

Abstract
We describe the development of a highly efficient catalytic asymmetric conjugate addition to α,β-unsaturated sulfones. Utilizing practical bifunctional organic catalysts and involving air- and moisture-tolerant conditions, conjugate additions of a wide range of Michael donors to α,β-unsaturated sulfones proceeded in excellent enantioselectivity/diastereoselectivity and high yield. This efficient and operationally simple new catalytic asymmetric reaction should provide a versatile approach for the asymmetric synthesis of chiral sulfones bearing all-carbon quaternary stereocenters.
Keywords: organocatalyst, hydrogen bonding, cinchona alkaloids, chiral sulfones, conjugate addition
1. Introduction
Sulfones are versatile intermediates in organic synthesis.1 Enantioselective C-C bond forming reactions generating chiral sulfones are of significant interest to asymmetric synthesis.2 In particular asymmetric conjugate additions with trisubstituted carbon-nucleophiles to α,β-unsaturated sulfones 4 (Scheme 1) provide a straightforward strategy to form optically active building blocks bearing the sulfone functionality and a quaternary stereocenter.3,4 In this full article, we document in detail our efforts on the development of such reactions catalyzed by practically accessible, cinchona alkaloid-based organic catalysts.3
Scheme 1.

Rational for the development of a catalytic asymmetric conjugate addition to α, β-unsaturated sulfones (4) with 6'-OH cinchona alkaloids 5
In 2004 we demonstrated that 6’-OH cinchona alkaloid derivatives such as 5, bearing a hydrogen bond acceptor in quinuclidine and a hydrogen bond donor in 6’-OH, could catalyze the conjugate additions of a broad range of enolizable carbon Michael donors 2 to nitroalkenes 1 with remarkably high enantioselectivity and diastereoselectivity (Scheme 1).5,6 These studies provided the first examples of cinchona alkaloids with rotational freedom around C8–C9 and C4’–C9 bonds serving as highly efficient acid-base bifunctional catalysts for asymmetric reactions. Mechanistic and conformational studies unambiguously established that 5 adopted a gauche-open active conformer (Scheme 1) in the transition state.5b With this key information in hand, we are able to construct a transition state model (6, Scheme 1) that is consistent with the stereochemistry outcome of the 5-catalyzed conjugate additions to nitroalkenes 1. In this model the C6’-OH cinchona alkaloids 5, adopting the gauche-open conformation, simultaneously activated and oriented the Michael donors 3 (generated by tautomerization of ketones 2) and the nitroalkenes 1 via a network of hydrogen bonding interactions. 5b We reasoned that if the replacement of the nitro with the sulfone functionality in the Michael donor did not disrupt or alter the pattern of hydrogen bonding network between the catalyst and the reacting substrates, 6’-OH cinchona alkaloaid 5 might be able to catalyze conjugate additions of enolizable Michael donors 2 to α,β-unsaturated sulfones with high diastereoselectivity and enantioselectivity. Thus we initiated an investigation of these conjugate additions to α,β-unsaturated sulfones with cinchona alkaloids 5.
2. Results and discussion
2.1. Catalyst discovery and reaction optimizations
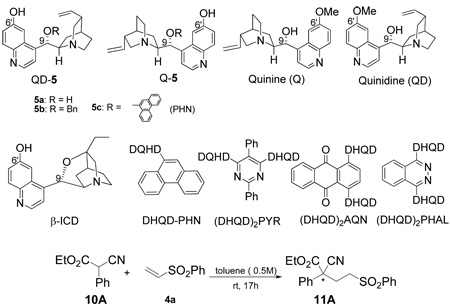
Our experiments began with a screening of a variety of cinchona alkaloid derivatives for their ability to promote enantioselective addition of α-phenyl α-cyanoacetate 10A to the commercially available phenyl vinyl sulfone (4a). As summarized in Table 1, the conjugate addition proceeded smoothly in toluene at room temperature in the presence of various natural and modified cinchona alkaloids. The enantioselectivity of the reaction was, however, found to be critically dependent on the structure of the cinchona alkaloid derivatives. Reactions with natural cinchona alkaloids such as quinidine and quinine generated the 1,4-adduct 11A as a nearly racemic mixture (entries 1 and 2). The enantioselectivity was in general improved with cinchona alkaloids bearing a C9-substituent (entries 3–7, 9–12). Importantly, in line with our hypothesis, the 6’-OH cinchona alkaloids 1 were found to be much more effective than those bearing C6’-OMe. The pronounced effect of the C9-substituent on the enantioselectivity of the C6’-OH cinchona alkaloids 1 is also noteworthy. While modest enantioselectivity was afforded by either the C6’-OH cinchona alkaloid bearing a C9-OH (entry 8) or a rigid C6’-OH cinchona alkaloid derivative (entry 7), significantly higher enantioselectivity could be attained with C6’-OH cinchona alkaloids bearing either an aryl or alkyl ether at C9 (entries 9 and 10). Thus the tunable group at C9 provided a crucial handle for the optimization of the catalytic enantioselectivity of 5. Our screening studies identified 5c as the best catalyst (entries 10 and 12).
Table 1.
Cinchona alkaloid-catalyzed addition of α-phenyl α-cyanoacetate 10A to vinyl phenyl sulfone (4a)
 | |||||||
|---|---|---|---|---|---|---|---|
| Entry | Catalyst | Conv.(%)b | ee(%)c | Entry | Cat. | Conv.(%)b | ee(%)c |
| 1 | Q | >98 | 0 | 7 | β-ICD | 98 | 42 |
| 2 | QD | >98 | 3 | 8 | QD-5a | 90 | 45 |
| 3 | DHQD-PHN | 79 | 24 | 9 | QD-5b | >98 | 63 |
| 4 | (DHQD)2PYR | >98 | 33 | 10 | QD-5c | 91 | 74 |
| 5 | (DHQR)2PHAL | 80 | 9 | 11 | Q-5b | 65 | 74 |
| 6 | (DHQR)2AQN | >98 | 4 | 12 | Q-5c | >98 | 84 |
Reactions were run with 10A (0.3 mmol), 4a (0.1 mmol) and the catalyst (0.02 mmol) in toluene (0.2 mL) at r.t. for 17h.
Determined by 1H NMR analysis.
Determined by HPLC analysis.
Next we examined the reaction in various solvents. The reaction was found to proceed at the fastest rate and in the highest enantioselectivtiy in toluene. It is noteworthy that such a non-polar solvent is known to favor the formation and stabilization of hydrogen bonding complexes between solvates. An excellent enantioselectivity could be achieved with Q-5c in toluene at −25 °C (entries 5 and 6, Table 2).
Table 2.
Solvent effect on the addition of α-phenyl α-cyanoacetates 10A to vinyl phenyl sulfone (4a) with Q-5ca
| entry | Time(h) | Sol. | Conv.(%)b | ee.(%)c |
|---|---|---|---|---|
| 1 | 17 | TBME | 9 | 58 |
| 2 | 17 | CHCl3 | 17 | 79 |
| 3 | 17 | CH2Cl2 | 17 | 74 |
| 4 | 17 | DCE | 22 | 81 |
| 5 | 17 | Toluene | 46 | 95 |
| 6 | 72 | Toluene | 95 | 95 |
Reactions were run 10A (0.3 mmol), 4a (0.1 mmol) and Q-5c (0.02 mmol) in toluene (0.2 mL) at −25°C.
Determined by 1H NMR analysis.
Determined by HPLC analysis.
2.2 Conjugate additions of α-substituted α-cyanoacetate to vinyl sulfones
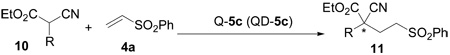
After establishing the optimal reaction conditions we began to investigate the substrate scope of the 5c-catalyzed conjugate addition to vinyl sulfones (4a). The α-cyanoacetates 10 bearing a range of α-aryl and – heteroaryl groups of varying electronic and steric properties underwent efficient enantioselective additions to phenyl vinyl sulfone (4a), providing the corresponding 1,4-adducts 11 bearing the all-carbon quaternary stereocenter in excellent enantioselectivity and good to excellent yield (Table 3).
Table 3.
 | |||||||
|---|---|---|---|---|---|---|---|
| Entry | R | T/°C | Time/h | Product | Yield/%d | ee/%e | |
| 1 | 10A | Ph- | −25 | 72(72) | 11A | 89(80) | 95(91) |
| 2 | 10B | 4-Me-Ph- | 0 | 48 | 11B | 96 | 93 |
| 3 | 10C | 4-MeO-Ph- | 0 | 70 | 11C | 92 | 93 |
| 4 | 10D | 4-F-Ph- | −25 | 72 | 11D | 90 | 94 |
| 5 | 10E | 4-Cl-Ph- | −25 | 69(72) | 11E | 95(94) | 94(89) |
| 6 | 10F | 4-Br-Ph- | −25 | 69(72) | 11F | 95(95) | 94(88) |
| 7 | 10G | 3-Cl-Ph- | −25 | 60 | 11G | 96 | 93 |
| 8 | 10H | 2-Naphtyyl- | −25 | 60(60) | 11H | 96(95) | 97(90) |
| 9 | 10I | 2-Thienyl | −25 | 48(48) | 11I | 95(91) | 93(88) |
Unless noted, reactions were run with 10 (0.5–0.6 mmol), 4a (0.2 mmol) and 5c (0.04 mmol).
The catalyst was recovered in greater than 95% yield after the reaction.
The results in parentheses were obtained with QD-5c to give the opposite enantiomer.
Isolated yield.
Determined by HPLC analysis.
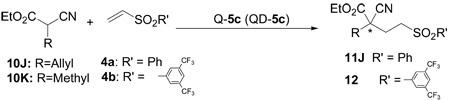
Having established a general scope with respect to α-aryl α-cyanoacetates, we attempted to extend the scope to α-alkyl α-cyanoacetates. We found that, compared to their aryl congeners, α-alkyl α-cyanoacetates were significantly less active as Michael donor for the cinchona alkaloid-catalyzed conjugate addition to vinyl sulfones. While addition of α-phenyl α-cyanoacetate 10A to phenyl vinyl sulfone 4a with Q-5c proceeded to completion at room temperature after 17 hours (entry 12, Table 1), the addition of α-allyl-α-cyanoacetate 10J to 4a proceeded to only 17% conversion during the same period (entry 1, Table 4). Importantly, both reactions proceeded in comparable enantioselectivity (87% ee vs 86% ee).
Table 4.
 | ||||||||
|---|---|---|---|---|---|---|---|---|
| Entry | 10 | 4 | T/°C | Time/h | Product | Conv./% | Yield/%d | ee/%e |
| 1 | 10J | 4a | 23 | 17 | 11J | 17 | N.D. | 87 |
| 2 | 10J | 4b | 23 | 17 | 12J | 88 | N.D. | 86 |
| 3 | 10J | 4b | 0 | 96 | 12J | 100 | 76 | 94f |
| 4 | 10K | 4b | 0 | 96(96) | 12K | 100(100) | 85(83) | 92(88) |
Unless noted, reactions were run with 10( 0.5-0.6 mmol) and 4a (0.2 mmol) with 5c (0.04 mmol).
The catalyst was recovered in greater than 95% yield after the reaction.
The results in parentheses were obtained with QD-5c to give the opposite enantiomer.
Isolated yield.
Determined by HPLC analysis.
The absolute configuration of adduct (+)-12J was determined to be “S” by X-ray structure analysis, see section 4.5 for details.
We reasoned that the cinchona alkaloid-catalyzed conjugate addition with α-alkyl α-cyanoacetate might be accelerated considerably by enhancing the electrophilicity of the vinyl sulfone, which could be implemented via the introduction of electron-withdrawing substituents on the aromatic ring of the phenyl vinyl sulfone 4. Based on our proposed transition state model outlined in Scheme 1, steric variations of the phenyl group of 4 should not disrupt how catalyst 5c interacts with the reacting substrates. With this consideration in mind, we investigated the conjugate addition of 10J to 3,5-bis(triflouromethyl)phenyl vinyl sulfone (4b)7with Q-5c at room temperature. We were pleased to find that the reaction was indeed significantly accelerated, proceeding to 88% conversion after 17 hours. Importantly, the corresponding 1,4-adduct 12 was obtained in 86% ee (entry 2, Table 4). Thus the ability of 6’-OH cinchona alkaloid catalysts to tolerate the structural change of the vinyl sulfone 4 allowed us to significantly improve the rate of the conjugate additions of a α-alkyl α-cyanoacetate by tuning the electronic property of the vinyl sulfone acceptor. Importantly, the high enantioselectivity could be readily extended to the conjugate addition of α-methyl α-cyanoacetate 10K to 4b (entry 4, Table 4).
2.3. Conjugate additions of α-substituted α-cyanoketones and β-ketoesters to vinyl sulfones
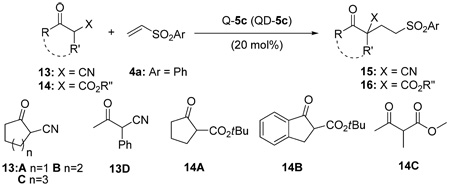
We also investigated the 5c-catalyzed additions of cyclic α-cyanoketones (13A–C) and cyclic β-ketoesters (14A and B) to vinyl sulfone 4a. As illustrated in Table 5 reactions with these Michael donors proceeded in high yield and excellent enantioselectivity. However, with either acyclic α-cyanoketones (13D) or acyclic β-ketoesters (14G), no conjugate addition product was obtained. Only starting materials were recovered.
Table 5.
Enantioselectivie conjugate addition of α-cyanoketones 13 and β-ketoesters 14 to vinyl sulfone 4a catalyzed by 5ca,b
 | |||||
|---|---|---|---|---|---|
| entry | Michael Donor | time/h | Product | yield /%c | ee /%d |
| 1 | 13A | 24 | 15A | 96 (90) | 95 (97) |
| 2 | 13B | 24 | 15B | 96 (98) | 95 (95) |
| 3 | 13C | 24 | 15C | 91(91) | 96e (97) |
| 4 | 13D | 24 | 15D | 0 | n.d |
| 5 | 14A | 48 | 16A | 93 (96) | 96 (96) |
| 6 | 14B | 48 | 16B | 96 (100) | 96 (97) |
| 7 | 14C | 24 | 16C | 0 | n.d |
Unless noted, reactions were run with 13 or 14 (0.40 mmol), 4a (0.20 mmol) in toluene (0.40 mL) with Q-5c (20 mol%).
the results in parentheses were obtained with QD-5c.
Isolated yield.
Determined by HPLC ananlysis.
The absolute configuration of (−)-15C was determined to be “S” by X-ray structure analysis, see section 4.5 for details.
2.4 Conjugate additions to β-substituted α,β -unsaturated sulfones
Adjacent quaternary and tertiary stereocenters are common structural motifs in natural products. Highly diastereoselective and enantioselective conjugate additions of prochiral trisubstituted carbon nucleophiles to prochiral β-substituted Michael acceptors with a chiral catalyst could provide a one-step and stereoselective construction of such complex motifs from simple precursors. Thus we began to investigate the conjugate additions of various nucleophiles to β-substituted α,β-unsaturated sulfones with Q-5c.
We first examined the Q-5c-catalyzed conjugate additions of β-ketoester 14D to β-methyl vinyl sulfone 17a and β-phenyl vinyl sulfone 17a’ in toluene at room temperature. Unfortunately, no addition product was found, and only starting materials were recovered. Trifluoromethanesulfonyl group (CF3SO2-) is one of the strongest neutral electron-withdrawing groups known. If the SO2Ph in 17 was replaced by SO2CF3, the resulting vinyl sulfones should be much more active toward conjugate additions. Following this thought, we began to investigate the QD-5c-catalyzed conjugate addition of β-ketoester 14D to β-substituted vinyl triflone 17b.8 We were pleased to find that, in contrast to reactions with 17a, the reaction proceeded smoothly to completion after 12 h at room temperature in toluene to afford the desired 1,4-adduct in excellent ee and promising dr (entry 2, Table 6). Further optimization revealed that CH2Cl2, ClCH2CH2Cl and CHCl3 were better solvents, and the conjugate addition proceeded in the highest enantioselectivity and diastereoselectivity in CH2Cl2 (entry 8, Table 6).
Table 6.
Additions of β-ketoester 14D to β -substituted vinyl triflone 17 with Q-5ca
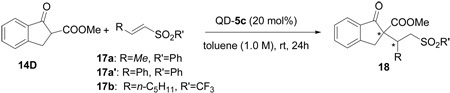 | ||||||
|---|---|---|---|---|---|---|
| entry | acceptor | solvent | conversion | drb | ee of major isomer/%c |
ee of minor isomer/%c |
| 1 | 17a | toluene | 0 | ND | ND | ND |
| 2 | 17b | toluene | 100 | 5.5 : 1 | 96 | 56 |
| 3 | 17b | THF | 100 | 3.7 : 1 | 81 | 6 |
| 4 | 17b | Et2O | 100 | 4.3 : 1 | 92 | 12 |
| 5 | 17b | EtOAc | 100 | 4.6 : 1 | 87 | 6 |
| 6 | 17b | ClCH2Cl2Cl | 100 | 7.0 : 1 | 94 | 23 |
| 7 | 17b | CHCl3 | 100 | 5.7 : 1 | 96 | 26 |
| 8 | 17b | CH2Cl2 | 100 | 8.0 : 1 | 95 | 20 |
Unless noted, reactions were run with 14D (0.025 mmol), 17 (0.0275 mmol) in a solvent (0.10 mL) with QD-5c (10 mol%).
Determined by 1H NMR analysis.
Determined by HPLC ananlysis.
Encouraged by these results, a variety of trisubstituted carbon Michael donors were subjected to the 5c-catalyzed conjugate additions to various β-substituted vinyl triflones 17b–d.8 Excellent diastereoselectivity and enantioselectivity were obtained with various α-aryl α-cyanoacetates 10, α-cyanoketones 13 and cyclic β-ketoesters 14 (Table 7).
Table 7.
Diastereoselective and enantioselective conjugate additions to β-alkyl vinyl triflones 17 catalyzed by 5ca
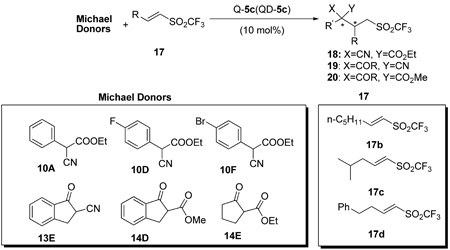 | ||||||||
|---|---|---|---|---|---|---|---|---|
| entry | donor | 17 | temp /°C |
time /h |
product | dr | ee /%b |
yield /%c |
| 1 | 10A | 17b | −50 | 96 | 18Ab | 5.6:1(4.6:1) | 93(87) | 87d(76)d |
| 2 | 10D | 17b | −50 | 96 | 18Db | 26:1 (12:1) | 92(84) | 94 d(91)d |
| 3 | 10F | 17b | −50 | 96 | 18Fb | 49:1 (26:1) | 81(86) | 92d(92)d |
| 4 | 13E | 17b | −50 | 24 | 19Eb | >49:1(>49: 1) | 99(94) | 95d(94) |
| 5 | 14D | 17b | −20 | 96 | 20Db | 32:1(9:1) | 98(97) | 90d(93)d |
| 6 | 14D | 17c | −20 | 72 | 20Dc | 24:1 (8:1) | 98(95) | 100(95) |
| 7 | 14E | 17d | −20 | 48 | 20Ed | >49:1 (>49:1) | 95(89) | 91d(80)d |
Unless noted, reactions were run with 10 or 13 or 14 (0.10 or 0.20 mmol),,17 (0.10 mmol) and Q-5c (0.02 mmol) in methylene chloride (0.20 mL), and the results in parentheses were obtained with QD-5c to give the opposite enantiomer.
Determined by HPLC ananlysis.
Isolated yield.
Yield of pure major diastereomer.
3. Conclusions
In summary, we have developed a highly efficient catalytic asymmetric conjugate addition to α,β-unsaturated sulfones. This reaction utilizes readily accessible and recyclable chiral organic catalysts and involves an air- and moisture-tolerant simple experimental protocol that avoids low temperature and low volumetric productivity. The general scopes with respect to both the donors and α,β-unsaturated sulfones attained by the bifunctional cinchona alkaloid catalysts are particularly noteworthy. A wide range of α-substituted β-carbonyl Michael donors, such as α-cyanoacetate, α-cyanoketone and β-ketoesters, could be employed in the reaction. The reaction also tolerates variations in the α,β-unsaturated sulfones. Specifically, vinyl sulfones bearing either no or a β-substituent could be applied in this reaction. Consequently, the reaction provides a highly versatile catalytic entry for the asymmetric synthesis of chiral sulfones containing all-carbon quaternary stereocenters.
4. Experimental
4.1 General
1H and 13C NMR spectra were recorded on a Varian instrument (400 MHz and 100 MHz, respectively) and internally referenced to SiMe4 signal. Low resolution mass spectra for all the new compounds were recorded on a Hewlett-Packard 5989A GC/MS, and exact mass spectra on a VG 7070 high resolution mass spectrometer. Infrared spectra were recorded on a Perkin Elmer FT-IR Spectrometer. Specific rotations were measured on a Jasco Digital Polarimeter. Liquid chromatography was performed using forced flow (flash chromatography) of the indicated solvent system on EM Science silica gel 60 (SiO2, 230–400 mesh). Analytical gas-liquid chromatography (GLC) was performed on a Hewlett-Packard 6890 Series instrument equipped with a split mode capillary injection system, a flame ionization detector, using a HP-5 GC column or a GC column with chiral stationary phase [Gamma cyclodextrin Trifluoroacetyl (30 m ×0.25 mm) or HP Chiral (20% Permethylated B-Cyclodextrin, 30 m × 0.25 mm)]. All the GC analyses of chiral cyanohydrin carbonates were carried out with both a crude reaction sample and a sample purified by silica gel chromatography. High performance liquid chromatography (HPLC) analyses were performed on a Hewlett-Packard 1100 Series instrument equipped with an isostatic pump, using a Daicel Chiralpak AS (250 × 4.6 mm), AD (250 × 4.6 mm) or Hypersil SI (200 × 4.6 mm) Column. UV detection was monitored at 254 nm or 220 nm.
4.2 Typical procedure for enantioselective addition of α-aryl (10A–I) or alkyl α-cyanoacetates (10J–K) to vinyl sulfones 4a or 4b with 5c
At −25°C or 0 °C, to a solution of α-cyanoacetate 10 (for 10A–C, using 0.6 mmol; for 10D–I, using 0.5mmol, for 10J–K, using 0.2 mmol) in toluene (0.4 mL) was added either Q- or QD-5c (20 mol%.) and vinyl phenyl sulfone 4a or 4b (0.2 mmol) as specified in Table 3 and Table 4. The resulting mixture was kept at the indicated temperature until sulfone is completely consumed. The reaction mixture was directly subjected to silica gel flash chromatography using the eluent specified below to afford the 1,4-adduct in the yields and enantiomeric excess summarized above. Finally, the column was washed with MeOH and the catalyst was recovered in greater than 95% yield. The recovered catalyst was shown to be identical to that before the reaction by NMR analysis.
4.2.1. (+)-11A (Table 3, entry 1)
This product was obtained as a colorless oil in 89 % yield after flash chromatography (elution gradient: ethyl acetate/hexane=1/8 to 6/1) and 95 % ee as determined by HPLC analysis [Daicel chiralcel OD, hexanes:IPA, 90:10, 1.0 ml/min, λ 220 nm, t (major) = 16.48 min, t (minor) = 14.93 min] from a reaction catalyzed by Q-5c (20 mol%) at −25 °C for 72 hours. [α]D 25 = +31.6 (c 1.0, CHCl3) ; 1H NMR (400 MHz, CDCl3) δ 7.89 (d, J = 8.0 Hz, 2H), 7.69 (t, J = 7.2 Hz, 1H), 7.59 (t, J = 8.0 Hz, 2H), 7.45–7.39 (m, 5H), 4.28-4.15 (m, 2H), 3.27 (dt, J = 4.4 Hz, 12.8 Hz, 1H), 3.04 (dt, J = 4.4 Hz, 12.8 Hz, 1H), 2.75-2.59 (m, 2H), 1.21 (t, J = 7.2 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 166.2, 138.3, 134.1, 132.6, 129.54, 129.48, 128.0, 125.8, 117.0, 63.7, 52.3, 52.1, 30.8, 13.7; HRMS (CI) m/z calcd. for (C19H19NO4S+H+): 358.1113, found: 358.1108; IR (CHCl3) ν 3064, 2984, 2939, 1745, 1448, 1321, 1237, 1087 cm−1.
(−)-11A
This product was obtained as a colorless oil in 80 % yield and 91 % ee from a reaction catalyzed by QD-5c (20 mol %) at −25 °C for 72 hours.
4.2.2. (+)-11B (Table 3, entry 2)
This product was obtained as a colorless oil in 96 % yield after flash chromatography (ethyl acetate/hexane=1/7) and 93 % ee as determined by HPLC analysis [Daicel Chiralpak AD, hexanes:IPA, 85:15, 1.0 ml/min, λ 220 nm, t(major) = 13.33 min, t(minor) = 22.06 min] from a reaction catalyzed by Q-4c (20 mol%) at 0 °C for 48 hours. [α]D 25 = +31.4 (c 1.02, CHCl3); 1H NMR (400 MHz, CDCl3) δ 7.89 (d, J = 8.0 Hz, 2H), 7.69 (t, J = 7.2 Hz, 1H), 7.58 (t, J = 8.0 Hz, 2H), 7.30 (d, J = 8.4 Hz, 2H), 7.20 (d, J = 8.4 Hz, 2H), 4.27–4.11 (m, 2H), 3.25 (dt, J = 5.6 Hz, 12.8 Hz, 1H), 3.03 (dt, J = 4.4 Hz, 12.8 Hz, 1H), 2.72-2.58 (m, 2H), 2.35 (s, 3H), 1.21 (t, J = 7.2 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ166.3, 139.5, 138.3, 134.0, 130.1, 129.5, 129.4, 127.9, 125.6, 117.2, 63.6, 52.06, 51.98, 30.7, 20.9, 13.6; HRMS (CI) m/z calcd. for (C20H21NO4S+H+): 372.1270, found: 372.1276; IR (CHCl3) ν 2983, 1744, 1447, 1321, 1235, 1148, 1087 cm−1.
4.2.3. (+)-11C (Table 3, entry 3)
This product was obtained as a colorless oil in 92 % yield after flash chromatography (ethyl acetate/hexane=1/6) and 93 % ee as determined by HPLC analysis [Daicel Chiralpak AD, hexanes:IPA, 85:15, 0.9 ml/min, λ 220 nm, t(major) = 28.93 min, t(minor) = 33.20 min] from a reaction catalyzed by Q-5c (20 mol%) at 0 °C for 70 hours. [α]D 25 = +31.0 (c 1.10, CHCl3); 1H NMR (400 MHz, CDCl3) δ 7.89 (d, J = 7.2 Hz, 2H), 7.69 (t, J = 7.2 Hz, 1H), 7.58 (t, 7.2 Hz, 2H), 7.34 (td, J = 2.4 Hz, 9.2 Hz, 2H), 6.90 (td, J = 2.4 Hz, 9.2 Hz, 2H), 4.27–4.14 (m, 2H), 3.81 (s, 3H), 3.25 (dt, J = 5.2 Hz, 12.0 Hz, 1H), 3.04 (dt, J = 4.0 Hz, 12.0 Hz, 1H), 2.72-2.57 (m, 2H), 1.21 (t, J = 6.8 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ166.4, 160.2, 138.3, 134.1, 129.4, 128.0, 127.1, 124.2, 117.2, 114.8, 63.6, 55.3, 52.1, 51.6, 30.7, 13.7; HRMS (CI) m/z calcd. for (C20H21NO5S+H+): 388.1219, found: 388.1210; IR (CHCl3) ν 2982, 1743, 1512, 1307, 1257, 1236, 1147 cm−1.
4.2.4. (+)-11D (Table 3, entry 4)
This product was obtained as a colorless oil in 90 % yield after flash chromatography (ethyl acetate/hexane = 1/5) and 94 % ee as determined by HPLC analysis [Daicel chiralcel OD, hexane:IPA, 90:10, 1.0 mL/min, λ 220 nm, t(major) = 21.42 min, t(minor) = 17.98min] from a reaction catalyzed by Q-5c (20 mol%) at −25 °C for 72 hours. [α]D 25 = +27.2 (c 1.13, CHCl3); 1H NMR (400 MHz, CDCl3) δ 7.90-7.88 (m, 2H), 7.70 (tt, J = 1.2 Hz, 7.6 Hz, 1H), 7.59 (t, J = 7.2 Hz, 2H), 7.45-7.41 (m, 2H), 7.11 (tt, J = 1.6 Hz, 8.0 Hz, 2H), 4.29-4.16 (m, 2H), 3.26 (dt, J = 4.8 Hz, 13.6 Hz, 1H), 3.03 (dt, J = 3.6 Hz, 12.8 Hz, 1H), 2.72 (dt, J = 4.4 Hz, 14.0 Hz, 1H), 2.61 (dt, J = 4.4 Hz, 14.0 Hz, 1H), 1.22 (t, J = 7.2 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 166.0, 163.0 (d, 1J C,F = 249.0 Hz), 138.3, 134.1, 129.5, 128.4 (d, 4J C,F= 3.0 Hz), 128.0, 127.8 (d, 3J C,F = 8.3 Hz), 116.9, 116.6(d, 2J C,F = 22.1 Hz), 63.9, 52.0, 51.7, 30.8, 13.6; HRMS (CI) m/z calcd for (C19H18FNO4S+H+): 376.1019, found: 376.1015; IR (CHCl3) ν 3070, 2985, 2940, 1745, 1604, 1510, 1447, 1307, 1237, 1087 cm−1.
4.2.5. (+)-11E (Table 3, entry 5)
This product was obtained as a colorless oil in 95 % yield after flash chromatograph (ethyl acetate/hexane=1/6) and 94 % ee as determined by HPLC analysis [Daicel chiralcel OD, hexane:IPA, 85:15, 1.0 mL/min, λ 220 nm, t(major) = 16.75 min, t(minor) = 13.57 min] from a reaction catalyzed by Q-5c (20 mol%) at −25 °C for 69 hours. [α]D 25 = +31.9 (c 1.10, CHCl3); 1H NMR (400 MHz, CDCl3) δ 7.85 (d, J = 8.8 Hz, 2H), 7.65 (t, J = 7.2 Hz, 1H), 7.55 (t, J = 6.4 Hz, 2H), 7.34 (s, 4H), 4.22–4.14 (m, 2H), 3.23 (dt, J = 4.4 Hz, 12.8 Hz, 1H), 2.99 (dt, J = 4.4 Hz, 12.8 Hz, 1H), 2.69 (dt, J = 4.4 Hz, 12.8 Hz, 1H), 2.56 (dt, J = 4.4 Hz, 12.8 Hz, 1H), 1.17 (t, J = 7.2 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 165.8, 138.2, 135.6, 134.1, 131.1, 129.6, 129.4, 127.9, 127.2, 116.6, 63.9, 51.9, 51.8, 30.6, 13.6; HRMS (CI) m/z calcd for (C19H18ClNO4S+H+): 392.0723, found: 392.0728; IR (CHCl3) ν 3066, 2984, 1745, 1493, 1447, 1322, 1236, 1148 cm−1.
(−)-11E
This product was obtained as a colorless oil in 94 % yield and 89 % ee from a reaction catalyzed by QD-5c (20 mol %) at −25 °C for 72 hours.
4.2.6. (+)-11F (Table 3, entry 6)
This product was obtained as a colorless oil in 95 % yield after flash chromatography (ethyl acetate/hexane=1/6) and 94 % ee as determined by HPLC analysis [Daicel chiralpak AD, hexane:IPA, 85:15, 1.0 mL/min, λ 220 nm, t(major) = 22.51 min, t(minor) = 26.82 min] from a reaction catalyzed by Q-5c (20 mol%) at −25 °C for 66 hours. [α]D 25 = +28.8 (c 1.10, CHCl3); 1H NMR (400 MHz, CDCl3) δ 7.89 (d, J = 8.0 Hz, 2H), 7.70 (t, J = 8.0 Hz, 1H), 7.60 (t, J = 8.0 Hz, 2H), 7.54 (d, J = 8.8 Hz, 2H), 7.32 (d, J = 8.8 Hz, 2H), 4.29-4.16 (m, 2H), 3.25 (dt, J = 4.4 Hz, 13.2 Hz, 1H), 3.01 (dt, J = 4.0 Hz, 13.2 Hz, 1H), 2.71 (dt, J = 4.4 Hz, 13.2 Hz, 1H), 2.60 (dt, J = 4.0 Hz, 13.2 Hz, 1H), 1.23(t, J = 7.2 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ165.8, 138.2, 134.2, 132.7, 131.6, 129.5, 128.0, 127.5, 123.9, 116.6, 64.0, 52.01, 51.96, 30.7, 13.7; HRMS (CI) m/z calcd for (C19H18BrNO4S+H+): 436.0218, found: 436.0208; IR (CHCl3) ν 2983, 1745, 1489, 1447, 1322, 1236, 1148 cm−1.
(−)-11F
This product was obtained as a colorless oil in 95 % yield and 88 % ee from a reaction catalyzed by QD-5c (20 mol %) at −25 °C for 72 hours.
4.2.7. (+)-11G (Table 3, entry 7)
This product was obtained as a colorless oil in 96 % yield after flash chromatography (ethyl acetate/hexane = 1/7) and 93 % ee as determined by HPLC analysis [Daicel chiralpak AD, hexane:IPA, 85:15, 1.0 mL/min, λ 220 nm, t (major) = 15.00 min, t (minor) = 12.58 min] from a reaction catalyzed by Q-5c (20 mol%) at −25 °C for 60 hours. [α]D 25 = +29.5 (c 1.00, CHCl3); 1H NMR (400 MHz, CDCl3) δ 7.91-7.88 (m, 2H), 7.73–7.68 (m, 1H), 7.60 (t, J = 7.2 Hz, 2H), 7.42-7.33 (m, 4H), 4.31-4.17 (m, 2H), 3.27 (dt, J = 4.4 Hz, 13.6 Hz, 1H), 3.04 (dt, J = 3.6 Hz, 12.8 Hz, 1H), 2.73 (dt, J = 4.8 Hz, 12.8 Hz, 1H), 2.58 (dt, J = 4.4 Hz, 12.8 Hz, 1H), 1.23 (t, J = 6.8 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 165.7, 138.2, 135.6, 134.5, 134.2, 130.8, 129.8, 129.5, 128.0, 126.1, 124.0, 116.5, 64.1, 52.0, 30.9, 13.7; HRMS (CI) m/z calcd for (C19H18ClNO4S+H+): 392.0723, found: 392.0721; IR (CHCl3) ν 2984, 1746, 1478, 1447, 1309, 1322, 1237, 1148 cm−1.
4.2.8. (+)-11H (Table 3, entry 8)
This product was obtained as a colorless oil in 96 % yield after flash chromatography (elution gradient: ethyl acetate / hexane = 1/6) and 98 % ee as determined by HPLC analysis [Daicel chiralcel OD, hexane:IPA, 90:10, 1.0 mL/min, λ 220 nm, t(major) = 23.90 min, t(minor) = 27.21 min] from a reaction catalyzed by Q-5c (20 mol%) at −25 °C for 60 hours. [α]D 25 = +46.7 (c 1.00, CHCl3);; 1H NMR (400 MHz, CDCl3) δ 7.97 (d, J = 1.6 Hz, 1H), 7.88-7.84 (m, 5H), 7.68-7.64 (m, 1H), 7.57 (m, 4H), 7.44 (dd, J = 2.4 Hz, 8.4 Hz, 1H), 4.29-4.15 (m, 2H), 3.32 (dt, J = 4.8 Hz, 13.6 Hz, 1H), 3.04 (dt, J = 4.4 Hz, 12.4 Hz, 1H), 2.87-2.71 (m, 2H), 1.20 (t, J = 7.6 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 166.2, 138.3, 134.0, 133.1, 132.9, 129.7, 129.6, 129.4, 128.2, 127.9, 127.6, 127.4, 127.2, 125.9, 122.0, 117.1, 63.8, 52.5, 52.1, 30.6, 13.6; HRMS (CI) m/z calcd for (C23H21NO4S+H+) : 408.1270, found: 408.1266; IR (CHCl3) ν 3061, 2983, 1744, 1321, 1234, 1151, 1087 cm−1.
(−)-11H
This product was obtained as a colorless oil in 95 % yield and 90 % ee from a reaction catalyzed by QD-5c (20 mol %) at −25 °C for 60 hours.
4.2.9. (+)-11I (Table 3, entry 9)
This product was obtained as a brown oil in 95 % yield after flash chromatography (ethyl acetate/hexane = 1/6) and 93% ee as determined by HPLC analysis [Daicel chiralcel OD, hexane:IPA, 90:10, 1.0 mL/min, λ 220 nm, t(major) = 20.54 min, t(minor) = 18.50 min] from a reaction catalyzed by Q-5c (20 mol%) at −25 °C for 48 hours. [α]D 25 = +20.2 (c 1.00, CHCl3); 1H NMR (400 MHz, CDCl3) δ 7.91-7.88 (m, 2H), 7.72-7.68 (m, 1H), 7.59 (t, J = 8.0 Hz, 2H), 7.36 (dd, J = 1.2 Hz, 4.8 Hz, 2H), 7.20 (dd, J = 1.2 Hz, 4.0 Hz, 1H), 6.99 (dd, J = 3.6 Hz, 4.8 Hz, 1H), 4.29-4.23 (m, 2H), 3.27 (dt, J = 4.8 Hz, 12.8 Hz, 1H), 3.14 (dt, J = 4.4 Hz, 12.4 Hz, 1H), 2.78-2.61 (m, 2H), 1.27 (t, J = 7.6 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 165.5, 138.2, 134.9, 134.1, 129.4, 127.9, 127.5, 127.4, 127.1, 116.3, 64.0, 52.0, 49.3, 32.1, 13.6; HRMS (CI) m/z calcd for (C17H17NO4S2+H+): 364.0677, found: 364.0669.
(−)-11I
This product was obtained as a brown oil in 91 % yield and 88 % ee from a reaction catalyzed by QD-5c (20 mol %) at −25 °C for 48 hours.
4.2.10. (+)-12J (Table 4, entry 3)
This product was obtained as a white solid in 76 % yield after flash chromatography (ethyl acetate: hexane= 1:10) and in 93% ee as determined by HPLC analysis [Daicel Chiralcel OD, hexane: IPA, 90:10, 1.0 ml/min, λ=220 nm, t(major) = 9.78 min, t(minor) = 13.73 min] from the reaction catalyzed by Q-5c (20 mol%) at 0 °C for 96 hours. [α]D 25 = +1.6 (c 1.5, CHCl3); m.p. 96–98 °C; 1H NMR (400 MHz, CDCl3) δ 8.36 (s, 2H), 8.18 (s, 1H), 5.66–5.78 (m, 2H) 5.20–5.30 (m, 1H), 4.18–4.32 (m, 2H), 3.36 (dt, J = 12.8 Hz, J = 4.4 Hz, 1H), 3.32(dt, J = 12.8 Hz, J = 4.4 Hz, 1H), 2.68 (dd, J=13.6 Hz, J=6.8 Hz, 1H), 2.65 (dd, J=13.6 Hz, J=6.8 Hz, 1H), 2.40 (dt, J = 12.4 Hz, J = 4.4 Hz, 1H), 2.24 (dt, J = 12.4 Hz, J = 4.4 Hz, 1H), 1.29 (t, J=6.8Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 166.8, 141.4, 133.5 (q, J = 34 Hz) 129.2, 128.5, 127.9, 122.3 (q, J = 273.0 Hz), 122.2, 117.2, 63.6, 52.3, 47.4, 41.3, 28.0, 13.9; IR (CHCl3) ν 3088, 2987, 1739, 1360, 1279, 1185, 1147 cm−1; HRMS (CI) m/z calcd for (C18H17F6NO4S + H+): 458.0861, found 458.0862.
The absolute configuration of adduct (+)-12J was determined to be “S” by X-ray structure analysis, see section 4.5 for details.
4.2.11. (−)-12K (Table 4, entry 4)
This product was obtained as a white solid in 85 % yield after flash chromatography (ethyl ether: hexane=1:5) and in 92% ee as determined by HPLC analysis [Daicel Chiralcel OD, hexane: IPA, 90:10, 1.0 ml/min, λ=220 nm, t(major) = 10.88 min, t(minor) = 17.64 min] from the reaction catalyzed by Q-5c (20 mol%) at 0 °C for 96 hours. [α]D 25 = −1.0 (c 2.0, CHCl3); m.p. 64–66 °C; 1H NMR (400 MHz, CDCl3) δ 8.40 (s, 2H), 8.21 (s, 1H), 4.28 (dq, J = 1.6 Hz, J = 7.2 Hz, 2H), 3.38 (dt, J = 13.6Hz, J = 4.4 Hz, 1H), 3.31(dt, J = 12.8 Hz, J = 4.4 Hz, 1H), 2.45 (dt, J = 12.8 Hz, J = 4.4 Hz, 1H), 2.27 (dt, J = 12.8 Hz, J = 4.4 Hz, 1H), 1.67 (s, 3H), 1.32 (t, J=6.8Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 167.6, 141.4, 133.5 (q, J = 30 Hz) 128.5, 128.0, 127.9, 122.1 (q, J = 272.4 Hz), 118.2, 63.1, 52.4, 42.4, 29.8, 23.7, 13.9; IR (CHCl3) ν 3088, 2990, 1745, 1281, 1186, 1147 cm−1; HRMS (CI) m/z calcd for (C16H15F6NO4S + H+): 432.0708, found 432.0712.
(+)-12K
This product was obtained as a white solid in 83% yield and 88% ee from a reaction catalyzed by QD-5c (20 mol %) at 0 °C for 96 hours.
4.3 Typical procedure for enantioselective additions of α-cyanoketones (13A–C) or β-ketoesters (14A–B) to vinyl sulfones 4a with 5c
To a mixture of 4a (0.20 mmol) and 5c (20 mol%) in toluene (0.40 mL) was added 13 (0.20 mmol) or 14 (0.40 mmol) in one portion. The resulting reaction mixture was kept at either room temperature or 0 °C from 24 to 96 h. The reaction mixture was purified by silica gel chromatography directly using the eluent specified below.
4.3.1. (−)-15A (Table 5, entry 1)
This product was obtained as a white solid in 96 % yield after flash chromatography (silica gel, Ethyl acetate: Hexanes = 1: 10) and in 95% ee as determined by HPLC [determined by HPLC, Daiso Chiralpak AD, Hexanes: IPA = 80: 20, 1.0 mL/min, λ = 220 nm, tr (major) = 32.66 min, tr (minor) = 27.03 min] from a reaction of 13A (43.6 mg, 0.40 mmol) and vinyl sulfone 4a (33.6 mg, 0.20 mmol) in toluene (400 µl) catalyzed by Q-5c (0.04 mmol) at room temperature for 24 h. [α]D 25 = −12.8 (c = 1.0, CHCl3); m.p. 113–115 °C; 1H NMR (400 MHz, CDCl3) δ 1.98–2.18 (m, 4H), 2.25 (dt, J = 4.4, 12.4 Hz, 1H), 2.36–2.46 (m, 1H), 2.46–2.58 (m, 2H), 3.265 (dt, J = 4.4, 12.4 Hz, 1H), 3.47 (dt, J = 4.4, 12.4 Hz, 1H), 7.61 (t, J = 7.6 Hz, 2H), 7.71 (d, J = 7.6 Hz, 1H), 7.90–7.96 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 19.2, 26.6, 35.0, 36.3, 46.7, 51.9, 117.5, 128.0, 129.5, 134.2, 138.4, 207.9; HRMS [M+H]+ Calcd for: C14H16O3SN: 278.0851 Found: 278.0854.
(+)-15A
This product was obtained as a white solid in 90 % isolated yield and in 95% ee from a reaction of 13A (43.6 mg, 0.40 mmol) and vinyl sulfone 4a (33.6 mg, 0.20 mmol) in toluene (400 µl) catalyzed by QD-5c (0.04 mmol) at room temperature for 24 h.
4.3.2. (−)-15B (Table 5, entry 2)
This product was obtained as a white solid in 96 % yield after flash chromatography and in 95% ee as determined by HPLC [determined by HPLC, Daiso Chiralcel OJ, hexanes: IPA = 70: 30, 1.0 mL/min, λ = 220 nm, tr (major) = 42.83 min, tr (minor) = 36.97 min] from a reaction of 13B (49.2 mg, 0.40 mmol) and vinyl sulfone 4a (33.6 mg, 0.20 mmol) in toluene (400 µl) catalyzed by Q-5c (0.04 mmol) at room temperature for 24 h. [α]D 25 = −97.1 (c = 1.1, CHCl3); m.p. 68–70 °C; 1H NMR (400 MHz, CDCl3) δ 1.64–1.76 (m, 2H), 1.86–1.96 (m, 1H), 1.98–2.10 (m, 2H), 2.10–2.18 (m, 1H), 2.28–2.36 (m, 2H), 2.46 (dt, J = 3.6, 14.0 Hz, 1H), 2.79 (dt, J = 6.4, 20.0 Hz, 1H), 3.22 (dt, J = 4.4, 13.2 Hz, 1H), 3.39 (dt, J = 4.4, 13.2 Hz, 1H), 7.61 (t, J = 7.6 Hz, 2H), 7.71 (m, 1H), 7.90–7.96 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 22.1, 26.8, 27.7, 39.0, 39.1, 49.9, 52.0, 118.3, 128.0, 129.4, 134.0, 138.5, 201.9; HRMS [M+H]+ Calcd for: C15H18O3SN: 292.1007 Found: 292.1008.
(+)-15B
This product was obtained as a white solid in 98 % isolated yield and in 95% ee from a reaction of 13B (49.2 mg, 0.40 mmol) and vinyl sulfone 4a (33.6 mg, 0.20 mmol) in toluene (400 µl) catalyzed by QD-5C (0.04 mmol) at room temperature for 24 h.
4.3.3. (−)-15C (Table 5, entry 3)
This product was obtained as a white solid in 91 % yield after flash chromatography and in 96% ee as determined by HPLC [determined by HPLC, Daiso Chiralcel OJ, Hexanes: IPA = 70: 30, 1.0 mL/min, λ = 220 nm, tr (major) = 16.27 min, tr (minor) = 22.27 min] from a reaction of 13C (54.8 mg, 0.40 mmol) and vinyl sulfone 4a (33.6 mg, 0.20 mmol) in toluene (400 µl) catalyzed by Q-5c (0.04 mmol) at room temperature for 24 h. [α]D 25 = −16.2 (c = 1.0, CHCl3); m.p. 153–156 °C; 1H NMR (400 MHz, CDCl3) δ 1.24–1.36 (m, 1H), 1.58–1.70 (m, 2H), 1.72–1.82 (m, 1H), 1.88–1.98 (m, 3H), 2.10–2.22 (m, 2H), 2.32 (dq, J = 4.4, 8.0 Hz, 1H), 2.61 (dq, J = 4.8, 10.8 Hz, 1H), 2.72 (dt, J = 5.2, 13.6 Hz, 1H), 3.16 (dt, J = 4.4, 13.6 Hz, 1H), 3.27 (dt, J = 4.4, 13.6 Hz, 1H), 7.61 (t, J = 7.6 Hz, 2H), 7.67–7.72 (m, 1H), 7.90–7.94 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 24.1, 25.8, 28.2, 28.9, 36.2, 40.6, 52.0, 53.2, 118.7, 127.9, 129.5, 134.1, 138.4, 204.0; HRMS [M+H]+ Calcd for: C16H20O3SN: 306.1163 Found: 306.1161.
The absolute configuration of (−)-15C was determined to be “S” by X-ray structure analysis, see section 4.5 for details.
(+)-15C
This product was obtained as a white solid in 91 % isolated yield and in 97% ee from a reaction of 3.7C (54.8 mg, 0.40 mmol) and vinyl sulfone (33.6 mg, 0.20 mmol) in toluene (400 µl) catalyzed by QD-2c (20 mol%) at rt for 24 h.
4.3.4. (−)-16A (Table 5, entry 5)
This product was obtained as a white solid in 93 % yield after flash chromatography (silica gel, ethyl ether: Hexanes = 1: 2) and in 96% ee as determined by HPLC [determined by HPLC, Daiso Chiralpak AD, Hexanes: IPA = 90: 10, 1.0 mL/min, λ = 220 nm, tr (major) = 15.64 min, tr (minor) = 19.24 min] from a reaction of 14a (73.6 mg, 0.40 mmol) and vinyl sulfone 4a (33.6 mg, 0.20 mmol) in toluene (400 µl) catalyzed by Q-5c (0.04 mmol) at room temperature for 72 h. [α]D 25 = −7.3 (c = 1.00, CHCl3); 1H NMR (400 MHz, CDCl3) δ 0.84 (t, J = 6.8 Hz, 3H), 1.04–1.54 (m, 8H), 1.56–1.78 (m, 1H), 2.84–2.96 (m, 1H), 3.20 (d, J = 17.6 Hz, 1H), 3.40 (dd, J = 6.8, 6.8 Hz, 1H), 3.70 (s, 3H), 3.87 (d, J = 17.6 Hz, 1H), 4.12 (dd, J = 6.8, 6.8 Hz, 1H), 7.43 (t, J = 7.2 Hz, 1H), 7.53 (d, J = 8.0 Hz, 1H), 7.67 (dt, J = 1.2, 8.0 Hz, 1H), 7.8 (d, J = 8.0 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ 19.5, 26.5, 27.7, 34.4, 33.7, 52.0, 58.6, 82.6, 128.0, 129.3, 133.7, 138.7, 169.6, 214.2; IR (neat) ν 2977, 1746, 1719, 1147; HRMS [M+Na]+ Calcd for: C18H24O5SNa: 375.1242 Found: 375.1233.
(+)-16A
This product was obtained as a white solid in 96 % isolated yield and in 96% ee from a reaction of 14A (73.6 mg, 0.40 mmol) and vinyl sulfone 4a (33.6 mg, 0.20 mmol) in toluene (400 µl) catalyzed by QD-5c (0.04 mmol) at room temperature for 72 h.
4.3.5. (−)-16B (Table 5, entry 6)
This product was obtained as a white solid in 100 % yield after flash chromatography (silica gel, ethyl ether: Hexanes = 2: 3) and in 97% ee as determined by HPLC [determined by HPLC, Daiso Chiralpak AD, Hexanes: IPA = 90: 10, 1.0 mL/min, λ = 220 nm, tr (major) = 28.57 min, tr (minor) = 24.85 min] from a reaction of 14B (93.0 mg, 0.40 mmol) and vinyl sulfone 4a (33.6 mg, 0.20 mmol) in toluene (400 µl) catalyzed by Q-5c (0.04 mmol) at room temperature for 36 h. [α]D 25 = −8.5 (c = 2.00, CHCl3); m.p. 114–116 °C1H NMR (400 MHz, CDCl3) δ 0.84 (t, J = 6.8 Hz, 3H), 1.04–1.54 (m, 8H), 1.56–1.78 (m, 1H), 2.84–2.96 (m, 1H), 3.20 (d, J = 17.6 Hz, 1H), 3.40 (dd, J = 6.8, 6.8 Hz, 1H), 3.70 (s, 3H), 3.87 (d, J = 17.6 Hz, 1H), 4.12 (dd, J = 6.8, 6.8 Hz, 1H), 7.43 (t, J = 7.2 Hz, 1H), 7.53 (d, J = 8.0 Hz, 1H), 7.67 (dt, J = 1.2, 8.0 Hz, 1H), 7.8 (d, J = 8.0 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ 26.6, 27.7, 37.86, 52.2, 59.1, 82.6, 124.9, 126.4, 128.1, 129.3, 133.8, 134.7, 135.6, 138.7, 140.0, 152.3, 169.3, 201.7; IR (neat) ν 2978, 1733, 1707, 1149; HRMS [M+Na]+ Calcd for: C22H24O5SNa: 423.1242 Found: 423.1245.
(+)-16B
This product was obtained as a white solid in 96 % isolated yield and in 96% ee from a reaction of 14B (93.0 mg, 0.40 mmol) and vinyl sulfone 4a (33.6 mg, 0.20 mmol) in toluene (400 µl) catalyzed by QD-5c (0.04 mmol) at room temperature for 36 h.
4.4 Typical procedure for enantioselective additions of α-cyanocyanoacetates 10, α-cyanoketones 13 and β-ketoesters 14 to β -substituted vinyl triflones 17 with 5c
To a mixture of 17 (0.10 mmol) and 5c (0.01 mmol) in dichloromethane (0.2 mL) was added 10 or 13 or 14 in one portion. The resulting reaction mixture was kept at −20 or −50 °C from 24 to 96 h. The reaction mixture was purified by silica gel chromatography directly using the eluent specified below.
4.4.1. (+)-18Ab (Table 7, entry 1)
This product was obtained from a Q-5c (4.8 mg, 0.01 mmol, 10 mol%) catalyzed reaction of 17b (23.0 mg, 0.10 mmol) and 10A (37.8 mg, 0.20 mmol) in methylene chloride (200 µl) at −50 °C for 96 h. The dr (5.6: 1) was determined by an analysis of the crude product via integrations of one set of 19F NMR (376 MHz, CF3COOH = −78.5 ppm) signals (s, δmajor = −81.86 ppm, δminor = −82.06 ppm)]. The crude product was purified by flash chromatography (silica gel, hexanes: ethyl ether = 20: 1) to give the major diastereomer of the 1,4-adduct as a colorless oil (36 mg, 85% yield, single diasteromer) and in 93% ee [determined by HPLC, Daiso Chiralpak AD, Hexanes: IPA = 98 : 2, 1.0 mL/min, λ = 220 nm, tr (major) = 5.70 min, tr (minor) = 6.75 min]. [α]D 25 = 12.0 (c = 1.00, CHCl3); 1H NMR (400 MHz, CDCl3) δ 0.75 (t, J = 7.2 Hz, 3H), 0.94–1.14 (m, 5H), 1.23 (t, J = 6.4 Hz, 3H), 1.22–1.34 (m, 1H), 1.42–1.52 (m, 2H), 3.20–3.30 (m, 1H), 3.40–3.48 (m, 2H), 4.18–4.42 (m, 2H), 7.38–7.44 (m, 3H); 7.50–7.58 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 13.9, 14.0, 22.2, 26.1, 31.1, 31.6, 39.5, 51.8, 59.3, 64.3, 116.9, 119.5 (q, J = 325 Hz), 126.7, 129.7, 130.0, 132.2, 166.5; 19F NMR (376 MHz, CF3COOH = −78.5 ppm) −81.9; IR (neat) ν 2959, 1746, 1510, 1374, 1228, 1122; HRMS (ESI) [M+H]+ Calcd for: C19H24F4O4S: 438.1362 Found: 438.1358.
(−)-18Ab
This product was obtained as an colorless oil in 76% yield (single diastereomer) and 87% ee from the corresponding QD-5c-catalyzed reaction of 17b (23.0 mg, 0.10 mmol) and 10A (37.8 mg, 0.20 mmol) in methylene chloride (200 µl) at −50 °C for 96 h. The dr (4.6: 1) of the reaction was determined by 1H NMR analysis of the crude product.
4.4.2. (+)-18Db (Table 7, entry 2)
This product was obtained from a Q-5c (4.8 mg, 0.01 mmol, 10 mol%) catalyzed reaction of 17b (23.0 mg, 0.10 mmol) and 10D (41.0 mg, 0.20 mmol) in methylene chloride (200 µl) at −50 °C for 96 h. The dr (26: 1) was determined by an analysis of the crude product via integration of one set of 19F NMR (376 MHz, CF3COOH = −78.5 ppm) signals (s, δmajor = −81.78 ppm, δminor = −82.11 ppm)]. The crude product was purified by flash chromatography (silica gel, Hexanes: ethyl ether = 30: 1) to give adduct 18Db as a colorless oil (41 mg, 94% yield, single diastereomer) and in 92% ee [determined by HPLC, Daiso Chiralpak AD, hexanes: IPA= 98: 2, 1.0 mL/min, λ = 220 nm, tr (major) = 5.94 min, tr (minor) = 8.01 min]. [α]D 25 = 11.0 (c = 1.00, CHCl3); 1H NMR (400 MHz, CDCl3) δ 0.79 (t, J = 6.8 Hz, 3H), 1.00–1.20 (m, 5H), 1.26 (t, J = 7.0 Hz, 3H), 1.28–1.38 (m, 1H), 1.42–1.52 (m, 2H), 3.20–3.30 (m, 1H), 3.40–3.48 (m, 2H), 4.20–4.36 (m, 2H), 7.16 (t, J = 8.4 Hz, 2H), 7.50–7.60 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 13.9, 14.0, 22.2, 26.1, 31.0, 31.5, 39.5, 51.6, 58.7, 64.5, 116.7, 116.8 (d, J = 22 Hz), 119.52 (q, J = 325 Hz), 128.00 (d, J = 3.0 Hz), 128.79 (d, J = 8.0 Hz), 163.50 (d, J = 118 Hz), 166.45; IR (neat) ν 2959, 1746, 1510, 1374, 1228, 1122; 19F NMR (376 MHz, CF3COOH = −78.5 ppm) −81.7, −113.8; HRMS(ESI) [M+H]+ Calcd for: C19H24F4O4S: 438.1362 Found: 438.1358.
(−)-18Db
This product was obtained as an colorless oil in 91% yield (single diastereomer) and 84% ee from the corresponding QD-5c-catalyzed reaction of 17b (23.0 mg, 0.10 mmol) and 10D (41.0 mg, 0.20 mmol) in methylene chloride (200 µl) at −50 °C for 96 h. The dr (12: 1) of the reaction was determined by 1H NMR analysis of the crude product.
4.4.3. (+)-18Fb (Table 7, entry 3)
This product was obtained from a Q-5c (4.8 mg, 0.010 mmol, 10 mol%) catalyzed reaction of 17b (23.0 mg, 0.10 mmol) and 10F (53.2 mg, 0.20 mmol, 2.0 eq.) in methylene chloride (200 µl) at −50 °C for 96 h. The dr (49: 1) was determined by an analysis of the crude product via integration of one set of 19F NMR (376 MHz, CF3COOH = −78.5 ppm) signals (s, δmajor = −81.69 ppm, δminor = −81.95 ppm)]. The crude product was purified by flash chromatography (silica gel, Hexanes: ethyl ether = 30: 1) to give adduct 18Fb as a colorless oil (46 mg, 92% yield, single diastereomer) and in 81% ee [determined by HPLC, Daiso Chiralpak AD, hexanes: IPA= 98: 2, 1.0 mL / min, λ = 220 nm, tr (major) = 5.92 min, tr (minor) = 9.91 min]. [α]D 25 = 4.6 (c = 2.00, CHCl3); 1H NMR (400 MHz, CDCl3) δ 0.79 (t, J = 6.8 Hz, 3H), 1.04–1.20 (m, 5H), 1.26 (t, J = 7.0 Hz, 3H), 1.30–1.38 (m, 1H), 1.41–1.52 (m, 2H), 3.20–3.30 (m, 1H), 3.40–3.50 (m, 2H), 4.20–4.36 (m, 2H), 7.46 (d, J = 9.2 Hz, 2H), 7.60 (d, J = 8.4 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 13.6, 13.7, 22.0, 25.8, 30.8, 31.3, 39.1, 51.3, 58.8, 64.4, 116.2, 119.3 (q, J = 327 Hz), 124.2, 128.2, 131.0, 132.7, 166.0; 19F NMR (376 MHz, CF3COOH = −78.5 ppm) −81.7; IR (neat) ν 2932, 1746, 1373, 1225, 1122.
(−)-18Fb
This product was obtained as an colorless oil in 92% yield (single diastereomer) and 86% ee from the corresponding QD-5c-catalyzed reaction of 17b (23.0 mg, 0.10 mmol) and 10F (53.2 mg, 0.20 mmol) in methylene chloride (200 μl) at −50 °C for 96 h. The dr (26: 1) of the reaction was determined by 1H NMR analysis of the crude product.
4.4.4. (+)-19Eb (Table 7, entry 4)
This product was obtained from a Q-5c (4.8 mg, 0.010 mmol, 10 mol%) catalyzed reaction of 13E (15.7 mg, 0.10 mmol) and 17b (25.3 mg, 0.11 mmol) in methylene chloride (400 µl) at −50 °C for 96 h. The dr (>49: 1) was determined by an analysis of the crude product via integration of one set of 19F NMR (376 MHz, CF3COOH = −78.5 ppm) signals (s, δmajor = −81.50 ppm, δminor = −81.24 ppm)]. The crude product was purified by flash chromatography (silica gel, hexanes:ethyl acetate = 3: 1) to give adduct 19Eb as a colorless oil (39 mg, 95% yield, single diastereomer) and in 99% ee [determined by HPLC, Daiso Chiralcel OJ, hexanes: IPA= 90: 10, 1.0 mL/min, λ = 220 nm, tr (major) = 19.15 min, tr (minor) = 24.02 min]. [α]D 25 = 75.0 (c = 1.00, CHCl3); 1H NMR (400 MHz, CDCl3) δ 0.89 (t, J = 6.8 Hz, 3H), 1.20–1.42 (m, 5H), 1.50–1.64 (m, 1H), 1.68–1.88 (m, 2H), 2.52–2.62 (m, 1H), 3.37 (dd, J = 6.8, 6.8Hz, 1H), 3.47 (d, J = 17.2 Hz, 1H), 3.73 (d, J = 7.2 Hz, 1H), 4.33 (dd, J = 3.6, 6.8 Hz, 1H), 7.52 (dd, J = 8.0, 8.0 Hz, 2H), 7.75 (dd, J = 8.0, 8.0 Hz, 1H), 7.87 (d, J = 8.0 Hz, 1H); 13C NMR (100 MHz, CDCl3 = 77.0 ppm) δ 14.1, 22.5, 26.4, 31.7, 32.6, 37.7, 38.0, 49.2, 50.7, 63.9, 118.2, 119.5(q, J = 325 Hz), 126.27, 126.66, 129.42, 133.44, 137.27, 150.09, 196.55; 19F NMR (376 MHz, CF3COOH = −78.5 ppm) −81.5. IR (neat) ν 2934, 1725, 1370, 1206, 1121; HRMS (ESI) [M+H]+ Calcd. for: C18H21NF3O5S: 388.1194 Found: 388.1188.
(−)-19Eb
This product was obtained as an colorless oil in 94% yield (single diastereomer) and 94% ee from the corresponding QD-5c-catalyzed reaction of 17b (25.3 mg, 0.11 mmol) and 13E (15.7 mg, 0.10 mmol) in methylene chloride (200 −l) at −50 °C for 96 h. The dr (>49: 1) of the reaction was determined by 1H NMR analysis of the crude product.
4.4.5. (+)-20Db (Table 7, entry 5)
This product was obtained from a Q-5c (4.8 mg, 0.010 mmol, 10 mol%) catalyzed reaction of 14D (19.0 mg, 0.10 mmol) and 17b (23 mg, 0.10 mmol) in methylene chloride (200 µl) at −20 °C for 96 h. The dr (32: 1) was determined by an analysis of the crude product via integration of one set of 19F NMR (376 MHz, CF3COOH = −78.5 ppm) signals (s, δmajor = −81.87 ppm, δminor = −81.79 ppm)]. The crude product was purified by flash chromatography (silica gel, hexanes: ethyl ether = 5: 1) to give adduct 20Db as a colorless oil (38mg, 90% yield, single diastereomer) and in 98% ee [determined by HPLC, Daiso Chiralcel OD + Regis (R, R)-whelk 1 O, hexanes: IPA = 99: 1, 1.0 mL/min, λ = 220 nm, t (major) = 20.15 min, t (minor) = 29.10min]. [α]D 25 = 116.4 (c = 1.00, CHCl3); 1H NMR (400 MHz, CDCl3) δ 0.84 (t, J = 6.8 Hz, 3H), 1.06–1.54 (m, 7H), 1.56–1.70 (m, 1H), 2.82–2.96 (m, 1H), 3.20 (d, J = 17.6 Hz, 1H), 3.40 (dd, J = 6.8, 6.8 Hz, 1H), 3.70 (s, 3H), 3.87 (d, J = 17.6 Hz, 1H), 4.12 (dd, J = 3.2, 14.4 Hz, 1H), 7.43 (t, J = 7.2 Hz, 1H), 7.53 (d, J = 8.0 Hz, 1H), 7.67 (dt, J = 1.2, 8.0 Hz, 1H), 7.8 (d, J = 8.0 Hz, 1H); 13C NMR (100 MHz, CDCl3 = 77.0ppm) δ 14.1, 22.5, 27.4, 31.7, 32.3, 35.8, 37,3, 50.4, 53.4, 63.9, 119.6(q, J = 325 Hz), 125.4, 126.6, 128.4, 135.0, 136.2, 152.9, 170.0, 200.4; 19F NMR (376 MHz, CF3COOH = −78.5 ppm) −81.8. IR (neat) ν 2957, 1742, 1717, 1367, 1200, 1122; HRMS (ESI) [M+H]+ Calcd. for: C19H24F3O5S: 421.1297 Found: 421.1289.
(−)-20Db
This product was obtained as an colorless oil in 93% yield (contains minor diastereomer, dr = 90:10) and 97% ee from the corresponding QD-5c-catalyzed reaction of 17b (23.0 mg, 0.10 mmol) and 10F (53.2 mg, 0.20 mmol) in methylene chloride (200 µl) at −50 °C for 96 h. The dr (9:1) of the reaction was determined by 1H NMR analysis of the crude product.
4.4.6. 20Dc (Table 7, entry 6)
This product was obtained from a Q-5c (4.8 mg, 0.010 mmol, 10 mol%) catalyzed reaction of 14D (38.0 mg, 0.20 mmol) and 17c (21.6 mg, 0.10 mmol) in methylene chloride (200 µl) at −20 °C for 72 h. The crude product was purified by flash chromatography (silica gel, hexanes: methylene chloride = 1: 1) to give the adduct 20Dc as a colorless oil and a diastereomeric mixture (41 mg, 100% yield, dr = 24: 1). The ee of the major diastereomer is determined to be 99% ee. Both dr and ee were determined by HPLC [Daiso Chiralcel AD + Chiralcel AD, hexanes: IPA = 98: 2, 1.0 mL/min, λ = 220 nm, tr (major) = 21.10 min, tr (minor) = 27.65 min]. [α]D 25 = 153.0 (c = 1.00, CHCl3); 1H NMR (400 MHz, CDCl3, major diastereomer was reported) δ 0.90 (d, J = 6.4 Hz, 3H), 0.91 (d, J = 6.4Hz, 3H), 1.02–1.12 (m, 1H), 1.60–1.76 (m, 2H), 2.92–3.06 (m, 1H), 3.18 (d, J = 17.2 Hz, 1H), 3.36 (dd, J = 6.8, 14.4 Hz, 1H), 3.71 (s, 3H), 3.88 (d, J = 17.2 Hz, 1H), 4.12 (dd, J = 6.8, 14.4 Hz, 1H), 7.44 (t, J = 7.2 Hz, 1H), 7.53 (d, J = 7.20 Hz, 1H), 7.68 (t, J = 6.8 Hz, 1H), 7.80 (d, J = 8.0 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ 21.1, 23.7, 25.1, 34.9, 35.7, 41.3, 50.2, 53.2, 63.6, 119.4 (q, J = 327 Hz), 125.2, 126.3, 128.2, 134.7, 134.0, 152.7, 169.7, 200.1; 19F NMR (376 MHz, CF3COOH = −78.5 ppm) −82.0; IR (neat) ν 2960, 1742, 1715, 1367, 1200, 1121; HRMS(ESI) [M+H]+ Calcd for: C18H22F3O5S: 407.1140 Found: 407.1150.
The QD-5c (4.8 mg, 0.010 mmol, 10 mol%) catalyzed reaction of 14E (38.0 mg, 0.20 mmol) and 17c (21.6 mg, 0.10 mmol) in methylene chloride (200 µl) was carried out at −20 °C for 72 h to furnish the crude product (dr = 8: 1). The crude product was purified by flash chromatography, to give the adduct 20Dc as a colorless oil and a diastereomeric mixture (40 mg, 98% yield, dr = 8: 1). The ee of the major diastereomer is determined to be 95%.
4.4.7. (−)-20Ed (Table 7, entry 7)
This product was obtained from a Q-5c (4.8 mg, 0.010 mmol, 10 mol%) catalyzed reaction of 14E (28.4 mg, 0.20 mmol) and 17d (26.4 mg, 0.10 mmol) in methylene chloride (100 µl) at −20 °C for 96 h the crude product (single diastereomer by 1H NMR analysis)[determined by integration of one set of 19F NMR (376 MHz, CF3COOH = −78.5ppm) signals (s, δmajor = −81.57 ppm)]. The crude product was purified by flash chromatography (silica gel, hexanes: methylene chloride = 2: 1) to give adduct 20Ed as a colorless oil (37 mg, 91% yield, single diastereomer) and in 95% ee [determined by HPLC, Daiso Chiralcel OJ, Hexanes: IPA= 90: 10, 1.0 mL/min, λ = 220 nm, t (major) = 17.45 min, t (minor) = 14.30 min]. [α]D 25 = −57.4 (c = 1.00, CHCl3); 1H NMR (400 MHz, CDCl3) δ 1.70–2.00 (m, 5H), 2.30–2.42 (m, 2H), 2.52–2.70 (m, 3H), 2.80–2.92 (m, 1H), 3.32 (dd, J = 6.4, 14.4 Hz, 1H), 3.71 (s, 3H), 4.25 (dd, J = 4.0, 14.4 Hz, 1H), 7.12–7.32 (m, 5H); 13C NMR (100 MHz, CDCl3 = 77.0 ppm) δ 19.3, 32.6, 34.0, 34.8, 35.4, 38.4, 50.6, 53.1, 63.2, 119.7 (q, J = 325 Hz), 126.5, 128.7, 128.8, 140.9, 170.0, 213.1; IR (neat) ν 1751, 1725, 1364, 1200, 1120; HRMS(ESI) [M+H]+ Calcd for: C18H22F3O5S: 407.1140 Found: 407.1145.
(+)-20Ed
The QD-5c (4.8 mg, 0.010 mmol, 10 mol%) catalyzed reaction of 14E (38.0 mg, 0.20 mmol) and 17c (21.6 mg, 0.10 mmol) in methylene chloride (200 µl) was carried out at −20 °C for 72 h to furnish the crude product as a single diastereomer (by 1H NMR analysis). The crude product was purified by flash chromatography, to give the adduct 20Ed as a colorless oil and a single diastereomer (33 mg, 80% yield) and in 89% ee.
4.5 X-Ray analysis for the determination of the abosolute configurations of (+)-12J and (−)-15C
X-Ray analysis was performed by Richard J. Staples at the department of chemistry and chemical biology, Harvard University. Crystallographic data (excluding structure factors) for the structures in this paper have been deposited with the Cambridge Crystallographic Data Centre as supplementary publication nos. CCDC 709230 [(+)-12J] and CCDC 709231 [(−)-15C]. The absolute configurations of compounds (+)-12J and (−)-15C were established using the anomalous scattering technique; Flack parameters were −0.06(10) and −0.06(6), respectively. Copies of the data can be obtained, free of charge, on application to CCDC, 12 Union Road, Cambridge CB2 1EZ, UK, (fax: +44-(0)1223-336033 or e-mail: deposit@ccdc.cam.ac.Uk).
Acknowledgment
We gratefully acknowledge the financial support of NIH (GM-61951) and Daiso Inc.
References
- 1.Simpkins NS. Sulphones in Organic Synthesis. Oxford: Pergamon Press; 1993. [Google Scholar]
- 2.For selective examples of asymmetric conjugate additions to α, β-unsaturated sulfones catalyzed by organic catalysts, see: Pinheiro S, Guingant A, Desmaële D, d’Angelo J. Tetrahedron: Asymmetry. 1992;3:1003. d’Angelo J, Revial G. Tetrahedron: Asymmetry. 1991;2:199. Enders D, Müller SF, Raabe G, Runsink J. Eur. J. Org. Chem. 2000:879. Mauleón P, Carretero JC. Org. Lett. 2004;6:3195. doi: 10.1021/ol048690p. Mauleón P, Carretero JC. Chem. Commun. 2005;39:4961. doi: 10.1039/b508142d. Mosse S, Alexakis A. Org. Lett. 2005;7:4361. doi: 10.1021/ol051488h. Sulzer-Mosse S, Tissot M, Alexakis A. Org. Lett. 2007;9:3749. doi: 10.1021/ol7015498. Zhu Q, Lu Y. Org. Lett. 2008 ASAP 10/4/08; for examples involving chiral metallic catalysts, see: Llamas T, Arrayas RG, Carretero JC. Angew. Chem., Int. Ed. 2007;46:3329. doi: 10.1002/anie.200700296. Desrosiers J-N, Carretero JC. Angew. Chem., Int. Ed. 2007;46:5955. Desrosiers J-N, Charette AB. Angew. Chem., Int. Ed. 2007;46:5955. doi: 10.1002/anie.200701367. Mauleón P, Alonso I, Rivero MR, Carretero JC. J. Org. Chem. 2007;72:9924. doi: 10.1021/jo7016197. Desrosiers J-N, Bechara WS, Charette AB. Org. Lett. 2008;10:2315. doi: 10.1021/ol800747v. Bos PH, Minnaard AJ, Feringa BL. Org. Lett. 2008;10:4219. doi: 10.1021/ol801566n.
- 3.For a preliminary communication of our studies of 5c-catalyzed conjugate additions to vinylsulfones, see: Li H, Song J, Liu X, Deng L. J. Am. Chem. Soc. 2005;127:8948. doi: 10.1021/ja0511063.
- 4.For select reviews of catalytic enantioselective construction of all carbon quaternary chiral centers, see: Douglas CJ, Overman LE. Proc. Natl. Acad. Sci. USA. 2004;101:5363. doi: 10.1073/pnas.0307113101. Christoffers J, Baro A. Angew. Chem., Int. Ed. 2003;42:1688. doi: 10.1002/anie.200201614. Christoffers J. Chem. Eur. J. 2003;9:4862. doi: 10.1002/chem.200304937. Christoffers J, Mann A. Angew. Chem.int. Ed. 2001;40:4591. doi: 10.1002/1521-3773(20011217)40:24<4591::aid-anie4591>3.0.co;2-v. Corey EJ, Guzman-Perez A. Angew. Chem., Int. Ed. 1998;37:388. doi: 10.1002/(SICI)1521-3773(19980302)37:4<388::AID-ANIE388>3.0.CO;2-V.
- 5. Li H, Wang Y, Tang L, Deng L. J. Am. Chem. Soc. 2004;126:9906. doi: 10.1021/ja047281l. Li H, Wang Y, Tang L, Wu F, Liu X, Guo C, Foxman BM, Deng L. Angew. Chem. Int. Ed. 2005;44:105. doi: 10.1002/anie.200461923. For other select examples of 5c-catalyzed other asymmetric reactions, see: Wu F, Hong R, Khan J, Liu X, Deng L. Angew. Chem., Int. Ed. 2006;45:4301. doi: 10.1002/anie.200600867. Wang Y, Liu X, Deng L. J. Am. Chem. Soc. 2006;128:3928. doi: 10.1021/ja060312n. Li H, Wang B, Deng L. J. Am. Chem. Soc. 2006;128:732. doi: 10.1021/ja057237l. Li H, Wang Y-Q, Deng L. Org. Lett. 2006;8:4063. doi: 10.1021/ol061552a. Wang Y, Li H, Wang Y-Q, Liu Y, Foxman BM, Deng L. J. Am. Chem. Soc. 2007;129:6364. doi: 10.1021/ja070859h.
- 6.For a minireview of asymmetric catalysis with C6’-OH cinchona alkaloids (cupreines and cupreidines) see: Marcelli T, van Maarseveen J, Hiemstra H. Angew. Chem. Int. Ed. 2006;45:7496. doi: 10.1002/anie.200602318. for a review of asymmetric catalysis by hydrogen-bond donors see: Doyle A, Jacobsen EN. Chem. Rev. 2007;107:5713. doi: 10.1021/cr068373r.
- 7.For the synthesis of 4b, see: Alonso DA, Nάjera C, Varea M. Synthesis. 2003:277.
- 8.For the synthesis of 17b–d, see Mahadevan A, Fuchs PL. Tetrahedron Lett. 1994;33:6025.