal/mol) was repeated for 29 out of 100 runs (29% probability), showing that the distance from the electrophilic C4 of shikonin to the OH of β5 Thr 1 was 2.64 Å (Fig. 1C, left). Another docking mode (28% probability; -9.28 kcal/mol) showed a distance of 2.69 Å from C1 to the OH of β5 Thr 1 (Fig. 1C, right). Since nucleophilic attack could occur within 4 Å,23 the docking data suggested that both C1 and C4 of shikonin could interact with the proteasomal β5, causing an inhibition of the chymotrypsin-like activity.
Figure 1. Computational modeling and cell-free proteasome-inhibitory activity of shikonin.
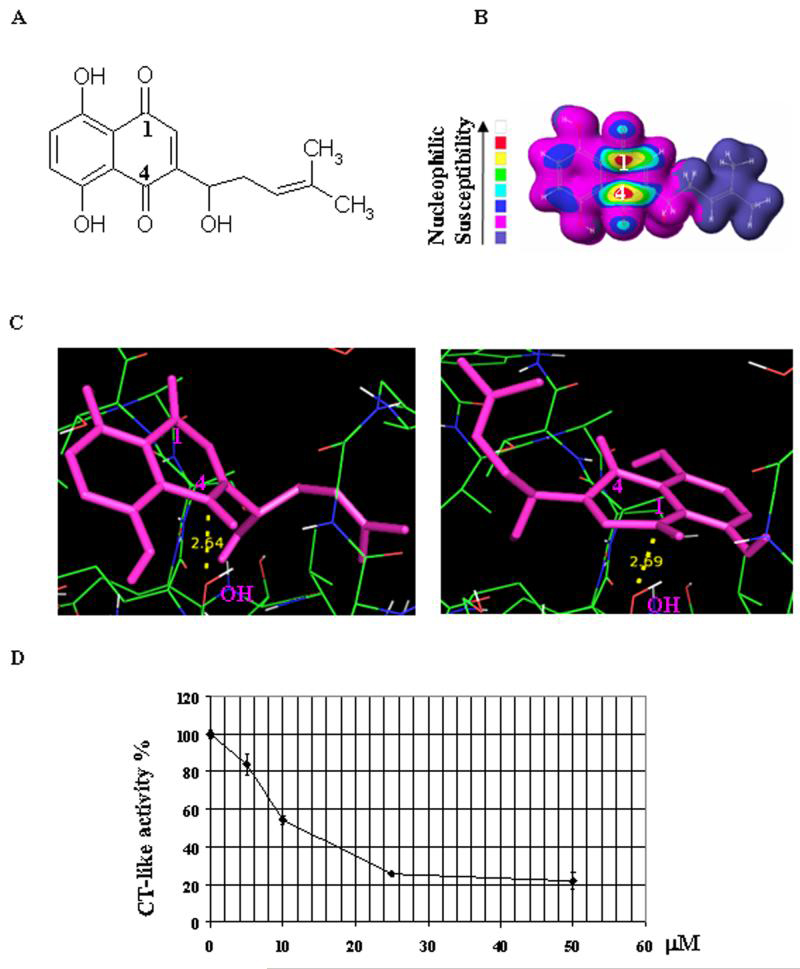
A, The chemical structure of shikonin was shown. B, Nucleophilic susceptibility of shikonin analyzed using CAChe software. Higher susceptibility was shown at the C1 and C4 positions of shikonin. C, Computational modeling of shikonin interacting with ²5 subunit of proteasome. Shikonin was shown in pink while the hydroxyl group (OH) of the N-terminal threonine (Thr 1) of ²5 subunit was shown in red and white. The distance of C4 to the OH of Thr 1 was 2.64 Å in one cluster with 29% possibility (C, left) while the distance of C1 to the OH of Thr 1 was 2.69 Å in another cluster with 28% possibility (C, right). D, Shikonin inhibits the chymotrypsin (CT)-like activity of a purified 20S proteasome. To verify computational modeling results, inhibition of CT-like activities of a purified 20S rabbit proteasome (35 ng) by shikonin was tested. The calculated IC50 value of shikonin was 12.5 μmol/L.
Shikonin inhibits the chymotrypsin-like activity of a purified rabbit 20S proteasome
To provide direct evidence for that shikonin inhibits the proteasomal chymotrypsin-like activity, as predicted by the computational model, we performed a cell-free proteasome activity assay. Purified rabbit 20S proteasome was incubated with up to 50 μmol/L shikonin for 2 h in the presence of a specific substrate for proteasomal chymotrypsin-like activity. As shown in Figure 1D, the chymotrysin-like activity of the purified 20S proteasome was significantly inhibited by shikonin with an IC50 value of 12.5 μmol/L. Shikonin at 25-50 μmol/L inhibited ~80% of chymotrypsin-like activity of the purified 20S proteasome (Fig. 1D).
Shikonin causes proteasome inhibition and cell death in human PC-3 prostate cancer and murine hepatoma H22 cell lines
To determine whether shikonin can also inhibit the cellular 26S proteasome activity, human androgen-independent PC-3 prostate cancer cells were treated with shikonin at 5, 15, 25 or 50 μmol/L for 6 h, followed by the measurement of proteasome activity in the cell lysates prepared. The proteasomal chymotrypsin-like activity was inhibited by shikonin in a dose-dependent manner (Fig. 2A): Shikonin caused 20, 45, 70 or 90% inhibition at 5, 15, 25 or 50 μmol/L, respectively, and the IC50 value calculated from this experiment was 16.5 μmol/L, comparable to that obtained by using the purified 20S proteasome (IC50 = 12.5 μmol/L; Fig. 1D). Accompanying with proteasome inhibition by shikonin, ubiquitinated proteins, which were tagged by polyubiquitins for the proteasome degradation29, were accumulated in a dose-dependent manner: slight accumulation by 5 μmol/L shikonin treatment and further accumulation by shikonin at 15-50 μmol/L (Fig. 2B). We also measured the levels of natural proteasome target proteins, such as IκB-α, Bax and p27. An ubiquitinated form of IκB-α30 was found to be accumulated by treatment of shikonin at 15 to 50 μmol/L (indicated by an arrow, Fig. 2B). Expression of Bax protein was also increased by 25-50 μmol/L shikonin, while p27 protein level was slightly increased by 15 μmol/L shikonin and further increased by 25-50 μmol/L shikonin (Fig. 2B).
Figure 2. Shikonin dosage effects on proteasome inhibition and cell death induction in PC-3 and H22 cells.
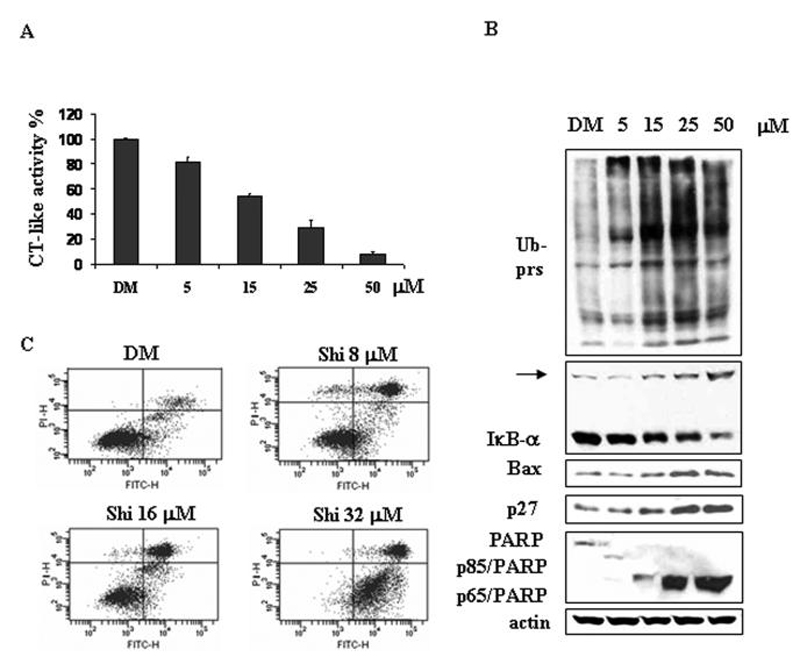
A-B, Human prostate cancer PC-3 cells were treated with either solvent DMSO (DM) or indicated concentrations of shikonin for 6 h, followed by measuring the inhibition of the protesomal CT-like activity using fluorescent substrate Suc-LLVY-AMC (A) and Western blotting analysis using specific antibodies against ubiquitinated proteins, IκB-α Bax, p27 and PARP (B). Columns, means of independent triplicate experiments; bars, SD. Molecular weight of IκB-α Bax and p27 is 37, 23 and 27 kDa, respectively. An ubiqutinated form of IºB-± (~56 kDa)30 was indicated by an arrow. Full length PARP is 116 kDa, the cleaved fragments of PARP are 85 kDa or 65 kDa. Actin was used as loading control. C, Murine hepatoma H22 cells were treated with either DMSO (DM) or 8, 16 or 32 μM shikonin for 24 h, followed by Annexin V-FITC binding assay. The lowerright part (Annexin V-FITC + / PI -) was considered as early stage of apoptotic cells and upright part (Annexin V-FITC + / PI +) was considered as late stage of apoptotic cells. The lowerleft part (Annexin-FITC -/PI -) was considered as viable cells and the upleft part (Annexin V-FITC - / PI +) was considered as necrotic cells.
Inhibition of the proteasomal chymotrypsin-like activity was reported to be associated with induction of tumor cell death.20 We then measured cell death by PARP cleavage and morphological changes in the same samples of PC-3 cells. The cleaved PARP fragment p85/PARP was detected after treatment of 5 μmol/L shikonin for 6 h, while p65/PARP fragment was generated by shikonin treatment at 15 to 50 μmol/L (Fig. 2B).
Similarly, treatment of murine hepatoma H22 cells with shikonin inhibited the proteasome in a dose-dependent manner (data not shown). In the same experiment, shikonin was also able to induce apoptotic cell death. As shown in Figure 2C, compared to the vehicle DMSO treatment, shikonin at 8-16 μmol/L induced apoptotic cell death in 51-56% of H22 cells, and it at 32 μmol/L induced 96% of H22 cells undergoing apoptotic cell death (Fig. 2C). Therefore, shikonin is able to inhibit the cellular proteasome and induce cell death in both PC-3 and H22 cancer cells.
Shikonin-induced proteasome inhibition occurs prior to tumor cell death
Next we performed kinetic experiments using both H22 and PC-3 cell lines to determine which event occurs first, proteasome inhibition or cell death induction. H22 cells were treated with 4 μmol/L shikonin for up to 48 h, followed by Western blotting and flow cytometry analysis. As shown in Fig. 3A, the levels of ubiquitinated proteins started to increase after 2 h treatment with shikonin, which was further increased afterwards (Fig. 3A). The level of IκB-α protein also started to increase after 2 h treatment and further increased in a similar kinetics as that of ubiquitinated proteins (Fig. 3A). Bax protein expression was increased slightly after ~8 h and significantly after 24 and 48 h treatment (Fig. 3A). Levels of p27 protein were found to increase apparently after 24 to 48 h treatment of shikonin (Fig. 3A). Hsp70, a major stress-inducible heat shock protein, was increased after 4 h treatment with shikonin and further increased afterwards in a time-dependent manner (Fig. 3A). In a sharp contrast to the proteasome inhibition at early hours, cell death occurred in later hours. PARP cleavage, an indicator of apoptotic cell death31 was detected only at 24-48 h treatment of shikonin (Fig. 3A). We also performed flow cytometry to measure the sub-G1 population, another cell death marker, in the cells after shikonin treatment. Compared to untreated control, sub-G1 population was increased by only 2-fold after 12 h and further increased by 4- to 5-fold after 24-48 h treatment (Fig. 3B).
Figure 3. Kinetic effect on proteasome inhibition and apoptosis induction by shikonin in H22 hepatoma cells.
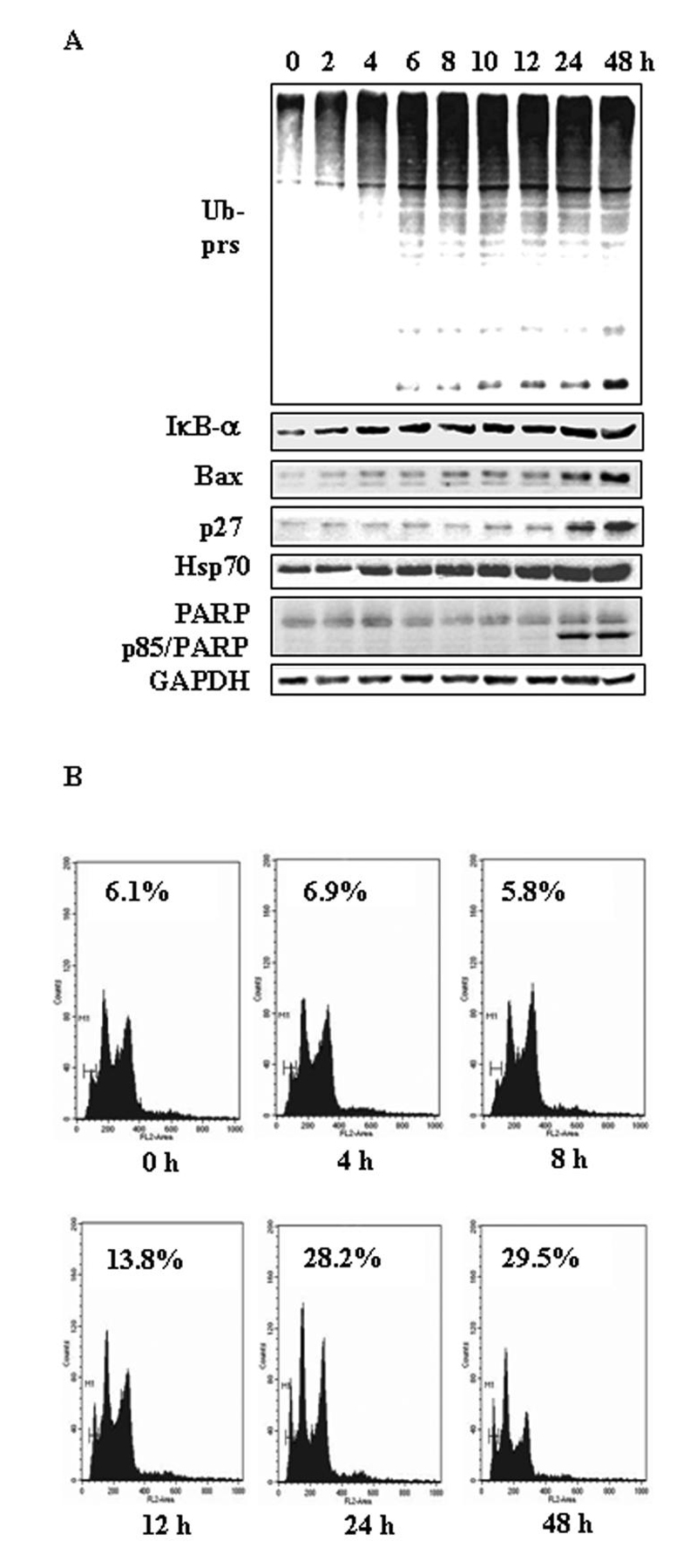
H22 cells were treated with 4 ¼mol/L of shikonin for indicated hours, followed by Western blotting analysis using specific antibodies against ubiquitinated proteins (Ub-prs), IκB-α Bax, p27, Hsp70, PARP, GAPDH (loading control, A) and flow cytometry analysis (B). Sub-G1 cell population, as indicated (%), was considered as apoptotic cells.
Then we treated PC-3 cells with 10 μmol/L shikonin for up to 24 h. The results showed that the proteasomal chymotrypsin-like activity in PC-3 cells was inhibited around 50% at as early as 0.5 h after addition of shikonin, which was lasted to 8 h and then further increased to 70% inhibition at 24 h (Fig. 4A). Consistently, accumulation of ubiquitinated proteins was detected from 1 h to 24 h, peaked at 8 h during the treatment of shikonin (Fig. 4B). Levels of Bax protein and the ubiquitinated form of IκB-α were slightly increased after 0.5 h treatment and greatly increased at 8 or 24 h, respectively (Fig. 4B). Expression of p27 protein was increased from 8 h to 24 h of shikonin treatment (Fig. 4B). In the same kinetic experiment, a band of PARP/p65 cleaved fragment was detected at 8 h (Fig. 4B), which became very abundant after 24 h treatment (Fig. 4B). Cell death was also observed under microscope after 8 h treatment (data not shown), consistent with the changes on molecular levels (Fig. 4B). Taken together, these results indicated that the proteasome inhibition by shikonin was followed by induction of cell death in both cancer cell lines, suggesting that the proteasome is a cellular target of shikonin for cell death.
Figure 4. Kinetic effect on proteasome inhibition and apoptosis induction by shikonin in PC-3 prostate cancer cells.
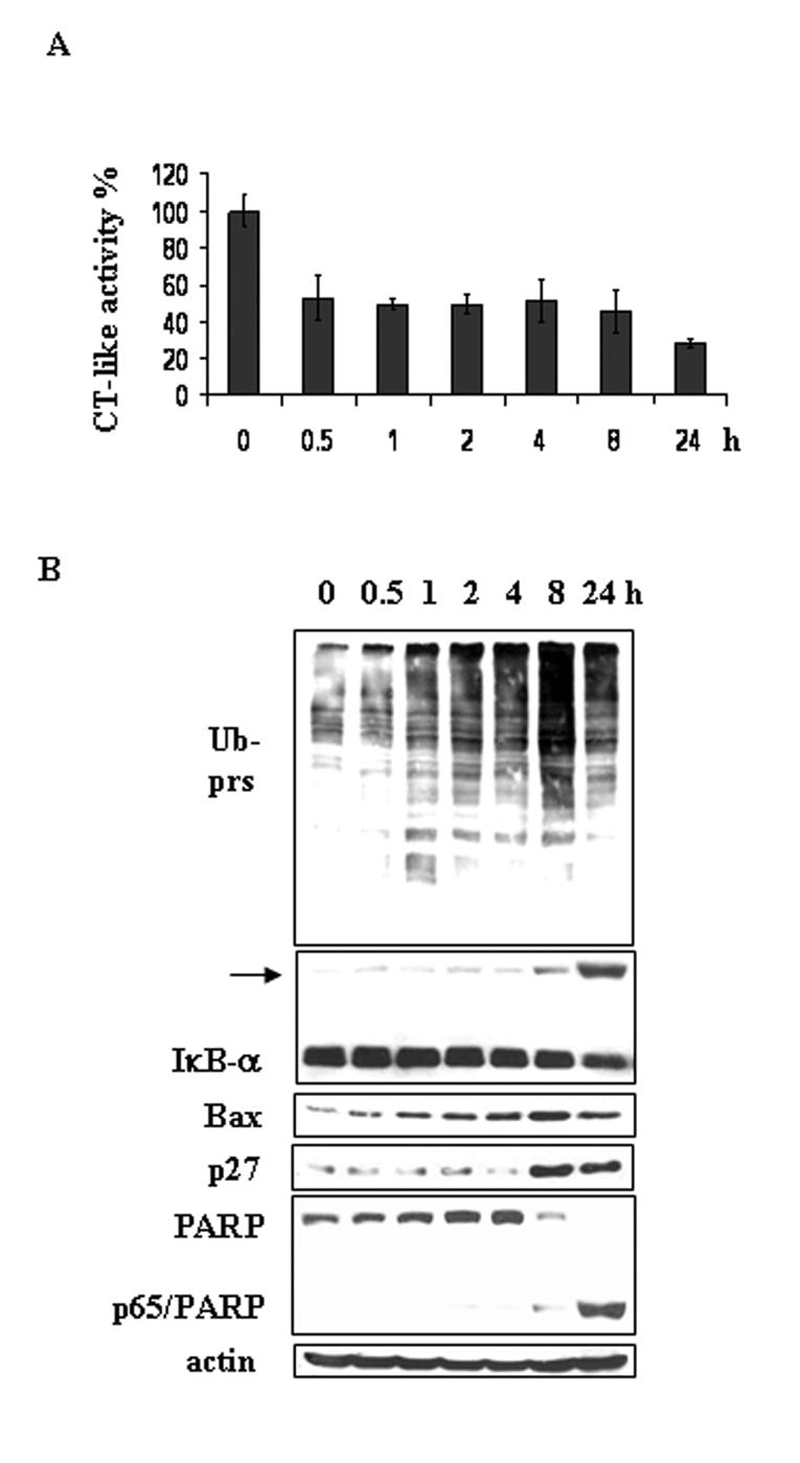
PC-3 cells were treated with 10 μmol/L of shikonin for different hours, followed by measurement of the protesomal CT-like activity using fluorescent substrate Suc-LLVY-AMC (A) and Western blotting analysis (B). Columns, means of independent triplicate experiments; bars, SD.
Cytotoxicity of shikonin to murine leukemia P388 cells
We then determined the cytotoxicity of shikonin in cultured murine leukemia P388 cells. Shikonin-induced dose-dependent inhibitory effects on the chymotryptic activity of the proteasome were observed in P388 cells, showing 17, 32, 56 and 75% inhibition at 1, 2, 4 and 6 μmol/L, respectively (Fig. 5A). Ubiquitinated proteins were also accumulated by shikonin treatment at 1-6 μmol/L (Fig. 5B). Accompanying with proteasome inhibition, cell death was induced by shikonin in a dose-dependent manner, as evident by appearance of cleaved PARP fragment (Fig. 5B). In the same experiment, Shikonin-induced cell death was quantified with Annexin-PI binding assay: 19.4, 40.4. 82.1 and 90.6% cell death detected in the cells treated with shikonin at 1, 2, 4 and 6 μmol/L, respectively (Fig. 5C), demonstrating dose-dependency.
Figure 5. Dosage effect on proteasome inhibition and cell death induction by shikonin in P388 leukemia cells.
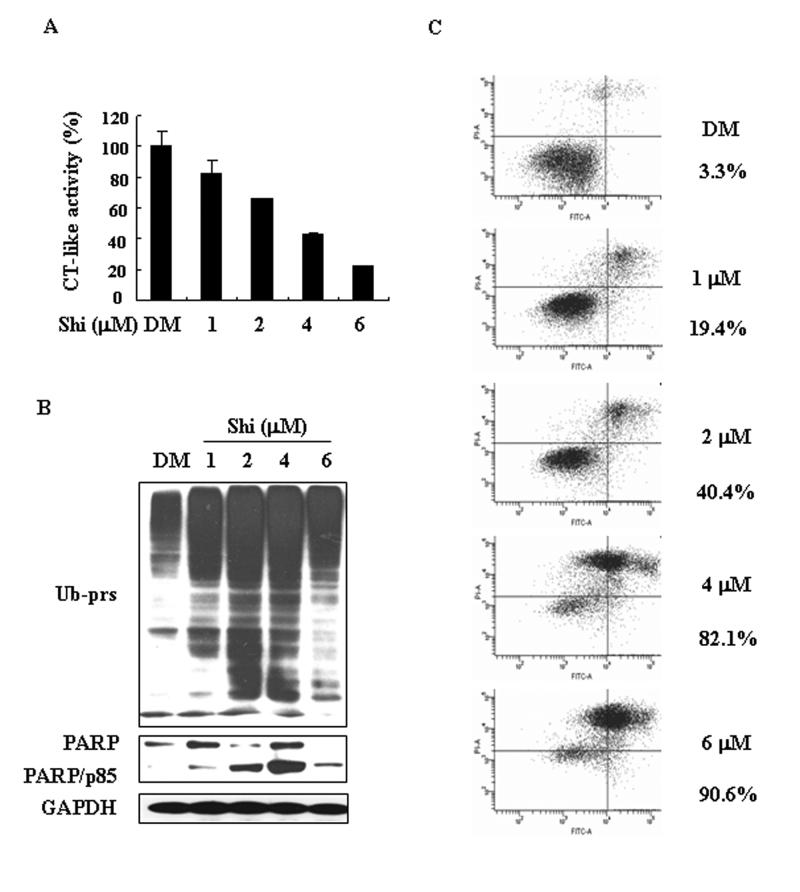
P388 cells were treated with various concentrations of shikonin (Shi) for 6 h, followed by the proteasome CT-like acitivity assay (A), Western blotting analysis using specific antibodies against ubiquitinated proteins (Ub-prs), PARP, GAPDH (loading control, B) and annexin-PI binding assay (C). Columns, means of independent triplicate experiments; bars, SD. Cell death rate (%) was shown.
In a kinetic experiment, shikonin-induced proteasome inhibition was followed by induction of cell death in P388 cells (Fig. 6), similar to what observed in H22 and PC-3 cell lines (Figures 3-4). After only 0.5 h treatment with 2 μmol/L shikonin, about 10% proteasome activity was already inhibited (Fig. 6A). This inhibition was further increased to 50% after 8-12 h treatment. Consistently, ubiquitinated proteins were also accumulated as early as 0.5 h treatment and further accumulated in a time-dependent manner (Fig. 6B). Following proteasome inhibition, cell death was detected at 1 or 2 h after addition of shikonin. A low level of cleaved PARP fragment was detected at 1 h which reached its peak at 8 h after shikonin treatment (Fig. 6B). Compared to control, cell death was not detected before 2 h treatment (Fig. 6C), which was increased by 3-fold at 2 h and by 7-8-fold after 8-12 h treatment (Fig. 6C). These data confirm that proteasome inhibition occurs prior to cell death induction by shikonin.
Figure 6. Kinetic effect on proteasome inhibition and cell death induction by shikonin in P388 leukimia cells.
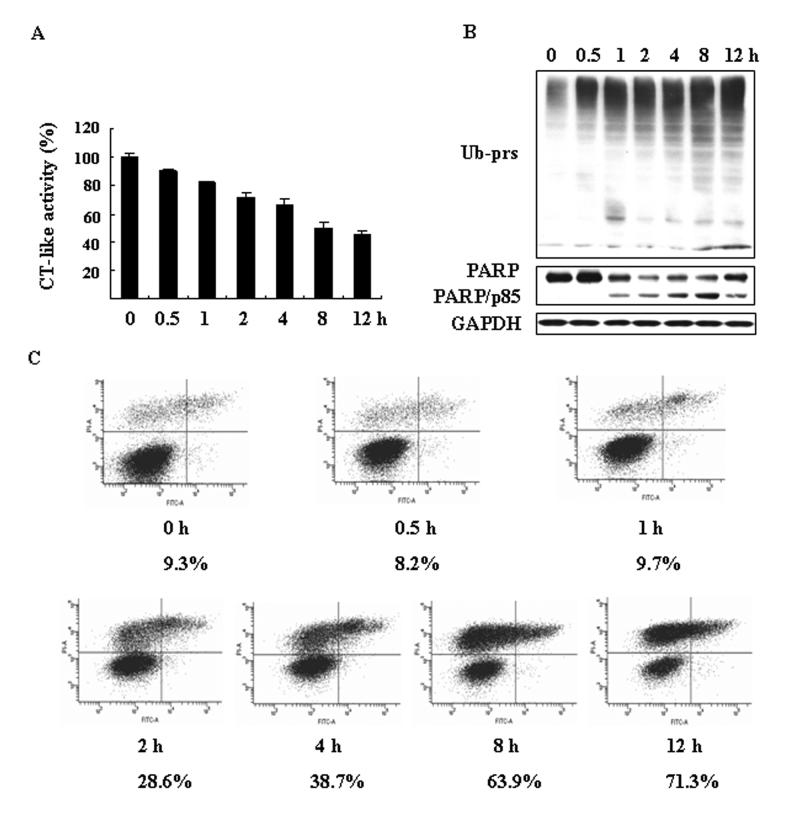
P388 cells were treated with 2 μmol/L of shikonin for different hours, followed by measurement of the protesomal CT-like activity (A) and Western blotting analysis using specific antibodies against ubiquitinated proteins (Ub-prs), PARP, GAPDH (loading control, B) and annexin-PI binding assay (C). Columns, means of independent triplicate experiments; bars, SD. Cell death rate (%) was shown.
Shikonin treatment significantly increased the survival rate of tumor-bearing mice
To study the in vivo effect of shikonin, we first determined effect of shikonin on survival of mice bearing tumors. Male KMF mice were inoculated by i.p. injection with P388 cells. After 24 h, mice were randomly divided into two groups (10 mice per group) and started i.p. bolus injections with either drug vehicle or 4 mg/kg/day shikonin for 7 consecutive days. After that, the effect of shikonin on mice survival was monitored daily for next 60 days (Fig. 7A). We found that all the mice in control group died within 23 days. In a sharp contrast, only one mouse in shikonin-treated group died on day 28 and all the others survived to the end of the experiment (for 60 days) (Fig. 7A). These data demonstrate that shikonin treatment greatly prolonged the survival period in leukemic mice.
Figure 7. Shikonin prolongs survival of mice beaing tumors and suppresses tumor growth.
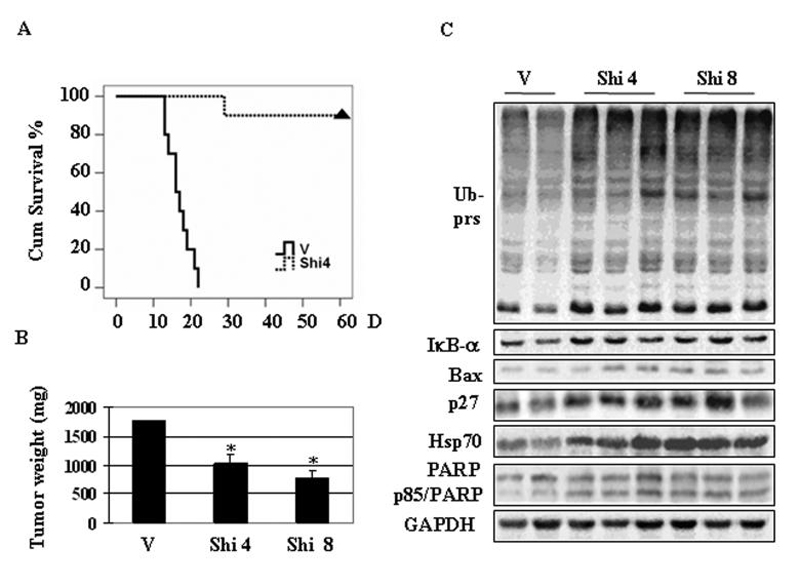
A, Shikonin effect on cumulative survival in KMF mice. Male KMF aged 5 weeks were i.p. inoculated with murine leukemia P388 cells. After 24 h, mice were randomly divided into two groups (10 mice per group) and treated with i.p. bolus injections of either drug vehicle (V) or 4 mg/kg/day shikonin (Shi 4) for 7 consecutive days. The mice were then kept for additional 60 days to determine the effect on mice survival of shikonin. Triangle indicates the end of the experiment. B, C, H22 allografts were generated in male KMF mice as described in Materials and Methods and treated with either vehicle (V) or shikonin at 4.0 mg/kg (Shi 4) or 8.0 mg/kg (Shi 8) (10 mice per group) for 7 consecutive days. Two days after that, the mice were sacrificed, and the tumor tissues were weighed (B), and tumor tissue samples were analyzed by either Western blotting using antibodies against proteasome target proteins and PARP (C). Bars, SD; *, P<0.01.
The antitumor property of shikonin was associated with its proteasome-inhibitory activity in vivo
Next we determined whether shikonin targets the tumor proteasome in vivo, as observed in cultured cancer cells. Male KMF mice were inoculated s.c. in the left armpit with H22 cells (1 × 107). After 24 h inoculation, mice were randomly divided into three groups (10 mice per group), which were i.p. daily treated with either the drug vehicle or shikonin at 4.0 mg/kg or 8.0 mg/kg for 7 consecutive days. Two days after that, all the mice were sacrificed, and the tumors were weighed. As shown in Fig. 7B, tumor weight in the vehicle-treated group reached to ~1800 mg in average. In comparison, tumor weight from groups treated with 4 mg/kg or 8 mg/kg shikonin was 1000 and 800 mg, respectively (Fig. 7B), demonstrating 45% and 56% inhibition. Therefore, shikonin at both doses could significantly inhibit the growth of H22 allografts (P<0.01).
Whether the proteasome was inhibited by shikonin in H22 tumors was then examined by Western blotting analysis using whole tissue extracts and immunohistochemistry. Compared with vehicle control, shikonin treatment caused accumulation of ubiqutinated proteins, as well as IκB-α, Bax, p27 and Hsp 70 proteins (Fig. 7C). In situ proteasome inhibition and increased p27 protein level in H22 tumors treated with shikonin was confirmed by tissue immunostaining. Increased expression of p27 protein was observed in groups treated with shikonin at 4 or 8 mg/kg compared to vehicle control (Fig. 8A). All these results suggested that shikonin could reach the proteasome and inhibit its activity in H22 tumor tissues. In the same H22 tumors treated with shikonin, apoptotic cell death was also induced, as evident by generation of p85/PARP cleavage fragment (Fig. 7C) and TUNEL positivity (Fig. 8A).
Figure 8. Immunostaining of p27 and TUNEL assay.
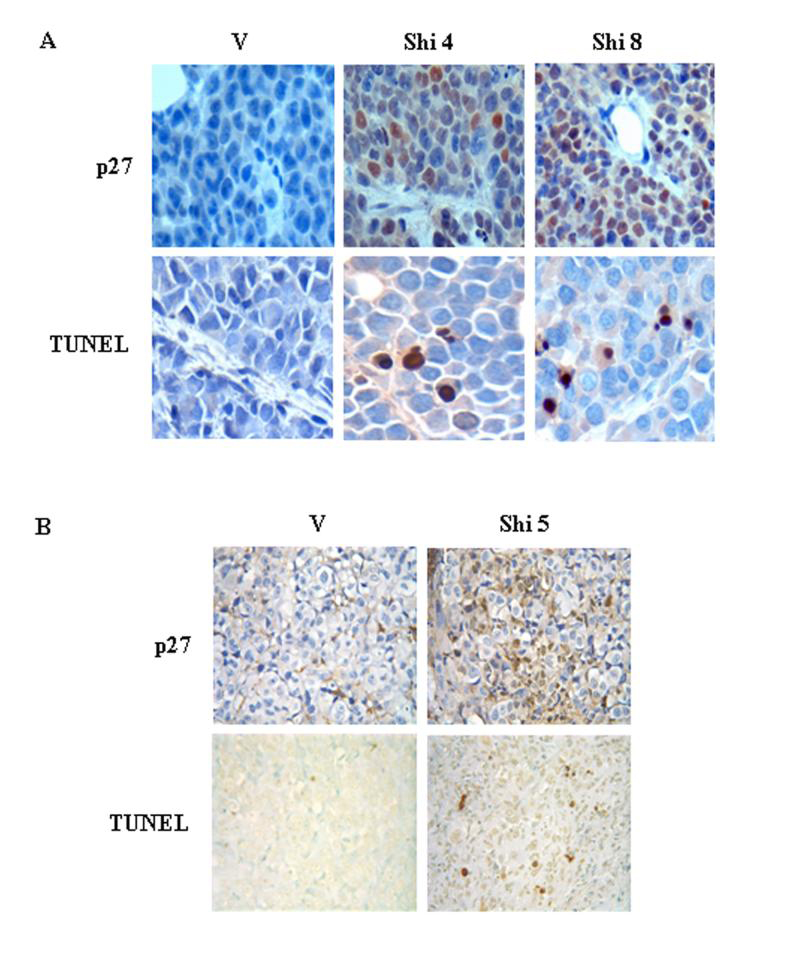
A, H22 tumors were generated and trerated with vehicle (V) or 4 mg/kg shikonin (Shi 4) or 8 mg/kg shikonin (Shi 8) as described in Figure 5. Tumor tissues were sectioned for immuostaining of p27 and TUNEL assay (A). B, PC-3 xenografts were generated in male athymic nude mice as described in Materials and Methods and treated with either vehicle (V) or 5 mg/kg shikonin (Shi 5) twice a week. By the end of the experiment, tumor tissues were removed for immunostaning of p27 and TUNEL assay. Pictures were taken under 400 magnitudes.
To confirm the H22 in vivo results, human prostate cancer PC-3 xenografts were also generated s.c. in male nude mice. Compared to the vehicle-treated group, shikonin at 5 mg/kg/day treatment induced ~20% growth inhibition in nude mice bearing PC-3 tumors, which is correlated with 30% inhibition of the chymotrypsin-like activity in the tumors (data not shown). Immunostaining of p27 and TUNEL assay were also performed in the tumor tissues from the same PC-3 xenografts. Both p27 expression and TUNEL-positive cell population were increased in shikonin treated PC-3 tumors compared to vehicle control (Fig. 8B). From these in vivo studies, we conclude that the antitumor property of shikonin is associated with its proteasome-inhibitory and cell death-inducing activities.
Discussion
Shikonin is the main component of Chinese herbal medicine Zi Cao (gromwell)1 that has antitumor activity.7 Although several in vitro molecular targets were found to be associated withn shikonin-induced apoptotic cell death,11,14,15 the in vivo cellular target of shikonin is still unknown. Here we report that shikonin targets the tumor proteasome and inhibits the proteasomal chymotrypsin subunit β5 in vitro and in vivo. Shikonin inhibits the chymotrypsin-like activity of a purified 20S proteasome and 26S proteasome in murine hepatoma and human prostate cancer cells and tumors, leading to induction of cell death and tumor growth inhibition.
Chemical structure analysis combined with computational modeling predicted shikonin might be a proteasome inhibitor. Shikonin contains two carbonyl carbons, C1 and (Fig. 1A), both of which are eletrophilic in nature that might therefore interact with Thr 1,22 the catalytic site of the proteasomal β5 subunit responsible for the chymotrypsin-like activity. This hypothesis was supported by the following lines of docking results: (i) C1 and C4 atoms of shikonin were highly susceptible towards nucleophilic attack by the β5 subunit of proteasome (Fig. 1B); (ii) there was a very high probability (29%+28%=57%, Fig. 1C) for C1 or C4 of shikonin to bind to the β5 subunit; (iii) these two potential carbons were oriented the effective range (4 Å)24 for interacting with the Thr 1of β5 (Fig. 1C); (iv) Theenergies of these docking modes were very low.
Consistent with the prediction from the computational modeling, our experimental data supported that shikonin targeted the proteasome. First, shikonin directly and potently inhibited the chymotrypsin-like activity of the purified proteasome with an IC50 value of 12.5 μmol/L (Fig. 1D). Secondly, shikonin inhibited the tumor cellular proteasome activity with an IC50 values between 2-16.5 μmol/L (Figs. 2, 4-6). Thirdly, treatment of both H22 and PC-3 cancer cell lines by shikonin caused accumulation of ubiquitinated and proteasome target proteins (Figs. 2-6). Finally, shikonin inhibited the proteasome in H22 allografts and PC-3 xenografts (Figs. 7, 8). It has been found that some natural compounds with proteasome inhibitory properties, such as EGCG and β-lactone, need higher concentration to reach the same inhibition to cellular 26S proteasome as it to purified 20S proteasome.32,33 We also found that shikonin showed less potency to inhibit 20S proteasome than that to inhibit 26S proteasome in P388 cells, although in PC-3 cells it showed comparable potency to inhibit purified 20S proteasome (IC50 = 12.5 μmol/L) and cellular 26S proteasome (50% inhibition of the proteasome at 10-16 μmol/L) (Figs. 1, 2, 4).
Proteasome inhibitors represent a novel class of anticancer drugs. Bortezomib (PS-341), the first proteasome inhibitor in clinical trial approved by FDA has been demonstrated to cause tumor cell death in vivo34, consistent with the hypothesis that inhibition of the proteasome causes induction of tumor cell death. In our dose-dependent studies, we found that shikonin inhibited the proteasome in different cancer cell lines, which was accompanied by cell death induction (Figs. 2, 5). Moreover, in our kinetic experiments, we found that proteasome inhibition by shikonin occurred before cell death induction. We determined cell death using multiple assays, all of which showed that cell death occurred after proteasome inhibition (Figs. 3, 4, 6). Therefore, proteasome inhibition by shikonin should contribute to the subsequent cell death.
Furthermore, we found that shikonin inhibited the tumor cellular proteasome and suppressed tumor growth in H22 allografts and PC-3 xenografts. We found inhibition of chymotrypsin-like activity (data not shown), accumulation of proteasome substrate proteins IκB-α, Bax and p27 as well as ubiquitinated proteins in shikonin-treated tumors compared to vehicle-treated tumors (Fig. 7). The proteasome inhibition was confirmed by the extensive accumulation of p27 in situ in shikonin-treated tumor tissues (Fig. 8). The same shikonin-treated tumor samples showed PARP cleavage and TUNEL positivity, indicating that in vivo proteasome inhibition by shikonin was associated with tumor cell death (Fig. 8). Finally, shikonin treatment at 4.0-8.0 mg/kg for 7 consecutive days caused 45-56% of tumor growth inhibition (Fig. 7B). All these results suggest that shikonin can reach the tumor cellular proteasome, causing tumor cell death and tumor growth inhibition.
We noticed that shikonin treatment resulted in Hsp 70 increase, which is implicated to protect cells from stress-induced cell death.35 The pleiotropic effects on activation of both apoptotic and antiapoptotic proteins have been found when other proteasome inhibitors were tested.36 Heat shock proteins are highly conserved proteins in response to stresses such as physical and chemical stresses.37 Hsp 70 is a predominant stress-inducible heat shock protein. Sikonin treatment (4 μmol/L) increased Hsp 70 protein in H22 cells (Fig. 3). There are at least two possible explanations for this Hsp 70 protein increase. i) In response to chemical stress introduced by shikonin treatment, Hsp 70 synthesis might be elevated. ii) Due to proteasome inhibition by shikonin, degradation of Hsp 70 protein was decreased. Regardless of the increase of Hsp 70 upon proteasome inhibition by shikonin, other pro-apoptotic proteins, IκB-α, Bax and p27 were also accumulated and cell death occured in the cultured hepatoma cells (Fig. 3). It has been suggested that cell proliferation and cell death is regulated by the balance between proapoptotic and anti-apoptotic proteins.35 Our results suggest that although shikonin activated both apoptotic and ant apoptotic pathways, as a net result, this treatment made the cells shift toward cell death.
A shikonin-containing mixture has been examined in 19 late-stage lung cancer patients who were not suitable for surgical or irradiation treatment, showing that one year survival rate was 47.3%.17 In accordance with this report, our cumulative survival experiments demonstrated that 4 mg/kg shikonin treatment significantly extended the survival period of KMF mice bearing murine leukemia (Fig. 7A). Within 23 days, the entire 10 control vehicle treated mice died. In a sharp contrast, the 9 out of 10 mice treated with 4 mg/kg shikonin survived by the end of the experiment (60 days). However, when shikonin was used at a higher concentration (8 mg/kg) in the same experiment, only 7 out 10 mice survived by the end of the experiment (60 days; data not shown). Therefore, high dose treatment of shikonin might have caused some toxicity although no apparent loss of body weight was found in these mice. We also found slight loss of body weight and activity in nude mice bearing human prostate cancer under our experimental conditions.
Natural compounds might have multiple cellular targets in order to achieve their biological beneficial effects such as tumor growth inhibition.38 Results of the present study, for the first time, show that the ²5 subunit of the proteasome is one of the targets of shikonin in vitro, inhibition of which leads to cell death in tumor cells. More importantly, our work also demonstrates that the tumor proteasomal chymotrypsin subunit is also an in vivo biological target of shikonin in tumor tissues, inhibition of which is associated with tumor growth inhibition. This study suggests that shikonin has a great potential to be used clinically for treatment of various human cancers.
Acknowledgements
We thank the Karmanos Cancer Institute Pathology Core Facility for assisting in TUNEL and immunohistochemistry assays. This work was supported by The National High Technology Research and Development Program of China 2006AA02Z4B5 (JL), Project 30770835 supported by National Natural Science Foundation of China (JL), Scientific Research Foundation for the Returned Overseas Chinese Scholars sponsored by State Education Ministry (JL). Karmanos Cancer Institute of Wayne State University (QPD), Department of Defense Breast Cancer Research Program Awards W81XWH-04-1-0688 and DAMD17-03-1-0175 (QPD),National Cancer Institute grant 1R01CA120009 (QPD), National Cancer Institute/NIH Cancer Center Support Grant (to Karmanos Cancer Institute).
References
- 1.Chen X, Yang L, Zhang N, Turpin JA, Buckheit RW, Osterling C, Oppenheim JJ, Howard OM. Shikonin, a component of Chinese herbal medicine, inhibits chemokine receptor function and suppresses human immunodeficiency virus type 1. Antimicrob Agents Chemother. 2003;47:2810–6. doi: 10.1128/AAC.47.9.2810-2816.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chen X, Yang L, Oppenheim JJ, Howard MZ. Cellular pharmacology studies of shikonin derivatives. Phytother Res. 2002;16:199–209. doi: 10.1002/ptr.1100. [DOI] [PubMed] [Google Scholar]
- 3.Staniforth V, Wang SY, Shyur LF, Yang NS. Shikonins, phytocompounds from Lithospermum erythrorhizon, inhibit the transcriptional activation of human tumor necrosis factor alpha promoter in vivo. J Biol Chem. 2004;279:5877–85. doi: 10.1074/jbc.M309185200. [DOI] [PubMed] [Google Scholar]
- 4.Chiu SC, Yang NS. Inhibition of tumor necrosis factor-alpha through selective blockade of Pre-mRNA splicing by shikonin. Mol Pharmacol. 2007;71:1640–5. doi: 10.1124/mol.106.032821. [DOI] [PubMed] [Google Scholar]
- 5.Shen CC, Syu WJ, Li SY, Lin CH, Lee GH, Sun CM. Antimicrobial activities of naphthazarins from Arnebia euchroma. J Nat Prod. 2002;65:1857–62. doi: 10.1021/np010599w. [DOI] [PubMed] [Google Scholar]
- 6.Sasaki K, Abe H, Yoshizaki F. In vitro antifungal activity of naphthoquinone derivatives. Biol Pharm Bull. 2002;25:669–70. doi: 10.1248/bpb.25.669. [DOI] [PubMed] [Google Scholar]
- 7.Sankawa U, Ebizuka Y, Miyazaki T, Isomura Y, Otsuka H. Antitumor activity of shikonin and its derivatives. Chem Pharm Bull (Tokyo) 1977;25:2392–5. doi: 10.1248/cpb.25.2392. [DOI] [PubMed] [Google Scholar]
- 8.Wu Z, Wu LJ, Li LH, Tashiro S, Onodera S, Ikejima T. Shikonin regulates HeLa cell death via caspase-3 activation and blockage of DNA synthesis. J Asian Nat Prod Res. 2004;6:155–66. doi: 10.1080/1028602032000169622. [DOI] [PubMed] [Google Scholar]
- 9.Yeh CC, Kuo HM, Li TM, Lin JP, Yu FS, Lu HF, Chung JG, Yang JS. Shikonin-induced apoptosis involves caspase-3 activity in a human bladder cancer cell line (T24) In Vivo. 2007;21:1011–9. [PubMed] [Google Scholar]
- 10.Yoon Y, Kim YO, Lim NY, Jeon WK, Sung HJ. Shikonin, an ingredient of Lithospermum erythrorhizon induced apoptosis in HL60 human premyelocytic leukemia cell line. Planta Med. 1999;65:532–5. doi: 10.1055/s-1999-14010. [DOI] [PubMed] [Google Scholar]
- 11.Han W, Li L, Qiu S, Lu Q, Pan Q, Gu Y, Luo J, Hu X. Shikonin circumvents cancer drug resistance by induction of a necroptotic death. Mol Cancer Ther. 2007;6:1641–9. doi: 10.1158/1535-7163.MCT-06-0511. [DOI] [PubMed] [Google Scholar]
- 12.Fujii N, Yamashita Y, Arima Y, Nagashima M, Nakano H. Induction of topoisomerase II-mediated DNA cleavage by the plant naphthoquinones plumbagin and shikonin. Antimicrob Agents Chemother. 1992;36:2589–94. doi: 10.1128/aac.36.12.2589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Min R, Tong J, Wenjun Y, Wenhu D, Xiaojian Z, Jiacai H, Jian Z, Wantao C, Chenping Z. Growth inhibition and induction of apoptosis in human oral squamous cell carcinoma Tca-8113 cell lines by shikonin was partly through the inactivation of NF-kappaB pathway. Phytother Res. 2008;22:407–15. doi: 10.1002/ptr.2340. [DOI] [PubMed] [Google Scholar]
- 14.Wu Z, Wu L, Li L, Tashiro S, Onodera S, Ikejima T. p53-mediated cell cycle arrest and apoptosis induced by shikonin via a caspase-9-dependent mechanism in human malignant melanoma A375-S2 cells. J Pharmacol Sci. 2004;94:166–76. doi: 10.1254/jphs.94.166. [DOI] [PubMed] [Google Scholar]
- 15.Gao D, Hiromura M, Yasui H, Sakurai H. Direct reaction between shikonin and thiols induces apoptosis in HL60 cells. Biol Pharm Bull. 2002;25:827–32. doi: 10.1248/bpb.25.827. [DOI] [PubMed] [Google Scholar]
- 16.Chen CH, Chern CL, Lin CC, Lu FJ, Shih MK, Hsieh PY, Liu TZ. Involvement of reactive oxygen species, but not mitochondrial permeability transition in the apoptotic induction of human SK-Hep-1 hepatoma cells by shikonin. Planta Med. 2003;69:1119–24. doi: 10.1055/s-2003-45193. [DOI] [PubMed] [Google Scholar]
- 17.Guo XP, Zhang XY, Zhang SD. Clinical trial on the effects of shikonin mixture on later stage lung cancer. Zhong Xi Yi Jie He Za Zhi. 1991;11:598–9. [PubMed] [Google Scholar]
- 18.Adams J. The development of proteasome inhibitors as anticancer drugs. Cancer Cell. 2004;5:417–21. doi: 10.1016/s1535-6108(04)00120-5. [DOI] [PubMed] [Google Scholar]
- 19.Nalepa G, Rolfe M, Harper JW. Drug discovery in the ubiquitin-proteasome system. Nat Rev Drug Discov. 2006;5:596–613. doi: 10.1038/nrd2056. [DOI] [PubMed] [Google Scholar]
- 20.Dou QP, Smith DM, Daniel KG, Kazi A. Interruption of tumor cell cycle progression through proteasome inhibition: implications for cancer therapy. Prog Cell Cycle Res. 2003;5:441–6. [PubMed] [Google Scholar]
- 21.Orlowski M, Wilk S. Catalytic activities of the 20 S proteasome, a multicatalytic proteinase complex. Arch Biochem Biophys. 2000;383:1–16. doi: 10.1006/abbi.2000.2036. [DOI] [PubMed] [Google Scholar]
- 22.Groll M, Bochtler M, Brandstetter H, Clausen T, Huber R. Molecular machines for protein degradation. Chembiochem. 2005;6:222–56. doi: 10.1002/cbic.200400313. [DOI] [PubMed] [Google Scholar]
- 23.Yang H, Chen D, Cui QC, Yuan X, Dou QP. Celastrol, a triterpene extracted from the Chinese "Thunder of God Vine," is a potent proteasome inhibitor and suppresses human prostate cancer growth in nude mice. Cancer Res. 2006;66:4758–65. doi: 10.1158/0008-5472.CAN-05-4529. [DOI] [PubMed] [Google Scholar]
- 24.Smith DM, Daniel KG, Wang Z, Guida WC, Chan TH, Dou QP. Docking studies and model development of tea polyphenol proteasome inhibitors: applications to rational drug design. Proteins. 2004;54:58–70. doi: 10.1002/prot.10504. [DOI] [PubMed] [Google Scholar]
- 25.Kazi A, Daniel KG, Smith DM, Kumar NB, Dou QP. Inhibition of the proteasome activity, a novel mechanism associated with the tumor cell apoptosis-inducing ability of genistein. Biochem Pharmacol. 2003;66:965–76. doi: 10.1016/s0006-2952(03)00414-3. [DOI] [PubMed] [Google Scholar]
- 26.An B, Dou QP. Cleavage of retinoblastoma protein during apoptosis: an interleukin 1 beta-converting enzyme-like protease as candidate. Cancer Res. 1996;56:438–42. [PubMed] [Google Scholar]
- 27.Li B, Dou QP. Bax degradation by the ubiquitin/proteasome-dependent pathway: involvement in tumor survival and progression. Proc Natl Acad Sci U S A. 2000;97:3850–5. doi: 10.1073/pnas.070047997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liu H, McPherson BC, Yao Z. Preconditioning attenuates apoptosis and necrosis: role of protein kinase C epsilon and -delta isoforms. Am J Physiol Heart Circ Physiol. 2001;281:H404–10. doi: 10.1152/ajpheart.2001.281.1.H404. [DOI] [PubMed] [Google Scholar]
- 29.Yang H, Landis-Piwowar KR, Chen D, Milacic V, Dou QP. Natural Proteasome Inhibitors in Cancer Prevention and Treatment. Curr Protein Pept Sci. 2008;9:227–39. doi: 10.2174/138920308784533998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chen D, Chen MS, Cui QC, Yang H, Dou QP. Structure-proteasome-inhibitory activity relationships of dietary flavonoids in human cancer cells. Front Biosci. 2007;12:1935–45. doi: 10.2741/2199. [DOI] [PubMed] [Google Scholar]
- 31.Duriez PJ, Shah GM. Cleavage of poly(ADP-ribose) polymerase: a sensitive parameter to study cell death. Biochem Cell Biol. 1997;75:337–49. [PubMed] [Google Scholar]
- 32.Nam S, Smith DM, Dou QP. Ester bond-containing tea polyphenols potently inhibit proteasome activity in vitro and in vivo. J Biol Chem. 2001;276:13322–30. doi: 10.1074/jbc.M004209200. [DOI] [PubMed] [Google Scholar]
- 33.Dick LR, Cruikshank AA, Grenier L, Melandri FD, Nunes SL, Stein RL. Mechanistic studies on the inactivation of the proteasome by lactacystin: a central role for clasto-lactacystin beta-lactone. J Biol Chem. 1996;271:7273–6. doi: 10.1074/jbc.271.13.7273. [DOI] [PubMed] [Google Scholar]
- 34.Orlowski RZ. Kuhn Proteasome inhibitors in cancer therapy: lessons from the first decade. Clin Cancer Res. 2008;14:1649–57. doi: 10.1158/1078-0432.CCR-07-2218. [DOI] [PubMed] [Google Scholar]
- 35.Jolly C, Morimoto RI. Role of the heat shock response and molecular chaperones in oncogenesis and cell death. J Natl Cancer Inst. 2000;92:1564–72. doi: 10.1093/jnci/92.19.1564. [DOI] [PubMed] [Google Scholar]
- 36.Robertson JD, Datta K, Biswal SS, Kehrer JP. Heat-shock protein 70 antisense oligomers enhance proteasome inhibitor-induced apoptosis. Biochem J. 1999;344:477–85. [PMC free article] [PubMed] [Google Scholar]
- 37.Burdon RH. Heat shock and the heat shock proteins. Biochem J. 1986;240:313–24. doi: 10.1042/bj2400313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cucciolla V, Borriello A, Oliva A, Galletti P, Zappia V, Della Ragione F. Resveratrol: from basic science to the clinic. Cell Cycle. 2007;6:2495–510. doi: 10.4161/cc.6.20.4815. [DOI] [PubMed] [Google Scholar]