

Abstract
Cisplatin is a chemotherapy drug that frequently causes auditory impairment due to the death of mechanosensory hair cells. Cisplatin ototoxicity may result from oxidative stress, DNA damage, and inflammatory cytokines. The transcription factor STAT1, an important mediator of cell death, can regulate all of these processes in other cell types. We used cultured utricles from mature Swiss Webster mice to investigate the role of STAT1 in cisplatin-induced hair cell death. We show that STAT1 phosphorylation is an early event in both hair cells and support cells after exposure of utricles to cisplatin. STAT1 phosphorylation peaked after 4 h of cisplatin exposure and returned to control levels by 8 h of exposure. The STAT1 inhibitor epigallocatechin gallate (EGCG) attenuated STAT1 phosphorylation in cisplatin-treated utricles and resulted in concentration-dependent increases in hair cell survival at 24 h postexposure. Furthermore, we show that utricular hair cells from STAT1-deficient mice are resistant to cisplatin toxicity. EGCG failed to provide additional protection from cisplatin in STAT1-deficient mice, further supporting the hypothesis that the protective effects of EGCG are due to its inhibition of STAT1. Treatment with IFN-γ, which also causes STAT1 activation, also induced hair cell death in wild-type but not STAT1-deficient mice. These results show that STAT1 is required for maximal cisplatin-induced hair cell death in the mouse utricle and suggest that treatment with EGCG may be a useful strategy for prevention of cisplatin ototoxicity.
Introduction
Cisplatin is a widely used chemotherapeutic agent with multiple adverse effects including nephrotoxicity, neurotoxicity, and ototoxicity. Cisplatin-induced sensorineural hearing loss primarily affects the hearing of high frequencies and corresponds to loss of mechanosensory hair cells and spiral ganglion cells in the basal turn of the cochlea. With higher doses or longer exposure, hearing loss extends to lower frequencies and can cause cell loss throughout most of the inner ear (Rybak et al., 2007).
Cisplatin induces death of malignant cells by creating inter- and intrastrand DNA crosslinks (Woźniak and Blasiak, 2002) and may also induce DNA damage in normal cells (Townsend et al., 2004a; Youlyouz-Marfak et al., 2008). Exposure of hair cells to cisplatin results in release of reactive oxygen species (ROS; Clerici et al., 1996), and antioxidants have been found to attenuate cisplatin ototoxicity, presumably by counteracting this response (Rybak and Kelly, 2003). Multiple studies have demonstrated secretion of inflammatory cytokines by auditory-like cell lines after exposure to cisplatin (Previati et al., 2007; So et al., 2007). Finally, a subset of cell signaling proteins appear to be involved in cisplatin ototoxicity, including the tumor suppressor p53 (Devarajan et al., 2002; Zhang et al., 2003), the MAP kinase protein ERK (So et al., 2007), caspases, and cytochrome c (Liu et al., 1998; Devarajan et al., 2002; Wang et al., 2004).
Signal transducers and activators of transcription (STAT) proteins are ubiquitous transcription factors involved in a diverse set of signaling cascades initiated by cytokines, growth factors, and cellular stress (Simon et al., 1998; Kim and Lee, 2007). Seven STAT proteins have been described. STAT1 has been recognized as an important mediator of cell death after various types of cellular stress (Battle and Frank, 2002). STAT1 activation occurs via phosphorylation at two conserved residues (tyrosine 701 and serine 727). Tyrosine phosphorylation occurs via Janus kinase proteins and has been associated with STAT1 dimerization and translocation to the nucleus in response to specific cytokines (Kim and Lee, 2007). Serine phosphorylation occurs via a diverse group of kinases and has been associated with exposure to ROS, UV, lipopolysaccharide (LPS), tumor necrosis factor α (TNF-α), and DNA damage (Kovarik et al., 1999; Stephanou et al., 2001, 2002; DeVries et al., 2004; Townsend et al., 2004a; Zykova et al., 2005; Kim and Lee, 2007). Serine phosphorylation of STAT1 may be required for maximal transcription and promotion of apoptosis after ischemia or other stress conditions (Stephanou et al., 2001).
Several of the events regulated by STAT1 have also been implicated in cisplatin ototoxicity, and STAT1 has been found to be activated in some other cell types after exposure to DNA-damaging chemotherapeutic drugs (DeVries et al., 2004; Townsend et al., 2004a; Youlyouz-Marfak et al., 2008). However, the role of STAT1 in cisplatin-induced death of hair cells has not yet been investigated. In the present study, we show that serine phosphorylation of STAT1 occurs after exposure of vestibular hair cells to cisplatin in vitro. We then show that pharmacologic and genetic manipulations of this event attenuate cisplatin-induced hair cell death.
Materials and Methods
Animals.
Swiss Webster mice at 4–6 weeks of age were obtained from Charles River. STAT1-deficient mice and wild-type mice of the same genetic background (129S6/SvEv) at 4 weeks of age were obtained from Taconic Farms. All animal procedures were approved by the Institutional Animal Care and Use Committee at the University of Washington.
Reagents and antibodies.
Cisplatin stock solution (1 mg/ml saline) was obtained from Bedford Laboratories. This stock solution has an acid pH (3.8–5.9). When kept at room temperature and away from light, this type of preparation is stable for months (Schuldes et al., 1997). (−)-Epigallocatechin gallate (EGCG) was obtained from Sigma-Aldrich and dissolved in sterile water to make a 5 mm stock solution. Recombinant mouse interferon-γ (IFN-γ) was obtained from R&D Systems and was dissolved in PBS with 0.1% bovine serum albumin (BSA).
Antibodies for labeling of mouse vestibular epithelium hair cells included a monoclonal mouse antibody against calmodulin (Sigma; diluted 1/150) and a polyclonal rabbit antibody against calbindin (Millipore; diluted 1/250). A polyclonal rabbit antibody against phospho-STAT1 (serine 727 residue) was obtained from Cell Signaling Technology (diluted 1/100). To label support cells in the vestibular epithelium, a polyclonal goat antibody against SOX2 was obtained from Santa Cruz Biotechnology (diluted 1/500). Secondary antibodies included Alexa 488 goat-anti-mouse, donkey-anti-mouse, and donkey-anti-goat; and Alexa 568 goat-anti-rabbit and donkey-anti-goat (Invitrogen; all diluted 1/500). Hoechst 33258 nuclear stain was obtained from Sigma.
Dissection and culture of mouse utricles.
Mice were killed and their temporal bones removed to expose the inner ear. Utricles were dissected using aseptic technique as previously described (Yamashita and Oesterle, 1995; Cunningham, 2006). Otoconia were removed before culture of utricles. Using a small syringe filled with DMEM (Sigma) and a 26 gauge needle, a gentle stream of DMEM was directed toward the exposed utricle until all otoconia were separated from the utricular epithelium. Utricles were then cultured free-floating in 4-well, nontreated culture plates (1–4 utricles per well) in DMEM supplemented with 1% fetal bovine serum (Invitrogen) and the non-ototoxic antibiotic ofloxacin (0.12%; Sigma). Utricles were cultured at 37°C and 5% CO2 for 1–24 h. Utricles were cultured in control medium (DMEM only), IFN-γ (125–10,000 international units/ml), cisplatin (10–80 μg/ml), the STAT1 inhibitor EGCG (10–200 μm), or cisplatin plus EGCG at all concentration combinations. All culture conditions and experimental manipulations were replicated in at least three independent experiments.
Immunocytochemistry.
At the end of each culture period, utricles were fixed for one h at room temperature with 4% paraformaldehyde followed by three rinses in PBS. Utricles were then incubated for 30 min at room temperature in blocking solution consisting of PBS, 10% normal goat or donkey serum, 2% BSA, 0.03% saponin, and 0.3% Triton X-100. Utricles were then incubated overnight (12–16 h) at 4°C in primary antibody diluted in solution consisting of PBS, 1% normal goat or donkey serum, and 0.1% Triton X-100. Tissue was then rinsed four times in PBS and then incubated in Alexa fluorophore-conjugated secondary antibodies (diluted in the same solution as primary antibodies) at room temperature for 2 h. For experiments investigating STAT1 phosphorylation, utricles were then incubated in 2 μm Hoechst 33258 for 20 min to label nuclei. All utricles were rinsed four additional times with PBS and whole-mounted in Fluoromount-G (Electron Microscopy Sciences).
Microscopy and photomicrograph analysis.
For STAT1 phosphorylation studies, utricles were labeled with an antibody against phospho-STAT1-serine (pSTAT1Ser) and an antibody against calmodulin to label hair cells. Utricles were then examined in their entirety at low power with an Olympus FluoView-1000 confocal microscope. A confocal z-axis series of randomly selected representative areas was then taken at high power with identical microscopy settings across all experimental and control tissue samples for the channel used to detect pSTAT1Ser.
For hair cell counts, utricles were double-labeled to distinguish two distinct populations of hair cells, extrastriolar and striolar, which show differential sensitivity to some ototoxic drugs (Yan et al., 1991; Cunningham and Brandon, 2006; Chiu et al., 2008). An antibody against calmodulin was used to label all hair cells, and a second antibody against calbindin was used to label only the striolar hair cells (Cunningham et al., 2002). Utricles were then imaged with a Zeiss Axioplan 2ie fluorescence microscope. Using ImageJ software, a 30 μm × 30 μm box was placed in eight different locations within the utricle: two in the medial extrastriolar region, two in the lateral extrastriolar region, and four evenly spaced across the striolar region. The hair cells within each box were counted, and the average number of hair cells per 900 μm2 box was calculated for the extrastriolar and striolar regions of each utricle, as previously described (Cunningham et al., 2002; Cunningham and Brandon, 2006). All resulting numbers were then multiplied by a factor of 1.111 to obtain the average number of hair cells per 1000 μm2.
SOX2 labels support cell nuclei as well as a small subset of hair cell nuclei (Oesterle et al., 2008). The few labeled hair cells were easily distinguished from the solid support cell nuclear layer. However, to avoid mistakenly counting hair cells as support cells, the microscope was advanced in the z-axis until all hair cells disappeared from view before obtaining photomicrographs of the support cell nuclear layer. Support cells were then counted in a similar manner to hair cells: four 30 μm × 30 μm boxes were evenly spaced across the utricle (two medially and two laterally) and support cells were counted in each box. The average number of support cells per 900 μm2 box was then calculated for each utricle. All resulting numbers were then multiplied by a factor of 1.111 to obtain the average number of support cells per 1000 μm2.
Statistical analysis.
Data were analyzed with Student's t test or with one- or two-way ANOVA and post hoc Tukey–Kramer or Bonferroni multiple comparison tests. GraphPad Prism Software was used for all statistical analyses. Results were considered significant for p values of <0.05.
Results
Utricular hair cells are lost in a concentration-dependent manner after 24 h treatment with cisplatin
Utricles were cultured in increasing concentrations of cisplatin for 24 h to determine the concentration–response relationships for its effects on extrastriolar and striolar hair cells (Fig. 1). Cisplatin treatment resulted in a concentration-dependent decrease in hair cell survival (p < 0.001 for both extrastriolar and striolar hair cells by one-way ANOVA). For both extrastriolar and striolar groups, post hoc tests showed that hair cell density decreased significantly vs control for all cisplatin concentrations at or >20 μg/ml (p < 0.001, Tukey–Kramer multiple comparison).
Figure 1.

Cisplatin treatment resulted in concentration-dependent decreases in hair cell survival (mean ± 1 SEM, p < 0.001). The concentration–response relationships of extrastriolar hair cells (solid line, squares) and striolar hair cells (dashed line, triangles) were determined in utricles treated for 24 h with increasing concentrations of cisplatin. Equivalent concentrations of cisplatin in μm are given in parentheses for comparison. Hair cell density was determined as the average number of hair cells per 1000 μm2 area. ***p < 0.001 compared with control.
STAT1 phosphorylation at Ser727 is an early event after exposure to cisplatin and is attenuated by EGCG
Immunocytochemistry was used to investigate whether STAT1 is phosphorylated at Ser727 after exposure of utricular hair cells to cisplatin and whether epigallocatechin gallate (EGCG) inhibits this event. Epigallocatechin gallate (EGCG) is one of several polyphenol catechins isolated from green tea extract and has been reported to inhibit phosphorylation and upregulation of STAT1 after a variety of cellular stress events (Menegazzi et al., 2001; Tedeschi et al., 2002; Townsend et al., 2004a; de Prati et al., 2005; Zykova et al., 2005).
Control cultures were maintained in culture medium only. Positive control cultures were treated with IFN-γ, a cytokine known to cause robust phosphorylation of STAT1, at a concentration of 250 international units (IU)/ml. Additional groups of cultures were treated with cisplatin at a concentration of 40 μg/ml, with or without 50 μm EGCG (after a 1 h pretreatment with EGCG alone). Utricles were cultured in these four treatment conditions for 1, 4, or 8 h, based on previous studies of STAT1 phosphorylation after cisplatin exposure in other cell types (Townsend et al., 2004a; Youlyouz-Marfak et al., 2008). Confocal photomicrographs of cultured utricles are shown in Figure 2. The results shown in this figure are representative examples from three to five independent experiments. Hair cells from untreated control cultures showed either undetectable or very faint nuclear labeling for pSTAT1Ser (e.g., Fig. 2 A). Cultures treated with IFN-γ exhibited more robust nuclear labeling of pSTAT1Ser at 1 h and strikingly increased labeling at 4 h (Figs. 2 B,F). Hair cells from cultures treated with cisplatin showed a similarly strong nuclear pattern for pSTAT1Ser at these time points (Figs. 2 C,G). In hair cells from utricles treated with cisplatin and the STAT1 inhibitor EGCG, a nuclear label for pSTAT1Ser was detected but appeared consistently weaker than in cultures treated with cisplatin alone (Figs. 2 D,H). By 8 h, pSTAT1Ser labeling was markedly diminished in all treatment groups (Figs. 2 J–L). Negative control cultures from all time points showed no detectable nuclear pSTAT1Ser pattern (data not shown).
Figure 2.

Serine phosphorylation of STAT1 is an early event after IFN-γ and cisplatin exposure. Mouse utricles were cultured for 1, 4, or 8 h in control medium (A, E, I), with IFN-γ as a positive control (B, F, J), with cisplatin at 40 μg/ml (133 μm; C, G, K), or with cisplatin at 40 μg/ml plus the STAT1 inhibitor EGCG at 50 μm (D, H, L). Hair cells were labeled with an antibody against calmodulin (green) and an antibody against pSTAT1Ser (red). Nuclei were counterstained with Hoechst 33258 (blue). Each inset shows an enlargement of one hair cell with its nucleus outlined (white dotted line). Very little pSTAT1Ser was noted in hair cells from control utricles (A, E, I). A faint pSTAT1Ser label was noted in the IFN-γ and cisplatin-treated utricles at 1 h (B, C), which peaked at 4 h (F, G) and was diminished by 8 h (J, K). In utricles treated with cisplatin plus EGCG, the pSTAT1Ser label consistently appeared diminished compared with utricles treated with cisplatin alone at 1 and 4 h (D, H). Examples shown are most representative of 6–8 utricles in each group from three to five independent experiments. Scale bars represent 10 μm.
We consistently saw a punctate labeling pattern for pSTAT1Ser. This was seen outside of the hair cell boundaries (presumably in the nuclei and apical processes of support cells) and to a lesser degree in the cytoplasm of hair cells. This pattern was seen in the majority of utricles treated with IFN-γ or cisplatin, and to a much lesser degree in untreated utricles or those treated with both cisplatin and EGCG. This was not seen in negative controls where the primary antibody was omitted but was seen when a different Alexa fluorophore or diaminobenzidine chromagen (DAB) were used (data not shown). This pattern may be consistent with localization of pSTAT1Ser to membrane-bound organelles known as signaling endosomes (Claudinon et al., 2007).
Pharmacologic inhibition of STAT1 attenuates cisplatin-induced death of utricular hair cells
We next asked whether pharmacologic inhibition of STAT1 could attenuate cisplatin-induced hair cell death. Utricles were cultured with a range of EGCG concentrations (10–200 μm) for one h, followed by continuous treatment with both EGCG and a moderate concentration of cisplatin (40 μg/ml) for 24 h. Figure 3 shows representative utricles after treatment with control medium, cisplatin plus EGCG, and cisplatin or EGCG alone. Cisplatin treatment resulted in loss of hair cells and morphologic changes suggestive of hair cell damage, including misshapen, swollen or shrunken cells. These changes were highly attenuated by the addition of EGCG.
Figure 3.
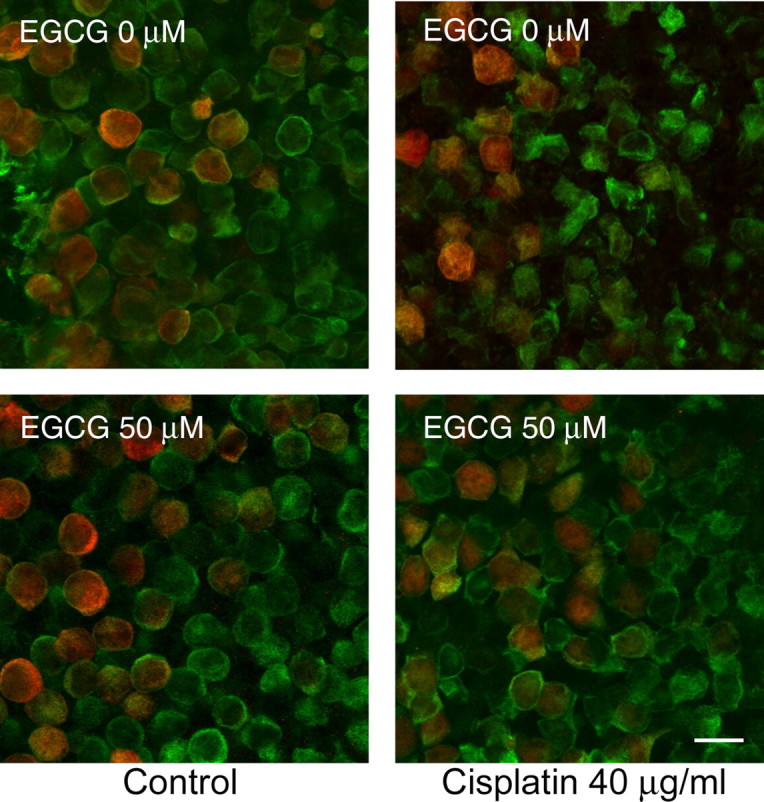
EGCG reduces hair cell loss and morphologic damage after cisplatin treatment. Utricles were cultured in control medium or a moderate concentration of cisplatin for 24 h with or without the STAT1 inhibitor EGCG. Confocal photomicrographs were taken at the junction of extrastriolar hair cells (green, labeled for calmodulin) and striolar hair cells (red and green, labeled for calbindin and calmodulin, respectively). Treatment with cisplatin alone resulted in hair cell loss and morphologic damage, including cellular shrinkage or swelling. When treated with cisplatin plus EGCG, utricular hair cells demonstrated near-normal density and morphology. Examples shown are representative utricles from at least three independent experiments. Scale bar represents 10 μm.
The effects of EGCG on hair cell survival are shown in more detail in Figure 4. EGCG treatment resulted in a significant concentration-dependent increase in survival of extrastriolar and striolar hair cells when exposed to 40 μg/ml cisplatin (Fig. 4 A; p < 0.001 and p < 0.01, respectively, one-way ANOVA). At concentrations of 10 μm and above, EGCG increased the survival of extrastriolar hair cells vs cisplatin alone (p < 0.05 and p < 0.001, Tukey–Kramer). Striolar hair cells were significantly protected from cisplatin at EGCG concentrations of 50 and 100 μm (p < 0.05). Treatment with EGCG alone did not result in a loss of hair cells at any of the concentrations used (data not shown).
Figure 4.

EGGC protects utricular hair cells from cisplatin. A, Utricles were treated for 24 h with or without a moderate concentration of cisplatin (40 μg/ml) and increasing concentrations of EGCG, which resulted in concentration-dependent increases in hair cell survival up to the optimal concentration of 50 μm EGCG (mean + 1 SEM, p < 0.001 for extrastriolar, p < 0.01 for striolar, n = 7–10 utricles per group). B, Utricles were treated for 24 h with increasing concentrations of cisplatin with or without the optimal concentration of EGCG (50 μm, based on results in A). Density of both extrastriolar hair cells (solid lines) and striolar hair cells (dashed lines) was increased by EGCG (mean ± 1 SEM, p < 0.001, n = 7–10 utricles per group). *p < 0.05, **p < 0.01, and ***p < 0.001 versus same dose of cisplatin without EGCG. Equivalent μm concentrations of cisplatin are shown in Figure 1.
Based on these results, we pretreated utricles with 50 μm EGCG for one h followed by treatment with both EGCG and escalating concentrations of cisplatin for 24 h. This concentration of EGCG significantly attenuated hair cell death induced by a wide range of cisplatin concentrations (Fig. 4 B; p < 0.001 for both extrastriolar and striolar hair cells by two-way ANOVA). Post hoc Bonferroni comparison tests showed that extrastriolar hair cells treated with EGCG were highly protected from all concentrations of cisplatin (p < 0.001). EGCG also protected striolar hair cells from cisplatin at concentrations of 40 μg/ml (p < 0.01) and 80 μg/ml (p < 0.001).
Interferon-γ results in a concentration-dependent loss of utricular hair cells
Because cisplatin-induced STAT1 phosphorylation appears to play a role in hair cell death, it is also possible that the classic STAT1-activating cytokine, IFN-γ, also induces hair cell death. IFN-γ has been reported to cause upregulation of the apoptosis-inducing protein Fas ligand in cochlear tissue from postnatal mice (Bodmer et al., 2003). However, to our knowledge the question of whether exposure to IFN-γ ultimately results in hair cell death has not been investigated.
Utricles from wild-type Swiss Webster mice were cultured with increasing concentrations of IFN-γ for 24 h, which resulted in loss of both extrastriolar and striolar hair cells in a concentration-dependent manner (Fig. 5; p < 0.001 and p < 0.05, respectively, by one-way ANOVA). For extrastriolar hair cells, a significant hair cell loss vs control was seen with concentrations at or >250 IU/ml (p < 0.001, Tukey–Kramer comparison). For striolar hair cells, a significant decrease in hair cell density vs control was seen with a concentration of 500 IU/ml (p < 0.05, Tukey–Kramer comparison). No additional loss of hair cells was noted with concentrations >500 IU/ml (data not shown).
Figure 5.

Effects of IFN-γ on hair cell survival. Utricles from wild-type Swiss Webster mice were treated with increasing concentrations of IFN-γ (0 – 500 IU/ml) for 24 h, which resulted in a concentration-dependent decrease in survival of both extrastriolar hair cells (solid line) and striolar hair cells (dashed line) (mean ± 1 SEM, p < 0.001 and p < 0.05 for extrastriolar and striolar, respectively; n = 7–9 utricles per group). *p < 0.05, ***p < 0.001 compared with control.
Utricular hair cells from STAT1−/− mice are highly resistant to cisplatin toxicity and appear unaffected by EGCG
We have demonstrated that EGCG attenuates cisplatin-induced hair cell death in the adult mouse utricle. We hypothesized that this result is due to the inhibition of STAT1. However, EGCG is also a powerful antioxidant (Tedeschi et al., 2002). This property alone could provide substantial protection of hair cells from cisplatin, as has been demonstrated in multiple previous studies using different antioxidants (Rybak and Kelly, 2003).
To further examine the hypothesis that STAT1 is necessary for cisplatin-induced hair cell death, we next cultured utricles from STAT1−/− and wild-type mice of the same genetic background with increasing concentrations of cisplatin for 24 h. Figure 6 shows photomicrographs from representative utricles of STAT1+/+ and STAT1−/− mice treated with control medium or the same medium with 40 μg/ml cisplatin. In utricles from STAT1+/+ mice treated with cisplatin, the majority of hair cells were misshapen, swollen, shrunken, or missing. Utricles from STAT1−/− mice treated with the same concentration of cisplatin demonstrated remarkable morphologic similarity to control utricles from mice of both genotypes. Quantitatively, utricular hair cells from STAT1−/− mice were found to be highly resistant to cisplatin (Fig. 7 A; p < 0.001 for both extrastriolar and striolar hair cells by two-way ANOVA). Extrastriolar hair cells from STAT1−/− mice showed resistance to all concentrations of cisplatin (p < 0.05 for 20 μg/ml and p < 0.001 for 40 and 80 μg/ml, Bonferroni comparison). Striolar hair cells from STAT1−/− mice also demonstrated highly significant resistance to a high concentration of cisplatin (80 μg/ml; p < 0.001). These results strongly suggest that STAT1 is indeed required for cisplatin-induced vestibular hair cell death under these in vitro conditions.
Figure 6.
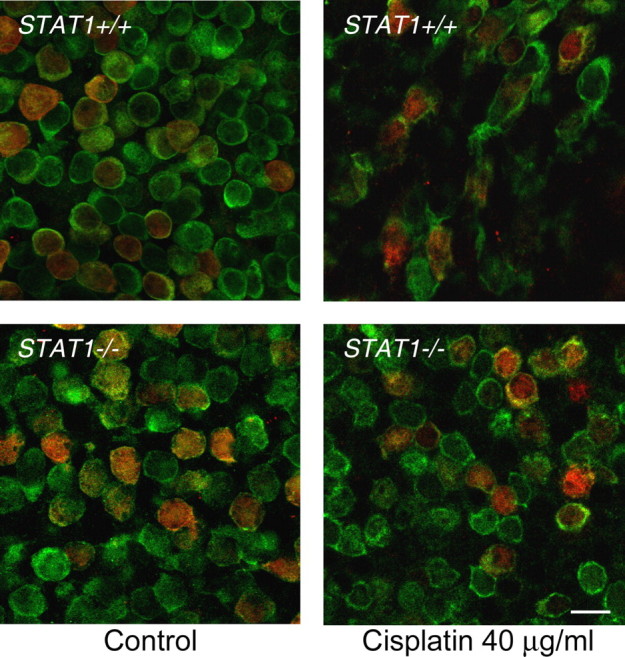
Utricles from STAT1−/− mice are resistant to hair cell loss and morphologic damage after cisplatin treatment. Utricles from STAT1+/+ and STAT1−/− mice were cultured in control medium or with a moderate concentration of cisplatin for 24 h. Confocal photomicrographs were taken at the junction of extrastriolar hair cells (green, labeled for calmodulin) and striolar hair cells (red and green, labeled for calbindin and calmodulin, respectively). Treatment of utricles from STAT1+/+ mice with cisplatin resulted in decreased hair cell density and substantial morphologic damage, including hair cell swelling or shrinkage. Utricles from STAT1−/− mice treated with cisplatin demonstrated near-normal density and morphology. Examples shown are representative utricles from three independent experiments. Scale bar represents 10 μm.
Figure 7.

Utricles from STAT1−/− mice are protected from cisplatin- and IFN-γ-induced hair cell death and are unaffected by EGCG. A, Utricles from STAT1+/+ and STAT1−/− mice were treated with increasing concentrations of cisplatin for 24 h. Death of both extrastriolar hair cells (solid lines) and striolar hair cells (dashed lines) was significantly decreased in utricles from STAT1−/− mice compared with utricles from STAT1+/+ mice treated with the same concentrations of cisplatin (mean ± 1 SEM, p < 0.001, n = 5–6 utricles per group). *p < 0.05, ***p < 0.001 vs same dose of cisplatin in utricles from STAT1+/+ mice. Equivalent μm concentrations of cisplatin are shown in Figure 1. B, Utricles from STAT1−/− mice were treated with a high concentration of cisplatin alone or cisplatin plus EGCG for 24 h. The addition of EGCG failed to increase hair cell survival (mean + 1 SEM, p > 0.9, n = 5–6 utricles per group). C, Utricles from STAT1−/− mice were cultured in control medium or a high concentration of IFN-γ (500 IU/ml) for 24 h. IFN-γ did not affect hair cell survival versus utricles treated with control medium (mean + 1 SEM, p > 0.4, n = 5–6 utricles per group).
We next asked whether EGCG could provide additional hair cell protection in utricles from STAT1−/− mice treated with a high concentration of cisplatin. Utricles were pretreated with EGCG at 50 μm for one h, followed by incubation for 24 h with this concentration of EGCG and cisplatin (80 μg/ml). There was no significant difference between hair cell densities in STAT1−/− utricles treated with cisplatin plus EGCG vs cisplatin alone (Fig. 7 B; p > 0.9 for both extrastriolar and striolar hair cells by two-tailed t test). The observation that this powerful antioxidant failed to provide additional protection from cisplatin toxicity in utricular hair cells from STAT1−/− mice further suggests that the primary mechanism of protection by EGCG is its inhibition of STAT1 phosphorylation.
Next, utricles from STAT1−/− mice were treated with a high concentration of IFN-γ (500 IU/ml), and there was no significant difference in hair cell density between control and IFN-γ-treated utricles (Fig. 7 C; for both extrastriolar and striolar hair cell density, p > 0.4 by two-tailed t test). This result is consistent with other studies demonstrating that cells from these mice are insensitive to interferons (Kim and Lee, 2007). The cytotoxic effects of IFN-γ in general are primarily due to its activation of STAT1. The observation that this cytokine can induce hair cell death in wild-type but not STAT1−/− mice strongly suggests that STAT1 activation is required for inducing hair cell death in vitro after IFN-γ exposure.
Support cells from utricles exposed to cisplatin demonstrate STAT1 serine phosphorylation but are not lost within 24 h
Throughout our experiments, we noted significant pSTAT1Ser in Hoechst-labeled areas near the basement membrane, presumably in the nuclear layer of support cells. To confirm this, additional utricles from wild-type Swiss Webster mice were treated with control medium or a moderate concentration of cisplatin (40 μg/ml) for 4 h and labeled for pSTAT1Ser and with an antibody against SOX2 (to label support cell nuclei). Confocal microscopy demonstrated scant, punctate pSTAT1Ser in the nuclei of support cells treated in control medium, but a marked increase in utricles treated with cisplatin (Fig. 8 A).
Figure 8.
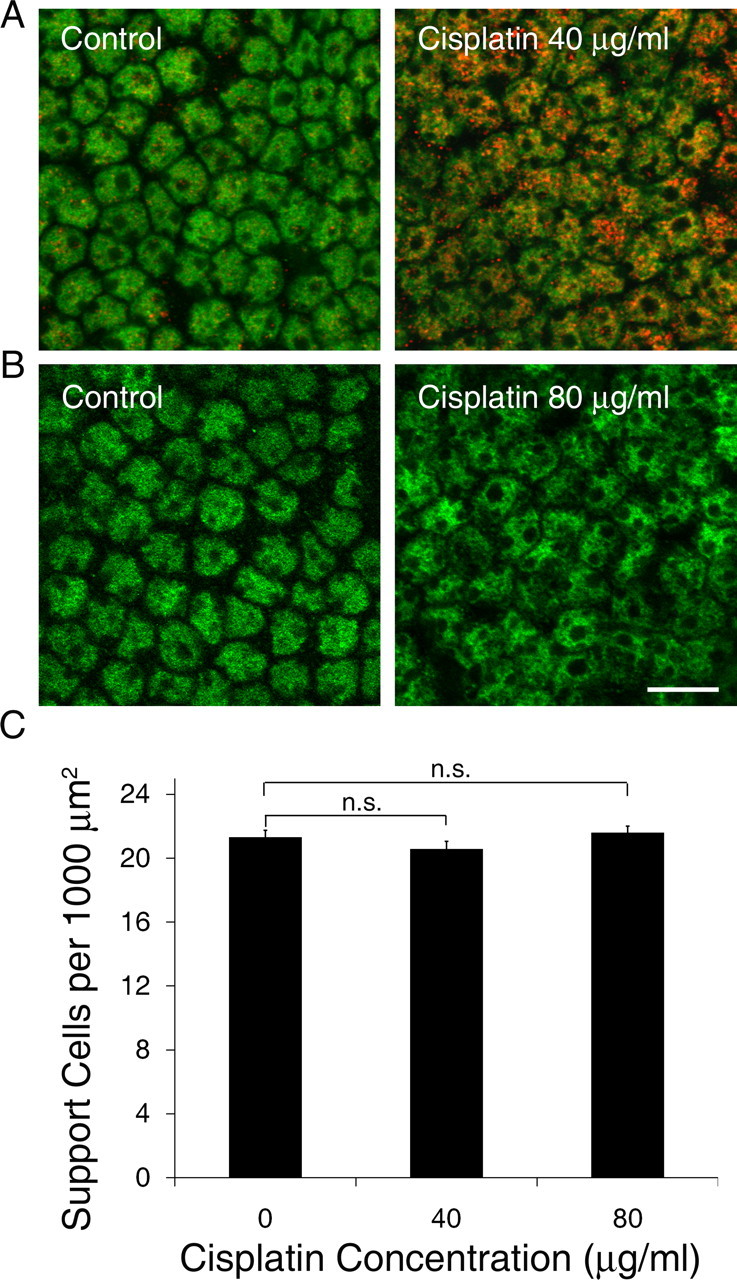
Effect of cisplatin on support cells. A, Utricles were treated with control medium or a moderate concentration of cisplatin for 4 h and labeled with pSTAT1Ser (red) and SOX2 (green) to visualize support cell nuclei. Support cells from control utricles demonstrated scant, punctate pSTAT1Ser (left), whereas pSTAT1Ser was abundant in nuclei of support cells from cisplatin-treated utricles (right). B, Utricles were treated with control medium or a high concentration of cisplatin for 24 h and labeled with SOX2 to visualize support cells. Cisplatin treatment resulted in mild disorganization but similar overall density of support cells (right). Scale bar corresponds to all panels and represents 10 μm. C, Utricles were treated with control medium or cisplatin for 24 h and support cell density was quantified. Cisplatin treatment did not result in a loss of support cells within 24 h (mean + 1 SEM, p > 0.2, n = 7–8 utricles per group). Equivalent μm concentrations of cisplatin are shown in Figure 1.
Given the parallel increase in serine-phosphorylated STAT1 after cisplatin exposure in support cells, we next cultured additional utricles in control medium or cisplatin for 24 h to determine whether the cisplatin-induced increase in STAT1 phosphorylation is sufficient to induce death of support cells in addition to hair cells. Confocal microscopy showed normal morphology but mild disorganization of the support cell nuclear layer after treatment with cisplatin (Fig. 8 B). This was not surprising, given that approximately half of the hair cells are lost and the overall utricular architecture is severely disrupted with a high concentration of cisplatin. However, the density of support cells did not change significantly after 24 h of continuous cisplatin exposure (Fig. 8 C; p > 0.2, one-way ANOVA).
Discussion
We show that treatment of vestibular hair cells with cisplatin results in serine-phosphorylation of STAT1, and that pharmacological or genetic inhibition of STAT1 attenuates cisplatin-induced hair cell death. STAT1 has increasingly been recognized as an important mediator of cell death. A variety of cell signaling cascades involved in cell death converge on this transcription factor (Kim and Lee, 2007).
STAT1 as a mediator of cisplatin-induced hair cell loss
Pathways implicated in cisplatin-induced hair cell death
Cisplatin is known to be genotoxic, and previous studies of cisplatin ototoxicity have implied the involvement of reactive oxygen species (ROS) and inflammatory cytokines. STAT1 is likely to be directly involved in the response to DNA damage, which has been reported in studies of other cell types and may involve p53 (Townsend et al., 2004a; Youlyouz-Marfak et al., 2008). STAT1 could also be involved in cisplatin-induced oxidative stress. Work by So et al. (2007) suggests that the release of ROS by a cisplatin-treated auditory-like cell line is downstream of inflammatory cytokines, which frequently activate STAT1. STAT1 may then participate in this release of ROS by regulating inducible nitric oxide synthase and other ROS-producing enzymes (Moriwaki et al., 2006; Kim and Lee, 2007; Lee et al., 2007; Stempelj et al., 2007). STAT1 can also be activated in response to ischemia/reperfusion injury and exogenous ROS such as H2O2 (Simon et al., 1998; Takagi et al., 2002; Terui et al., 2004; Townsend et al., 2004b), which can lead to a positive feedback loop of oxidative stress and cell damage. Finally, STAT1 induces constitutive expression of caspases and interacts directly with p53, both of which have been implicated in cisplatin-induced death of hair cells or auditory-like cells (Devarajan et al., 2002; Wang et al., 2004).
Other STAT1-related pathways that may be involved in cisplatin ototoxicity
We have not yet investigated which kinase(s) and downstream pathways are involved in the effects of pSTAT1Ser on cisplatin-treated utricular hair cells. It is possible that robust release of TNF-α (as noted in cisplatin-treated auditory-like cell lines by Previati et al., 2007, and So et al., 2007) is primarily responsible for STAT1 serine phosphorylation via its actions on p38 MAP kinase (Kim and Lee, 2007). STAT1 can also be serine-phosphorylated by protein kinase Cδ, which was observed in a study of HeLa cells treated with the genotoxic chemotherapy drug etoposide (DeVries et al., 2004). Finally, STAT1 can also be phosphorylated by the MAP kinase protein ERK (Haq et al., 2002), which was shown to be activated in an auditory cell line after cisplatin exposure (So et al., 2007).
Resistance of STAT1−/− hair cells to cisplatin
Cells from STAT1 gene-targeted mice have been found to be resistant to oxidative stress and a variety of other insults (Meraz et al., 1996; Kim and Lee, 2007). In the present study we demonstrate that utricular hair cells from these mice are highly resistant to cisplatin toxicity. STAT1−/− mice are homozygous viable and exquisitely sensitive to microbial infections due to a lack of sensitivity to interferons, which require STAT1 for their function (Meraz et al., 1996). These mice are also reported to have mildly increased bone density (Xiao et al., 2004), although this was not apparent in the otic capsule during microdissection of utricles. We also found these mice to have auditory brainstem response thresholds similar to wild-type mice of the same genetic background (data not shown). These mice do have low levels of STAT1, with a truncation of the N terminus that renders the protein dysfunctional (Meraz et al., 1996). The truncation does not affect the C-terminal domain that contains the phosphorylation sites (Kim and Lee, 2007), and a serine-phosphorylated, truncated form of STAT1 has been reported in stimulated cells from these mice (Stephanou et al., 2002). This observation suggests that either the N terminus or the total protein level of STAT1 may be important for in vitro cisplatin-induced hair cell death.
Prevention of cisplatin-induced hair cell death using green tea polyphenols
To our knowledge the effects of EGCG on hair cell injury have not been previously studied, but EGCG has been demonstrated to protect spiral ganglion neurons from exogenous H2O2 (Xie et al., 2004). A recent study also suggested protection from cisplatin toxicity in an auditory-like cell line and zebrafish lateral line hair cells by another green tea polyphenol, epigallocatechin (EC), presumably via its antioxidant effects (Kim et al., 2008). Of the several green tea polyphenols, only EGCG has been demonstrated to inhibit STAT1 activity and the expression of STAT1-inducible genes (Tedeschi et al., 2002). EGCG has also been specifically recognized as the polyphenol primarily responsible for the chemotherapeutic effects of green tea extract (Chen et al., 2008). Although it is possible that the protection of hair cells by EGCG in the present study is due in part to its antioxidant effects, the failure of EGCG to provide additional protection of cisplatin-treated hair cells from STAT1−/− mice strongly suggests that EGCG prevents cisplatin-induced hair cell death primarily by inhibiting STAT1.
Potential advantages of STAT1 inhibition over other proposed protective strategies
Prior animal studies have shown substantial protection from cisplatin using antioxidants (Rybak and Kelly, 2003). However, some antioxidants may hinder the chemotherapeutic effect of cisplatin (Reser et al., 1999). Clinical studies using antioxidants have been disappointing, and use of caspase or p53 inhibitors could have considerable side effects including enhanced tumor proliferation (Rybak et al., 2007). STAT1 inhibition may be more clinically acceptable, because some tumors demonstrate aberrant or constitutive activation of STAT1 (Klampfer, 2006). EGCG in particular has long been recognized as a chemopreventive and chemotherapeutic agent (Chen et al., 2008). Multiple studies have demonstrated that EGCG can protect normal cells from toxicity while enhancing the effect of chemotherapy drugs on malignant cells (Hsu et al., 2002; Yamamoto et al., 2004; Chen et al., 2008).
Limitations of STAT1 inhibition
Although inhibition of STAT1 appears to be a promising strategy for prevention of cisplatin ototoxicity, our results cannot be extrapolated to in vivo or clinical situations without further study. One group of investigators studying the role of STAT1 in cardiac ischemia/reperfusion injury noted that EGCG provided protection but STAT1−/− mice were more susceptible (Stephanou et al., 2002; Townsend et al., 2004b). This example and others in the literature demonstrate that manipulations of STAT1 can have dramatically different results depending on the cell type, species, and method of STAT1 manipulation.
Effects of cisplatin on support cells
We noted that cisplatin also induced serine phosphorylation of STAT1 in support cells of the utricular epithelium. However, unlike hair cells, the support cells were not lost within 24 h of continuous cisplatin exposure. The repeated observation that support cells exhibit several of the same responses as hair cells on exposure to toxic agents but do not die as readily is a prominent quandary in the research fields of hair cell death and regeneration. It may be that in support cells, phosphorylated STAT1 disproportionately activates downstream mediators that are protective, such as NF-κB or heat shock proteins. Heat shock can protect hair cells from cisplatin toxicity (Cunningham and Brandon, 2006), and STAT1 has been reported to play an anti-apoptotic role after exposure to oxidative stress via induction of HSP70 in hepatic cells (Terui et al., 2004). This is yet another example of the specificity of STAT1 signaling and function dependent on the cellular context.
Implications of the ototoxic effects of IFN-γ
Our results indicate that like other inflammatory cytokines, IFN-γ can also induce hair cell death. The observation that cisplatin and IFN-γ induce substantial hair cell death in wild-type but not in STAT1−/− mice strongly suggests that STAT1 is necessary for hair cell death induced by both agents. It is possible that IFN-γ is responsible for the cisplatin-induced STAT1 phosphorylation that we observed in hair cells, although one study of cisplatin nephrotoxicity showed that IFN-γ levels were not increased (Ramesh and Reeves, 2002). Given the observation that cisplatin can enhance IFN-γ induced apoptosis of cancer cells (Barton et al., 2005), it is more likely that these two agents converge on STAT1 or other common mediators.
Footnotes
This work was supported by National Institutes of Health Grants P30DC004661, T32DC00018, and F32DC009551 from the National Institute on Deafness and Other Communication Disorders as well as a seed grant from the Virginia Merrill Bloedel Hearing Research Center. We thank Dr. David Raible, Dr. Allison Coffin, Glen MacDonald, Dr. Jennifer Stone, and Dr. Elizabeth Oesterle for technical assistance.
References
- Barton C, Davies D, Balkwill F, Burke F. Involvement of both intrinsic and extrinsic pathways in IFN-γ-induced apoptosis that are enhanced with cisplatin. Eur J Cancer. 2005;41:1474–1486. doi: 10.1016/j.ejca.2005.03.022. [DOI] [PubMed] [Google Scholar]
- Battle TE, Frank DA. The role of STATs in apoptosis. Curr Mol Med. 2002;2:381–392. doi: 10.2174/1566524023362456. [DOI] [PubMed] [Google Scholar]
- Bodmer D, Brors D, Bodmer M, Pak K, Ryan AF. Fas ligand expression in the organ of Corti. Audiol Neurootol. 2003;8:243–249. doi: 10.1159/000071996. [DOI] [PubMed] [Google Scholar]
- Chen D, Milacic V, Chen MS, Wan SB, Lam WH, Huo C, Landis-Piwowar KR, Cui QC, Wali A, Chan TH, Dou QP. Tea polyphenols, their biological effects and potential molecular targets. Histol Histopathol. 2008;23:487–496. doi: 10.14670/hh-23.487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiu LL, Cunningham LL, Raible DW, Rubel EW, Ou HC. Using the zebrafish lateral line to screen for ototoxicity. J Assoc Res Otolaryngol. 2008;9:178–190. doi: 10.1007/s10162-008-0118-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claudinon J, Monier M-N, Lamaze C. Interfering with interferon receptor sorting and trafficking: impact on signaling. Biochimie. 2007;89:735–743. doi: 10.1016/j.biochi.2007.03.014. [DOI] [PubMed] [Google Scholar]
- Clerici WJ, Hensley K, DiMartino DL, Butterfield DA. Direct detection of ototoxicant-induced reactive oxygen species generation in cochlear explants. Hear Res. 1996;98:116–124. doi: 10.1016/0378-5955(96)00075-5. [DOI] [PubMed] [Google Scholar]
- Cunningham LL. The adult mouse utricle as an in vitro preparation for studies of ototoxic-drug-induced sensory hair cell death. Brain Res. 2006;1091:277–281. doi: 10.1016/j.brainres.2006.01.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunningham LL, Brandon CS. Heat shock inhibits both aminoglycoside- and cisplatin-induced sensory hair cell death. J Assoc Res Otolaryngol. 2006;7:299–307. doi: 10.1007/s10162-006-0043-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunningham LL, Cheng AG, Rubel EW. Caspase activation in hair cells of the mouse utricle exposed to neomycin. J Neurosci. 2002;22:8532–8540. doi: 10.1523/JNEUROSCI.22-19-08532.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Prati AC, Ciampa AR, Cavalieri E, Zaffini R, Darra E, Menegazzi M, Suzuki H, Mariotto S. STAT1 as a new molecular target of anti-inflammatory treatment. Curr Med Chem. 2005;12:1819–1828. doi: 10.2174/0929867054546645. [DOI] [PubMed] [Google Scholar]
- Devarajan P, Savoca M, Castaneda MP, Park MS, Esteban-Cruciani N, Kalinec G, Kalinec F. Cisplatin-induced apoptosis in auditory hair cells: roll of death receptor and mitochondrial pathways. Hear Res. 2002;174:45–54. doi: 10.1016/s0378-5955(02)00634-2. [DOI] [PubMed] [Google Scholar]
- DeVries TA, Kalkofen RL, Matassa AA, Reyland ME. Protein kinase Cδ regulates apoptosis via activation of STAT1. J Biol Chem. 2004;279:45603–45612. doi: 10.1074/jbc.M407448200. [DOI] [PubMed] [Google Scholar]
- Haq R, Halupa A, Beattie BK, Mason JM, Zanke BW, Barber DL. Regulation of erythropoietin-induced STAT serine phosphorylation by distinct mitogen-activated protein kinases. J Biol Chem. 2002;277:17359–17366. doi: 10.1074/jbc.M201842200. [DOI] [PubMed] [Google Scholar]
- Hsu SD, Singh BB, Lewis JB, Borke JL, Dickinson DP, Drake L, Caughman GB, Schuster GS. Chemoprevention of oral cancer by green tea. Gen Dent. 2002;50:140–146. [PubMed] [Google Scholar]
- Kim CH, Kang SU, Pyun J, Lee MH, Hwang HS, Lee H. Epicatechin protects auditory cells against cisplatin-induced death. Apoptosis. 2008;13:1184–1194. doi: 10.1007/s10495-008-0242-5. [DOI] [PubMed] [Google Scholar]
- Kim HS, Lee M-S. STAT1 as a key modulator of cell death. Cell Signal. 2007;19:454–465. doi: 10.1016/j.cellsig.2006.09.003. [DOI] [PubMed] [Google Scholar]
- Klampfer L. Signal transducers and activators of transcription (STATs): novel targets of chemopreventive and chemotherapeutic drugs. Curr Cancer Drug Targets. 2006;6:107–121. doi: 10.2174/156800906776056491. [DOI] [PubMed] [Google Scholar]
- Kovarik P, Stoiber D, Eyers PA, Menghini R, Neininger A, Gaestel M, Cohen P, Decker T. Stress-induced phosphorylation of STAT1 at Ser727 requires p38 mitogen-activated protein kinase whereas IFN-γ uses a different signaling pathway. Proc Natl Acad Sci U S A. 1999;96:13956–13961. doi: 10.1073/pnas.96.24.13956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HJ, Oh YK, Rhee M, Lim JY, Hwang JY, Park YS, Kwon Y, Choi KH, Jo I, Park SI, Gao B, Kim WH. The role of STAT1/IRF-1 on synergistic ROS production and loss of mitochondrial transmembrane potential during hepatic cell death induced by LPS/d-GalN. J Mol Biol. 2007;369:967–984. doi: 10.1016/j.jmb.2007.03.072. [DOI] [PubMed] [Google Scholar]
- Liu W, Staecker H, Stupak H, Malgrange B, Lefebvre P, Van de Water TR. Caspase inhibitors prevent cisplatin-induced apoptosis of auditory sensory cells. Neuroreport. 1998;9:2609–2614. doi: 10.1097/00001756-199808030-00034. [DOI] [PubMed] [Google Scholar]
- Menegazzi M, Tedeschi E, Dussin D, De Prati AC, Cavalieri E, Mariotto S, Suzuki H. Anti-interferon-γ action of epigallocatechin-3-gallate mediated by specific inhibition of STAT1 activation. FASEB J. 2001;15:1309–1311. doi: 10.1096/fj.00-0519fje. [DOI] [PubMed] [Google Scholar]
- Meraz MA, White JM, Sheehan KC, Bach EA, Rodig SJ, Dighe AS, Kaplan DH, Riley JK, Greenlund AC, Campbell D, Carver-Moore K, DuBois RN, Clark R, Aguet M, Schreiber RD. Targeted disruption of the Stat1 gene in mice reveals unexpected physiologic specificity in the JAK-STAT signaling pathway. Cell. 1996;84:431–442. doi: 10.1016/s0092-8674(00)81288-x. [DOI] [PubMed] [Google Scholar]
- Moriwaki K, Kiyomoto H, Hitomi H, Ihara G, Kaifu K, Matsubara K, Hara T, Kondo N, Ohmori K, Nishiyama A, Fukui T, Kohno M. Interferon-γ enhances superoxide production in human mesangial cells via the JAK-STAT pathway. Kidney Int. 2006;70:788–793. doi: 10.1038/sj.ki.5001639. [DOI] [PubMed] [Google Scholar]
- Oesterle EC, Campbell S, Taylor RR, Forge A, Hume CR. Sox2 and JAGGED1 expression in normal and drug-damaged adult mouse inner ear. J Assoc Res Otolaryngol. 2008;9:65–89. doi: 10.1007/s10162-007-0106-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Previati M, Lanzoni I, Astolfi L, Fagioli F, Vecchiati G, Pagnoni A, Martini A, Capitani S. Cisplatin cytotoxicity in organ of corti-derived immortalized cells. J Cell Biochem. 2007;101:1185–1197. doi: 10.1002/jcb.21239. [DOI] [PubMed] [Google Scholar]
- Ramesh G, Reeves WB. TNF-α mediates chemokine and cytokine expression and renal injury in cisplatin nephrotoxicity. J Clin Invest. 2002;110:835–842. doi: 10.1172/JCI15606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reser D, Rho M, Dewan D, Herbst L, Li G, Stupak H, Zur K, Romaine J, Frenz D, Goldbloom L, Kopke R, Arezzo J, Van De Water T. l- and d-methionine provide equivalent long term protection against CDDP-induced ototoxicity in vivo, with partial in vitro and in vivo retention of antineoplastic activity. Neurotoxicology. 1999;20:731–748. [PubMed] [Google Scholar]
- Rybak LP, Kelly T. Ototoxicity: bioprotective mechanisms. Curr Opin Otolaryngol Head Neck Surg. 2003;11:328–333. doi: 10.1097/00020840-200310000-00004. [DOI] [PubMed] [Google Scholar]
- Rybak LP, Whitworth CA, Mukherjea D, Ramkumar V. Mechanisms of cisplatin-induced ototoxicity and prevention. Hear Res. 2007;226:157–167. doi: 10.1016/j.heares.2006.09.015. [DOI] [PubMed] [Google Scholar]
- Schuldes H, Bade S, Knobloch J, Jonas D. Loss of in vitro cytotoxicity after storage of stock solution in cell culture medium at various temperatures. Cancer. 1997;79:1723–1728. [PubMed] [Google Scholar]
- Simon AR, Rai U, Fanburg BL, Cochran BH. Activation of the JAK-STAT pathway by reactive oxygen species. Am J Physiol Cell Physiol. 1998;275:1640–1652. doi: 10.1152/ajpcell.1998.275.6.C1640. [DOI] [PubMed] [Google Scholar]
- So H, Kim H, Lee JH, Park C, Kim Y, Kim E, Kim JK, Yun KJ, Lee KM, Lee HY, Moon SK, Lim DJ, Park R. Cisplatin cytotoxicity of auditory cells requires secretions of proinflammatory cytokines via activation of ERK and NF-kappaB. J Assoc Res Otolaryngol. 2007;8:338–355. doi: 10.1007/s10162-007-0084-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stempelj M, Kedinger M, Augenlicht L, Klampfer L. Essential role of the JAK/STAT1 signaling pathway in the expression of inducible nitric-oxide synthase in intestinal epithelial cells and its regulation by butyrate. J Biol Chem. 2007;282:9797–9804. doi: 10.1074/jbc.M609426200. [DOI] [PubMed] [Google Scholar]
- Stephanou A, Scarabelli TM, Brar BK, Nakanishi Y, Matsumura M, Knight RA, Latchman DS. Induction of apoptosis and Fas receptor/Fas ligand expression by ischemia/reperfusion in cardiac myocytes requires serine 727 of the STAT-1 transcription factor but not tyrosine 701. J Biol Chem. 2001;276:28340–28347. doi: 10.1074/jbc.M101177200. [DOI] [PubMed] [Google Scholar]
- Stephanou A, Scarabelli TM, Townsend PA, Bell R, Yellon D, Knight RA, Lachtman DS. The carboxyl-terminal activation domain of the STAT-1 transcription factor enhances ischemia/reperfusion-induced apoptosis in cardiac myocytes. FASEB J. 2002;13:1841–1843. doi: 10.1096/fj.02-0150fje. [DOI] [PubMed] [Google Scholar]
- Takagi Y, Harada J, Chiarugi A, Moskowitz MA. STAT1 is activated in neurons after ischemia and contributes to ischemic brain injury. J Cereb Blood Flow Metab. 2002;22:1311–1318. doi: 10.1097/01.WCB.0000034148.72481.F4. [DOI] [PubMed] [Google Scholar]
- Tedeschi E, Suzuki H, Menegazzi M. Antiinflammatory action of EGCG, the main component of green tea, through STAT-1 inhibition. Ann N Y Acad Sci. 2002;973:435–437. doi: 10.1111/j.1749-6632.2002.tb04678.x. [DOI] [PubMed] [Google Scholar]
- Terui K, Haga S, Enosawa S, Ohnuma N, Ozaki M. Hypoxia/reoxygenation-induced, redox-dependent activation of Stat1 confers resistance to apoptotic cell death via hsp70 induction. Biochem J. 2004;380:203–209. doi: 10.1042/BJ20031891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Townsend PA, Scarabelli TM, Davidson SM, Knight RA, Latchman DS, Stephanou A. STAT-1 interacts with p53 to enhance DNA damage-induced apoptosis. J Biol Chem. 2004a;279:5811–5820. doi: 10.1074/jbc.M302637200. [DOI] [PubMed] [Google Scholar]
- Townsend PA, Scarabelli TM, Pasini E, Gitti G, Menegazzi M, Suzuki H, Knight RA, Lachtman DS, Stephanou A. Epigallocatechin-3-gallate inhibits STAT-1 activation and protects cardiac myocytes from ischemia/reperfusion-induced apoptosis. FASEB J. 2004b;8:1621–1623. doi: 10.1096/fj.04-1716fje. [DOI] [PubMed] [Google Scholar]
- Wang J, Ladrech S, Pujol R, Brabet P, Van de Water T, Puel J-L. Caspase inhibitors, but not c-Jun NH2-terminal kinase inhibitor treatment, prevent cisplatin-induced hearing loss. Cancer Res. 2004;64:9217–9224. doi: 10.1158/0008-5472.CAN-04-1581. [DOI] [PubMed] [Google Scholar]
- Woźniak K, Blasiak J. Recognition and repair of DNA-cisplatin adducts. Acta Biochim Pol. 2002;49:583–596. [PubMed] [Google Scholar]
- Xiao L, Naganawa T, Obugunde E, Gronowicz G, Ornitz DM, Coffin JD, Hurley MH. Stat1 controls postnatal bone formation by regulating fibroblast growth factor signaling in osteoblasts. J Biol Chem. 2004;279:27743–27752. doi: 10.1074/jbc.M314323200. [DOI] [PubMed] [Google Scholar]
- Xie D, Liu G, Zhu G, Wu W, Ge S. (-)-Epigallocatechin-3-gallate protects cultured spiral ganglion cells from H2O2-induced oxidizing damage. Acta Otolaryngol. 2004;124:464–470. doi: 10.1080/00016480410018278. [DOI] [PubMed] [Google Scholar]
- Yamamoto T, Staples J, Wataha J, Lewis J, Lockwood P, Schoenlein P, Rao S, Osaki T, Dickinson D, Kamatani T, Schuster G, Hsu S. Protective effects of EGCG on salivary gland cells treated with γ-radiation or cis-platinum(II)diammine dichloride. Anticancer Res. 2004;24:3065–3074. [PubMed] [Google Scholar]
- Yamashita H, Oesterle EC. Induction of cell proliferation in mammalian inner ear sensory epithelia by transforming growth factor-alpha and epidermal growth factor. Proc Natl Acad Sci U S A. 1995;92:3152–3155. doi: 10.1073/pnas.92.8.3152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan HY, Saidel WM, Chang JS, Presson JC, Popper AN. Sensory hair cells of a fish ear: evidence of multiple types based on ototoxicity sensitivity. Proc R Soc Lond B Biol Sci. 1991;245:133–138. doi: 10.1098/rspb.1991.0099. [DOI] [PubMed] [Google Scholar]
- Youlyouz-Marfak I, Gachard N, Le Clorennec C, Najjar I, Baran-Marszak F, Reminieras L, May E, Bornkamm GW, Fagard R, Feuillard J. Identification of a novel p53-dependent activation pathway of STAT1 by antitumour genotoxic agents. Cell Death Differ. 2008;15:376–385. doi: 10.1038/sj.cdd.4402270. [DOI] [PubMed] [Google Scholar]
- Zhang M, Liu W, Ding D, Salvi R. Pifithrin-α suppresses p53 and protects cochlear and vestibular hair cells from cisplatin-induced apoptosis. Neuroscience. 2003;120:191–205. doi: 10.1016/s0306-4522(03)00286-0. [DOI] [PubMed] [Google Scholar]
- Zykova TA, Zhang Y, Zhu F, Bode AM, Dong Z. The signal transduction networks required for phosphorylation of STAT1 at Ser727 in mouse epidermal JB6 cells in the UVB response and inhibitory mechanisms of tea polyphenols. Carcinogenesis. 2005;26:331–342. doi: 10.1093/carcin/bgh334. [DOI] [PubMed] [Google Scholar]