

Abstract
The management of chronic hepatitis B currently rests with long-term therapy using oral nucleoside analogues. The major limitation of long-term therapy is antiviral resistance. Antiviral resistance is due to the high rate of mutations that can occur during hepatitis B virus (HBV) replication and the selection of these mutants due to a replication advantage in the presence of the antiviral agent. Indeed, high rates of antiviral resistance have been found with long-term use of lamivudine, in up to 76% of patients treated for 5 years or more. Rates of antiviral resistance are lower with adefovir therapy, ∼30% at 5 years. Newer more potent nucleoside analogues (tenofovir and entecavir) have proven to have much lower rates of antiviral resistance (<1% after 2 years in treatment-naïve subjects), but the long-term rates of resistance have yet to be fully defined. The appearance of these viral mutations (genotypic resistance) is usually followed by rises in HBV DNA levels (virological breakthrough) and then by rises in serum aminotransferase levels (biochemical breakthrough). The appearance of antiviral resistance can be accompanied by a transient but occasionally severe exacerbation of the underlying liver disease which in some instances have led to acute liver failure. Combinations of nucleoside analogues may offer an approach to preventing antiviral resistance, but the efficacy and safety of this approach has yet to be shown. A future research priority is to identify new agents active again HBV that target different steps in the viral life cycle and might provide effective means to circumvent the antiviral resistance of nucleoside analogues.
Keywords: Mutations, antiviral therapy, HBV DNA, virological breakthrough
Introduction
The management of chronic hepatitis B has improved in the last decade primarily because of the availability of oral nucleoside analogue therapy. These agents are well tolerated, very effective at suppressing viral replication and appear to be safe over our current five to ten year knowledge of experience. A major shortcoming of nucleoside analogue therapy is the high rate of virological relapse when treatment is discontinued. Therefore, treatment must be often administered long-term, if not indefinitely. Unfortunately, long-term therapy is associated with the development of antiviral drug resistance which frequently negates the benefits of therapy and sometimes may be associated with hepatitis flares and even death. Therefore, a major challenge in the management of hepatitis B is how to best use these agents, whether singly, sequentially or in combination, in order to minimize the risk of developing viral resistance. This review will focus on why antiviral resistance occurs, the nomenclature used to classify viral resistance, rates of viral resistance development to approved agents, management of established antiviral resistance and strategies to prevent antiviral resistance in chronic hepatitis B.
Why does Antiviral Resistance Occur?
Although the hepatitis B virus (HBV) has a DNA genome, it replicates via an RNA intermediate step.1 The lack of proof reading capacity of the viral encoded RNA dependent RNA polymerase coupled with the extremely high rate of HBV replication yields the potential for mutations to be generated at each nucleotide position within the entire genome on a daily basis.2 Under selection pressure created by the presence of an antiviral agent, viruses with mutations that confer a replication advantage are selected and ultimately become the predominant viral species.
Several factors are associated with the development of antiviral resistance, but the key ones based upon our current understanding, are viral fitness and the potency and genetic barrier to resistance of the antiviral agent.3 Viral fitness generally refers to the ability of a virus to replicate in a defined environment and is dependent on two primary components, the replication capacity which refers to the ability of mutant virus to replicate in the absence of a drug compared to a wild-type, drug-sensitive virus, and replication space, which refers to the ability of the environment to support replication-the hepatocyte in the case of HBV.4, 5 Usually, mutant viruses are “less fit” meaning they do not replicate as well as wild-type virus, but may have a survival advantage in the presence of an antiviral agent. Over time, compensatory or secondary mutations develop after the initial mutation, which restores functional defects in the viral polymerase caused by the primary mutations. These compensatory mutations enable the mutant virus to replicate at near wild-type levels leading to the development of antiviral resistance.
The potency of a nucleoside analogue acting as an antiviral agent is determined by its ability to serve as a competitive inhibitor of the HBV polymerase relative to that of the natural substrate.6 Clinically, potency of a drug is reflected by how rapidly it can suppress viral replication; the more rapid, the lower the risk of developing antiviral resistance.7 A drug with low antiviral potency exerts minimal pressure upon the viral population leading to a low probability of developing drug resistance. A highly potent drug will achieve rapid and complete suppression of viral replication, thus providing little opportunity for the emergence of resistant virus since mutagenesis is replication-dependent. An agent exerting modest antiviral activity that incompletely suppresses viral replication provides the greatest opportunity for selecting drug-resistant virus.
The genetic barrier to resistance refers to the number of mutations that the virus must accumulate in order to replicate efficiently in the presence of the antiviral agent. The genetic barrier to resistance is partly dependent upon the structure of the antiviral compound and the constraints imposed by the ability of the viral polymerase to tolerate compensatory mutations without significantly impairing its enzymatic activity. Thus, an agent with a high genetic barrier to accumulation of mutations will naturally have a lower likelihood of developing resistance.
A number of other host factors indirectly contribute to the development of antiviral resistance. Immune suppression, whether innate or acquired, can affect the rate of viral replication and, therefore, the rate of development of mutations. Achieving effective drug concentrations may be affected by obesity by altering the volume of distribution, by patient non-adherence and by the activity of host cellular enzymes necessary for phosphorylation of prodrugs.6 Finally, existing mutations due to prior use of antiviral agents can lower the genetic barrier to resistance and lead to a faster development of resistance, as is the case of the presence of the rtM204V/I mutation (conferring resistance to lamivudine) and the subsequent development of entecavir resistance.8
Nomenclature of Antiviral Resistance
Standardizing the nomenclature of antiviral resistance is important for several reasons relevant not only to clinical research but also to clinical practice. Defining which nucleotide changes constitute drug resistance and how new mutations are characterized will allow for the true incidence of antiviral resistance to be determined as well as permit direct comparison of the available antiviral agents. Furthermore, standardizing nomenclature allows for better characterization of the clinical effects of antiviral resistance and will improve strategies to manage resistance.
Primary Non-response
Primary non-response is defined as the inability of the antiviral agent to reduce serum HBV DNA by ≥1 log10 IU/mL within the first 6 months of treatment9 (Figure 1). Definitions vary among experts but this definition was chosen because it exceeds the variability within HBV DNA assays and reflects a true virological but not a clinical response. Primary non-response is related to the potency of the antiviral agent and perhaps to polymorphisms of host enzymes that are involved in converting prodrugs to their active moiety.6 There is no evidence that pre-existing antiviral mutations are associated with primary non-response.10 The observation of primary non-response is an indication to change therapy because a high residual viral level after 6 to 12 months of therapy has been associated with an increased risk for the development of resistance.11–14
Figure 1.
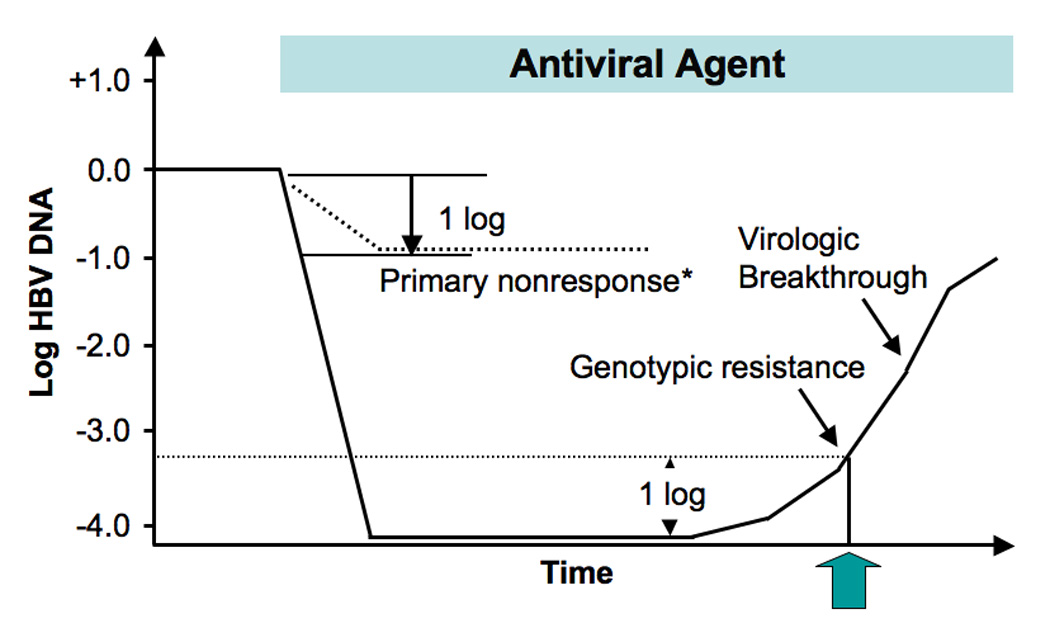
Definitions and timing of occurrence of virological forms of antiviral treatment failure during therapy of chronic hepatitis B, including primary non-response, genotypic resistance, and virological breakthrough. Abbreviations: HBV, hepatitis B virus. * Defined as < 1 log reduction in serum HBV DNA after 6 months of therapy. Adapted with permission from reference 9.
Virological Breakthrough
Virological breakthrough is the first clinical indication of the development of antiviral drug resistance. It is defined as an increase in serum HBV DNA by ≥1 log10 IU/mL above a nadir on two or more consecutive occasions at least one month apart while on treatment after achieving an initial response in a compliant patient9 (Figure 1 and Figure 2). Serum HBV DNA levels initially tend to be low because, as previously discussed, most antiviral-resistant mutants have lower replication fitness compared to wild-type virus.15, 16 However, with time compensatory mutations develop which restore replication fitness and the viral level returns to and sometimes exceeds pre-treatment levels.7, 17, 18 This phenomenon is termed viral rebound (Figure 2).
Figure 2.
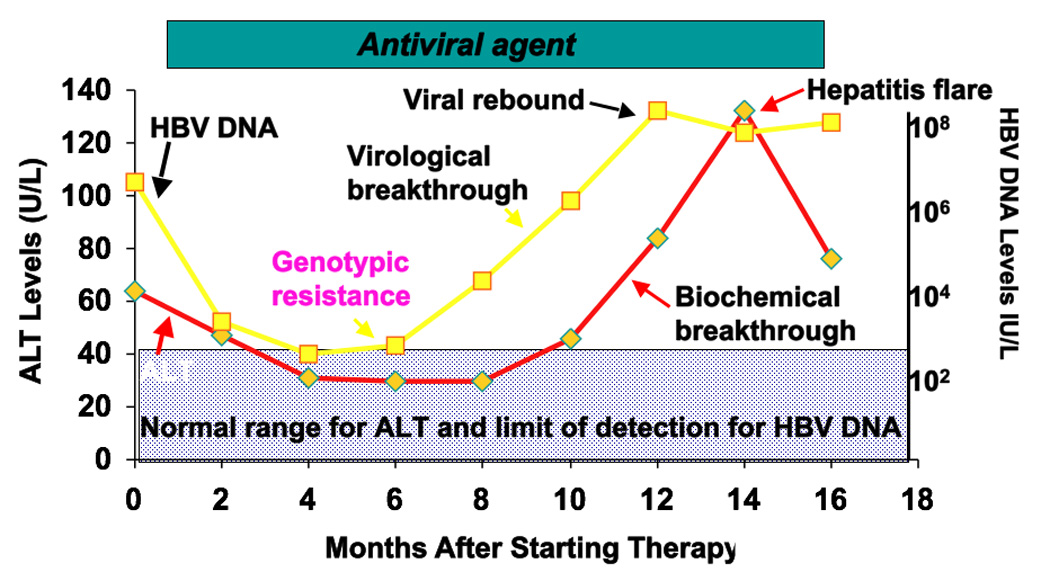
Course of a patient with chronic hepatitis B treated with an antiviral agent who develops genotypic resistance, followed by virological and biochemical breakthrough and viral rebound with a flare of hepatitis. Abbreviations: HBV, hepatitis B virus; ALT, alanine aminotransferase.
Biochemical Breakthrough
Biochemical breakthrough is defined as an elevation in serum alanine aminotransferase (ALT) level while on treatment after achieving normalization in a compliant patient9 (Figure 2). Biochemical breakthrough usually lags behind virological breakthrough, and serum ALT levels may remain normal for weeks to years after the development of antiviral resistance. Thus, serum ALT is not a sensitive indicator of antiviral resistance. On occasion, the serum ALT levels may increase markedly indicating a significant flare of disease (defined as a serum ALT level >5 times the upper limit of the normal range) and even place the patient at risk for hepatic decompensation.19, 20
Genotypic Resistance
Genotypic resistance refers to the detection of viral populations bearing amino acid substitutions in the reverse transcriptase region of the HBV genome that have been shown to confer resistance to antiviral drugs in a phenotypic assay during antiviral therapy (Figure 2).9 These mutations are usually detected in patients with virological breakthrough but they can also be present in low levels in patients whose virus levels plateau. Rarely these mutations can be detected before therapy.21
Rates of Resistance
Defining the rates of viral resistance in therapeutic clinical trials is problematic because of varying definitions used in the published literature (whether defined as genotypic or virological or biochemical breakthrough); different sensitivities of the assays used to assess genotypic resistance; and different patient populations tested across trials. Therefore, there is a need to standardize reporting regarding when to test and with which assays. For clinical trials, at a minimum, direct sequencing should be performed at baseline, annually on all HBV DNA positive patients and on all patients with primary treatment failure. Additionally, cumulative rates of virological and biochemical breakthrough should be reported.22 Standardized reporting in this fashion will allow for a better understanding not only of the true rates of resistance with antiviral agents but also their clinical significance, cross-resistance profiles, and ultimately improve patient management.
In clinical practice, it is unnecessary to perform resistance testing at baseline unless the patient is treatment-experienced. Resistance testing based on a commercially available assay (see below) should be performed in patients with a suboptimal antiviral response before changing therapy and in patients with virological and biochemical breakthrough.
The published average rates of resistance to approved agents in treatment-naïve subjects with chronic hepatitis B are shown in Figure 3. Rates of resistance are highest for the L-nucleoside class of antiviral agents (lamivudine, telbivudine) and lower for the acyclic phosphonates (adefovir and tenofovir) and the cyclopentane class (entecavir).
Figure 3.
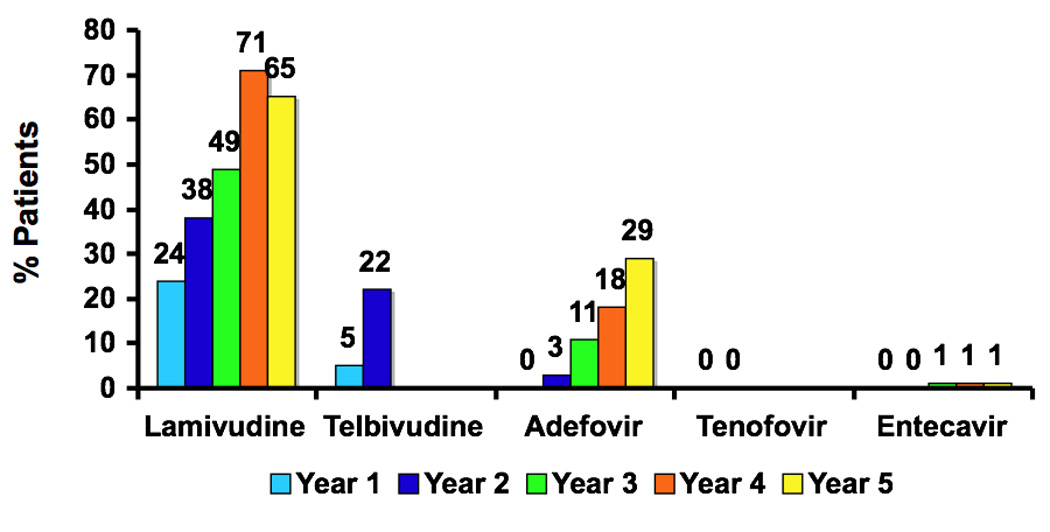
Cumulative rates of antiviral resistance to the major nucleoside analogues in current use as therapy of chronic hepatitis B in treatment naïve HBeAg-positive subjects (with the exception of adefovir data which was derived from HBeAg negative subjects). Abbreviations: Lam, lamivudine; ADV, adefovir dipivoxil; ETV, entecavir; LdT, telbivudine; HBeAg, hepatitis B e antigen. Data from references 12, 13, 31–33, 38, 43, 45, 46, 52 and 53.
L-nucleosides
Lamivudine and telbivudine are the only L-nucleosides approved for use in the United States. Clevudine is approved for use in Korea and emtricitabine is used off label in combination with tenofovir disoproxil fumarate. All L-nucleosides have a similar molecular structure and target site of action, and therefore similar patterns of antiviral resistance mutations.7, 23 Consequently, intra-class cross-resistance exists. L-nucleosides act as chain terminators inhibiting negative and positive HBV DNA strand synthesis.17 The target for these agents is the reverse transcriptase domain of the HBV polymerase. The specific primary mutations conferring resistance to lamivudine are the rtM204V/I substitutions17 and for telbivudine, only the rtM204I substitution.24 Case reports have also documented primary resistance to lamivudine occurring with rtA181T/S.25–27 From a structural perspective, primary lamivudine resistance mutations induce steric hindrance with decreased binding of lamivudine to the viral polymerase.17 As a consequence of the primary resistance mutation, catalytic activity of the polymerase is reduced due to altered alignment of the incoming nucleotide with respect to the template and primer in the active site.17, 28, 29 In vitro studies have shown that these mutations result in a >1000 fold decreased susceptibility of the viral polymerase for lamivudine and reduced functional capacity of the viral polymerase.16, 17 Compensatory mutations including rtV173L, rtL180M and rtL80I restore the viral polymerase function to near wild-type levels and are almost always associated with the primary resistance mutations.16, 18, 30 In isolation, compensatory mutations are associated with very low level resistance in vitro.
In clinical trials the rate of resistance to lamivudine is relatively high, with 14% to 24% of HBeAg positive and 6–18% of HBeAg negative patients developing genotypic resistance at one year and >70% after 8 years of continuous therapy.31–35 The somewhat different reported rates reflect differences in the studies: whether all subjects were tested or only those with virological breakthrough and whether testing was performed by direct sequencing or restriction fragment length polymorphism (RFLP) analysis.
Telbivudine is associated with a lower rate of resistance compared to lamivudine in HBeAg positive subjects at one year, 5%, but the rate jumps to 22% at two years, suggesting that the development of resistance may become a significant problem with longer duration of therapy.12 Rates of resistance are lower in HBeAg negative subjects are lower at one year 2% but increase to 11% at 2 years.12, 13
Patients with high baseline HBV DNA, slower rates of clearance of HBV DNA (defined as detectable virus after 6 months of continuous therapy) and extended duration of therapy are more prone to develop lamivudine and telbivudine resistance.11, 13, 36 In one study 60% of patients receiving lamivudine with an HBV DNA level >200 copies/mL at week 24 developed resistance compared to only 8% in those whom the HBV DNA level was <200 copies/mL as measured by Cobas Amplicor HBV Monitor assay.11 Similarly, with telbivudine, approximately 80% of patients with an HBV DNA level >3 log10 copies/mL at week 24 developed resistance by week 104 compared to only 4% of patients with undetectable HBV DNA measured by Cobas Amplicor HBV Monitor assay.13 However, it is possible that these differences may disappear with long-term therapy.
Acyclic Phosphonates
Adefovir and tenofovir are acylic phosphonates approved for therapy of chronic hepatitis B. Structurally, instead of a phosphate group, these agents possess a phosphonate group that cannot be cleaved by host esterases and act as chain terminators.37 The structural similarity of these compounds to the natural substrate dATP and the small, flexible phosphonate linker allow these compounds greater access to the HBV polymerase active site. Mutations that confer resistance to adefovir reside outside of the YMDD motif, in the B and D domains of the HBV polymerase. There are two primary adefovir resistant mutations, the rtA181T and rtN236T substitutions in the viral polymerase.38 These mutations result in only a modest (2 to 9-fold) increase in the EC50.38–40 Nevertheless, virological breakthrough occurs with this subtle change in the binding of adefovir to the mutant HBV polymerase. A report of a mutation associated with primary non-response in three patients treated with adefovir has not been substantiated in a subsequent study.41, 42
Long-term follow-up studies in hepatitis B e antigen (HBeAg)-negative subjects treated with adefovir reported a rate of genotypic resistance of 3% at 2 years and a cumulative rate of 29% at 5 years.43 Interestingly, the rate of virological breakthrough was only 20% and biochemical breakthrough, 11% at year 5, probably because of the small change in EC50 with the resistance mutations. The long-term rate of resistance in HBeAg-positive subjects is unclear. A 20% rate of resistance was reported after a median of 235 weeks,44 however, this result should be interpreted with caution and may be higher because the duration of adefovir therapy varied greatly among subjects due to multiple dose interruptions. As with lamivudine, residual virus during the first year of therapy was predictive of future resistance. Development of resistance to adefovir can be predicted by the viral level at week 48. Forty-nine percent of patients with a serum HBV DNA level greater than 1000 copies/ml at week 48 subsequently developed resistance by week 192, compared to 6% of patients with serum HBV DNA levels <1000 copies/ml at week 48.43 This longer duration of 48 instead of 24 weeks used with other antiviral agents is likely due to the lower antiviral potency associated with the suboptimal dose of adefovir.
In vitro studies indicated that tenofovir is of equal potency to adefovir but is more effective clinically due to the fact that the administered dose is 30 times higher than that of adefovir. Virological breakthrough has been observed during therapy with tenofovir in both HBeAg-positive and -negative subjects but no genotypic resistance mutations were detected in the HBV reverse transcriptase after 2 years of continuous tenofovir use.45, 46 Virological breakthrough was attributed mostly to non-compliance.
Cyclopentane Group
Entecavir is a carbocyclic analogue of 2’deoxyguanosine with potent activity against HBV. Entecavir acts at three stages in the viral replication cycle by inhibiting priming as well as inhibiting synthesis of both negative and positive strand HBV DNA.47 Entecavir has a high genetic barrier to resistance, which means that the virus must accumulate several mutations before virological breakthrough occurs.8 The rtM204V/I mutations classically associated with lamivudine resistance are necessary for the development of virological breakthrough to entecavir.8 In the absence of the primary lamivudine-resistant mutation, the entecavir associated resistant mutations results in only a modest change in EC50 (<10 fold); however, with the presence of the lamivudine-resistant mutation, there is an over 1000-fold change in susceptibility for the viral polymerase.8, 48 Two patterns of mutations have been reported. One pattern includes rtI169T + rtL180M + rtM204V + rtM250V and the other rtL180M + rtT184G + rtS202I and rtM204V.8, 48 Mutations at rt169 and rt250 impact on the primer binding region of the reverse transcriptase whereas, the rt184 and rt202 mutations alter the geometry of the polymerase nucleotide binding pocket of the catalytic site.49–51
A very low rate of antiviral resistance in nucleoside-naïve patients has been reported with entecavir, 0% at one year and 1.2% at 5 years.52, 53 As predicted by the in-vitro data, entecavir resistance develops at a significantly higher rate in patients with pre-existing lamivudine-resistance mutations rtM204V/I ± rtL180M. The cumulative incidence of genotypic resistance to entecavir in lamivudine-resistant patients increases from 1% at one year to 51% at 5 years.53, 54
Clinical Consequences of Antiviral Resistance
The development of antiviral resistance is generally associated with worse clinical outcomes. In a landmark trial that randomized patients with bridging fibrosis or cirrhosis to either lamivudine or placebo to prevent liver disease progression, lamivudine was shown to delay the development of liver disease progression by 50 percent during a median period of 32 months of treatment.55 Patients who developed lamivudine resistance had less benefit compared to those without resistance, but they still experienced fewer adverse outcomes compared to placebo-treated patients.55 Whether patients with resistance would have developed worse outcomes with longer duration of follow-up remains to be determined. In another study evaluating the long-term histological benefits of lamivudine, the effectiveness of therapy was negated by the development of lamivudine resistance.56 Patients who received lamivudine continuously for 3 years and who had no evidence of drug resistance were more likely to demonstrate histological improvement (77% versus 44%) and less likely to show deterioration (5% versus 15%) compared to those who developed resistance.56 Patients with lamivudine resistance for more than 2 years were least likely to demonstrate histological improvement (36%). Lower rates of HBeAg loss and seroconversion were reported in patients receiving lamivudine treatment following the development of resistance.57
Of particular concern have been reports of hepatitis flares, hepatic decompensation and death following the development of antiviral resistance. In a retrospective analysis of 998 HBeAg-positive patients with compensated liver disease who received lamivudine for a median of 4 years, both the risk and severity of hepatitis flares increased with the duration of lamivudine resistance, such that 80% patients who had lamivudine resistance for more than 4 years experienced at least one flare of hepatitis.19 The risk of hepatic decompensation was extremely low in this cohort, <1%, but was increased for those with lamivudine resistance for more than 4 years.19 The generally favorable short-term clinical outcome of this cohort may be related to their young age (mean 32 years) and the fact that only 10% had cirrhosis at the onset of treatment.
Particularly problematic with the development of antiviral resistance is the limitation it may place on future treatment options. For instance, entecavir has a very low rate of resistance in treatment-naïve patients, ∼1% at 5 years, but is associated with high failure rates in lamivudine-resistant patients, 51% at 5 years, due to the cross-resistance between entecavir and lamivudine.8, 53 This problem becomes a clinically relevant issue given the numbers of patients who are lamivudine-experienced. Finally, the transmission of antiviral resistant mutants to treatment naïve persons poses a potential public health problem. Fortunately, this clinical scenario has yet to be found to be a significant problem.
Monitoring for Antiviral Resistance
Detection of antiviral resistance requires the implementation of an adequate monitoring schedule during therapy as recommended by professional society guidelines and expert opinion. Recommendations on what constitutes an appropriate monitoring schedule or what is the most appropriate viral assay to monitor for resistance differ among the various guidelines.22, 58–61 Guidelines and expert opinion recommend monitoring for antiviral resistance by periodic assessment of serum HBV DNA using a sensitive assay. 22, 58–61 Surveillance for resistance using genotypic testing is not practical due to the high costs, the lack of standardized assays and the fact that the genotypic resistance profile of the antiviral agent must first be known. Similarly, monitoring serum ALT alone is neither sensitive nor specific for the detection of antiviral resistance. Therefore, HBV DNA levels should be monitored in all patients receiving nucleoside analogues to document an initial virological response and to monitor for treatment failure during therapy in those who achieved an initial virological response. A reasonable monitoring schedule would be at baseline and every 3 months until HBV DNA becomes undetectable and then every 3 to 6 months while on treatment. Despite the low specificity for detection of resistance, serum ALT should also be monitored for biochemical breakthrough.
A rise in serum HBV DNA level from a nadir is suggestive of treatment failure or medication non-compliance. Thus it is advisable to repeat the HBV DNA test in one month to confirm the result, unless the initial rise in HBV DNA level occurred simultaneously with a rise in serum ALT in a compliant patient. If available, testing for genotypic resistance should be ordered at the time of virological breakthrough to allow the clinician to more precisely select alternative therapeutic options and to prevent an inappropriate change in therapy.
No specific assay for monitoring HBV DNA levels is recommended. Rather, the following characteristics are desired: a test with excellent sensitivity, such as a lower limit of detection of 10 IU/mL, so as to be able to detect virological breakthrough early; a broad dynamic range to cover both off and on-treatment viral levels and one that is accurate across all genotypes. Thus a real time PCR assay is recommended because of its sensitivity and a dynamic range of 7–8 logs10. It is important to use the same assay during treatment monitoring so results can be accurately compared.
The role of genotypic testing in clinical practice is still being defined. Currently, resistance testing prior to initiating therapy in treatment-naïve patients is not recommended because of the low rate of resistance mutations in untreated patients and the unknown clinical significance of minor mutant populations (<5%) detected with ultra-sensitive assays. If available, testing should be performed in treatment-experienced patients to guide rescue therapy because of cross-resistance among different agents. Two types of commercial tests are available for genotypic assessment: direct sequencing (TRUGENE HBV -Siemens Health Care Diagnostic Solutions, Tarrytown, NY and Affigene HBV DE/3TC Assay -Sangtec Molecular Diagnostics AB) and reverse hybridization (INNO-LiPA DR Version 2.0 [Inogenetics]). Advantages and limitations exist with both techniques. Direct sequencing can identify all existing and emerging mutations but cannot detect mutations that are present at low proportions (<20%) of the viral population. Furthermore, interpretation of sequencing data can be problematic and confirmation of genotypic resistance requires either in vitro phenotypic analysis confirming that the mutation within HBV polymerase decreases susceptibility to treatment or extensive correlation with clinical data. Hybridization-based methods are more sensitive and can detect mutant populations that represent 5% of the overall population. However, a limitation of this approach is that only previously identified mutations can be detected and individual probes have to be incorporated into the assay in order to identify new mutations. More sensitive technologies are being developed such as mass spectrometry and ultradeep pyrosequencing but currently these are research tools.62, 63
Management of Antiviral Resistance
Management of antiviral resistance has been shaped by several important concepts which have emerged in recent years. Earlier alteration of therapy after the emergence of virological breakthrough has been associated with better long-term virological outcome. In a study to assess whether the timing of administration of rescue therapy influenced virological outcome, adefovir rescue therapy was initiated either at the time of detection of just genotypic resistance or at the time of genotypic and biochemical breakthrough in patients treated with lamivudine.64 At 2 years after instituting adefovir rescue therapy, the virological response was 100% in patients who were treated at the time of detection of genotypic resistance but only 78% in those who presented with both genotypic and biochemical breakthrough suggesting a more favorable clinical outcome with early implementation of rescue therapy.64
The next emerging concept is that add-on therapy rather than switching therapy appears to be a superior approach with regard to preventing subsequent multi-drug resistance. In a retrospective analysis of 588 patients with lamivudine resistance, 303 were switched to adefovir and 285 had adefovir added in combination with lamivudine.65 The rates of virological response were similar in both the switched and combination groups after three years of therapy, (71% versus 78%, respectively). However, the rate of virological breakthrough (30% versus 6%), and genotypic resistance to adefovir (16% versus 0%) were significantly higher in the switched group as opposed to the add-on group, respectively, underscoring the benefit of combination therapy in preventing the development of multi-drug resistance.65 Further support for the add-on approach is evident from a small randomized trial in HBeAg-negative patients with confirmed genotypic resistance to lamivudine and biochemical breakthrough, who were managed by being switched to adefovir monotherapy or addition of adefovir to ongoing lamivudine therapy.66 The virological and biochemical response rates were similar with both strategies but adefovir resistance occurred in 21% of patients who were switched to adefovir monotherapy within 15 to 18 months from the start of treatment compared to 0% in patients receiving add-on combination therapy.66
Therapy should be altered immediately in patients with cirrhosis if they develop drug resistance because of a higher risk for hepatic decompensation should a hepatitis flare occur. Finally, the choice of rescue therapy for a patient with drug-resistant HBV should be based on the cross-resistant profile of the mutations present, the potency of available agents against these mutations and the presence of other co-morbid conditions such as renal insufficiency.
Options for Management of Antiviral Resistance
The management options for patients with lamivudine resistance are evolving and based upon available data from clinical trials and in vitro testing. Therapeutic options include adding adefovir, switching to drugs with higher potency and higher genetic barriers to resistance such as entecavir or tenofovir or the combination of tenofovir plus emtricitabine (off label use) (Table 1).7 54, 65–70 However, emerging data suggests that entecavir is associated with an unacceptably high rate of resistance in patients with lamivudine resistance, 51% with extended duration therapy, and therefore may not be a viable option for management of lamivudine resistance.71 For HBeAg-positive patients, rates of undetectable HBV DNA (using a sensitive polymerase chain reaction [PCR]-based assay) at week 48 in those treated with adefovir 10 mg daily as add on therapy, or who were switched to entecavir 1.0 mg daily or tenofovir 300 mg daily were 35%, 21% and 91%, respectively.69, 72, 73 Although these results were not directly comparable because of different baseline patient characteristics, the one-year rate of resistance to adefovir add-on, or switching to entecavir or tenofovir were 0%, 1% and 0% respectively.69, 72, 73 These data would support any of these strategies at least in the short-term, but longer-term data is necessary before one strategy can be recommended over the other.
Table 1.
Primary resistance mutations, frequency of resistance at one year and preferred management strategies to resistance with the approved nucleoside analogues.
| Antiviral Agent |
Primary antiviral resistant mutation |
Frequency of resistance at 1 year |
Preferred Management |
|
|---|---|---|---|---|
| HBeAg (+) | HBeAg(−) | |||
| Lamivudine | rtM204V/I rtA181V/T |
15–30%31, 32 | 11–27%81, 82 | Add adefovir Add or switch to tenefovir |
| Telbivudine | rtM204I | 5%12 | 2%12 | Add adefovir Add or switch to tenefovir |
| Adefovir | rtA181V/T rtN236T |
0%83 | 0%84 | Add lamivudine or telbivudine Switch to tenofovir Switch to entecavir |
| Tenofovir | None | 0%45 | 0%45 | |
| Entecavir | rtL180M & rtM204V plus rtI169T & rtM250V or rtT184G & rtS202I |
0%52 | 0%34 | ? Add adefovir Add or switch to tenofovir |
For HBeAg-negative patients with lamivudine resistance the virological response to salvage therapy is better compared to HBeAg-positive patients with resistance as is the case in treatment-naïve patients.65, 66 In one study, 66% and 64% of patients treated by switching or adding on adefovir therapy, respectively achieved a virological response. However, add-on therapy was associated with a lower rate of subsequent adefovir resistance compared to switching therapy, 0% versus 16%, respectively. 65 Currently, only case reports are available on the use of entecavir or tenofovir as salvage therapy in HBeAg-negative chronic hepatitis B with lamivudine resistance and therefore no evidenced-based recommendations can be made about these agents.
For the management of adefovir resistance, evidence is based upon small case reports and in vitro testing.68, 74, 75 These reports suggested that management should be based upon the pattern of adefovir resistant mutations present.38, 40 For patients with the rtN236T mutation, options include switching to or adding entecavir; adding lamivudine; switching to tenofovir; or switching to the combination of tenofovir plus emtricitabine (off label use). For the scenario where the rtA181T mutation has developed, options are fewer and include switching to or adding entecavir; or switching to tenofovir; or switching to the combination of tenofovir plus emtricitabine (off label use). Lamivudine should not be used in this setting due to cross-resistance with the rtA181T mutation.25
Management of entecavir resistance is based on data derived largely from case reports and in vitro phenotypic testing. On the basis of this limited evidence, three approaches are available: to switch or add adefovir; or to switch or add tenofovir; or switch to tenofovir plus emtricitabine (off label).76, 77
There is limited information on management of resistance to telbivudine, however, the recommendations would be similar to those for lamivudine resistance based upon the similarities of these two compounds. Scant data is available on the management of multi-drug resistant HBV.78 Every precaution should be taken to prevent this clinical scenario from occurring because of limited management options left when it occurs. Peginterferon may have a role in carefully selected cases and expert advice should be sought in managing these patients.
Prevention of Antiviral Resistance
The prevention of antiviral resistance is a major goal of future management strategies. This should begin with proper patient selection and judicious use of antiviral agents in patients with chronic hepatitis B. One should avoid inappropriate antiviral therapy, such as HBeAg-positive patients in the immunotolerant phase of the disease (those with extremely high HBV DNA levels in conjunction with normal serum ALT levels) where the benefit has not been demonstrated. If therapy is indicated, then an antiviral agent with the highest potency and a high genetic barrier to resistance should be selected. This is especially the case in HBeAg-positive patients with high viral levels in order to prevent the emergence of drug-resistant mutants. Whether initiating treatment with combination therapy will achieve the goal of minimizing the development of antiviral resistance is currently being investigated. Since all available antiviral agents have the same target of action, the utility of this approach would have to be proven in clinical trials as resistance has been reported in a study of de novo combination therapy.79 Certainly, the low rate of resistance, 1.2% at 5 years in treatment naïve patients receiving entecavir would suggest that monotherapy remains a viable option.53 Other unanswered questions related to combination therapy include which agents to combine and whether an agent could be withdrawn after HBV DNA is fully suppressed.
It is important to minimize sequential monotherapy which can lead to multi-dug resistance and to avoid use of agents with similar cross-resistance profiles. Monitoring the antiviral response is crucial for the early detection of virological breakthrough thus permitting early intervention with the prospect of better outcomes. Monitoring viral kinetics is emerging as a useful adjunct to preventing the development of antiviral resistance. Studies indicate that early, profound viral suppression is associated with lower rates of development of resistance.13, 43, 80 A retrospective analysis of the telbivudine registration trial served to highlight this point. Serum HBV DNA level following 24 weeks of telbivudine treatment was a predictive marker for the development of telbivudine resistance mutations after 2 years of therapy.13 In HBeAg-positive patients, only 6% of patients who were HBV DNA negative developed resistance at the end of 2 years. On the other hand, approximately 50% of all patients with HBV DNA levels above 10,000 copies/mL (> 2000 IU/mL) at the end of 6 months of telbivudine therapy developed resistance by 2 years of treatment.13 In HBeAg-negative chronic hepatitis B, the rate of resistance was only 5% of patients after 2 years in those who were HBV DNA negative (by PCR) after 6 months of telbivudine treatment. In contrast, more than two thirds of patients with HBV DNA levels above 10,000 copies/mL after 6 months developed resistance to telbivudine by 2 years of treatment.13 Based on these results, consideration should be given to changing therapy early for patients who do not achieve rapid viral clearance with the one exception if agents with a high genetic barrier to resistance are being used. Finally, reinforcement of compliance with a prescribed regimen is of paramount importance to preventing resistance.
Conclusions and Needs for Future Research
Preventing and managing antiviral resistance during nucleoside therapy is an important goal. The most effective way to achieve this is not clear. Well-designed studies of long-term safety and efficacy of combination versus monotherapy need to be conducted. A critical assessment of the role of genotypic resistance testing in clinical practice needs to be performed to determine if inclusion of this test in clinical practice would improve outcomes. Management algorithms based on resistance testing need to be developed and validated. What are most needed are other antiviral agents with different sites of action, a paradigm that has worked well for HIV management. A relatively untouched area is experimental immunological approaches to therapy of hepatitis B. Understanding and effectively harnessing the immune system to control or eradicate HBV infection remains a formidable, though important challenge for the clinical management of chronic hepatitis B.
Acknowledgments
Financial Support: This research was supported by the Intramural Research Program of the NIH, NIDDK.
List of Abbreviations
- HBV
hepatitis B virus
- ALT
alanine aminotransferase
- HBeAg
hepatitis B e antigen
Footnotes
Potential Conflicts of Interest: None
References
- 1.Nassal M. Hepatitis B viruses: reverse transcription a different way. Virus Res. 2008;134:235–249. doi: 10.1016/j.virusres.2007.12.024. [DOI] [PubMed] [Google Scholar]
- 2.Murray JM, Purcell RH, Wieland SF. The half-life of hepatitis B virions. Hepatology. 2006;44:1117–1121. doi: 10.1002/hep.21364. [DOI] [PubMed] [Google Scholar]
- 3.Richman DD. The impact of drug resistance on the effectiveness of chemotherapy for chronic hepatitis B. Hepatology. 2000;32:866–867. doi: 10.1053/jhep.2000.18194. [DOI] [PubMed] [Google Scholar]
- 4.Zhang YY, Summers J. Enrichment of a precore-minus mutant of duck hepatitis B virus in experimental mixed infections. J Virol. 1999;73:3616–3622. doi: 10.1128/jvi.73.5.3616-3622.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhang YY, Summers J. Low dynamic state of viral competition in a chronic avian hepadnavirus infection. J Virol. 2000;74:5257–5265. doi: 10.1128/jvi.74.11.5257-5265.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hulgan T, Haas DW. Toward a pharmacogenetic understanding of nucleotide and nucleoside analogue toxicity. J Infect Dis. 2006;194:1471–1474. doi: 10.1086/508550. [DOI] [PubMed] [Google Scholar]
- 7.Locarnini S, Hatzakis A, Heathcote J, Keeffe EB, Liang TJ, Mutimer D, Pawlotsky JM, Zoulim F. Management of antiviral resistance in patients with chronic hepatitis B. Antivir Ther. 2004;9:679–693. [PubMed] [Google Scholar]
- 8.Tenney DJ, Levine SM, Rose RE, Walsh AW, Weinheimer SP, Discotto L, Plym M, Pokornowski K, Yu CF, Angus P, Ayres A, Bartholomeusz A, Sievert W, Thompson G, Warner N, Locarnini S, Colonno RJ. Clinical emergence of entecavir-resistant hepatitis B virus requires additional substitutions in virus already resistant to Lamivudine. Antimicrob Agents Chemother. 2004;48:3498–3507. doi: 10.1128/AAC.48.9.3498-3507.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lok AS, Zoulim F, Locarnini S, Bartholomeusz A, Ghany MG, Pawlotsky JM, Liaw YF, Mizokami M, Kuiken C. Antiviral drug-resistant HBV: standardization of nomenclature and assays and recommendations for management. Hepatology. 2007;46:254–265. doi: 10.1002/hep.21698. [DOI] [PubMed] [Google Scholar]
- 10.Carrouee-Durantel S, Durantel D, Werle-Lapostolle B, Pichoud C, Naesens L, Neyts J, Trepo C, Zoulim F. Suboptimal response to adefovir dipivoxil therapy for chronic hepatitis B in nucleoside-naive patients is not due to pre-existing drug-resistant mutants. Antivir Ther. 2008;13:381–388. [PubMed] [Google Scholar]
- 11.Yuen MF, Sablon E, Hui CK, Yuan HJ, Decraemer H, Lai CL. Factors associated with hepatitis B virus DNA breakthrough in patients receiving prolonged lamivudine therapy. Hepatology. 2001;34:785–791. doi: 10.1053/jhep.2001.27563. [DOI] [PubMed] [Google Scholar]
- 12.Lai CL, Gane E, Liaw YF, Hsu CW, Thongsawat S, Wang Y, Chen Y, Heathcote EJ, Rasenack J, Bzowej N, Naoumov NV, Di Bisceglie AM, Zeuzem S, Moon YM, Goodman Z, Chao G, Constance BF, Brown NA. Telbivudine versus lamivudine in patients with chronic hepatitis B. N Engl J Med. 2007;357:2576–2588. doi: 10.1056/NEJMoa066422. [DOI] [PubMed] [Google Scholar]
- 13.Liaw YF, Gane E, Leung N, Zeuzem S, Wang Y, Lai CL, Heathcote EJ, Manns M, Bzowej N, Niu J, Han SH, Hwang SG, Cakaloglu Y, Tong MJ, Papatheodoridis G, Chen Y, Brown NA, Albanis E, Galil K, Naoumov NV. 2-Year GLOBE Trial Results: Telbivudine Is Superior to Lamivudine in Patients With Chronic Hepatitis B. Gastroenterology. 2008 doi: 10.1053/j.gastro.2008.10.026. [DOI] [PubMed] [Google Scholar]
- 14.Locarnini SQX, Arterburn S, Snow A, Brosgart CL, Currie G, et al. Incidence and predictors of emergence of Adefovir resistant HBV during four years of adefovir dipivoxil (ADV) therapy for patients with chronic hepatitis B (CHB) J Hepatol. 2005;42:17. [Google Scholar]
- 15.Zhou T, Saputelli J, Aldrich CE, Deslauriers M, Condreay LD, Mason WS. Emergence of drug-resistant populations of woodchuck hepatitis virus in woodchucks treated with the antiviral nucleoside lamivudine. Antimicrob Agents Chemother. 1999;43:1947–1954. doi: 10.1128/aac.43.8.1947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ono SK, Kato N, Shiratori Y, Kato J, Goto T, Schinazi RF, Carrilho FJ, Omata M. The polymerase L528M mutation cooperates with nucleotide binding-site mutations, increasing hepatitis B virus replication and drug resistance. J Clin Invest. 2001;107:449–455. doi: 10.1172/JCI11100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Allen MI, Deslauriers M, Andrews CW, Tipples GA, Walters KA, Tyrrell DL, Brown N, Condreay LD. Identification and characterization of mutations in hepatitis B virus resistant to lamivudine. Lamivudine Clinical Investigation Group. Hepatology. 1998;27:1670–1677. doi: 10.1002/hep.510270628. [DOI] [PubMed] [Google Scholar]
- 18.Delaney WEt, Yang H, Westland CE, Das K, Arnold E, Gibbs CS, Miller MD, Xiong S. The hepatitis B virus polymerase mutation rtV173L is selected during lamivudine therapy and enhances viral replication in vitro. J Virol. 2003;77:11833–11841. doi: 10.1128/JVI.77.21.11833-11841.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lok AS, Lai CL, Leung N, Yao GB, Cui ZY, Schiff ER, Dienstag JL, Heathcote EJ, Little NR, Griffiths DA, Gardner SD, Castiglia M. Long-term safety of lamivudine treatment in patients with chronic hepatitis B. Gastroenterology. 2003;125:1714–1722. doi: 10.1053/j.gastro.2003.09.033. [DOI] [PubMed] [Google Scholar]
- 20.Nafa S, Ahmed S, Tavan D, Pichoud C, Berby F, Stuyver L, Johnson M, Merle P, Abidi H, Trepo C, Zoulim F. Early detection of viral resistance by determination of hepatitis B virus polymerase mutations in patients treated by lamivudine for chronic hepatitis B. Hepatology. 2000;32:1078–1088. doi: 10.1053/jhep.2000.19619. [DOI] [PubMed] [Google Scholar]
- 21.Colonno RJ, Rose R, Baldick CJ, Levine S, Pokornowski K, Yu CF, Walsh A, Fang J, Hsu M, Mazzucco C, Eggers B, Zhang S, Plym M, Klesczewski K, Tenney DJ. Entecavir resistance is rare in nucleoside naive patients with hepatitis B. Hepatology. 2006;44:1656–1665. doi: 10.1002/hep.21422. [DOI] [PubMed] [Google Scholar]
- 22.Pawlotsky JM, Dusheiko G, Hatzakis A, Lau D, Lau G, Liang TJ, Locarnini S, Martin P, Richman DD, Zoulim F. Virologic monitoring of hepatitis B virus therapy in clinical trials and practice: recommendations for a standardized approach. Gastroenterology. 2008;134:405–415. doi: 10.1053/j.gastro.2007.11.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ghany M, Liang TJ. Drug targets and molecular mechanisms of drug resistance in chronic hepatitis B. Gastroenterology. 2007;132:1574–1585. doi: 10.1053/j.gastro.2007.02.039. [DOI] [PubMed] [Google Scholar]
- 24.Seifer M, Patty A, Serra I, Li B, Standring DN. Telbivudine, a nucleoside analog inhibitor of HBV polymerase, has a different in vitro cross-resistance profile than the nucleotide analog inhibitors adefovir and tenofovir. Antiviral Res. 2008 doi: 10.1016/j.antiviral.2008.10.008. [DOI] [PubMed] [Google Scholar]
- 25.Villet S, Pichoud C, Billioud G, Barraud L, Durantel S, Trepo C, Zoulim F. Impact of hepatitis B virus rtA181V/T mutants on hepatitis B treatment failure. J Hepatol. 2008;48:747–755. doi: 10.1016/j.jhep.2008.01.027. [DOI] [PubMed] [Google Scholar]
- 26.Yatsuji H, Noguchi C, Hiraga N, Mori N, Tsuge M, Imamura M, Takahashi S, Iwao E, Fujimoto Y, Ochi H, Abe H, Maekawa T, Tateno C, Yoshizato K, Suzuki F, Kumada H, Chayama K. Emergence of a novel lamivudine-resistant hepatitis B virus variant with a substitution outside the YMDD motif. Antimicrob Agents Chemother. 2006;50:3867–3874. doi: 10.1128/AAC.00239-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yeh CT, Chien RN, Chu CM, Liaw YF. Clearance of the original hepatitis B virus YMDD-motif mutants with emergence of distinct lamivudine-resistant mutants during prolonged lamivudine therapy. Hepatology. 2000;31:1318–1326. doi: 10.1053/jhep.2000.7296. [DOI] [PubMed] [Google Scholar]
- 28.Lee K, Chu CK. Molecular modeling approach to understanding the mode of action of L-nucleosides as antiviral agents. Antimicrob Agents Chemother. 2001;45:138–144. doi: 10.1128/AAC.45.1.138-144.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Das K, Xiong X, Yang H, Westland CE, Gibbs CS, Sarafianos SG, Arnold E. Molecular modeling and biochemical characterization reveal the mechanism of hepatitis B virus polymerase resistance to lamivudine (3TC) and emtricitabine (FTC) J Virol. 2001;75:4771–4779. doi: 10.1128/JVI.75.10.4771-4779.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gutfreund KS, Williams M, George R, Bain VG, Ma MM, Yoshida EM, Villeneuve JP, Fischer KP, Tyrrel DL. Genotypic succession of mutations of the hepatitis B virus polymerase associated with lamivudine resistance. J Hepatol. 2000;33:469–475. doi: 10.1016/s0168-8278(00)80284-6. [DOI] [PubMed] [Google Scholar]
- 31.Lai CL, Chien RN, Leung NW, Chang TT, Guan R, Tai DI, Ng KY, Wu PC, Dent JC, Barber J, Stephenson SL, Gray DF. A one-year trial of lamivudine for chronic hepatitis B. Asia Hepatitis Lamivudine Study Group. N Engl J Med. 1998;339:61–68. doi: 10.1056/NEJM199807093390201. [DOI] [PubMed] [Google Scholar]
- 32.Dienstag JL, Schiff ER, Wright TL, Perrillo RP, Hann HW, Goodman Z, Crowther L, Condreay LD, Woessner M, Rubin M, Brown NA. Lamivudine as initial treatment for chronic hepatitis B in the United States. N Engl J Med. 1999;341:1256–1263. doi: 10.1056/NEJM199910213411702. [DOI] [PubMed] [Google Scholar]
- 33.Yuen MF, Seto WK, Chow DH, Tsui K, Wong DK, Ngai VW, Wong BC, Fung J, Yuen JC, Lai CL. Long-term lamivudine therapy reduces the risk of long-term complications of chronic hepatitis B infection even in patients without advanced disease. Antivir Ther. 2007;12:1295–1303. [PubMed] [Google Scholar]
- 34.Lai CL, Shouval D, Lok AS, Chang TT, Cheinquer H, Goodman Z, DeHertogh D, Wilber R, Zink RC, Cross A, Colonno R, Fernandes L. Entecavir versus lamivudine for patients with HBeAg-negative chronic hepatitis B. N Engl J Med. 2006;354:1011–1020. doi: 10.1056/NEJMoa051287. [DOI] [PubMed] [Google Scholar]
- 35.Marcellin P, Lau GK, Bonino F, Farci P, Hadziyannis S, Jin R, Lu ZM, Piratvisuth T, Germanidis G, Yurdaydin C, Diago M, Gurel S, Lai MY, Button P, Pluck N. Peginterferon alfa-2a alone, lamivudine alone, and the two in combination in patients with HBeAg-negative chronic hepatitis B. N Engl J Med. 2004;351:1206–1217. doi: 10.1056/NEJMoa040431. [DOI] [PubMed] [Google Scholar]
- 36.Manolakopoulos S, Bethanis S, Elefsiniotis J, Karatapanis S, Triantos C, Sourvinos G, Touloumi G, Economou M, Vlachogiannakos J, Spandidos D, Avgerinos A, Tzourmakliotis D. Lamivudine monotherapy in HBeAg-negative chronic hepatitis B: prediction of response-breakthrough and long-term clinical outcome. Aliment Pharmacol Ther. 2006;23:787–795. doi: 10.1111/j.1365-2036.2006.02806.x. [DOI] [PubMed] [Google Scholar]
- 37.De Clercq E, Holy A. Acyclic nucleoside phosphonates: a key class of antiviral drugs. Nat Rev Drug Discov. 2005;4:928–940. doi: 10.1038/nrd1877. [DOI] [PubMed] [Google Scholar]
- 38.Angus P, Vaughan R, Xiong S, Yang H, Delaney W, Gibbs C, Brosgart C, Colledge D, Edwards R, Ayres A, Bartholomeusz A, Locarnini S. Resistance to adefovir dipivoxil therapy associated with the selection of a novel mutation in the HBV polymerase. Gastroenterology. 2003;125:292–297. doi: 10.1016/s0016-5085(03)00939-9. [DOI] [PubMed] [Google Scholar]
- 39.Qi X, Xiong S, Yang H, Miller M, Delaney WEt. In vitro susceptibility of adefovir-associated hepatitis B virus polymerase mutations to other antiviral agents. Antivir Ther. 2007;12:355–362. [PubMed] [Google Scholar]
- 40.Villeneuve JP, Durantel D, Durantel S, Westland C, Xiong S, Brosgart CL, Gibbs CS, Parvaz P, Werle B, Trepo C, Zoulim F. Selection of a hepatitis B virus strain resistant to adefovir in a liver transplantation patient. J Hepatol. 2003;39:1085–1089. doi: 10.1016/j.jhep.2003.09.022. [DOI] [PubMed] [Google Scholar]
- 41.Schildgen O, Sirma H, Funk A, Olotu C, Wend UC, Hartmann H, Helm M, Rockstroh JK, Willems WR, Will H, Gerlich WH. Variant of hepatitis B virus with primary resistance to adefovir. N Engl J Med. 2006;354:1807–1812. doi: 10.1056/NEJMoa051214. [DOI] [PubMed] [Google Scholar]
- 42.Curtis M, Zhu Y, Borroto-Esoda K. Hepatitis B virus containing the I233V mutation in the polymerase reverse-transcriptase domain remains sensitive to inhibition by adefovir. J Infect Dis. 2007;196:1483–1486. doi: 10.1086/522521. [DOI] [PubMed] [Google Scholar]
- 43.Hadziyannis SJ, Tassopoulos NC, Heathcote EJ, Chang TT, Kitis G, Rizzetto M, Marcellin P, Lim SG, Goodman Z, Ma J, Brosgart CL, Borroto-Esoda K, Arterburn S, Chuck SL. Long-term therapy with adefovir dipivoxil for HBeAg-negative chronic hepatitis B for up to 5 years. Gastroenterology. 2006;131:1743–1751. doi: 10.1053/j.gastro.2006.09.020. [DOI] [PubMed] [Google Scholar]
- 44.Marcellin P, Chang TT, Lim SG, Sievert W, Tong M, Arterburn S, Borroto-Esoda K, Frederick D, Rousseau F. Long-term efficacy and safety of adefovir dipivoxil for the treatment of hepatitis B e antigen-positive chronic hepatitis B. Hepatology. 2008;48:750–758. doi: 10.1002/hep.22414. [DOI] [PubMed] [Google Scholar]
- 45.Marcellin P, Heathcote EJ, Buti M, Gane E, de Man RA, Krastev Z, Germanidis G, Lee SS, Flisiak R, Kaita K, Manns M, Kotzev I, Tchernev K, Buggisch P, Weilert F, Kurdas OO, Shiffman ML, Trinh H, Washington MK, Sorbel J, Anderson J, Snow-Lampart A, Mondou E, Quinn J, Rousseau F. Tenofovir disoproxil fumarate versus adefovir dipivoxil for chronic hepatitis B. N Engl J Med. 2008;359:2442–2455. doi: 10.1056/NEJMoa0802878. [DOI] [PubMed] [Google Scholar]
- 46.Marcellin P, Buti M, Krastev Z, Gurel S, Balabanska R, et al. Two Year Tenofovir Disoproxil Fumarate (TDF) Treatment and Adefovir Dipivoxil (ADV) Switch Data in HBeAg-Negative Patients with Chronic Hepatitis B (Study 102), Preliminary Analysis. Hepatology. 2008;48:370A. [Google Scholar]
- 47.Innaimo SF, Seifer M, Bisacchi GS, Standring DN, Zahler R, Colonno RJ. Identification of BMS-200475 as a potent and selective inhibitor of hepatitis B virus. Antimicrob Agents Chemother. 1997;41:1444–1448. doi: 10.1128/aac.41.7.1444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Baldick CJ, Tenney DJ, Mazzucco CE, Eggers BJ, Rose RE, Pokornowski KA, Yu CF, Colonno RJ. Comprehensive evaluation of hepatitis B virus reverse transcriptase substitutions associated with entecavir resistance. Hepatology. 2008;47:1473–1482. doi: 10.1002/hep.22211. [DOI] [PubMed] [Google Scholar]
- 49.Langley DR, Walsh AW, Baldick CJ, Eggers BJ, Rose RE, Levine SM, Kapur AJ, Colonno RJ, Tenney DJ. Inhibition of hepatitis B virus polymerase by entecavir. J Virol. 2007;81:3992–4001. doi: 10.1128/JVI.02395-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Warner N, Locarnini SA, Colledge D, Edwards R, Angus P, et al. Molecular Modelling of Entecavir Resistant Mutations in the Hepatitis B Virus Polymerase Selected During Therapy. Hepatology. 2005;40:245A. [Google Scholar]
- 51.Tenney DJLD, Oliver AJ, Rose RE, Levine SE, et al. Hepatitis B Virus Resistance to Entecavir Involves Novel Changes in the Viral Polymerase. Hepatology. 2005;40:245A. [Google Scholar]
- 52.Chang TT, Gish RG, de Man R, Gadano A, Sollano J, Chao YC, Lok AS, Han KH, Goodman Z, Zhu J, Cross A, DeHertogh D, Wilber R, Colonno R, Apelian D. A comparison of entecavir and lamivudine for HBeAg-positive chronic hepatitis B. N Engl J Med. 2006;354:1001–1010. doi: 10.1056/NEJMoa051285. [DOI] [PubMed] [Google Scholar]
- 53.Tenney DJPK, Rose RE, Baldick CJ, Eggers BJ, et al. Entecavir at five years shows long-term maintenance of high genetic barrier to hepatitis B virus resistance. Hepatol Int. 2008:s76–s77. [Google Scholar]
- 54.Sherman M, Yurdaydin C, Sollano J, Silva M, Liaw YF, Cianciara J, Boron-Kaczmarska A, Martin P, Goodman Z, Colonno R, Cross A, Denisky G, Kreter B, Hindes R. Entecavir for treatment of lamivudine-refractory, HBeAg-positive chronic hepatitis B. Gastroenterology. 2006;130:2039–2049. doi: 10.1053/j.gastro.2006.04.007. [DOI] [PubMed] [Google Scholar]
- 55.Liaw YF, Sung JJ, Chow WC, Farrell G, Lee CZ, Yuen H, Tanwandee T, Tao QM, Shue K, Keene ON, Dixon JS, Gray DF, Sabbat J. Lamivudine for patients with chronic hepatitis B and advanced liver disease. N Engl J Med. 2004;351:1521–1531. doi: 10.1056/NEJMoa033364. [DOI] [PubMed] [Google Scholar]
- 56.Dienstag JL, Goldin RD, Heathcote EJ, Hann HW, Woessner M, Stephenson SL, Gardner S, Gray DF, Schiff ER. Histological outcome during long-term lamivudine therapy. Gastroenterology. 2003;124:105–117. doi: 10.1053/gast.2003.50013. [DOI] [PubMed] [Google Scholar]
- 57.Lai CL, Dienstag J, Schiff E, Leung NW, Atkins M, Hunt C, Brown N, Woessner M, Boehme R, Condreay L. Prevalence and clinical correlates of YMDD variants during lamivudine therapy for patients with chronic hepatitis B. Clin Infect Dis. 2003;36:687–696. doi: 10.1086/368083. [DOI] [PubMed] [Google Scholar]
- 58.Lok AS, McMahon BJ. Chronic hepatitis B. Hepatology. 2007;45:507–539. doi: 10.1002/hep.21513. [DOI] [PubMed] [Google Scholar]
- 59.European Association For The Study Of The Liver. EASL Clinical Practice Guidelines: Management of chronic hepatitis B. J Hepatol. 2009;50:227–242. doi: 10.1016/j.jhep.2008.10.001. [DOI] [PubMed] [Google Scholar]
- 60.Liaw YF, Leung N, Guan R, Lau GK, Merican I, McCaughan G, Gane E, Kao JH, Omata M. Asian-Pacific consensus statement on the management of chronic hepatitis B: a 2005 update. Liver Int. 2005;25:472–489. doi: 10.1111/j.1478-3231.2005.01134.x. [DOI] [PubMed] [Google Scholar]
- 61.Keeffe EB, Dieterich DT, Han SH, Jacobson IM, Martin P, Schiff ER, Tobias H. A treatment algorithm for the management of chronic hepatitis B virus infection in the United States: 2008 update. Clin Gastroenterol Hepatol. 2008;6:1315–1341. doi: 10.1016/j.cgh.2008.08.021. quiz 1286. [DOI] [PubMed] [Google Scholar]
- 62.Hong SP, Kim NK, Hwang SG, Chung HJ, Kim S, Han JH, Kim HT, Rim KS, Kang MS, Yoo W, Kim SO. Detection of hepatitis B virus YMDD variants using mass spectrometric analysis of oligonucleotide fragments. J Hepatol. 2004;40:837–844. doi: 10.1016/j.jhep.2004.01.006. [DOI] [PubMed] [Google Scholar]
- 63.Mitsuya Y, Varghese V, Wang C, Liu TF, Holmes SP, Jayakumar P, Gharizadeh B, Ronaghi M, Klein D, Fessel WJ, Shafer RW. Minority human immunodeficiency virus type 1 variants in antiretroviral-naive persons with reverse transcriptase codon 215 revertant mutations. J Virol. 2008;82:10747–10755. doi: 10.1128/JVI.01827-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lampertico P, Vigano M, Manenti E, Iavarone M, Lunghi G, Colombo M. Adefovir rapidly suppresses hepatitis B in HBeAg-negative patients developing genotypic resistance to lamivudine. Hepatology. 2005;42:1414–1419. doi: 10.1002/hep.20939. [DOI] [PubMed] [Google Scholar]
- 65.Lapertico P, Marzano A, Levero M, Santantonio T, Di Marco V, et al. Adefovir and Lamivudine Combination Therapy is Superior to Adefovir Monotherapy for Lamivudine Resistant Patients with HBeAg Negative Chronic Hepatitis B. Hepatology. 2006;44:693A. [Google Scholar]
- 66.Rapti I, Dimou E, Mitsoula P, Hadziyannis SJ. Adding-on versus switching-to adefovir therapy in lamivudine-resistant HBeAg-negative chronic hepatitis B. Hepatology. 2007;45:307–313. doi: 10.1002/hep.21534. [DOI] [PubMed] [Google Scholar]
- 67.Chang TT, Gish RG, Hadziyannis SJ, Cianciara J, Rizzetto M, Schiff ER, Pastore G, Bacon BR, Poynard T, Joshi S, Klesczewski KS, Thiry A, Rose RE, Colonno RJ, Hindes RG. A dose-ranging study of the efficacy and tolerability of entecavir in Lamivudine-refractory chronic hepatitis B patients. Gastroenterology. 2005;129:1198–1209. doi: 10.1053/j.gastro.2005.06.055. [DOI] [PubMed] [Google Scholar]
- 68.Yang H, Qi X, Sabogal A, Miller M, Xiong S, Delaney WEt. Cross-resistance testing of next-generation nucleoside and nucleotide analogues against lamivudine-resistant HBV. Antivir Ther. 2005;10:625–633. [PubMed] [Google Scholar]
- 69.Peters MG, Hann Hw H, Martin P, Heathcote EJ, Buggisch P, Rubin R, Bourliere M, Kowdley K, Trepo C, Gray Df D, Sullivan M, Kleber K, Ebrahimi R, Xiong S, Brosgart CL. Adefovir dipivoxil alone or in combination with lamivudine in patients with lamivudine-resistant chronic hepatitis B. Gastroenterology. 2004;126:91–101. doi: 10.1053/j.gastro.2003.10.051. [DOI] [PubMed] [Google Scholar]
- 70.van Bommel F, Wunsche T, Mauss S, Reinke P, Bergk A, Schurmann D, Wiedenmann B, Berg T. Comparison of adefovir and tenofovir in the treatment of lamivudine-resistant hepatitis B virus infection. Hepatology. 2004;40:1421–1425. doi: 10.1002/hep.20464. [DOI] [PubMed] [Google Scholar]
- 71.Bristol Meyers Squibb. Baraclude (Entecavir) Data Continue To Demonstrate Low Incidence Of Resistance Through Five Years Of Treatment In Nucleoside-Naive Chronic Hepatitis B Patients. Press release. 2008 March 24; [Google Scholar]
- 72.Sherman M, Yurdaydin C, Simsek H, Silva M, Liaw YF, Rustgi VK, Sette H, Tsai N, Tenney DJ, Vaughan J, Kreter B, Hindes R. Entecavir therapy for lamivudine-refractory chronic hepatitis B: improved virologic, biochemical, and serology outcomes through 96 weeks. Hepatology. 2008;48:99–108. doi: 10.1002/hep.22323. [DOI] [PubMed] [Google Scholar]
- 73.Van Bommel F, De Man RA, Erhardt A, Huppe D, Stein K, et al. First Multicenter Evaluation of the Efficacy of Tenofovir in Nucleoside Analogue Experienced Patients with HBV Monoinfection. Hepatology. 2007;46:270A. [Google Scholar]
- 74.Brunelle MN, Jacquard AC, Pichoud C, Durantel D, Carrouee-Durantel S, Villeneuve JP, Trepo C, Zoulim F. Susceptibility to antivirals of a human HBV strain with mutations conferring resistance to both lamivudine and adefovir. Hepatology. 2005;41:1391–1398. doi: 10.1002/hep.20723. [DOI] [PubMed] [Google Scholar]
- 75.van Bommel F, Zollner B, Sarrazin C, Spengler U, Huppe D, Moller B, Feucht HH, Wiedenmann B, Berg T. Tenofovir for patients with lamivudine-resistant hepatitis B virus (HBV) infection and high HBV DNA level during adefovir therapy. Hepatology. 2006;44:318–325. doi: 10.1002/hep.21253. [DOI] [PubMed] [Google Scholar]
- 76.Villet S, Ollivet A, Pichoud C, Barraud L, Villeneuve JP, Trepo C, Zoulim F. Stepwise process for the development of entecavir resistance in a chronic hepatitis B virus infected patient. J Hepatol. 2007;46:531–538. doi: 10.1016/j.jhep.2006.11.016. [DOI] [PubMed] [Google Scholar]
- 77.Leemans WF, Niesters HG, van der Eijk AA, Janssen HL, Schalm SW, de Man RA. Selection of an entecavir-resistant mutant despite prolonged hepatitis B virus DNA suppression, in a chronic hepatitis B patient with preexistent lamivudine resistance: successful rescue therapy with tenofovir. Eur J Gastroenterol Hepatol. 2008;20:773–777. doi: 10.1097/MEG.0b013e3282f793d6. [DOI] [PubMed] [Google Scholar]
- 78.Yim HJ, Hussain M, Liu Y, Wong SN, Fung SK, Lok AS. Evolution of multi-drug resistant hepatitis B virus during sequential therapy. Hepatology. 2006;44:703–712. doi: 10.1002/hep.21290. [DOI] [PubMed] [Google Scholar]
- 79.Sung JJ, Lai JY, Zeuzem S, Chow WC, Heathcote EJ, Perrillo RP, Brosgart CL, Woessner MA, Scott SA, Gray DF, Gardner SD. Lamivudine compared with lamivudine and adefovir dipivoxil for the treatment of HBeAg-positive chronic hepatitis B. J Hepatol. 2008;48:728–735. doi: 10.1016/j.jhep.2007.12.026. [DOI] [PubMed] [Google Scholar]
- 80.Yuen MF, Fong DY, Wong DK, Yuen JC, Fung J, Lai CL. Hepatitis B virus DNA levels at week 4 of lamivudine treatment predict the 5-year ideal response. Hepatology. 2007;46:1695–1703. doi: 10.1002/hep.21939. [DOI] [PubMed] [Google Scholar]
- 81.Tassopoulos NC, Volpes R, Pastore G, Heathcote J, Buti M, Goldin RD, Hawley S, Barber J, Condreay L, Gray DF. Efficacy of lamivudine in patients with hepatitis B e antigen-negative/hepatitis B virus DNA-positive (precore mutant) chronic hepatitis B. Lamivudine Precore Mutant Study Group. Hepatology. 1999;29:889–896. doi: 10.1002/hep.510290321. [DOI] [PubMed] [Google Scholar]
- 82.Di Marco V, Marzano A, Lampertico P, Andreone P, Santantonio T, Almasio PL, Rizzetto M, Craxi A. Clinical outcome of HBeAg-negative chronic hepatitis B in relation to virological response to lamivudine. Hepatology. 2004;40:883–891. doi: 10.1002/hep.20381. [DOI] [PubMed] [Google Scholar]
- 83.Marcellin P, Chang TT, Lim SG, Tong MJ, Sievert W, Shiffman ML, Jeffers L, Goodman Z, Wulfsohn MS, Xiong S, Fry J, Brosgart CL. Adefovir dipivoxil for the treatment of hepatitis B e antigen-positive chronic hepatitis B. N Engl J Med. 2003;348:808–816. doi: 10.1056/NEJMoa020681. [DOI] [PubMed] [Google Scholar]
- 84.Hadziyannis SJ, Tassopoulos NC, Heathcote EJ, Chang TT, Kitis G, Rizzetto M, Marcellin P, Lim SG, Goodman Z, Wulfsohn MS, Xiong S, Fry J, Brosgart CL. Adefovir dipivoxil for the treatment of hepatitis B e antigen-negative chronic hepatitis B. N Engl J Med. 2003;348:800–807. doi: 10.1056/NEJMoa021812. [DOI] [PubMed] [Google Scholar]