

SUMMARY
West Nile virus (WNV), a mosquito-transmitted single-stranded RNA (ssRNA) flavivirus, causes human disease of variable severity. We investigated Toll-like receptor 7-deficient (Tlr7−/−) and myeloid differentiation factor 88-deficient (Myd88−/−) mice, which both have defective recognition of ssRNA, and found increased viremia and susceptibility to lethal WNV infection. Despite increased tissue concentrations of most innate cytokines, CD45+ leukocytes and CD11b+ macrophages failed to home to WNV-infected cells and infiltrate into target organs of Tlr7−/− mice. Tlr7−/− mice and macrophages had reduced interleukin-12 (IL-12) and IL-23 responses after WNV infection, and mice deficient in IL-12 p40 and IL-23 p40 (Il12b−/−) or IL-23 p19 (Il23a−/−), but not IL-12 p35 (Il12a−/−), responded similarly to Tlr7−/− mice, with increased susceptibility to lethal WNV encephalitis. Collectively, these results demonstrate that TLR7 andIL-23-dependent WNV responses representa vital host defense mechanism that operates by affecting immune cell homing to infected target cells.
INTRODUCTION
West Nile virus (WNV) is a mosquito-transmitted single-stranded RNA (ssRNA) virus that has emerged as the most common cause of epidemic viral encephalitis in North America and has recently become a worldwide public health concern (Campbell et al., 2002; Debiasi and Tyler, 2006; Gould and Fikrig, 2004; Gubler, 2007). Although infection in humans is typically asymptomatic, the elderly and immunocompromised are particularly at risk for life-threatening neurological disease, including meningitis and encephalitis (Campbell et al., 2002; Davis et al., 2006). An approved therapy for use in humans does not currently exist, and viral pathogenesis is incompletely understood. In mice, WNV induces systemic infection and then crosses the blood-brain barrier, leading to encephalitis and death (Wang et al., 2001, 2004). Induction of type-1 interferons (IFNs) (Anderson and Rahal, 2002; Brinton, 2001; Gilfoy and Mason, 2007) and humoral immunity (Diamond et al., 2003a, 2003b) provide front-line defense against WNV pathogenesis, and cellular immunity, including γδ T cells (Wang et al., 2003a), CD4+ (Kulkarni et al., 1991; Sitati and Diamond, 2006), and CD8+ (Shrestha and Diamond, 2004, 2007; Shrestha et al., 2006; Sitati et al., 2007; Wang et al., 2003b) αβ T cells participate in host recovery from WNV infection.
Toll-like receptors (TLRs) are a set of phylogenetically conserved, germline-encoded molecules that play an essential role in initiating innate immunity. Mammalian TLRs consist of 11–12 members that function as pattern recognition receptors (PRRs) to sense pathogen-associated molecular patterns (PAMPs) (Qureshi and Medzhitov, 2003; Yamamoto et al., 2004). A large growing body of evidence implicates TLR recognition of a diverse set of pathogens, including bacteria (by TLR2, TLR4, TLR6, and TLR9), flagellated protozoans (by TLR5), and pathogenic fungi (by TLR2, TLR4, and TLR6) (Roeder et al., 2004; Yamamoto et al., 2004). TLR2 and TLR4 recognize viral proteins (Bieback et al., 2002; Kurt-Jones et al., 2000; Rassa et al., 2002), and our data demonstrate that TLR3 mediates host recognition of viral components (including double-stranded RNA [dsRNA] and the dsRNA analog poly(I:C)) and intact virus including Lang reovirus and WNV (Alexopoulou et al., 2001; Edelmann et al., 2004; Town et al., 2006; Wang et al., 2004). Further, TLR7 and TLR8 are implicated in MyD88-dependent recognition of ssRNA and ssRNA-producing viruses including vesicular stomatitis virus, influenza virus, human parechoviris 1, and human immunodeficiency virus (Diebold et al., 2004; Heil et al., 2004; Lund et al., 2004; Schlaepfer et al., 2006; Triantafilou et al., 2005).
A functional role for TLR7 in recognizing viral infection in vivo is still unclear. Because WNV is a positive ssRNA virus and may therefore be detected by host TLR7 (Lund et al., 2004), we sought to elucidate the role of TLR7 in controlling viral infection.
RESULTS
Tlr7−/− and Myd88−/− Mice Are More Susceptible to Lethal WNV Infection
We used the mouse model of WNV encephalitis to examine the role of TLR7 and MyD88 in viral recognition in vivo. Mice were challenged with 2 × 103 plaque-forming units (p.f.u.) of WNV corresponding to a dose at which approximately 50% of wild-type (C57BL/6J) animals survive (LD50) and were monitored twice daily for mortality. TLR7-deficient (Tlr7−/−) mice were significantly more susceptible (9% survival) to lethal WNV infection than were wild-type control mice (50% survival, p < 0.05; Figure 1). The adaptor molecule MyD88 is required for TLR7 signaling (Diebold et al., 2004; Hemmi et al., 2002; Lund et al., 2004), and our data indicated a similar pattern of survival results after WNV infection of MyD88-deficient (Myd88−/−) mice (15% survival) compared to wild-type controls (p < 0.05; Figure 1). TLR9 recognizes bacterial DNA containing unmethylated CpG motifs (Hemmi et al., 2000) and, like TLR7, requires MyD88 for signaling (Bauer et al., 2001; Hemmi et al., 2003). Furthermore, it has recently been suggested that TLR9 might cooperate with TLR7 in recognizing viral nucleic acid associated with murine cytomegalovirus (Zucchini et al., 2008). However, our results suggested that increased susceptibility of Tlr7−/− mice to WNV infection was specific, because TLR9-deficient (Tlr9−/− −/−) mice infected with WNV at LD50 (43% survival) were not significantly different from controls (50% survival, p > 0.10; Figure 1).
Figure 1. Increased Susceptibility of Tlr7−/− and Myd88−/− Mice, but Not Tlr9−/− Mice, after West Nile Virus Challenge.
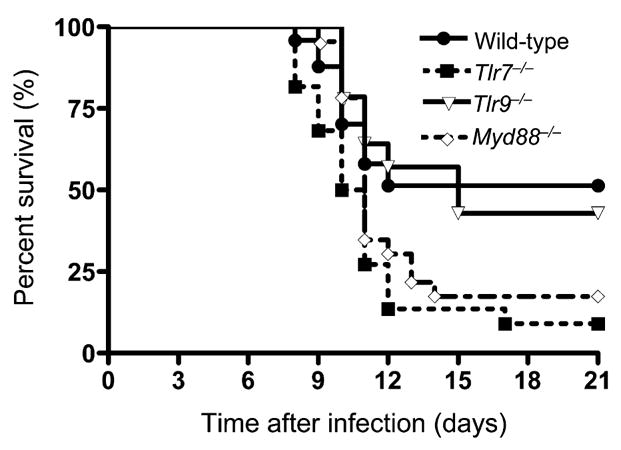
Wild-type mice (n = 24), Tlr7−/− mice (n = 22), Myd88−/− mice (n = 13), and Tlr9−/− mice (n = 14) were i.p. infected with WNV (LD50) and monitored twice daily for 21 days for mortality. Data shown are represented as time after infection (days) on the x axis and percent survival (%) on the y axis. Kaplan-Meier survival analysis revealed significant differences between wild-type and Tlr7−/− or Myd88−/− mice, but not between wild-type and Tlr9−/− mice. Data shown are pooled from 2–4 independent experiments.
Viral Load and Cytokines in Tlr7−/− and Myd88−/− Mice after WNV Infection
We next examined viral load in the brain as well as systemically. WNV was detected at high amounts in the spleen, liver, and blood at days 2–3 postinfection (p.i.) and in the brain at day 6 p.i. (Wang et al., 2004). Quantitative real-time polymerase chain reaction (Q-PCR) measuring WNV envelope gene (WNVE) revealed approximate 3-fold increased RNA abundance compared to control mice in Tlr7−/− mice (p < 0.05; Figure 2A) and Myd88−/− mice (p < 0.05; Figure 2B) in blood at days 2–3 p.i. There was also a modest (2-fold) but significant (p < 0.05) increase in Myd88−/− splenic WNVE RNA expression (Figure 2B) at day 3 p.i., which did not reach significance in Tlr7−/− mice at day 3 (data not shown) but was significantly higher at day 6 p.i. (p < 0.01; Figure 2A). Strikingly, WNVE RNA expression was markedly (8-fold) elevated in Tlr7−/− brains at day 6 p.i. (p < 0.05; Figure 2A) and was also significantly (3-fold) increased in Myd88−/− brains at day 6 p.i. (p < 0.05; Figure 2B).
Figure 2. Tlr7- and Myd88-Dependent Viral Load and Innate Immune Cytokine Responses after West Nile Virus Challenge.

Wild-type, Tlr7−/−, or Myd88−/− mice were i.p. challenged with West Nile virus (LD50).
(A) Quantitative real-time PCR (Q-PCR; mean unit-less ratio + 1 SEM) was performed for WNVE on day (D) 3 postinfection peripheral blood, D6 perfused brain, or D6 perfused spleen samples from wild-type versus Tlr7−/− mice.
(B) Q-PCR results (mean unitless ratio + 1 SEM) for WNVE on D3 peripheral blood, D3 spleen, or D6 perfused brain samples from wild-type versus Myd88−/− mice.
(C) Q-PCR results (mean unitless ratio + 1 SEM) for blood innate immune cytokines in wild-type and Tlr7−/− mice on D3 after infection.
(D) ELISA results (pg/mL; mean + 1 SEM) for IL-23 (left bars) or IL-12 p40 (right bars) in blood samples from wild-type, Myd88−/−, or Tlr7−/− mice on D2 Myd88−/− mouse experiment) or D3 (Tlr7−/− mouse experiment) after infection.
(E) Q-PCR results (mean unitless ratio (+ 1 SEM) for brain innate immune cytokines in wild-type and Tlr7−/− mice on D6 after infection.
Data are pooled results from 2–4 similar independent experiments, with at least n = 3 (and up to n = 22) per group for each experiment. ***p < 0.001, **p < 0.01, and *p < 0.05 compared to wild-type mice.
Furthermore, infectious viral plaque formation assay was carried out on Tlr7−/− and control brains at day 6 p.i. with WNV (LD50). Consistent with WNVE Q-PCR results, there was a striking increase in infectious virus recovered from Tlr7−/− brains (n = 4; 1.33 × 107 pfu/g of brain ± 0.9 × 107 SEM) versus wild-type brains (n = 5; 0.04 × 107 pfu/g of brain ± 0.03 × 107 SEM) that trended toward statistically significant (p = 0.07). Additionally, cytokine abundance was measured in blood and brain. Surprisingly, RNA expression of interferon-α (IFN-α), IFN-β, interleukin (IL)-1β, IL-6, and tumor necrosis factor-α (TNF-α) were all significantly (*p < 0.05, **p < 0.01, ***p < 0.001) increased in blood from WNV-infected Tlr7−/− mice versus controls at day 3 p.i. (Figure 2C).
Despite a generalized increase in systemic innate cytokine RNAs after WNV infection of Tlr7−/− mice, IL-12 p40 (the shared cytokine chain with IL-23) RNA (Il12b) was significantly (p < 0.05) reduced in blood samples from Tlr7−/− mice versus controls (Figure 2C), and a similar pattern of results was noted for secreted heterodimeric IL-23 protein in Myd88−/− mice compared with controls early after infection (Supplemental Experimental Procedures available online) (p < 0.05; Figure 2D). Also, in concert with reduced IL-12 p40 RNA in blood from Tlr7−/− mice versus controls, there was significant (p < 0.05) reduction in IL-12 p40 protein concentrations in blood plasma (Figure 2D). Quantification of IL-12 p35, IL-12 p40, and IL-23 p19 RNAs in brains of Tlr7−/− mice versus controls at day 6 p.i. disclosed significant (*p < 0.05, **p < 0.01) reductions in IL-12 p35 and IL-23 p19 RNAs in Tlr7−/− mice (Figure 2E).
Immune Cell Homing to WNV-Infected Cells In Vivo depends on TLR7 and MyD88
Next, we performed immunofluorescence analysis of WNV antigen and CD11b (microglial and macrophage marker) or CD45 (leukocyte marker) in brains of Tlr7−/− versus control mice at day 6 p.i. Our focus was on the olfactory bulb, because this brain region is most sensitive to WNV infection and brain inflammation (Wang et al., 2004). Consistent with WNVE Q-PCR, these analyses revealed increased WNV antigen immunoreactivity in brains of infected Tlr7−/− mice compared with infected wild-type animals (Figure 3A). Similar results were obtained in other brain regions, including cerebral cortex, brainstem, cerebellum, and striatum (data not shown). In wild-type mice, CD11b+ microglia and macrophages and CD45+ leukocytes were found in close apposition to (often in direct contact with) infected neurons. However, despite increased WNV burden, CD11b+ and CD45+ immune cells failed to home to infected brain cells in Tlr7−/− mice, although they were detected in these mice at a distance from WNV-infected brain cells (Figure 3A). As expected, uninfected (control) brain sections from wild-type or Tlr7−/− mice did not display signal for WNV antigen, CD45, or CD11b (Figure 3A). A more severe phenotype was observed in infected Myd88−/− brains, where CD45+ cells were nearly absent (Figure S1A), despite increased WNV burden (Figure S1B).
Figure 3. In Vivo Leukocyte Homing to West Nile-Infected Cells Is Tlr7 Dependent.

Wild-type (WT) or Tlr7−/− mice were i.p. challenged with WNV (LD50). Uninfected WT and Tlr7−/− mice were euthanized and processed side-by-side as negative controls.
(A) Perfused brains were isolated on day 6 postinfection, and WNV antigen (green signal) and CD45 (leukocyte common antigen, red signal) or CD11b (macrophage and microglia marker, red signal) were detected by immunofluorescence with a Zeiss ApoTome-equipped epifluorescence microscope (original magnification 63×).
(B) Perfused livers were isolated on day 3 postinfection, and WNV antigen (green signal) and CD45 (red signal) were imaged with a Zeiss Apo-Tome-equipped epifluorescence microscope (original magnification 20×). DAPI (blue signal) was used as a nuclear counterstain, and representative images are shown. Numbers of CD45+ or CD11b+ cells per image colocalized with WNV antigen+ areas (first number) and total CD45+ or CD11b+ cells per image (second number) are shown in the bottom right in (A) and (B).
(C) Quantitative PCR for WNVE in wild-type or Tlr7−/− livers at day 3 p.i. Graph shows means + 1 SEM.
(D) Quantitative PCR for Il23a in wild-type or Tlr7−/− livers at day 3 p.i. Graph shows means + 1 SEM. Similar results were observed in 2–4 independent experiments with at least n = 4 per group for each experiment. **p < 0.01 and *p < 0.05 compared to wild-type mice.
To determine whether impaired immune cell homing in infected Tlr7−/− mice was specific to the brain, we also analyzed livers (another target organ of WNV infection) (Venter et al., 2005) from wild-type versus Tlr7−/− mice at day 3 p.i.. In wild-type mice, confocal microscopy revealed numerous CD45+ leukocytes in close vicinity of infected hepatocytes. However, a different pattern of results was evident in Tlr7−/− mice. Similar to observations in brain, CD45+ cells were present, but were often found at a distance from WNV infected hepatocytes (Figure 3B). This phenotype was even more striking when considering that infected Tlr7−/− mouse livers had nearly 6-fold higher abundance of WNVE RNA copies compared with wild-type controls (p < 0.01; Figure 3C). Furthermore, this effect was associated with significantly (p < 0.05) reduced IL-23 p19 RNA in Tlr7−/− versus wild-type mouse livers (Figure 3D). To determine whether this effect was owed to a generalized defect in Tlr7−/− immune cells, macrophages were recovered from the peritoneal cavity at 4 days after thioglycollate injection and enumerated. There was a modest nonsignificant trend for more cells in the Tlr7−/− mice (n = 4 mice per group, means ± SD for wild-type versus Tlr7−/− mice: 7.16 × 106 ± 1.94 × 106 versus 9.14 × 106 ± 1.61 × 106; p = 0.17), indicating that macrophage recruitment, locomotion, and homing were intact in these animals. When taken together, these data suggested that TLR7 mediated immune cell homing to WNV-infected target cells in vivo and that this effect was associated with IL-12 and IL-23 responses.
Recognition of WNV Triggers TLR7 and IL-23-Dependent Macrophage Chemotaxis In Vitro
To determine whether reduced immune cell homing to WNV-infected cells in Tlr7−/− mice could be recapitulated in vitro, we established a transwell chemotaxis assay. Because CD11b+ macrophage cell homing to infected target cells was inhibited in Tlr7−/− mice, peripheral macrophages were elicited from Tlr7−/− and wild-type mice for in vitro chemotaxis analyses. Lower chambers of transwell plates (containing a glass coverslip) were loaded with a dose range of the TLR7 small molecule agonist loxoribine (loxO), supernatants from WNV-infected neuroblastoma-2a (N2a) lysates, or macrophage chemoattractant protein-1 (MCP-1, as a positive control), and 1 × 105 macrophages were placed in the upper chamber. After 6 hr, lower-chamber glass coverslips were recovered and immunolabeled with CD11b antibody for confocal microscopy. Wild-type macrophage migration was increased in response to loxO, infected N2a lysate supernatants, and MCP-1. Yet, although Tlr7−/− macrophages increased migration toward MCP-1, indicating an intact chemotactic response, they were refractive to loxO or infected N2a lysate supernatants (Figure 4A). Quantitation revealed significantly (*p < 0.05, **p < 0.01, ***p < 0.001) reduced chemotaxis of Tlr7−/− macrophages toward loxO (at 50 or 100 μM) or infected N2a lysate supernatants (multiplicity of infection [MOI] = 0.5, from undiluted to 1:50), but not toward MCP-1 (Figure 4B). We also infected N2a cells with MOI = 1 of WNV and prepared cell lysate supernatants and noted similar effects with this material on Tlr7−/− versus wild-type macrophages transwell migration (data not shown).
Figure 4. Tlr7-Dependent Macrophage Homing In Vitro.

(A) Peritoneal thioglycollate-elicited macrophages were prepared from wild-type or Tlr7−/− mice and placed in the upper chamber of transwell plates. In the lower chamber, the TLR7 agonist loxoribine (loxO, from 0 to 100 μM), supernatants from WNV-infected N2a cell lysates (MOI = 0.5, diluted from 1:1 to 1:50), or macrophage chemoattractant protein-1 (MCP-1, 1000 μg/mL) was added for 6 hr. Glass coverslips placed in the lower chamber were recovered for confocal microscopy for CD11b (green signal). DAPI (blue signal) was used as a nuclear counterstain (original magnification 63×), and representative images are shown. (B) CD11b+ cells in high-power fields (original magnification 63×) were counted (n = 3 per condition), and data are presented as means + 1 SD. Similar results were obtained in 3–4 independent experiments. **p < 0.01 and *p < 0.05 compared to wild-type macrophages.
Next, we determined whether the inhibition of migration of Tlr7−/− macrophages in response to WNV infection was associated with reduced IL-12 and IL-23 amounts. Tlr7−/− and wild-type macrophages were infected with WNV (MOI = 1) and assayed for IL-12 p40 RNA and IL-23 p19 protein by Q-PCR and immunoblot, respectively. Infected Tlr7−/− macrophages produced significantly (p < 0.01) less IL-12 p40 RNA compared with wild-type cells, and WNV-induced IL-23 p19 protein was also clearly reduced in infected Tlr7−/− macrophages (Figure 5A). Additionally, Tlr7−/− macrophages were completely nonresponsive to a dose-range of loxO when measuring IL-12 p40 or TNF-α, which further suggested that the above effect was TLR7 dependent (Figure S2).
Figure 5. Tlr7 and IL-23 Signaling-Dependent Macrophage Responses.

(A) Peritoneal thioglycollate-elicited macrophages were prepared from wild-type (WT) or Tlr7−/− mice (key applies to [A] and [C]) and infected with WNV at MOI = 1. 24 hr thereafter, cell lysates were prepared for Il12b Q-PCR analysis (left, unitless ratio + 1 SEM) or immunoblot for IL-23 p19 and actin as a loading control (middle). Densitometry (ratio of IL-23 p19 to actin signal) is shown in right panel.
(B) Macrophages were stimulated with the TLR7 ligand loxoribine (loxO, from 0 to 250 μM as indicated) for 24 hr, and cell lysates were immunoblotted for IL-12Rβ1, IL-12Rβ2, IL-23R, or actin (left). Densitometry (ratio of IL-12Rβ1 to actin signal) is shown in right panel (n = 3 for each condition).
(C) Macrophages (left) were infected with WNV (from 1.0 to 2.0 MOI), and cell lysates were harvested 24 hr later for immunoblot analysis of IL-12Rβ1, IL-12Rβ2, IL-23R, or actin. Densitometry (ratio of target signal to actin) is shown in graphs below each immunoblot. Macrophages (right) were placed in the upper chamber of transwell plates, and IL-23 (from 0 to 10 ng/mL) was placed in the lower chamber. 6 hr later, CD11b+ cells in high-power fields were counted on glass coverslips in the lower chamber by confocal microscopy (n = 3 per condition). **p < 0.01 and *p < 0.05 compared to wild-type macrophages at the same dose. Means ± 1SD are shown in (B) and (C). Similar results were observed in 2–4 independent experiments.
In our model, we proposed that brain-resident macrophages (microglia) initially produced IL-12 and IL-23 upon TLR7 recognition of brain-penetrating WNV and that infiltrating macrophages and other leukocytes would then migrate in response to this signal. If this were the case, then one might expect reduced IL-12 receptor (R) and IL-23R expression to underlie hyporesponsiveness of Tlr7−/− macrophages to WNV-induced chemotaxis. In an effort to address this possibility, Tlr7−/− or wild-type macrophages were challenged with a dose-range of loxO, and protein expression of IL-12R and IL-23R was determined. Much as IL-12 and IL-23 share the IL-12 p40 subunit (Cooper and Khader, 2007), their receptors also form heterodimers sharing the common chain IL-12Rβ1 subunit (van de Vosse et al., 2003). Although IL-12Rβ2 and IL-23R were not further inducible in Tlr7−/− and wild-type macrophages after loxO challenge, IL-12Rβ1 was induced in wild-type macrophages, but Tlr7−/− macrophages were nonresponsive (Figure 5B). It was next determined whether WNV infection of wild-type versus Tlr7−/− macrophages could produce a similar effect. Similar to loxO challenge, WNV at MOI = 1 or 2 induced IL-12Rβ1 expression in wild-type macrophages, but this response was inhibited in Tlr7−/− macrophages (Figure 5C). Finally, we examined whether IL-23 or IL-12 could directly affect TLR7-dependent macrophage chemotaxis in vitro by using our transwell assay. Results showed that IL-23 dose dependently (from 2.5 to 10 ng/mL) augmented chemotaxis of wild-type but not Tlr7−/− macrophages (Figure 5C). However, both wild-type and Tlr7−/− macrophages were refractive to IL-12 p70 chemotaxis (n = 3 transwells per group, means ± SD for wild-type versus Tlr7−/− macrophages for cells per high power field, 0 ng: 4 cells ± 1 cell versus 4.33 cells ±.58 cells; 2.5 ng: 4.67 cells ±.58 cells versus 5.33 cells ±.58 cells; 5.0 ng: 4.67 cells ±.58 cells versus 5.33 cells ±.58 cells; 10 ng: 4.33 cells ±.58 cells versus 4.67 cells ±.58 cells). Thus, IL-23 (and not IL-12) was responsible for TLR7-dependent macrophage chemotaxis in vitro.
To determine whether the effects we observed in Tlr7−/− macrophages might be due to a compensatory Tlr response, Tlr Q-PCR array was performed on WNV-infected macrophages, and only Tlr7 RNA was substantially altered after infection in Tlr7−/− macrophages (data not shown). Further, Q-PCR arrays were carried out for chemokines and chemokine receptors, and cytokines and cytokine receptors, to identify any additional targets of TLR7 after WNV infection, but there were no obvious alterations (data not shown). Finally, supernatants from infected macrophages were assayed by protein cytokine array and we did not observe obvious differences between wild-type and Tlr7−/− macrophages (data not shown). Thus, according to unbiased approaches, additional TLR7-dependent targets of WNV were not identified in macrophages.
Immune Cell Homing to WNV-Infected Cells Is TLR7-IL-23 Dependent In Vivo
Our data thus far suggested that recognition of WNV by TLR7 caused IL-23-dependent immune cell homing. To directly test this hypothesis in vivo, wild-type, IL-12 p35-deficient (Il12a−/−), IL-12 p40-deficient (Il12b−/−), or IL-23 p19-deficient (Il23a−/−) mice were infected with WNV and we assessed infected brains for presence of WNV and infiltrating leukocytes by confocal microscopy on day 6 p.i.. Wild-type and Il12a−/− mice demonstrated CD11b+ macrophages and microglia (Figure 6) and CD45+ leukocytes (Figure S3A) in close apposition to infected brain cells. However, in both Il12b−/− and Il23a−/− mice, CD11b+ macrophages and microglia (Figure 6) and CD45+ leukocytes (Figure S3A) were not clearly colocalized to foci of WNV-infected cells. Further, Il23a−/− mice had fewer infiltrating CD11b+ macrophages and microglia (Figure 6) that were not clearly associated with WNV-infected brain cells. Thus, proper infiltration and homing of immune cells to target WNV-infected cells required IL-23.
Figure 6. Macrophage Homing to West Nile Virus Is IL-23 Signaling Dependent.

Wild-type (n = 6), Il12a−/− (n = 4), Il12b−/− (n = 4), and Il23a−/− (n = 3) mice were infected with West Nile virus (LD50). Brains were isolated on day 6 after infection and immunostained for confocal microscopy with antibodies against CD11b (green signal) and WNV antigen (red signal) to reveal microglia and infiltrating macrophages in WNV-infected brain regions. TOPRO3 was used as a nuclear counterstain (blue signal) and merged images are shown to the right. Numbers of CD11b+ cells per image colocalized with WNV antigen+ areas (first number) and total CD11b+ cells per image (second number) are shown in the bottom right. Similar results were obtained in 2–4 independent experiments.
To better define and enumerate brain immune cells, wild-type, Tlr7−/−, Myd88−/−, Il12a−/−, Il23a−/−, or Il12b−/− mice were infected with WNV (LD50) and brains were isolated on day 6 p.i.. We applied flow cytometry methodology to single-cell suspensions of harvested brains to characterize numbers of brain-infiltrating macrophages (based on CD45hiCD11b+ status; Juedes and Ruddle, 2001; Town et al., 2008), brain-resident microglia (by CD45intCD11b+F4/80 Ag+; Juedes and Ruddle, 2001; Town et al., 2008), and brain-infiltrating CD45hiCD4+ and CD45hiCD8+ T cells in these mice. In concert with our confocal microscopic analyses, brain-infiltrating macrophages were reduced by 59% to as much as 98% when comparing wild-type to Tlr7−/−, Myd88−/−, Il12b−/−, or Il23a−/− mice, whereas Il12a−/− mice appeared similar to wild-type WNV-infected brains (Figure 7). A generally similar pattern of results was evident when considering CD4+ and CD8+ T cells, which were attenuated between 55% and 89% when comparing wild-type to Tlr7−/−, Myd88−/−, or Il12b−/− mice, whereas Il12a−/− or Il23a−/− mice appeared similar to wild-type WNV-infected brains (Figure 7). However, a different pattern of results emerged when considering brain-resident microglia, which did not obviously differ from wild-type WNV-infected brains except for an apparent 75% reduction in Il12b−/− mice (Figure 7). We also analyzed B cells by CD19 expression, but did not detect consistent differences between these mouse genotypes (data not shown).
Figure 7. Characterization of Brain Immune Cells in Wild-Type, Tlr7−/− −/−, Myd88−/−, Il12−/−, and Il23−/− Mice after West Nile Virus Infection.

Brain flow cytometry results are shown from 2–3 independent experiments on day 6 post-WNV infection (LD50; n = 1–5 mice per genotype and numbers represent percentages ± SD for the indicated gates). The 37:70% Percoll interface was collected and stained for CD45, CD4, CD8, CD11b, and F4/80 antigen as indicated in (A) (right, applies for all panels). A minimum of 100,000 events were collected for flow cytometry analysis, and dot plots show side scatter (SSC) on the x axis and CD45 log fluorescence intensity on the y axis. Numbers represent percentages of positive cells within gated regions.
(A) Brain flow cytometry results are shown from wild-type vs. Tlr7−/− mice.
(B) Brain flow cytometry data are shown for wild-type compared to Myd88−/− or Il12a−/− mice.
(C) Brain flow cytometry data are shown for wild-type compared to Il23a−/− or Il12b−/− mice.
To determine whether impaired leukocyte homing to WNV-infected brains was due to a general defect in leukocyte expansion or differentiation, or cell death in the periphery, we analyzed spleens from the same animals as above and did not detect any differences between genotypes (data not shown). Interestingly, similar effects are reported in other neuroinflammatory conditions where peripheral leukocytes infiltrate into the central nervous system (CNS) of (1) experimental autoimmune encephalomyelitis-induced mice or (2) Alzheimer’s mouse models in the absence of innate immune TGF-β signaling, but populations of these cells in the periphery are essentially unaltered (Laouar et al., 2008; Town et al., 2008). Taken together, these brain flow cytometry results corroborated our confocal microscopic analyses and strengthened our conclusion that brain infiltration or homing of leukocytes (specifically, macrophages and T cells) to WNV-infected CNS was dependent on TLR7-MyD88-IL-23 signaling.
Finally, we determined whether survival after lethal WNV encephalitis was dependent on IL-12, IL-23, or both. Thus, wild-type, Il12a−/−, Il12b−/−, or Il23a−/− mice were infected with WNV (LD50) and monitored twice daily for survival. Based on our findings of IL-23-dependent immune cell homing to WNV-infected brain cells, we hypothesized reduced survival in Il12b−/− and Il23a−/− but not Il12a−/− mice after lethal WNV challenge. Results were consistent with this hypothesis. Specifically, although Il12a−/− mice did not differ from wild-type controls (42% survival for both groups, p > 0.10; Figure S3B), both Il12b−/− mice (27% survival versus 53% for wild-type controls, p < 0.05; Figure S3B) and Il23a−/− mice (0% survival versus 25% for wild-type controls, p < 0.01; Figure S3B) were more susceptible to lethal WNV infection. Collectively, these results showed that survival after lethal WNV challenge required intact IL-23 as opposed to IL-12 responses.
DISCUSSION
Mammalian innate immune responses are a vital first line of defense against invading pathogens. Notably, TLRs are indispensable sensors of PAMPs and function to drive innate immune responses that fine-tune adaptive immunity (Qureshi and Medzhitov, 2003; Yamamoto et al., 2004). It is known that TLR3 recognizes dsRNA (Alexopoulou et al., 2001; Town et al., 2006). WNV is a positive ssRNA flavivirus that produces dsRNA during its life cycle, and Tlr3−/− mice have reduced susceptibility to lethal WNV encephalitis (Wang et al., 2004). This finding implicates TLR3 as a host immune receptor that recognizes WNV dsRNA. Furthermore, the virus uses TLR3 to promote infectivity of the host by eliciting systemic TNF-α that increases permeability of the blood-brain barrier. Yet, most WNV infections in humans are asymptomatic (Campbell et al., 2002; Davis et al., 2006), highlighting that host immune mechanisms must exist to suppress WNV infection. In this report, we showed that Tlr7−/− mice had increased susceptibility to lethal WNV encephalitis; this finding pointed to TLR7 as the host’s first line of defense against WNV infection, by recognizing ssRNA present immediately upon viral entry into the host—even before the first round of replication. TLR7 is thought to exclusively rely on MyD88 as an adaptor molecule, and, consistent with this notion, we found that Myd88−/− mice phenocopy Tlr7−/− mice on reduced survival after lethal WNV infection.
We sought to understand the cellular mechanism responsible for increased sensitivity of Tlr7−/− and Myd88−/− mice to lethal WNV encephalitis. Interestingly, viral burden was increased both systemically and in brains of Tlr7−/− and Myd88−/− mice, suggesting that loss of these innate immune molecules caused a failure of the host to control viral infectivity. Surprisingly, Tlr7−/− mice had increased systemic concentrations of most innate proinflammatory cytokines after WNV challenge including IFN-α, IFN-β, IL-1β, IL-6, and TNF-α, thought to be key mediators of host antiviral immunity (Anderson and Rahal, 2002; Brinton, 2001; Gilfoy and Mason, 2007). This paradoxical result is most likely owing to other innate immune viral PRRs, such as TLR3, RIG-I, and MDA-5, which would still be expected to operate in Tlr7−/− mice upon WNV infection. Further, WNV infection studies in Il6−/− and TNF-soluble receptor type I-deficient (Tnfsr1−/−) mice show that this reduced susceptibility is dependent on TNF-α (but not IL-6) signaling through the type I receptor (Tnfsr1), resulting in increased blood-brain-barrier permeability and consequent enhanced entry of the virus into the CNS (Wang et al., 2004). Importantly however, recent reports using a different WNV strain show a protective role of TLR3 and Tnfsr1, suggesting that host antiviral innate immunity may be WNV strain specific (Daffis et al., 2008; Shrestha et al., 2008). Plasmacytoid dendritic cells (pDCs) are thought to be the primary source of IFN-α and IFN-β produced in response to viral infection via the TLR7 signaling pathway (Cella et al., 1999; Hornung et al., 2002). However, in response to WNV challenge, Tlr7−/− mice paradoxically produced more IFN-α and IFN-β than did wild-type mice. Therefore, it seemed unlikely to us that dysfunction of pDCs was the underlying cause of increased susceptibility to WNV in Tlr7−/− mice.
In contrast to the increased amounts of IFN-α, IFN-β, IL-1β, IL-6, and TNF-α, there was reduction of systemic IL-12 p40 and brain IL-12 p35 and IL-23 p19 after lethal WNV infection of Tlr7−/− mice. Additionally, Myd88−/− mice had reduced systemic abundance of IL-23. Further, homing of leukocytes and microglia and macrophages to WNV-infected cells was impaired in Tlr7−/− and Myd88−/− mice. This effect could be recapitulated in vitro, because we found that whereas wild-type macrophages migrated toward either the TLR7 agonist loxO or infected supernatants from WNV-infected neuron-like cells, migration of Tlr7−/− macrophages was significantly inhibited. However, it should be noted that thioglycollate-elicited peritoneal macrophages do not necessarily reflect the behavior of monocytes or tissue macrophages such as microglia and Kupffer cells. We went on to hypothesize that TLR7 recognition of WNV promotes IL-12-IL-23-dependent immune cell homing to infected target cells. Interestingly, TLR7 cooperates with heat-shock protein 70 to promote macrophage phagocytosis (Wang et al., 2006), raising the possibility that TLR7 may promote both macrophage homing to and clearance of WNV-infected cells.
The IL-12-IL-23 axis has recently emerged as a key immunomodulatory pathway. Interestingly, it is postulated that “tipping the balance” between IL-12 and IL-23 controls the outcome of inflammatory responses, with the IL-23 arm of the scale leading to production of IL-17 via T helper 17 (Th17) cells (Goriely et al., 2008). IL-17 plays a “certain but subdominant role” in IL-23-mediated resistance to vaccinia virus (Kohyama et al., 2007), demonstrating that IL-17 (likely induced by IL-23 produced by innate immune cells) may link innate immune TLR-IL-23 signaling with adaptive immune Th17 cells. In support of this, it was recently demonstrated that IL-17 is secreted into supernatants from human peripheral blood mononuclear cells treated with TLR7 and TLR8 agonists (Kattah et al., 2008). Based on results reported here linking TLR7 and IL-23 signaling, future studies are warranted to evaluate the putative role of IL-17 in WNV encephalitis.
Because IL-12 and IL-23 share the p40 subunit and both utilize the common IL-12Rβ1 chain for signaling, it was initially difficult to tease apart effects from these two cytokines. For example, although it was once thought that IL-12 mediated the inflammatory, autoaggressive response in experimental autoimmune encephalomyelitis, it was later shown in a seminal report that IL-23 was the responsible cytokine (Cua et al., 2003). TLR ligands differentially promote IL-12 and IL-23 production in innate immune cells. Specifically, TLR2 recognition of Gram-positive bacteria and fungi by dendritic cells leads to induction of p40 and p19 subunits (but not p35), whereas TLR3 stimulation by viruses or TLR4 engagement by Gram-negative bacteria induces all three subunits (Bekeredjian-Ding et al., 2006; Gautier et al., 2005; Goriely et al., 2006; Napolitani et al., 2005; Re and Strominger, 2001). We found IL-12 p40 RNA and IL-23 p19 protein induction in wild-type macrophages after WNV infection, and this was significantly abrogated in Tlr7−/− macrophages. Additionally, the IL-12 and IL-23 common receptor chain, IL-12Rβ1, was induced in a TLR7-dependent fashion after loxO challenge or WNV infection. It was noteworthy that IL-12Rβ2 (which pairs with IL-12Rβ1 to promote IL-12 signaling) was constitutively reduced in Tlr7−/− macrophages, suggesting a compensatory mechanism whereby these cells were “biased” in favor of IL-23 as opposed to IL-12 responses.
To further determine whether TLR7-dependent immune responses were dependent on IL-12 or IL-23 (or both) pathways, we (1) evaluated wild-type versus Tlr7−/− macrophage homing both in vitro in response to IL-12 or IL-23 and in vivo in WNV-infected wild-type versus Il12a−/−, Il12b−/−, or Il23a−/− mice, and (2) performed survival analysis after WNV infection of wild-type versus Il12a−/−, Il12b−/−, or Il23a−/− mice. Exogenous addition of IL-23 (but not IL-12) was able to promote chemotaxis of wild-type macrophages, whereas Tlr7−/− macrophages were refractive to stimulation. Consistently, WNV infection led to impaired immune cell homing to infected brain cells in Il12b−/− and Il23a−/− mice, but not in Il12a−/− mice. If IL-23-dependent immune cell homing to infected target cells were a mitigating factor against lethal WNV encephalitis, one would expect reduced survival after challenge with a lethal dose of WNV in Il12b−/− and Il23a−/− mice, but not in Il12a−/− animals. A pattern of results consistent with this hypothesis was observed. Interestingly, a previous report has shown that the chemokine receptor CCR5 is required for leukocyte trafficking to the brain and mitigation of lethal WNV encephalitis (Glass et al., 2005), further highlighting the role of immune cell infiltration and homing as an essential host defense mechanism.
Our results suggest a model of host-WNV pathogen interaction that requires intact TLR7 and IL-23 signaling to promote effective viral clearance. Specifically, we postulate in this model that resident tissue macrophages in WNV target organs such as Kupffer cells in liver and microglia in brain sense tissue-invading WNV by way of TLR7 recognition of viral ssRNA. These resident macrophages then secrete IL-23, which promotes infiltration and homing of peripheral immune cells including blood-borne macrophages or monocytes, which in turn upregulate IL-12Rβ1 in order to receive the IL-23 signal. Once infiltrated into WNV target organs, these peripheral immune cells can then affect neutralization and clearance of the virus by both TLR7-IL-23-dependent and -independent mechanisms. The importance of TLR7 in promoting immune cell activation is further underscored by inflammatory responses and autoimmunity in TLR7-overex-pressing mice (Deane et al., 2007). Taken together, our results show that TLR7 is a critical host sensor of WNV required for IL-23-dependent immune cell homing to infected target cells and suggest that pharmacotherapy aimed at promoting TLR7-IL-23 signaling will be beneficial for treatment of WNV and perhaps other encephalitides.
EXPERIMENTAL PROCEDURES
Mice
Tlr7−/− (Lund et al., 2004), Myd88−/− (Adachi et al., 1998), and Tlr9−/−(Hemmi et al., 2003) mice were bred to the C57BL/6 background by backcrossing for 10 successive generations. Il12a−/− (Mattner et al., 1996) and Il12b−/− (Magram et al., 1996) mice on a C57BL/6 background were obtained from Jackson Laboratories. Il23a−/− mice (Cua et al., 2003) on a mixed C57BL/6 × 129 background were obtained from the Mutant Mouse Regional Resource Center (MMRRC). We performed all experiments on 8- to 12-week-old mice, and mouse groups were rigorously age- and sex-matched for each infection experiment. All experiments were performed with approval from the Yale Animal Resources Center Institutional Animal Care and Use Committee.
Virus Infection
We inoculated mice intraperitoneally with 2000 plaque-forming units (p.f.u.) (LD50) of WNV isolate 2741 in 100 μl of PBS with 5% gelatin as previously described (Wang et al., 2004). Mice were observed for up to 21 days after infection and we checked them twice daily for morbidity (including lethargy, anorexia, and difficulty ambulating) and mortality.
Quantitative PCR
Ribonucleic acid was extracted from blood, spleen, liver, and brain tissue with the RNeasy kit (QIAGEN). RNA was used to synthesize complementary (c) DNA with the ProSTAR First-strand RT-PCR kit (Stratagene). The flurogenic probes and primers that we used have been described elsewhere (Wang et al., 2004). Probes contained a 5′ reporter, FAM, and a 3′ quencher, TAMRA (Applied Biosystems). The assay was performed on an iCycler (Bio-Rad). The thermal cycling consisted of 95°C for 3.5 min and 48 cycles of 95°C for 30 s and 60°C for 1 min. To normalize the samples, the same amount of cDNA was used in the Actb Q-PCR. The ratio of the amount of amplified gene compared with the amount of Actb cDNA represented the relative amounts in each sample.
Immunofluorescence Imaging
Brains and livers were rapidly isolated, fixed in 4% paraformaldehyde (PFA) overnight at 4°C, and cryoprotected in a graded series of sucrose (10%, 20%, and 30%, each overnight at 4°C). Spleens from infected mice sacrificed day 3 postinfection were used as a positive control to ensure specificity of WNV antibodies (data not shown). Para-median sagital sections were cut at 25 μm with a cryostat. Tissue sections were PAP pen (Zymed Laboratories) applied and preblocked in serum-free protein block (Dakocytomation) for 30 min at ambient temperature. Sections were then reacted overnight at 4°C with various combinations of primary antibodies against CD11b (Serotec; 1:200), CD45 (Serotec, 1:200), or WNV antigen (from J.F. Anderson; 1:250). After three rinses in PBS, sections were reacted with appropriate secondary antibodies conjugated with Alexa Fluor 488, 594, or 647 for 1 hr at ambient temperature. After three additional rinses in PBS, sections were then nuclear counterstained with DAPI or TOPRO3 (Invitrogen) and mounted in fluorescence mounting medium (ProLong Gold). Images were acquired in independent channels with a Zeiss ApoTome-equipped fluorescence microscope or a Zeiss LSM510 META confocal microscope. Immune cells in brain and liver and numbers of immune cells colocalized with WNV-infected target cells were counted in a blind fashion with Zeiss Axiovision software.
Statistical Analysis
We calculated standard errors of the means (SEM) and standard deviations (SD) and analyzed data by nonpaired Student’s t test for single mean comparisons or ANOVA and post-hoc testing for multiple comparisons of the means. Survival curve comparisons were conducted with the log-rank test (equivalent to the Mantel-Haenszel test, Prism software).
Supplementary Material
Acknowledgments
We are grateful to A. Ferrandino, D. Beck, X. Liu, and D. Gate for expert technical assistance and F. Manzo for assistance with preparing this manuscript. This work was supported by NIH awards AI055749 and AI50031. T.T. is supported by an Alzheimer’s Association grant and a National Institutes of Health, National Institute on Aging “Pathway to Independence” award (1K99AG029726-01 and 4R00AG029726-02). F.B. is supported by a Career Development Award from the Northeast Biodefense Center (U54-AI057158-Lipkin). T.W. is supported by a grant from the National Institutes of Health (5R03AI067409). R.A.F. and E.F. are Investigators of the Howard Hughes Medical Institute. T.T. is the inaugural holder of the Ben Winters Endowed Chair in Regenerative Medicine.
Footnotes
Supplemental Data include Supplemental Experimental Procedures and three figures and can be found with this article online at http://www.immunity.com/supplemental/S1074-7613(09)00064-8.
References
- Adachi O, Kawai T, Takeda K, Matsumoto M, Tsutsui H, Sakagami M, Nakanishi K, Akira S. Targeted disruption of the MyD88 gene results in loss of IL-1- and IL-18-mediated function. Immunity. 1998;9:143–150. doi: 10.1016/s1074-7613(00)80596-8. [DOI] [PubMed] [Google Scholar]
- Alexopoulou L, Holt AC, Medzhitov R, Flavell RA. Recognition of double-stranded RNA and activation of NF-kappaB by Toll-like receptor 3. Nature. 2001;413:732–738. doi: 10.1038/35099560. [DOI] [PubMed] [Google Scholar]
- Anderson JF, Rahal JJ. Efficacy of interferon alpha-2b and ribavirin against West Nile virus in vitro. Emerg Infect Dis. 2002;8:107–108. doi: 10.3201/eid0801.010252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauer S, Kirschning CJ, Hacker H, Redecke V, Hausmann S, Akira S, Wagner H, Lipford GB. Human TLR9 confers responsiveness to bacterial DNA via species-specific CpG motif recognition. Proc Natl Acad Sci USA. 2001;98:9237–9242. doi: 10.1073/pnas.161293498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bekeredjian-Ding I, Roth SI, Gilles S, Giese T, Ablasser A, Hornung V, Endres S, Hartmann G. T cell-independent, TLR-induced IL-12p70 production in primary human monocytes. J Immunol. 2006;176:7438–7446. doi: 10.4049/jimmunol.176.12.7438. [DOI] [PubMed] [Google Scholar]
- Bieback K, Lien E, Klagge IM, Avota E, Schneider-Schaulies J, Duprex WP, Wagner H, Kirschning CJ, Ter Meulen V, Schneider-Schaulies S. Hemagglutinin protein of wild-type measles virus activates toll-like receptor 2 signaling. J Virol. 2002;76:8729–8736. doi: 10.1128/JVI.76.17.8729-8736.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brinton MA. Host factors involved in West Nile virus replication. Ann N Y Acad Sci. 2001;951:207–219. doi: 10.1111/j.1749-6632.2001.tb02698.x. [DOI] [PubMed] [Google Scholar]
- Campbell GL, Marfin AA, Lanciotti RS, Gubler DJ. West Nile virus. Lancet Infect Dis. 2002;2:519–529. doi: 10.1016/s1473-3099(02)00368-7. [DOI] [PubMed] [Google Scholar]
- Cella M, Jarrossay D, Facchetti F, Alebardi O, Nakajima H, Lanzavecchia A, Colonna M. Plasmacytoid monocytes migrate to inflamed lymph nodes and produce large amounts of type I interferon. Nat Med. 1999;5:919–923. doi: 10.1038/11360. [DOI] [PubMed] [Google Scholar]
- Cooper AM, Khader SA. IL-12p40: an inherently agonistic cytokine. Trends Immunol. 2007;28:33–38. doi: 10.1016/j.it.2006.11.002. [DOI] [PubMed] [Google Scholar]
- Cua DJ, Sherlock J, Chen Y, Murphy CA, Joyce B, Seymour B, Lucian L, To W, Kwan S, Churakova T, et al. Interleukin-23 rather than interleukin-12 is the critical cytokine for autoimmune inflammation of the brain. Nature. 2003;421:744–748. doi: 10.1038/nature01355. [DOI] [PubMed] [Google Scholar]
- Daffis S, Samuel MA, Suthar MS, Gale M, Jr, Diamond MS. Toll-like receptor 3 has a protective role against West Nile virus infection. J Virol. 2008;82:10349–10358. doi: 10.1128/JVI.00935-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis LE, DeBiasi R, Goade DE, Haaland KY, Harrington JA, Harnar JB, Pergam SA, King MK, DeMasters BK, Tyler KL. West Nile virus neuroinvasive disease. Ann Neurol. 2006;60:286–300. doi: 10.1002/ana.20959. [DOI] [PubMed] [Google Scholar]
- Deane JA, Pisitkun P, Barrett RS, Feigenbaum L, Town T, Ward JM, Flavell RA, Bolland S. Control of toll-like receptor 7 expression is essential to restrict autoimmunity and dendritic cell proliferation. Immunity. 2007;27:801–810. doi: 10.1016/j.immuni.2007.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Debiasi RL, Tyler KL. West Nile virus meningoencephalitis. Nat Clin Pract Neurol. 2006;2:264–275. doi: 10.1038/ncpneuro0176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diamond MS, Shrestha B, Marri A, Mahan D, Engle M. B cells and antibody play critical roles in the immediate defense of disseminated infection by West Nile encephalitis virus. J Virol. 2003a;77:2578–2586. doi: 10.1128/JVI.77.4.2578-2586.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diamond MS, Sitati EM, Friend LD, Higgs S, Shrestha B, Engle M. A critical role for induced IgM in the protection against West Nile virus infection. J Exp Med. 2003b;198:1853–1862. doi: 10.1084/jem.20031223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diebold SS, Kaisho T, Hemmi H, Akira S, Reis e Sousa C. Innate antiviral responses by means of TLR7-mediated recognition of single-stranded RNA. Science. 2004;303:1529–1531. doi: 10.1126/science.1093616. [DOI] [PubMed] [Google Scholar]
- Edelmann KH, Richardson-Burns S, Alexopoulou L, Tyler KL, Flavell RA, Oldstone MB. Does Toll-like receptor 3 play a biological role in virus infections? Virology. 2004;322:231–238. doi: 10.1016/j.virol.2004.01.033. [DOI] [PubMed] [Google Scholar]
- Gautier G, Humbert M, Deauvieau F, Scuiller M, Hiscott J, Bates EE, Trinchieri G, Caux C, Garrone P. A type I interferon autocrine-paracrine loop is involved in Toll-like receptor-induced interleukin-12p70 secretion by dendritic cells. J Exp Med. 2005;201:1435–1446. doi: 10.1084/jem.20041964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilfoy FD, Mason PW. West Nile virus-induced interferon production is mediated by the double-stranded RNA-dependent protein kinase PKR. J Virol. 2007;81:11148–11158. doi: 10.1128/JVI.00446-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glass WG, Lim JK, Cholera R, Pletnev AG, Gao JL, Murphy PM. Chemokine receptor CCR5 promotes leukocyte trafficking to the brain and survival in West Nile virus infection. J Exp Med. 2005;202:1087–1098. doi: 10.1084/jem.20042530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goriely S, Molle C, Nguyen M, Albarani V, Haddou NO, Lin R, De Wit D, Flamand V, Willems F, Goldman M. Interferon regulatory factor 3 is involved in Toll-like receptor 4 (TLR4)- and TLR3-induced IL-12p35 gene activation. Blood. 2006;107:1078–1084. doi: 10.1182/blood-2005-06-2416. [DOI] [PubMed] [Google Scholar]
- Goriely S, Neurath MF, Goldman M. How microorganisms tip the balance between interleukin-12 family members. Nat Rev Immunol. 2008;8:81–86. doi: 10.1038/nri2225. [DOI] [PubMed] [Google Scholar]
- Gould LH, Fikrig E. West Nile virus: a growing concern? J Clin Invest. 2004;113:1102–1107. doi: 10.1172/JCI21623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gubler DJ. The continuing spread of West Nile virus in the western hemisphere. Clin Infect Dis. 2007;45:1039–1046. doi: 10.1086/521911. [DOI] [PubMed] [Google Scholar]
- Heil F, Hemmi H, Hochrein H, Ampenberger F, Kirschning C, Akira S, Lipford G, Wagner H, Bauer S. Species-specific recognition of single-stranded RNA via toll-like receptor 7 and 8. Science. 2004;303:1526–1529. doi: 10.1126/science.1093620. [DOI] [PubMed] [Google Scholar]
- Hemmi H, Takeuchi O, Kawai T, Kaisho T, Sato S, Sanjo H, Matsumoto M, Hoshino K, Wagner H, Takeda K, Akira S. A Toll-like receptor recognizes bacterial DNA. Nature. 2000;408:740–745. doi: 10.1038/35047123. [DOI] [PubMed] [Google Scholar]
- Hemmi H, Kaisho T, Takeuchi O, Sato S, Sanjo H, Hoshino K, Horiuchi T, Tomizawa H, Takeda K, Akira S. Small anti-viral compounds activate immune cells via the TLR7 MyD88-dependent signaling pathway. Nat Immunol. 2002;3:196–200. doi: 10.1038/ni758. [DOI] [PubMed] [Google Scholar]
- Hemmi H, Kaisho T, Takeda K, Akira S. The roles of Toll-like receptor 9, MyD88, and DNA-dependent protein kinase catalytic subunit in the effects of two distinct CpG DNAs on dendritic cell subsets. J Immunol. 2003;170:3059–3064. doi: 10.4049/jimmunol.170.6.3059. [DOI] [PubMed] [Google Scholar]
- Hornung V, Rothenfusser S, Britsch S, Krug A, Jahrsdorfer B, Giese T, Endres S, Hartmann G. Quantitative expression of toll-like receptor 1–10 mRNA in cellular subsets of human peripheral blood mononuclear cells and sensitivity to CpG oligodeoxynucleotides. J Immunol. 2002;168:4531–4537. doi: 10.4049/jimmunol.168.9.4531. [DOI] [PubMed] [Google Scholar]
- Juedes AE, Ruddle NH. Resident and infiltrating central nervous system APCs regulate the emergence and resolution of experimental autoimmune encephalomyelitis. J Immunol. 2001;166:5168–5175. doi: 10.4049/jimmunol.166.8.5168. [DOI] [PubMed] [Google Scholar]
- Kattah MG, Wong MT, Yocum MD, Utz PJ. Cytokines secreted in response to Toll-like receptor ligand stimulation modulate differentiation of human Th17 cells. Arthritis Rheum. 2008;58:1619–1629. doi: 10.1002/art.23497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohyama S, Ohno S, Isoda A, Moriya O, Belladonna ML, Hayashi H, Iwakura Y, Yoshimoto T, Akatsuka T, Matsui M. IL-23 enhances host defense against vaccinia virus infection via a mechanism partly involving IL-17. J Immunol. 2007;179:3917–3925. doi: 10.4049/jimmunol.179.6.3917. [DOI] [PubMed] [Google Scholar]
- Kulkarni AB, Mullbacher A, Blanden RV. Functional analysis of macrophages, B cells and splenic dendritic cells as antigen-presenting cells in West Nile virus-specific murine T lymphocyte proliferation. Immunol Cell Biol. 1991;69:71–80. doi: 10.1038/icb.1991.12. [DOI] [PubMed] [Google Scholar]
- Kurt-Jones EA, Popova L, Kwinn L, Haynes LM, Jones LP, Tripp RA, Walsh EE, Freeman MW, Golenbock DT, Anderson LJ, Finberg RW. Pattern recognition receptors TLR4 and CD14 mediate response to respiratory syncytial virus. Nat Immunol. 2000;1:398–401. doi: 10.1038/80833. [DOI] [PubMed] [Google Scholar]
- Laouar Y, Town T, Jeng D, Tran E, Wan Y, Kuchroo VK, Flavell RA. TGF-beta signaling in dendritic cells is a prerequisite for the control of autoimmune encephalomyelitis. Proc Natl Acad Sci USA. 2008;105:10865–10870. doi: 10.1073/pnas.0805058105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lund JM, Alexopoulou L, Sato A, Karow M, Adams NC, Gale NW, Iwasaki A, Flavell RA. Recognition of single-stranded RNA viruses by Toll-like receptor 7. Proc Natl Acad Sci USA. 2004;101:5598–5603. doi: 10.1073/pnas.0400937101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magram J, Connaughton SE, Warrier RR, Carvajal DM, Wu CY, Ferrante J, Stewart C, Sarmiento U, Faherty DA, Gately MK. IL-12-deficient mice are defective in IFN gamma production and type 1 cytokine responses. Immunity. 1996;4:471–481. doi: 10.1016/s1074-7613(00)80413-6. [DOI] [PubMed] [Google Scholar]
- Mattner F, Magram J, Ferrante J, Launois P, Di Padova K, Behin R, Gately MK, Louis JA, Alber G. Genetically resistant mice lacking interleukin-12 are susceptible to infection with Leishmania major and mount a polarized Th2 cell response. Eur J Immunol. 1996;26:1553–1559. doi: 10.1002/eji.1830260722. [DOI] [PubMed] [Google Scholar]
- Napolitani G, Rinaldi A, Bertoni F, Sallusto F, Lanzavecchia A. Selected Toll-like receptor agonist combinations synergistically trigger a T helper type 1-polarizing program in dendritic cells. Nat Immunol. 2005;6:769–776. doi: 10.1038/ni1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qureshi ST, Medzhitov R. Toll-like receptors and their role in experimental models of microbial infection. Genes Immun. 2003;4:87–94. doi: 10.1038/sj.gene.6363937. [DOI] [PubMed] [Google Scholar]
- Rassa JC, Meyers JL, Zhang Y, Kudaravalli R, Ross SR. Murine retroviruses activate B cells via interaction with toll-like receptor 4. Proc Natl Acad Sci USA. 2002;99:2281–2286. doi: 10.1073/pnas.042355399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Re F, Strominger JL. Toll-like receptor 2 (TLR2) and TLR4 differentially activate human dendritic cells. J Biol Chem. 2001;276:37692–37699. doi: 10.1074/jbc.M105927200. [DOI] [PubMed] [Google Scholar]
- Roeder A, Kirschning CJ, Rupec RA, Schaller M, Weindl G, Korting HC. Toll-like receptors as key mediators in innate antifungal immunity. Med Mycol. 2004;42:485–498. doi: 10.1080/13693780400011112. [DOI] [PubMed] [Google Scholar]
- Schlaepfer E, Audige A, Joller H, Speck RF. TLR7/8 triggering exerts opposing effects in acute versus latent HIV infection. J Immunol. 2006;176:2888–2895. doi: 10.4049/jimmunol.176.5.2888. [DOI] [PubMed] [Google Scholar]
- Shrestha B, Diamond MS. Role of CD8+ T cells in control of West Nile virus infection. J Virol. 2004;78:8312–8321. doi: 10.1128/JVI.78.15.8312-8321.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shrestha B, Diamond MS. Fas ligand interactions contribute to CD8+ T-cell-mediated control of West Nile virus infection in the central nervous system. J Virol. 2007;81:11749–11757. doi: 10.1128/JVI.01136-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shrestha B, Samuel MA, Diamond MS. CD8+ T cells require perforin to clear West Nile virus from infected neurons. J Virol. 2006;80:119–129. doi: 10.1128/JVI.80.1.119-129.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shrestha B, Zhang B, Purtha WE, Klein RS, Diamond MS. Tumor necrosis factor alpha protects against lethal West Nile virus infection by promoting trafficking of mononuclear leukocytes into the central nervous system. J Virol. 2008;82:8956–8964. doi: 10.1128/JVI.01118-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sitati EM, Diamond MS. CD4+ T-cell responses are required for clearance of West Nile virus from the central nervous system. J Virol. 2006;80:12060–12069. doi: 10.1128/JVI.01650-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sitati E, McCandless EE, Klein RS, Diamond MS. CD40–CD40 ligand interactions promote trafficking of CD8+ T cells into the brain and protection against West Nile virus encephalitis. J Virol. 2007;81:9801–9811. doi: 10.1128/JVI.00941-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Town T, Jeng D, Alexopoulou L, Tan J, Flavell RA. Microglia recognize double-stranded RNA via TLR3. J Immunol. 2006;176:3804–3812. doi: 10.4049/jimmunol.176.6.3804. [DOI] [PubMed] [Google Scholar]
- Town T, Laouar Y, Pittenger C, Mori T, Szekely CA, Tan J, Duman RS, Flavell RA. Blocking TGF-beta-Smad2/3 innate immune signaling mitigates Alzheimer-like pathology. Nat Med. 2008;14:681–687. doi: 10.1038/nm1781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Triantafilou K, Vakakis E, Orthopoulos G, Ahmed MA, Schumann C, Lepper PM, Triantafilou M. TLR8 and TLR7 are involved in the host’s immune response to human parechovirus 1. Eur J Immunol. 2005;35:2416–2423. doi: 10.1002/eji.200526149. [DOI] [PubMed] [Google Scholar]
- van de Vosse E, Lichtenauer-Kaligis EG, van Dissel JT, Ottenhoff TH. Genetic variations in the interleukin-12/interleukin-23 receptor (beta1) chain, and implications for IL-12 and IL-23 receptor structure and function. Immunogenetics. 2003;54:817–829. doi: 10.1007/s00251-002-0534-9. [DOI] [PubMed] [Google Scholar]
- Venter M, Myers TG, Wilson MA, Kindt TJ, Paweska JT, Burt FJ, Leman PA, Swanepoel R. Gene expression in mice infected with West Nile virus strains of different neurovirulence. Virology. 2005;342:119–140. doi: 10.1016/j.virol.2005.07.013. [DOI] [PubMed] [Google Scholar]
- Wang T, Anderson JF, Magnarelli LA, Wong SJ, Koski RA, Fikrig E. Immunization of mice against West Nile virus with recombinant envelope protein. J Immunol. 2001;167:5273–5277. doi: 10.4049/jimmunol.167.9.5273. [DOI] [PubMed] [Google Scholar]
- Wang T, Scully E, Yin Z, Kim JH, Wang S, Yan J, Mamula M, Anderson JF, Craft J, Fikrig E. IFN-gamma-producing gamma delta T cells help control murine West Nile virus infection. J Immunol. 2003a;171:2524–2531. doi: 10.4049/jimmunol.171.5.2524. [DOI] [PubMed] [Google Scholar]
- Wang Y, Lobigs M, Lee E, Mullbacher A. CD8+ T cells mediate recovery and immunopathology in West Nile virus encephalitis. J Virol. 2003b;77:13323–13334. doi: 10.1128/JVI.77.24.13323-13334.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T, Town T, Alexopoulou L, Anderson JF, Fikrig E, Flavell RA. Toll-like receptor 3 mediates West Nile virus entry into the brain causing lethal encephalitis. Nat Med. 2004;10:1366–1373. doi: 10.1038/nm1140. [DOI] [PubMed] [Google Scholar]
- Wang R, Town T, Gokarn V, Flavell RA, Chandawarkar RY. HSP70 enhances macrophage phagocytosis by interaction with lipid raft-associated TLR-7 and upregulating p38 MAPK and PI3K pathways. J Surg Res. 2006;136:58–69. doi: 10.1016/j.jss.2006.06.003. [DOI] [PubMed] [Google Scholar]
- Yamamoto M, Takeda K, Akira S. TIR domain-containing adaptors define the specificity of TLR signaling. Mol Immunol. 2004;40:861–868. doi: 10.1016/j.molimm.2003.10.006. [DOI] [PubMed] [Google Scholar]
- Zucchini N, Bessou G, Traub S, Robbins SH, Uematsu S, Akira S, Alexopoulou L, Dalod M. Cutting edge: overlapping functions of TLR7 and TLR9 for innate defense against a herpesvirus infection. J Immunol. 2008;180:5799–5803. doi: 10.4049/jimmunol.180.9.5799. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.