

Abstract
BimEL the most abundant Bim splice variant, is subject to ERK1/2-catalysed phosphorylation, which targets it for ubiquitination and proteasome-dependent destruction. In contrast, inactivation of ERK1/2, following withdrawal of survival factors, promotes stabilization of BimEL. It has been proposed that the RING finger protein Cbl binds to BimEL and serves as its E3 ubiquitin ligase. However, this is controversial since most Cbl substrates are tyrosine phosphoproteins and yet BimEL is targeted for destruction by ERK1/2-catalysed serine phosphorylation. Consequently, a role for Cbl could suggest a second pathway for BimEL turnover, regulated by direct tyrosine phosphorylation, or could suggest that BimEL is a coincidence detector, requiring phosphorylation by ERK1/2 and a tyrosine kinase. Here we show that degradation of BimEL does not involve its tyrosine phosphorylation; indeed, BimEL is not a tyrosine phosphoprotein. Furthermore, BimEL fails to interact with Cbl and growth factor-stimulated, ERK1/2-dependent BimEL turnover proceeds normally in Cbl-/- fibroblasts. These results indicate that Cbl is not required for ERK1/2-dependent BimEL turnover in fibroblasts and epithelial cells and any role it has in other cell types is likely to be indirect.
Keywords: Bim, Cbl, ERK1/2, proteasome, ubiquitin ligase
1. Introduction
Bax and Bak proteins promote the release of cytochrome c from the mitochondria, but in viable cells are restrained by their binding to pro-survival Bcl-2 proteins [1]. Binding of pro-apoptotic ‘BH3-only proteins’ (BOPs, including Bim, Puma and Bad) to the pro-survival Bcl-2 proteins neutralizes them, thereby allowing Bax and/or Bak to initiate cell death [2-4]. Bim is one of the most effective BOPs for cell killing [5] and plays a major role in promoting cell death following withdrawal of trophic survival factors [6-9]. Bim is expressed de novo following withdrawal of survival factors [7-12] due to inactivation of PKB [10,12] and ERK1/2 [11], or activation of JNK [7]. Alternative splicing gives rise to multiple isoforms that differ in pro-apoptotic potency [13] due to differences in their post-translational regulation [14]. Of the short, long and extra-long Bim proteins (BimS, BimL & BimEL), BimEL is the most abundant and exhibits a pronounced increase in expression following withdrawal of survival factors. This seems to be because BimEL stability is subject to post-translational regulation [14]. BimEL contains an ERK1/2 docking domain [15] and phosphorylation sites that promote the proteasomal turnover of BimEL turnover [15-19]. Consequently, withdrawal of survival factors results in the stabilization of the BimEL protein as well as de novo transcription of the Bim gene.
Proteins are ‘flagged’ for proteasomal degradation by the covalent attachment of a polyubiquitin (polyUb) chain, catalysed by the coordinated action of an E1 Ub-activating enzyme, an E2 Ub-conjugating enzyme and a substrate-specific E3 Ub-ligase [20]. Substrate recognition by the E3 is the critical regulatory step and in the case of phosphorylation-dependent degradation the E3 Ub-ligase often has a phosphorylation-specific recognition domain [21]. It has been suggested that the RING finger protein Cbl serves as the E3 Ub-ligase for degradation of BimEL in osteoclasts [22]. However, this suggestion has proved controversial since another study reported increased levels of BimEL in the testis of c-Cbl null mice but failed to detect direct binding of BimEL to Cbl, concluding that the effect of Cbl on BimEL expression was not direct [23]. Furthermore, Cbl catalyses the ubiquitination of the EGF receptor and other tyrosine kinases to promote their endosomal trafficking and interacts with such tyrosine-phosphorylated proteins via a tyrosine kinase-binding domain (TKB) [24-26]. However, to date the only signal that has been implicated in BimEL degradation is its ERK1/2-dependent phosphorylation [16-19], raising the possibility that BimEL degradation requires the coincident phosphorylation of serine and tyrosine residues or that BimEL interacts with Cbl in a unique manner.
Here we have tested the hypothesis that Cbl is required for ERK1/2-dependent BimEL degradation. We show that degradation of BimEL proceeds without tyrosine phosphorylation; indeed, we find no evidence for BimEL being a tyrosine phosphoprotein. Furthermore, BimEL fails to interact with Cbl under conditions in which it is targeted for turnover. Finally, growth factor-stimulated BimEL turnover proceeds normally in Cbl-/- fibroblasts indicating that Cbl is not required for ERK1/2-dependent BimEL turnover.
2. Materials and Methods
2.1 Materials
U0126 was purchased from Promega, and MG132 from Calbiochem. AZD6244 [27] was provided by AstraZeneca. The following antibodies were used throughout this study: HA-probe beads, Myc and Cbl were from Santa Cruz Biotechnology; Phospho-ERK and ERK were from Cell Signalling Technology; Bim (Rat) was from Calbiochem; Bim (Rabbit) was from Chemicon, and HA (mouse) was provided by the Babraham Institute Monoclonal Antibody Facility. Horseradish peroxidise-conjugated secondary antibodies were from Bio-Rad. All other chemicals were purchased from Sigma, and were of the highest grade available.
2.2 Cell Culture
Cell culture reagents were purchased from Invitrogen. Culture of HR1 cells [28] and c-Cbl-/- and HA-Cbl iMEFs [29,30] was as previously described.
2.3 Cell Treatments
Cycling HR1 cells were switched to serum free medium 30 minutes prior to stimulation. In the case of treatment with UO126 (20μM), AZD6244 (10μM) or MG132 (10μM), drug was included in this 30 minute incubation. Cells were stimulated with 100nM 4-hydroxytamoxifen (4-HT), 10nM EGF or Pervanadate for the times indicated. Pervanadate was prepared by combining 100ul 10mM sodium orthovanadate, 34ul of 1% hydrogen peroxide and 866ul PBS; this was then diluted 1: 10 onto cells in PBS, for 10 minutes. Cycling iMEFs were serum starved overnight and pre-treated with 10μM emetine, 10μM MG132 or 10μM AZD6244 for 30 minutes prior to stimulation.
2.4 Western Blot and Immunoprecipitation analysis
Following treatment, cells were lysed in TG lysis buffer and analysed by Western Blot as described previously [11]. TG lysates were normalised for protein content and used in HA immunoprecipitations using the HA-probe beads. Lysates were incubated with HA-probe for 90 minutes, end-over-end at 4°C, washed 3 times in TG lysis buffer and boiled in sample buffer. Samples were then analysed by SDS-PAGE and immunoblotting.
2.5 Plasmids and Transfections
The pcDNA3-HA-BimEL construct used was described previously [16]. The pBK CMV Myc-c-Cbl was kindly provided by Roland Baron, Yale University School of Medicine. The pcDNA3-HA-BimEL phosphotyrosine mutant (BimEL ΔTyr) was generated by site directed mutagenesis to make the following alterations: Y92F, Y123F, Y161F and Y170F, which were confirmed by DNA sequencing. HR1 cells were transfected 16 hours prior to treatment with these plasmids by the calcium phosphate precipitation technique [31].
3. Results
3.1 BimEL does not undergo tyrosine phosphorylation
The suggestion that Cbl serves as an E3 Ub ligase for BimEL raises several predictions that are readily testable. The first of these is that BimEL is phosphorylated on tyrosine residues. The majority of proteins that are substrates for the E3 ligase activity of Cbl are either tyrosine kinases or substrates of tyrosine kinases [24-26]. Cbl proteins possess a highly conserved N-terminal tyrosine kinase-binding domain (TKB), which mediates binding to phosphotyrosine residues [24] and is thought to be important for their specific E3 Ub ligase activity by facilitating substrate binding. However, to date the only signal that has been implicated in BimEL ubiquitylation and degradation is its ERK1/2-dependent phosphorylation on serine residues [16-19]. This raised the possibility that BimEL degradation might require the coincident phosphorylation of serine and tyrosine residues, with ERK1/2-dependent phosphorylation perhaps facilitating phosphorylation of BimEL by a tyrosine kinase. Consequently we examined whether BimEL was phosphorylated on tyrosine residues under conditions in which it was targeted for degradation.
We used HR1 cells [28], a clone of HEK293 cells expressing the conditional protein kinase ΔRaf-1:ER*, which selectively activates ERK1/2 in response to treatment with 4-hydroxytamoxifen (4-HT). HR1 cells were transfected with HA-BimEL for 16 hours and then treated with 4-HT to activate ERK1/2, a treatment that normally promotes degradation of BimEL [16-19]. HA-BimEL was then immunoprecipitated and probed by western blot with the PY20 anti-phosphotyrosine antibody (Figure 1A). These results revealed that whilst activation of ERK1/2 by ΔRaf-1:ER* promoted the multi-site phosphorylation of BimEL, as indicated by its reduced electrophoretic mobility on SDS-PAGE, it failed to promote tyrosine phosphorylation of BimEL. This is perhaps not surprising since activation of ΔRaf-1:ER* bypasses receptor and non-receptor tyrosine kinases at the plasma membrane and selectively activates ERK1/2. However, stimulation with EGF, which also promoted the phosphorylation of BimEL as judged by SDS-PAGE mobility, also failed to promote any tyrosine phosphorylation of BimEL (Figure 1A) despite the fact that the EGFR is a receptor tyrosine kinase and can also activate Src family non-receptor tyrosine kinases.
Figure 1. BimEL does not undergo tyrosine phosphorylation under conditions known to target it for degradation.
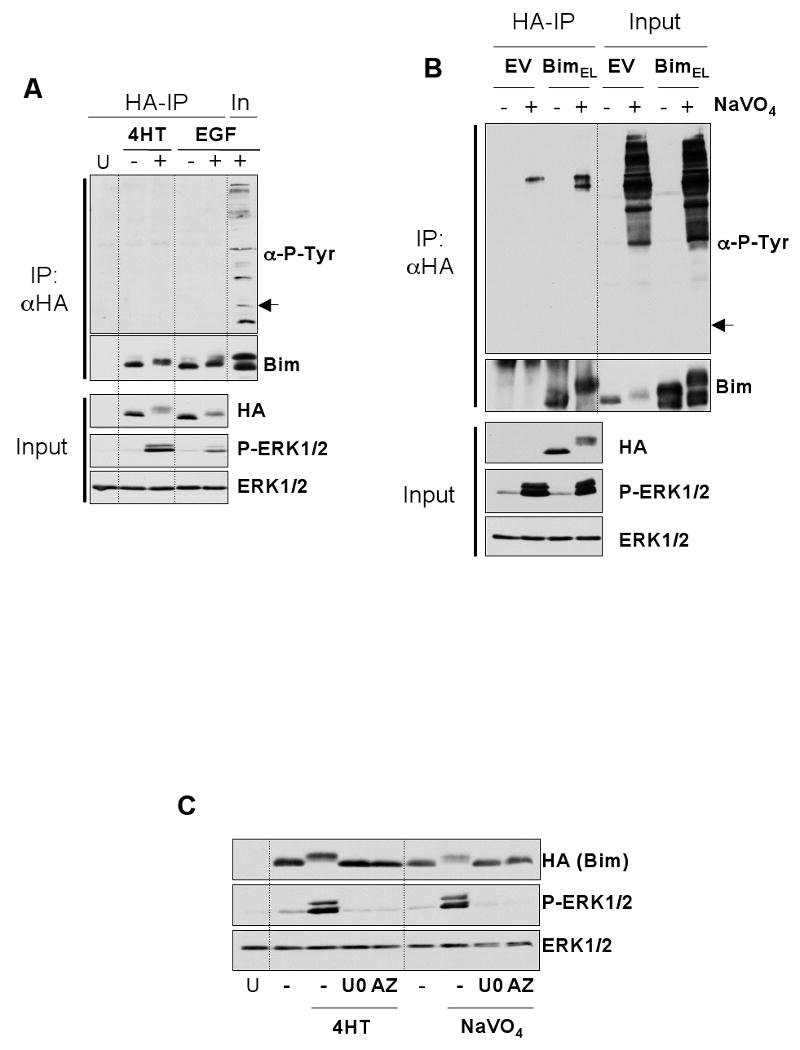
HR1 cells were transfected with plasmids encoding HA-BimEL or empty vector. After 16 hours, cells were serum starved for 30 minutes and then stimulated with 100nM 4-hydroxytamoxifen (4-HT) for 1 hour or 10nM EGF for 5 minutes (A) or pervanadate (see materials and methods) for 10 minutes (B). Whole cell extracts were then prepared, normalised for protein content, and boiled directly (input) or used for immunoprecipitation of HA-BimEL. Input and IP samples were then immunoblotted with antibodies to anti-phosphotyrosine, Bim, HA (Bim), P-ERK1/2 and ERK1/2. Input lysates were included with the IP samples to act as a positive control for the anti-phosphotyrosine immunoblot. Note: the arrow in (A) & (B) indicates the position at which Bim resolves and would appear if it reacted with the anti-P-Tyr antibody. (C) Phosphorylation of BimEL after treatment with 4-HT or pervanadate is ERK1/2 dependent. HR1 cells were transfected with HA-BimEL for 16 hours, then switched to serum free medium ± 20μM UO126 or 10μM AZD6244. Cells were stimulated with 100nM 4-HT for 1 hour or pervanadate for 10 minutes (see materials and methods) and whole cell extracts prepared for immunoblot analysis using antibodies to HA (Bim), P-ERK1/2 and ERK1/2. Results are from single representative experiments; similar results were obtained in a total of three experiments.
One possible explanation for these results was that BimEL was being targeted by a particular tyrosine kinase that was not EGF responsive. Therefore, we again transfected HR1 cells with HA-BimEL and treated them with pervanadate, a general inhibitor of phosphotyrosine phosphatases. This led to a striking increase in global protein tyrosine phosphorylation (Figure 1B, Input lysate) and led to the multi-site phosphorylation of BimEL; however, we were still unable to detect any tyrosine phosphorylated BimEL in the anti-HA IPs. The pervanadate-dependent phosphorylation of BimEL, monitored by changes in mobility on SDS-PAGE gels, was actually abolished by U0126 and AZD6244 [27], two distinct MEK1/2 inhibitors, indicating that this was due to pervanadate-dependent activation of ERK1/2 (Figure 1C). These results indicate that BimEL does not undergo tyrosine phosphorylation either under conditions that are known to target it for degradation via the proteasome or in response to global increases in tyrosine kinase activation.
Our inability to detect tyrosine phosphorylation of BimEL could be because the relevant site was not accessible to anti-phosphotyrosine antibodies even after denaturation. To address this we constructed a mutant of BimEL in which all four tyrosine residues were changed to non-phosphorylatable phenylalanine residues by site-directed mutagenesis (BimELΔTyr). Stimulation with pervanadate caused the same reduction in SDS-PAGE mobility in the WT and BimELΔTyr constructs (Figure 2A) indicating that the pervanadate-dependent phosphorylation of BimEL was not due to tyrosine phosphorylation. We also monitored the ERK1/2-dependent turnover of HA-BimEL and HA-BimELΔTyr in response to activation of ΔRaf-1:ER*. HR1 cells were transfected with the relevant BimEL plasmids, serum starved and then re-stimulated with 4-HT in the presence of emetine to block new protein synthesis. These experiments revealed that activation of ERK1/2 resulted in the identical phosphorylation and turnover of both HA-BimEL and HA-BimELΔTyr (Figure 2B). In summary, these results indicated that BimEL was not phosphorylated on tyrosine residues under conditions in which it was targeted for degradation and tyrosine residues in BimEL play no role in its ERK1/2-dependent turnover.
Figure 2. Mutation of all four tyrosine residues does not affect the ability of BimEL to undergo ERK1/2-dependent phosphorylation and degradation.
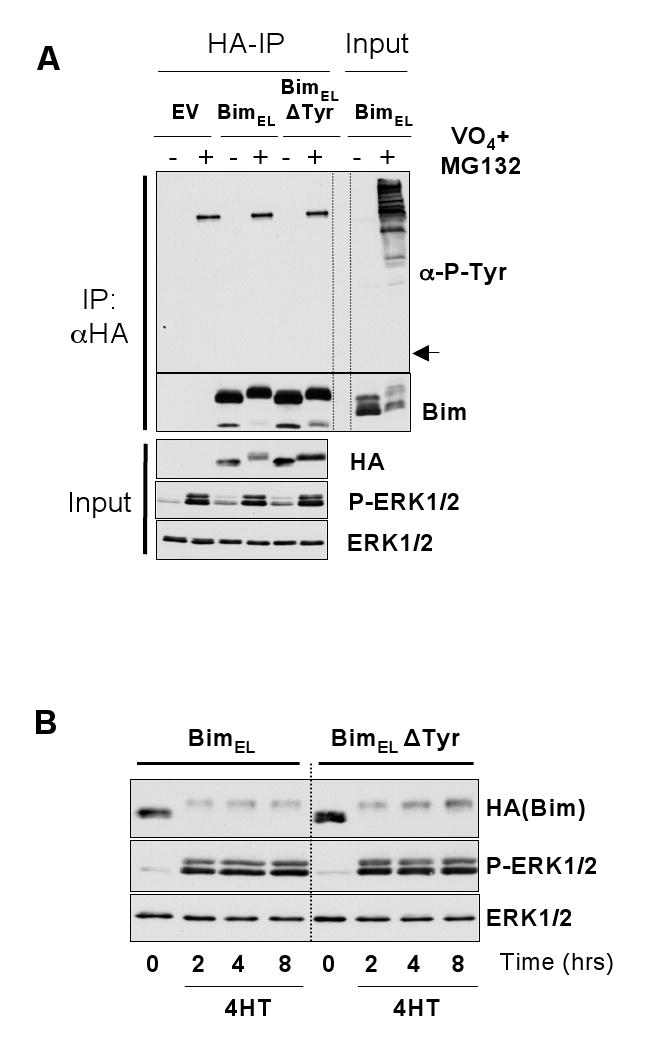
(A) HR1 cells were transfected with HA-BimEL, HA-BimELΔTyr or empty vector (EV) for 16 hours. Cells were then serum starved for 30 minutes in the presence of MG132 and subsequently treated with pervanadate for 10 minutes (see materials and methods). Whole cell extracts were then prepared, normalised for protein content, and boiled directly (input) or used for immunoprecipitaion of HA-BimEL. Input and IP samples were then immunoblotted with antibodies to anti-phosphotyrosine, Bim, HA (Bim), P-ERK1/2 and ERK1/2. Input lysates were included with the IP samples to act as a positive control for the anti-phosphotyrosine immunoblot. Note: the arrow in (A) indicates the position at which Bim resolves and would appear if it reacted with the anti-P-Tyr antibody. (B) HR1 cells were transfected with HA-BimEL or HA-BimELΔTyr for 16 hours, then switched to serum free medium in the presence of emetine for a 30 minute pre-treatments. Cells were then stimulated with 100nM 4-HT for 2, 4 or 8 hours after which whole cell lysates were prepared for immunoblot analysis using antibodies to HA (Bim), P-ERK1/2 and ERK1/2.
3.2 BimEL does not interact with Cbl under conditions that promote BimEL degradation
For Cbl to act as an E3 ligase it must directly or indirectly interact with its substrates. The fact that BimEL was not phosphorylated on tyrosine residues suggested that the interaction between BimEL and Cbl must be independent of tyrosine phosphorylation. There is precedent for this since inactivation of the Cbl tyrosine kinase binding domain does not affect the ability of Cbl to bind and negatively regulate the Src-family tyrosine kinase Fyn [29]. Furthermore, Cbl can clearly play a non-catalytic adaptor role in various aspects of signal transduction [25,26]. Whilst a previous report had shown that BimEL and Cbl could indeed associate [22], this was not confirmed in a separate study[23]. Consequently we examined the association between BimEL and Cbl following activation of ERK1/2 as we reasoned that this should facilitate their association. HR1 cells were transfected with HA-BimEL and Myc-Cbl and treated with 4-HT or pervanadate in the presence or absence of MG132. HA-BimEL was then immunoprecipitated and probed for the presence of Myc-Cbl. Activation of ΔRaf-1:ER* caused activation of ERK1/2 and phosphorylation of HA-BimEL, but did not induce the formation of a HA-BimEL:Myc-Cbl complex, as judged by co-immunoprecipitation, despite the fact that we readily immunoprecipitated HA-BimEL (Figure 3). Even the inclusion of MG132 to capture BimEL that was targeted for degradation failed to identify HA-BimEL:Myc-Cbl complexes. Similar results were obtained in pervanadate treated cells. For example, pervanadate activated ERK1/2, induced BimEL phosphorylation and also promoted the phosphorylation of Cbl as seen by an electrophoretic mobility shift, consistent with reports that Cbl is phosphorylated on tyrosine residues [25,26]. Despite this, pervanadate failed to promote the formation of HA-BimEL:Myc-Cbl complexes. Thus, in contrast to the study by Akiyama et al [22] we were unable to identify BimEL:Cbl complexes under conditions in which BimEL is known to be degraded, even when over-expression would tend to favour such associations. Rather, our results accord with El Chami et al [23] who failed to detect BimEL:Cbl interactions.
Figure 3. BimEL and Cbl fail to associate under conditions that target BimEL for degradation.
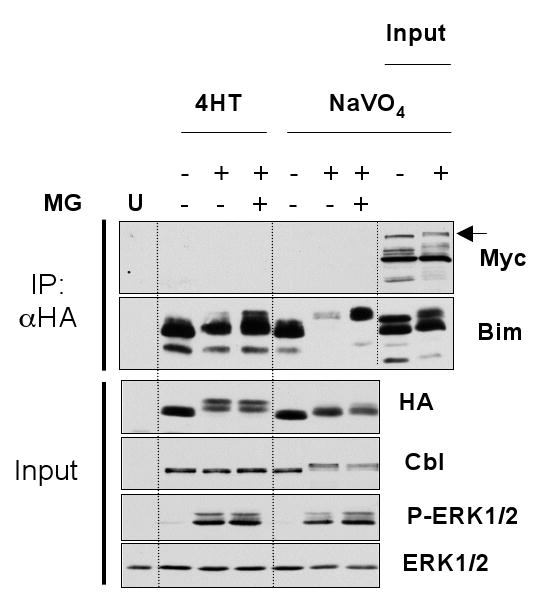
Cycling HR1 cells were transfected with HA-BimEL and Myc-c-Cbl and 16 hours later switched to serum free media with or without MG132 for 30 minutes. Cells were then treated for 1 hour with 100nM 4-hydroxytamoxifen (4-HT) or for 10 minutes with pervanadate. Whole cell extracts were used for immunoprecipitation of BimEL and input and IP samples were then fractionated by SDS-PAGE and immunoblotted with antibodies to HA, Myc (Cbl), Cbl, ERK1/2 and P-ERK1/2. Input lysates were included with the IP samples to act as a positive control for the immunoblot and to highlight where c-Cbl would run (indicated by the arrow).
3.3 Cbl is not required for ERK1/2-dependent, proteasomal degradation of BimEL
The fact that Cbl failed to interact with BimEL suggested that Cbl could play an important but indirect role in BimEL turnover, though this might be less consistent with a role as an E3 ligase. Alternatively, it suggested that Cbl was simply not required for BimEL degradation. To address these possibilities we examined the turnover of BimEL in Cbl-deficient cells, again testing the simple prediction that if Cbl was acting as the relevant E3 ubiquitin ligase, then turnover of BimEL should be compromised in the absence of Cbl. We examined the growth factor-stimulated turnover of BimEL in immortalized MEFs (iMEFs) from c-Cbl-/- mice [29,30], making comparisons with Cbl-/- iMEFs that had been reconstituted with wild type HA-Cbl. As a primary observation, we noted that the basal level of BimEL was actually 1.51±0.32 (mean ± s.d, n=3 experiments) fold higher in the Cbl-/-+HA-Cbl iMEFs compared to the Cbl-/- iMEFs (Figure 4A, t=0 in both cell lines), the opposite of what one might expect if Cbl was promoting BimEL turnover. This did however correlate with a slight reduction in the basal level of P-ERK1/2 in the +HA-Cbl iMEFs (Figure 4A). We then serum starved iMEFs overnight to increase the amount of endogenous BimEL and re-stimulated them with FBS in the presence of emetine to measure turnover of the pre-existing BimEL. FBS promoted the phosphorylation and turnover of BimEL and these responses were virtually identical in the Cbl-/- and Cbl-/-+HA-Cbl iMEFs (Figure 4A). Indeed, when we quantified BimEL turnover from three independent experiments we found there was no significant difference between the turnover of BimEL in the presence or absence of Cbl (Figure 4B, p>0.05 at all time points). These results indicate that Cbl is not required for the growth factor-dependent turnover of BimEL.
Figure 4. ERK1/2-dependent BimEL turnover proceeds normally in the presence or absence of Cbl.
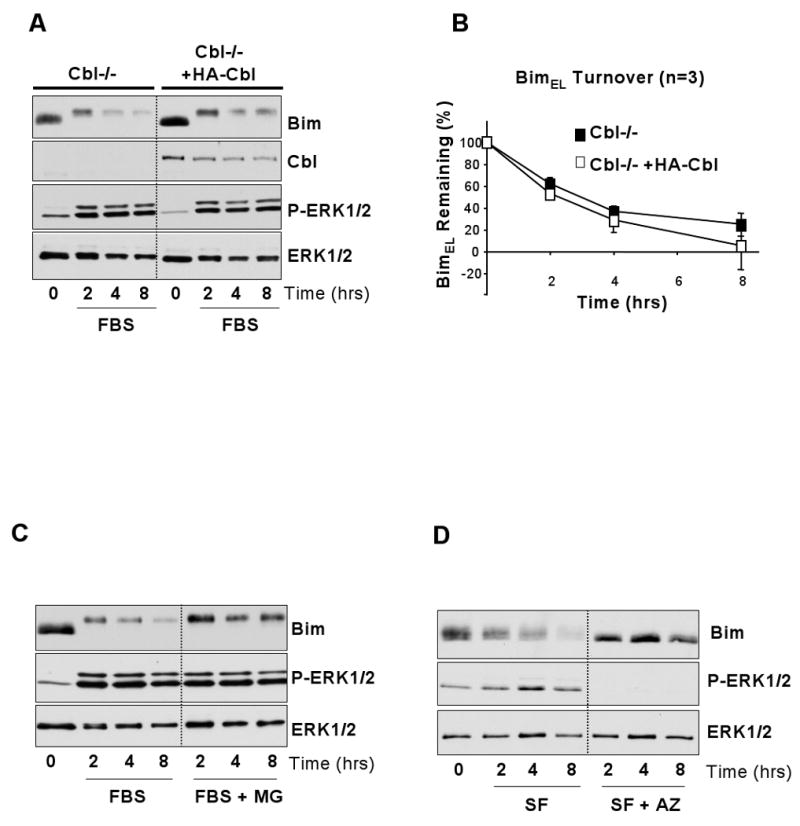
(A) Cycling c-Cbl-/- and c-Cbl-/- HA-c-Cbl reconstituted iMEFs were serum starved overnight to elevate levels of BimEL. Cells were then pre-treated with emetine for 30 minutes before being stimulated with FBS and harvested after 2, 4 or 8 hours. Whole cell extracts were prepared, normalised for protein content and boiled directly in sample buffer. Lysates were fractionated by SDS-PAGE and immunoblotted with antobodies to Bim, Cbl, P-ERK1/2 and ERK1/2, in which ERK1/2 served as a loading control. The immunoblots shown are from a single experiment representative of three giving identical results. (B) The amount of BimEL in the lysates relative to the ERK1/2 loading control was quantified by densitometric analysis. Data from three independent experiments was combined and plotted, confirming that the turnover of BimEL in the c-Cbl-/- MEFs did not differ significantly from that in the HA-c-Cbl addback cell line. (C) Cycling c-Cbl-/- iMEFs were serum starved overnight to elevate levels of BimEL. Cells were then pre-treated with emetine and MG132 for 30 minutes before being treated with FBS and harvested at set time intervals. (D) Cycling c-Cbl-/- were serum starved overnight to elevate levels of BimEL. Cells were then pre-treated with emetine and AZD6244 for 30 minutes before harvested at set time intervals. In both (C) & (D) whole cell extracts were prepared, normalised for protein content and boiled directly in sample buffer. Lysates were fractionated by SDS-PAGE and immunoblotted with antibodies to Bim, P-ERK1/2 and ERK1/2, the latter serving as a loading control. Results are taken from a single representative experiment; similar results were obtained in three experiments.
One explanation for these results is that the absence of Cbl revealed the existence of an alternative non-proteasomal pathway for BimEL degradation. To address this directly, serum starved Cbl-/- iMEFs were re-stimulated with FBS and emetine in the presence or absence of MG132 (Figure 4C). MG132 had no effect on the activation of ERK1/2 or the phosphorylation of BimEL but inhibited the FBS-dependent degradation of BimEL, suggesting that turnover of BimEL in the Cbl-/- MEFS does indeed proceed via the proteasome. The same results were obtained in the +HA-Cbl MEFs (data not shown). In addition, the primary signal for degradation of BimEL in the c-Cbl-/- iMEFs was the ERK1/2 pathway, since the MEK1/2 inhibitor AZD6244 prevented BimEL phosphorylation and degradation of BimEL. Taken together, these results indicate that the turnover of BimEL in iMEFs and HEK293 cells proceeds through the same ERK1/2- and proteasome-dependent pathway that has been described in a variety of other cell systems [9,14,16-19] and this does not require Cbl.
4. Discussion
Bim is involved in promoting apoptosis in response to a variety of insults including withdrawal of survival factors and is of interest because it plays important roles in regulating cell death in the immune system [6] and CNS [7]. In addition, Bim exhibits many of the properties expected of a tumour suppressor gene; loss of Bim accelerates tumour development in a mouse model of Myc-induced leukaemia [32], and up to 17% of human mantle cell lymphomas exhibit loss of the Bim locus [33]. Thus, cellular mechanisms that antagonise Bim function or expression are of great interest. Indeed, Bim transcription is negatively regulated by the PKB-dependent phosphorylation and inhibition of FOXO3A [10,12], whilst ERK1/2 activation can also repress Bim mRNA levels [11]. BimEL, the most abundant Bim splice variant, is phosphorylated by ERK1/2 at Ser residues, including Ser65 (Ser69 in human), and this promotes BimEL ubiquitination and proteasomal degradation [16-19]. By targeting BimEL for destruction, the E3 ligase responsible for ubiquitination of BimEL will promote cell survival and could therefore be considered a candidate oncogene. In this sense the suggestion that Cbl is the E3 Ub ligase for BimEL [22] is attractive since c-Cbl is known to be a proto-oncogene [34]. However, consideration of this proposal raises some fundamental questions, which we have sought to address here.
The first of these relates to the role of phosphorylation in BimEL turnover. The role of Cbl proteins as E3 Ub ligases and adaptor proteins has been extensively studied and to date over 150 proteins are known to interact with Cbl [26]. Cbl regulates the endocytosis of receptor and non-receptor tyrosine kinases and interacts with such tyrosine-phosphoproteins via its N-terminal phospho-tyrosine recognition domain, the so-called tyrosine kinase-binding (TKB) domain [24]. By analogy with other Cbl substrates, a role for Cbl in BimEL degradation suggested that BimEL might be regulated by tyrosine phosphorylation. In addition, since BimEL is targeted for proteasomal degradation by ERK1/2-catalysed serine phosphorylation [16-19], this raised the possibility that BimEL ubiquitination might require coincident serine and tyrosine phosphorylation. We tested this hypothesis directly using anti-phosphotyrosine antibodies but failed to detect any tyrosine phosphorylation of BimEL, following activation of ΔRaf-1:ER* or stimulation with EGF; two treatments which promote the ERK1/2-dependent degradation of BimEL. Furthermore, treatment of cells with pervanadate caused a global increase in cellular tyrosine phosphorylation but again failed to promote tyrosine phosphorylation of BimEL. Finally, a ΔTyr mutant of BimEL, lacking all tyrosine residues, expressed normally, underwent ERK1/2-dependent phosphorylation in cells and turned over at the same rate as wild type BimEL. These results show that BimEL is not phosphoryated upon tyrosine residues, consistent with a previous study that found BimEL to be exclusively phosphorylated at serine/threonine residues upon phospho-amino acid analysis [35]. More importantly, the results show that tyrosine phosphorylation is not required for BimEL turnover.
The second key question related to the physical association between BimEL and Cbl. If Cbl served as the relevant E3 ligase then it should associate with BimEL. Proteins that interact with Cbl have been identified by immunoprecipitation and pull-down assays [26]. Whilst many such interactions are phosphotyrosine-dependent, there are examples where Cbl substrates interact in manner independent of the Cbl TKB domain. For example, the TKB domain of Cbl is not required for interaction with the Src-family protein kinase Fyn, which rather takes place through an interaction between the SH3 domain of Fyn and the proline-rich region of Cbl [29]. Thus it seemed likely that Cbl would interact with BimEL in a non-phosphotyrosine-dependent fashion. However, we failed to detect binding between Myc-Cbl and HA-BimEL, even when both proteins were over-expressed and cells were treated with the proteasome inhibitor MG132, conditions that would tend to favour such associations.
The final prediction we tested was the requirement for Cbl in turnover of BimEL. Whilst our failure to show an association between BimEL and Cbl suggested that the effect of Cbl on the proteasomal turnover of BimEL was indirect, we would still expect BimEL to accumulate in the absence of Cbl if Cbl was indeed a negative regulator of BimEL. However, BimEL turnover proceeded normally in the presence or absence of Cbl. There are several potential explanations for this result. The first is that BimEL degradation proceeds through an alternative non-proteasomal pathway in the absence of Cbl. However, we found that BimEL was degraded by the same ERK1/2-dependent and proteasome-dependent pathway in Cbl-/- iMEFs as has been described in many other cell types. A second explanation is that Cbl is just one E3 ligase capable of targeting BimEL for destruction. For example, different BimEL E3 ligases may have distinct tissue distributions so that Cbl is important in osteoclasts [22] but is not the relevant E3 in fibroblasts (iMEFs) or epithelial cells (HEK293s). However, it is worth noting that even in osteoclasts the loss of Cbl caused only a very modest reduction in BimEL turnover [22] suggesting a relatively minor role for Cbl. In addition to cell type specificity, Cbl may be important in mediating turnover of BimEL in response to other, ERK1/2-independent pathways. However, to date only the RAF-MEK-ERK1/2 pathway has been shown to be involved in BimEL turnover so the significance of any alternative pathway is at best a matter of conjecture.
It is worth noting that several studies have shown that loss of Cbl causes enhanced activation of various receptor and non-receptor tyrosine kinases, resulting in enhanced downstream signalling to ERK1/2 and other effector pathways [36,37]. This includes the EGF receptor (EGFR/ErbB1), whose ligand-induced down-regulation requires Cbl [30]. Thus, the decrease in basal P-ERK1/2 and the increase in basal BimEL that we observed upon re-expression of HA-Cbl in Cbl-/- iMEFs (Figure 4) may reflect the re-imposition of negative regulation of such tyrosine kinases when Cbl is re-expressed. In this sense, there may be role for Cbl in the regulation of BimEL expression but this may be as an indirect positive regulator rather than a direct negative regulator.
5. Conclusion
In summary, we find no evidence to support a role for Cbl in mediating the growth factor-inducible, ERK1/2-dependent degradation of BimEL in fibroblasts and HEK293 epithelial cells; a role for Cbl may be confined to certain specific cell types or stimuli. We believe the identity of the E3 ubiquitin ligase responsible for the ERK1/2-dependent proteasomal degradation of BimEL is still unknown. It has long been recognised that the ability to target and modulate the activity of the proteasome may prove therapeutically useful [38]. Proteasome inhibitors have been shown to induce apoptosis and have anti-tumour effects in vivo, with malignant cells showing greater sensitivity [39] so the ability to inhibit the proteasome and promote apoptosis may provide an effective anti-cancer strategy. In tumours where the initiation of apoptosis is dependent on Bim, the ability to modulate levels of this protein could be of benefit. For example, it has been shown that Bim expression is required for cooperative cell killing and tumour regression in response to combined treatment with paclitaxel and proteasome inhibitors [40]. Consequently, the identification of the E3 ligase responsible for the degradation of BimEL is not only of academic interest, but may also prove useful in the development of new therapeutics for certain cancers.
Acknowledgments
We are grateful to Roland Baron for providing the Cbl expression construct and to Katherine Ewings and members of the Cook lab for advice and encouragement. We thank Becky Gilley for comments on the manuscript. CW was supported by a BBSRC Quota studentship. This work was supported by a BBSRC Competitive Strategic Grant to the Babraham Institute and BBSRC Project Grant BB/E02162X/1 (SJC) and the NIH grants CA 87986, CA 76118, CA 99900 and CA99163 (HB).
Abbreviations
- Bim
Bcl-2-interacting mediator of cell death
- BOP
BH3-only protein
- Cbl
Casitas B-lineage lymphoma oncogene
- EGF
Epidermal growth factor
- ERK1/2
extracellular signal-regulated protein kinases 1 & 2
- iMEF
immortalized mouse embryo fibroblast
- JNK
c-Jun N-terminal kinase
- MEK
MAPK or ERK Kinase
- PKB
protein kinase B
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Cory S, Adams JM. Nat Rev Cancer. 2002;2:647. doi: 10.1038/nrc883. [DOI] [PubMed] [Google Scholar]
- 2.Willis SN, Adams JM. Curr Opin Cell Biol. 2005;17:617. doi: 10.1016/j.ceb.2005.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Willis SN, Fletcher JI, Kaufmann T, van Delf MF, Chen L, Czabotar PE, Ierino H, Lee EF, Fairlie WD, Bouillet P, Strasser A, Kluck RM, Adams JM, Huang DCS. Science. 2007;315:856. doi: 10.1126/science.1133289. [DOI] [PubMed] [Google Scholar]
- 4.Puthalakath H, Strasser A. Cell Death Differ. 2002;9:505. doi: 10.1038/sj.cdd.4400998. [DOI] [PubMed] [Google Scholar]
- 5.Chen L, Willis SN, Wei A, Smith BJ, Fletcher JI, Hinds MG, Colman PM, Day CL, Adams JM, Huang DCS. Mol Cell. 2005;17:393. doi: 10.1016/j.molcel.2004.12.030. [DOI] [PubMed] [Google Scholar]
- 6.Bouillet P, Metcalf D, Huang DC, Tarlington DM, Kay TW, Kontgen F, Adams JM, Strasser A. Science. 1999;286:1735. doi: 10.1126/science.286.5445.1735. [DOI] [PubMed] [Google Scholar]
- 7.Whitfield J, Neame SJ, Paquet L, Bernard O, Ham J. Neuron. 2001;29:629. doi: 10.1016/s0896-6273(01)00239-2. [DOI] [PubMed] [Google Scholar]
- 8.Wang P, Gilmore AP, Streuli CH. J Biol Chem. 2004;279:41280. doi: 10.1074/jbc.C400248200. [DOI] [PubMed] [Google Scholar]
- 9.Ewings KE, Hadfield-Moorhouse K, Wiggins CM, Wickenden JA, Balmanno K, Gilley R, Degenhardt K, White E, Cook SJ. EMBO J. 2007;26:2856. doi: 10.1038/sj.emboj.7601723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dijkers PF, Medema RH, Lammers JW, Koenderman L, Coffer PJ. Curr Biol. 2000;10:1201. doi: 10.1016/s0960-9822(00)00728-4. [DOI] [PubMed] [Google Scholar]
- 11.Weston CR, Balmanno K, Chalmers C, Hadfield K, Molton SA, Ley R, Wagner EF, Cook SJ. Oncogene. 2003;22:1281. doi: 10.1038/sj.onc.1206261. [DOI] [PubMed] [Google Scholar]
- 12.Gilley J, Coffer PJ, Ham J. J Cell Biol. 2003;162:613. doi: 10.1083/jcb.200303026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.O'Connor L, Strasser A, O'Reilly LA, Hausmann G, Adams JM, Cory S, Huang DC. EMBO J. 1998;17:384. doi: 10.1093/emboj/17.2.384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ley R, Ewings KE, Hadfield K, Cook SJ. Cell Death Differ. 2005;12:1008. doi: 10.1038/sj.cdd.4401688. [DOI] [PubMed] [Google Scholar]
- 15.Ley R, Hadfield K, Howes E, Cook SJ. J Biol Chem. 2005;280:17657. doi: 10.1074/jbc.M412342200. [DOI] [PubMed] [Google Scholar]
- 16.Ley R, Balmanno K, Hadfield K, Weston C, Cook SJ. J Biol Chem. 2003;278:18811. doi: 10.1074/jbc.M301010200. [DOI] [PubMed] [Google Scholar]
- 17.Luciano F, Jaquel A, Colosetti P, Herrant M, Cagnol S, Pages G, Auberger P. Oncogene. 2003;22:6785. doi: 10.1038/sj.onc.1206792. [DOI] [PubMed] [Google Scholar]
- 18.Ley R, Ewings KE, Hadfield K, Howes E, Balmanno K, Cook SJ. J Biol Chem. 2004;279:8837. doi: 10.1074/jbc.M311578200. [DOI] [PubMed] [Google Scholar]
- 19.Marani M, Hancock D, Lopes R, Tenev T, Downward J, Lemoine NR. Oncogene. 2004;23:2431. doi: 10.1038/sj.onc.1207364. [DOI] [PubMed] [Google Scholar]
- 20.Ciechanover A. Nat Rev Mol Cell Biol. 2005;6:79. doi: 10.1038/nrm1552. [DOI] [PubMed] [Google Scholar]
- 21.Willems AR, Schwab M, Tyers M. Biochim Biophys Acta. 2004;1695:133. doi: 10.1016/j.bbamcr.2004.09.027. [DOI] [PubMed] [Google Scholar]
- 22.Akiyama T, Bouillet P, Miyazaki T, Kadono Y, Chikuda H, Chung UI, Fukuda A, Hikita A, Seto H, Okada T, Inaba T, Sanjay A, Baron R, Kawaguchi H, Oda H, Nakamura K, Strasser A, Tanaka S. EMBO J. 2003;22:6653. doi: 10.1093/emboj/cdg635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.El Chami N, Ikhlef F, Kaszas K, Yakoub S, Tabone E, Siddeek B, Cunha S, Beaudoin C, Morel L, Benahmed M, Regnier DC. J Cell Biol. 2005;171:651. doi: 10.1083/jcb.200507076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Meng W, Sawasdikosol S, Burakoff SJ, Eck MJ. Nature. 1999;398:84. doi: 10.1038/18050. [DOI] [PubMed] [Google Scholar]
- 25.Thien CB, Langdon WY. Biochem J. 2005;391:153. doi: 10.1042/BJ20050892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schmidt MH, Dikic I. Nat Rev Mol Cell Biol. 2005;6:907. doi: 10.1038/nrm1762. [DOI] [PubMed] [Google Scholar]
- 27.Yeh TC, Marsh V, Bernat BA, Ballard J, Colwell H, Evans RJ, Parry J, Smith D, Brandhuber BJ, Gross S, Marlow A, Hurley B, Lyssikatos J, Lee PA, Winkler JD, Koch K, Wallace E. Clin Cancer Res. 2007;13:1576. doi: 10.1158/1078-0432.CCR-06-1150. [DOI] [PubMed] [Google Scholar]
- 28.Boughan PK, Argent RH, Body-Malapel M, Park JH, Ewings KE, Bowie AG, Ong SJ, Cook SJ, Sorensen OE, Manzo BA, Inohara N, Klein NJ, Nuñez G, Atherton JC, Bajaj-Elliott M. J Biol Chem. 2006;281:11637. doi: 10.1074/jbc.M510275200. [DOI] [PubMed] [Google Scholar]
- 29.Andoniou CE, Lill NL, Thien CB, Lupher ML, Jr, Ota S, Bowtell DD, Scaife RM, Langdon WY, Band H. Mol Cell Biol. 2000;20:851. doi: 10.1128/mcb.20.3.851-867.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Duan L, Miura Y, Dimri M, Majumder B, Dodge IL, Reddi AL, Ghosh A, Fernandes N, Zhou P, Mullane-Robinson K, Rao N, Donoghue S, Rogers RA, Bowtell D, Naramura M, Gu H, Band V, Band H. J Biol Chem. 2003;278:28950. doi: 10.1074/jbc.M304474200. [DOI] [PubMed] [Google Scholar]
- 31.Sambrook J. Molecular Cloning: A laboratory manual. Cold Spring Harbour Laboratory Press; 1989. [Google Scholar]
- 32.Egle A, Harris AW, Bouillet P, Cory S. Proc Natl Acad Sci USA. 2004;101:6164. doi: 10.1073/pnas.0401471101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tagawa H, Karnan S, Suzuki R, Matsuo K, Zhang X, Ota A, Morishima Y, Nakamura S, Seto M. Oncogene. 2005;24:1348. doi: 10.1038/sj.onc.1208300. [DOI] [PubMed] [Google Scholar]
- 34.Langdon WY, Hartley JW, Klinken SP, Ruscetti SK, Morse HC. Proc Natl Acad Sci USA. 1989;86:1168. doi: 10.1073/pnas.86.4.1168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Seward RJ, von Haller PD, Aebersold R, Huber BT. Mol Immunol. 2003;39:983. doi: 10.1016/s0161-5890(03)00047-6. [DOI] [PubMed] [Google Scholar]
- 36.Murphy MA, Schnall RG, Venter DJ, Barnett L, Bertoncello I, Thien CB, Langdon WY, Bowtell DD. Mol Cell Biol. 1998;18:4872. doi: 10.1128/mcb.18.8.4872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Naramura M, Kole HK, Hu RJ, Gu H. Proc Natl Acad Sci U S A. 1998;95:15547. doi: 10.1073/pnas.95.26.15547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kisselev AF, Goldberg AL. Chemistry and Biology. 2001;8:739. doi: 10.1016/s1074-5521(01)00056-4. [DOI] [PubMed] [Google Scholar]
- 39.Voorhees PM, Dees EC, O'Neil B, Orlowski RZ. Clin Cancer Res. 2003;9:6316. [PubMed] [Google Scholar]
- 40.Tan TT, Degenhardt K, Nelson DA, Beaudoin B, Nieves-Neira W, Bouillet P, Villunger A, Adams JM, White E. Cancer Cell. 2005;7:227. doi: 10.1016/j.ccr.2005.02.008. [DOI] [PubMed] [Google Scholar]