

Abstract
Background and purpose:
Nitroxyl (HNO) is emerging as an important regulator of vascular tone as it is potentially produced endogenously and dilates conduit and resistance arteries. This study investigates the contribution of endogenous HNO to endothelium-dependent relaxation and hyperpolarization in resistance arteries.
Experimental approach:
Rat and mouse mesenteric arteries were mounted in small vessel myographs for isometric force and smooth muscle membrane potential recording.
Key results:
Vasorelaxation to the HNO donor, Angeli's salt, was attenuated in both species by the soluble guanylate cyclase inhibitor (ODQ, 1H-[1,2,4]oxadiazolo[4,3-a]quinoxaline-1-one), the voltage-dependent K+ channel inhibitor, 4-aminopyridine (4-AP) and the HNO scavenger, l-cysteine. In mouse mesenteric arteries, nitric oxide (NO) synthase inhibition (with l-NAME, Nω-Nitro-L-arginine methyl ester) markedly attenuated acetylcholine (ACh)-mediated relaxation. Scavenging the uncharged form of NO (NO•) with hydroxocobalamin (HXC) or HNO with l-cysteine, or 4-AP decreased the sensitivity to ACh, and a combination of HXC and l-cysteine reduced ACh-mediated relaxation, as did l-NAME alone. ACh-induced hyperpolarizations were significantly attenuated by 4-AP alone and in combination with l-NAME. In rat mesenteric arteries, blocking the effects of endothelium-derived hyperpolarizing factor (EDHF) (charybdotoxin and apamin) decreased ACh-mediated relaxation 10-fold and unmasked a NO-dependent component, mediated equally by HNO and NO•, as HXC and l-cysteine in combination now abolished vasorelaxation to ACh. Furthermore, ACh-evoked hyperpolarizations, resistant to EDHF inhibition, were virtually abolished by 4-AP.
Conclusions and implications:
The factors contributing to vasorelaxation in mouse and rat mesenteric arteries are NO• = HNO > EDHF and EDHF > HNO = NO• respectively. This study identified HNO as an endothelium-derived relaxing and hyperpolarizing factor in resistance vessels.
British Journal of Pharmacology (2009) 157, 540–550; doi:10.1111/j.1476-5381.2009.00150.x; published online 26 March 2009
This article is commented on by Martin, pp. 537–539 of this issue and is part of a themed section on Endothelium in Pharmacology. For a list of all articles in this section see the end of this paper, or visit: http://www3.interscience.wiley.com/journal/121548564/issueyear?year=2009
Keywords: nitroxyl, vasorelaxation, endothelial factors, nitric oxide, redox signalling, endothelium-derived relaxing factor
Introduction
The endogenous release of nitric oxide (NO) plays an integral role in the regulation of vascular tone. NO, produced by NO synthase (NOS) from the conversion of L-arginine to L-citrulline, activates the haem protein, soluble guanylate cyclase (sGC), resulting in an accumulation of cyclic guanosine monophosphate (cGMP) and subsequent vasorelaxation (Moncada et al., 1991). Traditionally, it has been the uncharged form of NO (NO•), which was thought to mediate these effects. Reduced congeners of NO such as nitroxyl (HNO, Irvine et al., 2008), the one-electron reduced sibling of NO may be of physiological importance as there is evidence to suggest that it is formed endogenously (Pufahl et al., 1995; Schmidt et al., 1996; Hobbs, 1997; Rusche et al., 1998; Adak et al., 2000) and it can mediate vasorelaxation (Wanstall et al., 2001; Irvine et al., 2003).
Indeed, the endogenous production of HNO may occur via a number of distinct pathways. Biochemical studies have shown that HNO can be formed by NOS itself (Hobbs et al., 1994; Schmidt et al., 1996; Rusche et al., 1998), particularly in the absence of the NOS co-factor tetrahydrobiopterin (BH4) or after oxidation of the NOS intermediates Nω-hydroxy-L-arginine (Fukuto et al., 1992b; Pufahl et al., 1995) and hydroxylamine (Donzelli et al., 2008). Moreover HNO can be produced from non-NOS sources, including the reduction of NO• by mitochondrial cytochrome c (Sharpe and Cooper, 1998), xanthine oxidase (Saleem and Ohshima, 2004) and haemoglobin (Gow and Stamler, 1998). Similarly, the reaction of S-nitrosothiols with other thiol species can yield HNO at physiological pH (Arnelle and Stamler, 1995; Wong et al., 1998).
HNO generated from Angeli's salt (AS; sodium trioxodinitrate) serves as a vasorelaxant in both large conduit (Fukuto et al., 1992a; Ellis et al., 2000; Wanstall et al., 2001; Irvine et al., 2007) and small resistance arteries (Irvine et al., 2003; Favaloro and Kemp-Harper, 2007), and like NO•, its response is mediated via the stimulation of sGC (Wanstall et al., 2001; Irvine et al., 2003; Favaloro and Kemp-Harper, 2007; Irvine et al., 2007) and the resulting accumulation of cGMP (Fukuto et al., 1992a; Irvine et al., 2007). However, in contrast to NO•, which has been shown to activate calcium-activated K+ channels (KCa) in mesenteric arteries (Mistry and Garland, 1998; Sampson et al., 2001), we have previously demonstrated that HNO instead activates voltage-dependent K+ (Kv) channels in the same vascular bed (Irvine et al., 2003).
The potential for HNO to be produced endogenously and its ability to mediate vasorelaxation suggests that it may serve as an endothelium-derived relaxing factor (EDRF), accounting at least in part for the responses previously attributed to NO•. Indeed it has been suggested that in conduit arteries, the endogenous production of HNO contributes at least in part to the endothelium-dependent relaxation previously attributed to NO• (Ellis et al., 2000; Wanstall et al., 2001). Yet the role of HNO in the resistance vasculature remains unknown. Furthermore, given that HNO can be derived from NOS-independent sources (Arnelle and Stamler, 1995; Sharpe and Cooper, 1998; Wong et al., 1998), exert sGC-independent actions (Irvine et al., 2003) and activate KV channels to hyperpolarize vascular smooth muscle (J. L. Favaloro and B. K. Kemp-Harper, unpubl. obs.), the additional possibility that HNO may also serve as an endothelium-derived hyperpolarizing factor (EDHF), remains to be explored.
Thus, the aim for the current study was to investigate the possible contribution of HNO to endothelium-dependent relaxation and hyperpolarization in resistance arteries from two different species: the mouse small mesenteric artery (MMA) where, under conditions of low resting tone, NO-mediated vasorelaxation is prominent and the rat small mesenteric artery (RMA) where EDHF and NO are the major EDRFs. This study identifies HNO as an endothelium-derived relaxing and hyperpolarizing factor in resistance vessels.
Methods
All animal care and experimental procedures were approved by the Animal Ethics Committee at RMIT University or the Pharmacology Animal Ethics Committee, Monash University, Australia and were in accordance with the NH&MRC Australian code of practice for the care and use of animals for scientific purposes and with the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH Publication No. 85-23, revised 1996).
Isolation and study of rat mesenteric arteries
Male Wistar Kyoto rats (16–17 weeks; 359 ± 4 g, n = 38) were killed by means of stunning and exsanguination and the mesenteric bed removed. Small mesenteric arteries (2 mm segments of second order branch of the superior mesenteric artery) were dissected free of fat and connective tissue and mounted in Mulvany-style isometric myographs (Danish Myo Technology A/S, Inc., Skejbyparken, Denmark) (Kemp and Cocks, 1997). Vessels were maintained at 37°C in physiological Krebs solution containing 114 mmol·L−1 NaCl, 4.7 mmol·L−1 KCl, 0.8 mmol·L−1 KH2PO4, 1.2 mmol·L−1 MgCl2, 11 mmol·L−1 D-glucose, 25 mmol·L−1 NaHCO3, 2.5 mmol·L−1 CaCl2 and 0.026 mmol·L−1 EDTA that was bubbled with carbogen (95% O2, 5% CO2) to maintain the buffer at pH 7.4. Data were captured through the use of the CVMS data acquisition system (World Precision Instruments, USA). After a 30 min equilibration period, vessel diameters were normalized to an equivalent transmural pressure of 100 mmHg (D100) (McPherson, 1992).
Isolation and study of mouse mesenteric arteries
The mesenteric bed was removed from C57BL/6J mice (12–16 weeks, 32 ± 3 g, n = 42) that had been killed by cervical dislocation. Small mesenteric arteries (2 mm segments of second order branch of the superior mesenteric artery) were dissected free of fat and connective tissue and mounted in Mulvany-style isometric myographs. Vessels were maintained at 37°C in physiological Krebs' buffer that was bubbled with a carbogen (95% O2, 5% CO2) to maintain the buffer at pH 7.4. Data were captured through the use of the Myodaq data acquisition system. After a 30 min equilibration period, vessel tension was increased to 1 mN.
Vasorelaxation experiments
A further 30 min after resting tension was established, rat and mouse small mesenteric arteries were maximally contracted with a K+-depolarizing solution (KPSS: 123 mmol·L−1 KCl, 1.17 mmol·L−1 MgSO4, 2.37 mmol·L−1 KH2PO4, 2.5 mmol·L−1 CaCl2, 11.1 mmol·L−1 D-glucose and 0.026 mmol·L−1 EDTA). Responses to vasodilators were then examined in arteries pre-contracted to ∼50% KPSS with cirazoline (10–100 nmol·L−1) or in some cases 30 mmol·L−1 K+, for both rat and mouse. Cumulative concentration–response curves to either, the HNO donor AS (0.001–10 µmol·L−1), the NO• donor diethylamine NONOate (DEA/NO; 0.001–30 µmol·L−1), or acetylcholine (ACh; 0.001–30 µmol·L−1) were constructed. Responses to AS and DEA/NO were obtained in the absence and presence of the HNO scavenger l-cysteine (3 mmol·L−1, 3 min) or the NO• scavenger hydroxocobalamin (HXC; 100 µmol·L−1, 15 min). In subsequent experiments in the mouse, vasorelaxation responses to AS were always conducted in the presence of HXC (100 µmol·L−1). Responses to AS and DEA/NO were also examined in the absence or presence of either ODQ (1H-[1,2,4]oxadiazolo[4,3-a]quinoxaline-1-one; 3 or 10 µmol·L−1, 30 min) or 4-aminopyridine (4-AP; 1 mmol·L−1, 30 min). Vasorelaxation responses to ACh were always conducted in the presence of indomethacin (10 µmol·L−1, 30 min) and in the presence or absence of differing combinations of (i) l-cysteine (3 mmol·L−1, 3 min); (ii) HXC (100 µmol·L−1, 15 min); (iii) l-NAME (Nω-Nitro-L-arginine methyl ester; 100 µmol·L−1, 30 min), (iv) ODQ (10 µmol·L−1, 30 min); (v) charybdotoxin (ChTx; 100 nmol·L−1, 30 min); (vi) apamin (100 nmol·L−1, 30 min); (vii) K+ (30 mmol·L−1, 30 min); and (viii) 4-AP (1 mmol·L−1, 30 min). Maximal relaxation was ensured by using isoprenaline (1 µmol·L−1) at the conclusion of each concentration–response curve.
Electrophysiological experiments
Mouse and rat mesenteric arteries were isolated and mounted in myographs, and the same normalization procedures were carried out as described above. Arteries were continuously superfused with warmed, oxygenated physiological Krebs' buffer (as above). Smooth muscle membrane potential was recorded simultaneously with tension by using conventional intracellular glass microelectrodes filled with 1 mol·L−1 KCl and having resistances of 60–100 MΩ. Data were recorded by using a Digidata-Axoscope data acquisition system (Molecular Devices). Arteries were submaximally constricted (∼50% KPSS) and depolarized with cirazoline. The endothelium was stimulated with increasing concentrations of ACh (MMA: 0.1–30 µmol·L−1; RMA: 3–300 nmol·L−1), each applied for 1 min with washout between each dose to allow membrane potential and tension to return to pre-stimulatory levels as described previously (Tare et al., 2000). All experiments were conducted in the presence of 10 µmol·L−1 indomethacin.
Data analysis and statistical procedures
Relaxation responses are expressed as a percentage reversal of the cirazoline pre-contraction. Individual relaxation curves were fitted to a sigmoidal logistic equation (GraphPad Prism 4.0) and pEC50 values (concentration of agonist causing a 50% relaxation) calculated and expressed as −Log mol·L−1. Statistical comparisons between the experimental groups' mean pEC50 and maximum relaxation (Rmax) values were made by using a one-way anova with Bonferroni's post hoc comparisons (Graphpad Prism 4.0). Where pEC50 values could not be obtained, concentration–response curves were compared by means of a two-way anova (GraphPad Prism 5.0). For the electrophysiological data, hyperpolarization responses were expressed as a percentage of the maximal control hyperpolarization to 30 µmol·L−1 and 300 nmol·L−1 ACh for MMA and RMA respectively. Statistical significance was tested with a two-way anova with Bonferroni's post hoc comparisons (Graphpad Prism 5.0). Unless otherwise stated, data represent the mean ± SEM (error bars on graphs; n = number of animals), and statistical significance was accepted at a level of P < 0.05. Receptor nomenclature conforms to the guidelines described by Alexander et al., 2008.
Drugs
Drugs and their sources were AS (sodium trioxodinitrate), ODQ (Sapphire Bioscience, Crows Nest, NSW, Australia), diethylamine NONOate [Diethylammonium (Z)-1-(N,N-diethylamino)diazen-1-ium-1,2-diolate], cirazoline hydrochloride [2-[(2-Cyclopropylphenoxy)methyl]-4,5-dihydro-1H-imidazole], l-cysteine hydrochloride, ACh, HXC, indomethacin, l-NAME, ChTx, apamin, 4-AP (Sigma-Aldrich, St. Louis, MO, USA). Stock solutions of AS and DEA/NO (10 mmol·L−1) were constituted in 0.01 mol·L−1 NaOH, as were all subsequent dilutions. Stock solutions of ODQ (10 mmol·L−1) were made up in absolute ethanol (EtOH). Stock solutions of ChTx (10 µmol·L−1) and apamin (100 µmol·L−1) were made up in 0.1% bovine serum albumin in 0.9% saline. Stock solutions of indomethacin (100 mmol·L−1) were made up in 0.1 mol·L−1 Na2CO3 solution. All subsequent dilutions of stock solutions were in distilled water. All other drugs were made up in distilled water, and all dilutions were prepared fresh daily.
Results
Hydroxocobalamin and l -cysteine distinguish between HNO and NO•
The NO• donor DEA/NO and the HNO donor AS caused concentration-dependent vasorelaxation of both MMA and rat RMA (Table 1). The NO• scavenger, HXC (100 µmol·L−1) decreased the sensitivity to DEA/NO in both MMA and RMA, by threefold (P < 0.05) and fivefold (P < 0.05), respectively without altering maximum relaxation (Rmax). In contrast, HXC had no significant effect on AS-mediated relaxation in RMA. In MMA, treatment with HXC resulted in a significant sevenfold (P < 0.05) rightward shift of the concentration–response curve to AS, indicative of extracellular oxidation of HNO to NO•. As a consequence, all further experiments with AS in MMA were conducted in the presence of HXC (100 µmol·L−1). The HNO scavenger, l-cysteine (3 mmol·L−1) failed to alter DEA/NO-mediated relaxation in mesenteric arteries from either species. However, in both MMA and RMA, l-cysteine decreased the potency of AS up to 40-fold (P < 0.01) and eightfold (P < 0.01) respectively (Table 1).
Table 1.
Effect of modulators on AS- and DEA/NO-induced vasorelaxation in MMA and RMA
| Nitrovasodilator |
DEA/NO |
DEA/NO |
AS |
AS |
||||
|---|---|---|---|---|---|---|---|---|
| Vessel |
MMA |
RMA |
MMA |
RMA |
||||
| Treatment | pEC50 | Rmax | pEC50 | Rmax | pEC50 | Rmax | pEC50 | Rmax |
| Control | 6.8 ± 0.1 | 99 ± 1 | 7.8 ± 0.1 | 98 ± 1 | 7.7 ± 0.2 | 94 ± 3 | 7.2 ± 0.2 | 96 ± 1 |
| HXC (100 µmol·L−1) | 6.3 ± 0.2* | 97 ± 1 | 7.1 ± 0.1* | 99 ± 1 | 6.9 ± 0.2* | 98 ± 1 | 7.5 ± 0.1 | 92 ± 3 |
| L-cysteine (3 mmol·L−1) | 6.9 ± 0.2 | 96 ± 2 | 7.7 ± 0.2 | 97 ± 1 | NCφ(∼6) | 59 ± 13φ# | 6.4 ± 0.1** | 92 ± 2 |
| 4-AP (1 mmol·L−1) | 6.8 ± 0.1 | 99 ± 1 | 7.8 ± 0.1 | 93 ± 2 | 5.9 ± 0.2†φ | 88 ± 8φ | 6.5 ± 0.3*** | 91 ± 3 |
| ODQ (10 µmol·L−1) | 6.1 ± 0.1* | 97 ± 1 | 6.3 ± 0.3** | 93 ± 2 | NCφ | 15±5ΨΨΨφ | NC(∼6) | 48 ± 3## |
| 4-AP (1 mmol·L−1) & ODQ (10 µmol·L−1) | – | – | – | – | NCφ | 10±6ΨΨΨφ | NC | 2 ± 1## |
pEC5O values are expressed as –Log M and Rmax values as percentage reversal of the level of pre-contraction in response to 10 µmol·L−1 of the nitrovasodilator. Values are given as mean ± SEM. n = 4–7 per group.
NC = pEC50 value could not be calculated as a plateau was not obtained or vasorelaxation was less than 20% reversal of the level of pre-contraction. In some instances pEC50 values have been approximated (∼pEC50).
4-AP, 4-aminopyridine; AS, Angeli's salt; HXC, hydroxocobalamin; MMA, mouse mesenteric arteries; ODQ, 1H-[1,2,4]oxadiazolo[4,3-a]quinoxaline-1-one; RMA, rat mesenteric arteries.
Responses to AS obtained in the presence of HXC (100 µmol·L−1).
P < 0.05,
P < 0.01,
P < 0.001 for pEC50 versus untreated control (one-way anova).
P < 0.05,
P < 0.01 for response at 10 µmol·L−1 versus untreated control (one-way anova).
P < 0.05 for pEC50 versus HXC (one-way anova).
P < 0.001 for response at 10 µmol·L−1 versus HXC (one-way anova).
4-AP discriminates between HNO and NO•
In agreement with our previous studies (Irvine et al., 2003), vasorelaxation to AS in RMA was significantly attenuated in the presence of the sGC inhibitor, ODQ (10 µmol·L−1) and the Kv channel blocker, 4-AP (1 mmol·L−1) and abolished when ODQ and 4-AP were combined (Table 1). A role for sGC and Kv channels in AS-mediated vasorelaxation was also evident in MMA, with 4-AP decreasing the sensitivity to AS 10-fold (P < 0.05) and the response virtually abolished in the presence of ODQ (P < 0.001, Table 1). In MMA, 4-AP in combination with ODQ had no further inhibitory effect on the response to AS as compared with ODQ alone.
In contrast, vasorelaxation to DEA/NO in MMA and RMA was unchanged in the presence of 4-AP (1 mmol·L−1; Table 1). Furthermore, relaxation responses to DEA/NO were more resistant to the inhibitory effects of ODQ as compared with AS with 10 µmol·L−1 ODQ causing a fivefold (P < 0.05, MMA) and 30-fold (P < 0.01, RMA) decrease in sensitivity yet not changing the maximum response to DEA/NO (Table 1).
Differential contribution of NO and EDHF to endothelium-dependent vasorelaxation in mouse and rat mesenteric arteries
Although vasorelaxation responses to ACh in MMA and RMA were unaffected by the cyclooxygenase inhibitor indomethacin (10 µmol·L−1; data not shown), indomethacin was included in all subsequent experiments to block this pathway. Under the resting tension conditions utilized in the present study for MMA, NO served as the primary EDRF. Thus inhibition of EDHF with the KCa inhibitors, ChTx (100 nmol·L−1) and apamin (100 nmol·L−1) in combination, did not alter the concentration–response curve to ACh (pEC50 = 6.3 ± 0.3, Rmax = 86 ± 4%, n = 4; Figure 1A) when compared with control (pEC50 = 6.7 ± 0.2, Rmax = 86 ± 4%, n = 6). However, ACh-mediated relaxation was markedly attenuated by the combination of ODQ (10 µmol·L−1) and the NOS inhibitor l-NAME (100 µmol·L−1; Rmax = 44 ± 11%, n = 5, P < 0.01). Combining l-NAME and ODQ with either, ChTx and apamin (Rmax = 47 ± 3%, n = 4) or 30 mmol·L−1 K+ (Rmax = 28 ± 16%, n = 4) to block all K+ channels did not attenuate the relaxation any further.
Figure 1.
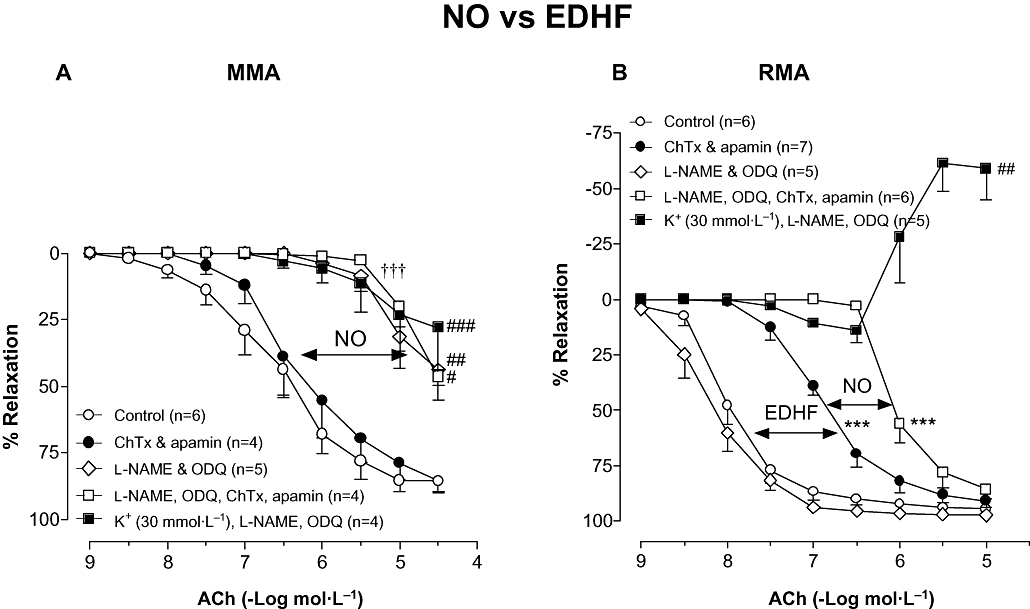
Concentration–response curves to ACh in (A) MMA and (B) RMA in the absence (Control) or presence of ChTx (100 nmol·L−1) & apamin (100 nmol·L−1) in combination, l-NAME (100 µmol·L−1) & ODQ (100 µmol·L−1) in combination, l-NAME, ODQ, ChTx & apamin in combination and K+ (30 mmol·L−1), l-NAME & ODQ in combination. Indomethacin (10 µmol·L−1) was present throughout. Values are expressed as % reversal of pre-contraction and given as mean ± SEM, where n = number of animals. ***P < 0.001 for pEC50 versus untreated control (one-way anova), †††P < 0.001 versus untreated control (two-way anova), #P < 0.05, ##P < 0.01, ###P < 0.001 for response at 10 µmol·L−1 or 30 µmol·L−1 versus untreated control (one-way anova). ACh, acetylcholine; ChTx, charybdotoxin; EDHF, endothelium-derived hyperpolarizing factor; l-NAME, Nω-Nitro-L-arginine methyl ester; MMA, mouse mesenteric arteries; NO, nitric oxide; ODQ, 1H-[1,2,4]oxadiazolo[4,3-a]quinoxaline-1-one; RMA, rat mesenteric arteries.
In contrast, in RMA a contribution of NO to endothelium-dependent relaxation was only observed once the effects of EDHF had been blocked. Thus l-NAME alone (data not shown) or l-NAME and ODQ together had no effect on ACh-mediated vasorelaxation (pEC50 = 8.1 ± 0.2, Rmax = 97 ± 1%, n = 5) when compared with control (pEC50 = 8.0 ± 0.1, Rmax = 95 ± 1%, n = 6; Figure 1B) while ChTx and apamin combined reduced the sensitivity to ACh 13-fold (pEC50 = 6.9 ± 0.1, P < 0.001, Rmax = 97 ± 1%, n = 7). However, in the presence of ChTx and apamin, l-NAME and ODQ resulted in a further eightfold (P < 0.001, n = 6) rightward shift of the concentration–response curve to ACh. Raising the extracellular K+ to 30 mmol·L−1, in the presence of l-NAME and ODQ, virtually abolished vasorelaxation and unmasked a concentration-dependent contraction to ACh at concentrations >0.3 µmol·L−1 (10 µmol·L−1 ACh = +59 ± 14% level pre-contraction, n = 5, P < 0.01).
NO• contributes to endothelium-dependent vasorelaxation in resistance arteries
Having identified a NO-mediated component to the relaxation evoked by ACh in both MMA and RMA we next sought to identify the redox form of NO responsible. In MMA, the NO• scavenger HXC (100 µmol·L−1) reduced the sensitivity to ACh up to 15-fold (P < 0.01) without suppressing the maximum response (Rmax = 81 ± 5%, n = 9; Figure 2A) when compared with control (pEC50 = 6.9 ± 0.3, Rmax = 92 ± 2%, n = 6). In the presence of HXC, l-NAME further decreased the sensitivity to ACh (P < 0.001, n = 6) and attenuated the maximum response (Rmax = 50 ± 7%, n = 6, P < 0.001; Figure 2A).
Figure 2.
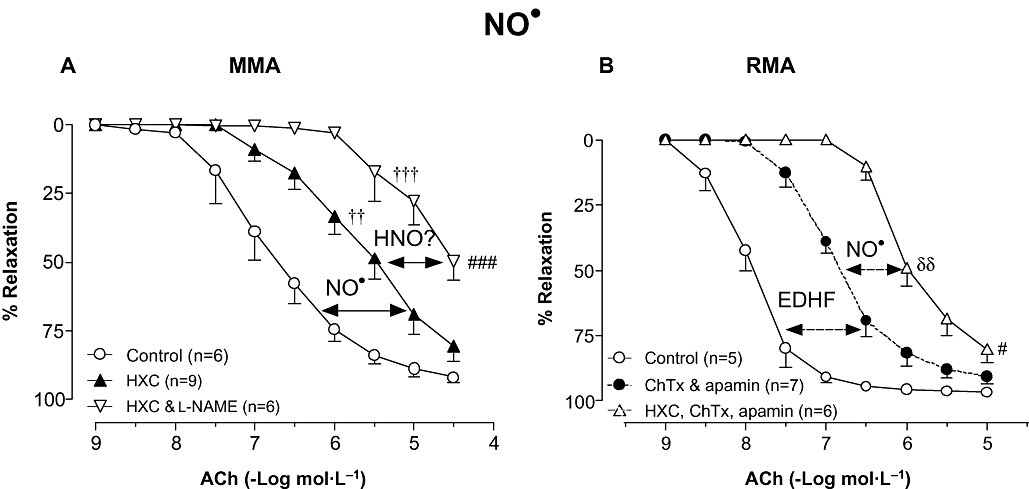
Concentration–response curves to ACh in either (A) MMA in the absence (Control) or presence of HXC (100 µmol·L−1), and l-NAME (100 µmol·L−1) & HXC in combination or (B) RMA in the absence (Control) or presence of ChTx (100 nmol·L−1) & apamin (100 nmol·L−1) in combination (reproduced from Figure 1B for comparison), and ChTx, apamin & HXC in combination. Indomethacin (10 µmol·L−1) was present throughout. Values are expressed as % reversal of pre-contraction and given as mean ± SEM, where n = number of animals. δδP < 0.01 for pEC50 versus ChTx & apamin treatment (one-way anova), ††P < 0.01, †††P < 0.001 versus untreated control (two-way anova), #P < 0.05, ###P < 0.001 for response at 10 µmol·L−1 or 30 µmol·L−1 versus untreated control (one-way anova). ACh, acetylcholine; ChTx, charybdotoxin; HNO, nitroxyl; HXC, hydroxocobalamin; l-NAME, Nω-Nitro-L-arginine methyl ester; MMA, mouse mesenteric arteries; NO, nitric oxide; RMA, rat mesenteric arteries.
In contrast to the findings in MMA, HXC had no effect on ACh-induced relaxation in RMA (pEC50 = 8.1 ± 0.1, Rmax = 97 ± 1%, n = 5). However, following the inhibition of EDHF with ChTx and apamin, a contribution of NO• to endothelium-dependent vasorelaxation was apparent with HXC shifting the concentration–response curve to ACh to the right by eightfold (pEC50 = 6.1 ± 0.1, P < 0.01; Figure 2B) when compared with ChTx and apamin alone.
HNO contributes to endothelium-dependent vasorelaxation and hyperpolarization in resistance arteries
In MMA, inhibiting the effects of HNO with either l-cysteine (3 mmol·L−1) or 4-AP (1 mmol·L−1) impaired the response to ACh to a similar extent such that its potency was decreased up to 25-fold (P < 0.001) and the maximum response tended to be reduced (l-cysteine: Rmax = 69±9%, n = 8; 4-AP: Rmax = 87 ± 5%, n = 5; Figure 3A), when compared with control (Rmax = 98 ± 1%, n = 6) but not significantly. Inhibition of both HNO and NO• through the use of HXC and l-cysteine in combination markedly impaired vasorelaxation to ACh (Rmax = 21 ± 10%, n = 4, P < 0.001).
Figure 3.
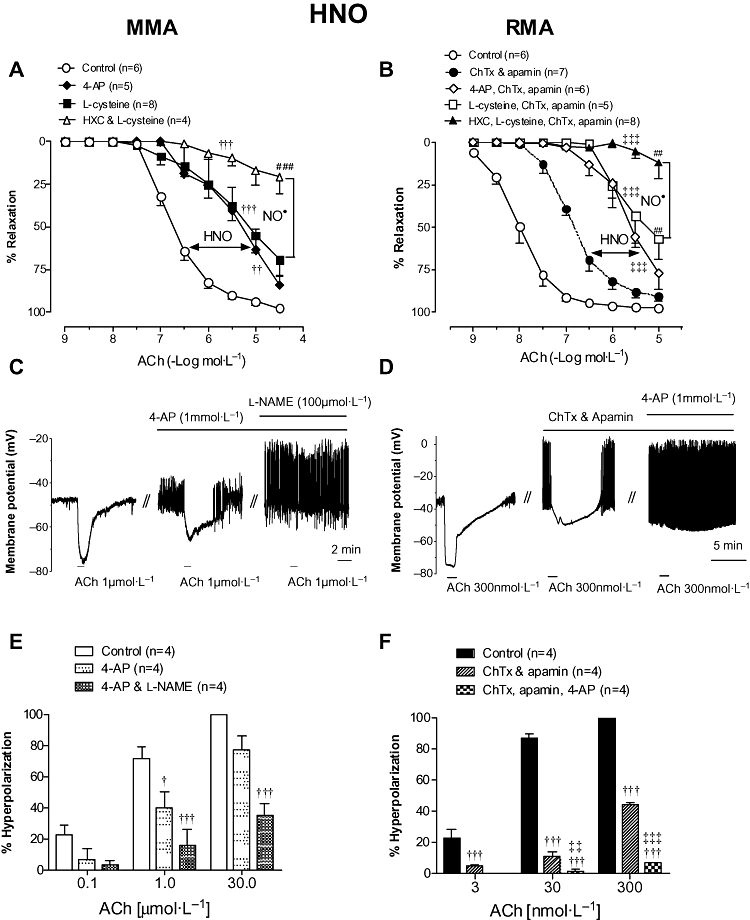
Vasorelaxation responses to ACh in either (A) MMA in the absence (Control) or presence of 4-AP (1 mmol·L−1), l-cysteine (3 mmol·L−1) and l-cysteine & HXC (100 µmol·L−1) in combination, or (B) RMA in the absence (Control; reproduced from Figure 1B for comparison) or presence of ChTx (100 nmol·L−1) & apamin (100 nmol·L−1) alone (reproduced from Figure 1B for comparison), or combined with 4-AP (1 mmol·L−1), l-cysteine (3 mmol·L−1) or l-cysteine & HXC (100 µmol·L−1). Smooth muscle membrane potential recordings illustrating ACh-evoked hyperpolarization in MMA (C) and RMA (D) following depolarization with cirazoline. Hyperpolarizations were recorded in the absence and presence of 4-AP alone (C, E) or combined with l-NAME (C, E); or ChTx and apamin alone (D, F) or combined with 4-AP (D, F). In (C) and (D) // indicates a break in a continuous recording made in the same cell. Indomethacin (10 µmol·L−1) was present throughout. Values are expressed as (A, B) % reversal of contraction or (E, F) % of maximal control hyperpolarization and given as mean ± SEM, where n = number of animals. †P < 0.05, ††P < 0.01, †††P < 0.001 versus untreated control (two-way anova), ‡‡P < 0.01, ‡‡‡P < 0.001 versus ChTx & apamin treatment (two-way anova), ##P < 0.01, ###P < 0.001, for response at 10 µmol·L−1 or 30 µmol·L−1 versus untreated control (one-way anova). 4-AP, 4-aminopyridine; ACh, acetylcholine; ChTx, charybdotoxin; HNO, nitroxyl; HXC, hydroxocobalamin; l-NAME, Nω-Nitro-L-arginine methyl ester; MMA, mouse mesenteric arteries; NO, nitric oxide; RMA, rat mesenteric arteries.
Having identified a contribution of HNO to ACh-mediated vasorelaxation in MMA, electrophysiological recordings were made to determine if HNO also contributes to ACh-mediated smooth muscle cell hyperpolarization. In the presence of indomethacin, the resting membrane potential (RMP) of MMA smooth muscle cells was −60 ± 2 mV (n = 4) and after depolarization with cirazoline (by 17 ± 2 mV, n = 4), ACh produced a concentration-dependent hyperpolarization (Figure 3C and E) with a maximal response at 30 µmol·L−1 ACh (19 ± 4mV, n = 4). The addition of either 4-AP (1 mmol·L−1) or 4-AP and l-NAME (100 µmol·L−1) had no significant effect on either RMP or the extent of the depolarization to cirazoline (data not shown). In the presence of 4-AP, the hyperpolarization to 1 µmol·L−1 ACh was reduced by ∼45% (P < 0.05, n = 4; Figure 3C and E) and was further attenuated in the presence of 4-AP and l-NAME in combination (P < 0.001, n = 4). Similarly, 4-AP and l-NAME together significantly reduced the hyperpolarization to 30 µmol·L−1 ACh (36 ± 7% of initial response, P < 0.001, n = 4; Figure 3E).
In RMA, neither l-cysteine (pEC50 = 7.6 ± 0.1, Rmax = 94 ± 2%, n = 5) nor 4-AP (pEC50 = 7.5 ± 0.1, Rmax = 95 ± 1%, n = 4) alone altered ACh-mediated vasorelaxation, yet in the absence of EDHF a contribution of HNO to endothelium-dependent relaxation was evident. Thus, in the presence of ChTx and apamin, l-cysteine and 4-AP reduced the sensitivity to ACh (P < 0.001, Figure 3B), to a similar degree as observed with the MMA. l-cysteine also reduced the response to 10 µmol·L−1 ACh (Rmax = 57 ± 12%, n = 5, P < 0.01) yet 4-AP had no significant effect (Rmax = 77 ± 9%, n = 6; Figure 3D). Applied together, ChTx, apamin, HXC and l-cysteine virtually abolished ACh-mediated relaxation (Rmax = 12 ± 9%, n = 8, P < 0.001).
Following depolarization of RMA smooth muscle cells with cirazoline (RMP: −60 ± 1 mV; cirazoline depolarization: 23 ± 1 mV, n = 4), ACh evoked hyperpolarization (Figure 3D and F), which was maximal at 300 nmol·L−1 (44 ± 0.4 mV, n = 4). Blocking the effects of EDHF with ChTx and apamin markedly reduced (ACh 30 nmol·L−1: 11.3 ± 2.8 %; ACh 300 nmol·L−1: 44.6 ± 1.6% of initial response, P < 0.001, n = 4), but did not abolish, ACh-mediated hyperpolarization (Figure 3D and F). The ChTx and apamin-resistant hyperpolarization was further attenuated by 4-AP (P < 0.001; Figure 3D and F) such that at 30 and 300 nmol·L−1 ACh there was a slowing of the action potential frequency without breakthrough hyperpolarization, probably due to complex conductance changes (Figure 3D).
HNO derived from NOS-dependent and independent sources
The NOS inhibitor, l-NAME was used to identify the source of HNO and NO• in MMA and RMA. In MMA, l-NAME (100 µmol·L−1) markedly attenuated vasorelaxation to ACh (Rmax = 42 ± 12%, P < 0.01, n = 6; Figure 4A). Scavenging NO• (with HXC) or HNO (with l-cysteine), together with NOS inhibition did not impair the response any further.
Figure 4.
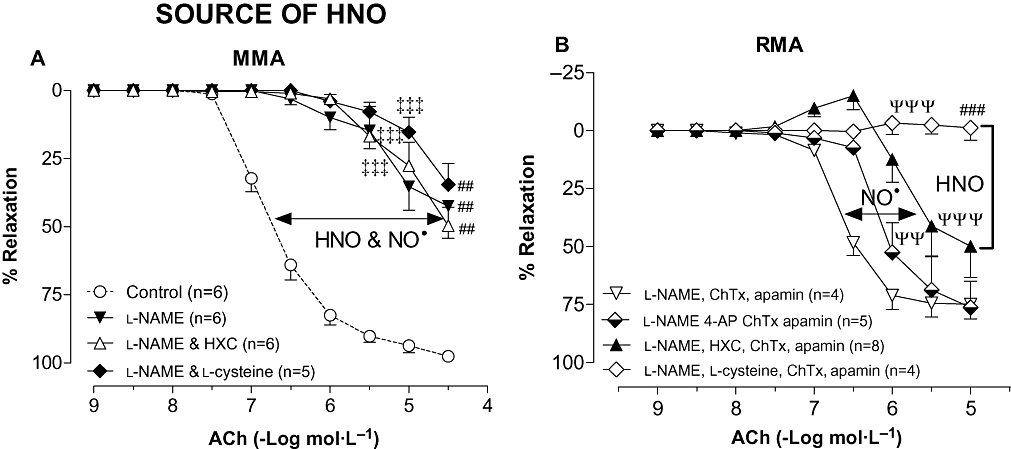
Concentration–response curves to ACh in either (A) MMA in the absence (Control; reproduced from Figure 3A for comparison) or presence of l-NAME (100 µmol·L−1), l-NAME & HXC (100 µmol·L−1), and l-NAME & l-cysteine (3 mmol·L−1), or (B) RMA in the presence of l-NAME (100 µmol·L−1), ChTx (100 nmol·L−1) & apamin (100 nmol·L−1) combined with either: 4-AP (1 mmol·L−1), HXC (100 µmol·L−1) or l-cysteine (3 mmol·L−1). Indomethacin (10 µmol·L−1) was present throughout. Values are expressed as % reversal of contraction and given as mean ± SEM, where n = number of animals. (A) ‡‡‡P < 0.001 versus control (two-way anova), ##P < 0.01 for response at 30 µmol·L−1 versus Control (one-way anova). (B) ΨΨP < 0.01, ΨΨΨP < 0.001 versus l-NAME, ChTx & apamin treatment (two-way anova), ###P < 0.001, for response at 10 µmol·L−1 versus l-NAME, ChTx & apamin treatment (one-way anova). 4-AP, 4-aminopyridine; ACh, acetylcholine; ChTx, charybdotoxin; HNO, nitroxyl; HXC, hydroxocobalamin; l-NAME, Nω-Nitro-L-arginine methyl ester; MMA, mouse mesenteric arteries; NO, nitric oxide; RMA, rat mesenteric arteries.
In RMA, in the presence of l-NAME, ChTx and apamin, the subsequent addition of HXC (100 µmol·L−1) reduced the sensitivity to ACh further (P < 0.001) and tended to attenuate the maximal response (Rmax = 58 ± 12%, n = 8) when compared with ChTx, apamin and l-NAME alone (Figure 4B). A NOS-independent source of HNO also appeared to contribute to endothelium-dependent relaxation such that in the presence of l-NAME, ChTx and apamin, 4-AP (1 mmol·L−1) shifted the concentration response curve to ACh sixfold to the right (P < 0.01, n = 5) while l-cysteine (3 mmol·L−1) completely abolished ACh-mediated relaxation (Rmax = −1 ± 5%, n = 4, P < 0.01; Figure 4B).
Discussion and conclusions
This study has provided functional pharmacological and electrophysiological evidence consistent with the concept that HNO is produced endogenously and serves as an endothelium-derived relaxing and hyperpolarizing factor in both mouse and rat isolated small mesenteric resistance-like arteries. Under the experimental conditions used in this study, the predominant endothelium-derived vasodilators in MMA and RMA are NO and EDHF, respectively, with a contribution of NO in RMA evident following loss of EDHF. Interestingly in both MMA and RMA, NO• and HNO contribute equally to the NO-mediated component of endothelium-dependent relaxation. Furthermore, HNO appears to be derived wholly from NOS in MMA, yet in the RMA it may in addition also be released from a non-NOS-derived source, possibly from S-nitrosothiol stores.
To distinguish between the redox congeners of NO that may contribute to endothelium-dependent relaxation, we employed the well-established HNO scavenger, l-cysteine (at mmol·L−1 concentrations, Pino and Feelisch, 1994; Ellis et al., 2000; Wanstall et al., 2001; Irvine et al., 2003; Irvine et al., 2007) and the NO• scavenger, HXC (Li and Rand, 1993; Wanstall et al., 2005). The specificity of these inhibitors was confirmed in RMA, where l-cysteine attenuated the response to the HNO donor AS but not the NO• donor DEA/NO, and conversely HXC attenuated DEA/NO but not AS. Although extracellular oxidation of HNO to NO• was limited via the inclusion of the Cu2+ chelator EDTA, in MMA, HXC decreased the potency of AS. These findings suggest that the MMA may have a different oxidative environment as compared with RMA, resulting in some extracellular conversion of HNO to NO• (Murphy and Sies, 1991; Nelli et al., 2000). To prevent this, all subsequent experiments in the MMA with AS were performed in the presence of HXC.
Previous studies with RMA from our laboratory suggested that AS-induced vasorelaxation was mediated via the activation of sGC/cGMP and KV channels (Irvine et al., 2003). We have confirmed those findings in the present study and also demonstrated for the first time that this mechanism of action is not species-specific as the sGC inhibitor, ODQ and the KV channel inhibitor, 4-AP also reduced AS-mediated relaxation in the MMA. Whether HNO activation of Kv channels is direct or cGMP-dependent remains to be elucidated. Interestingly, in both species, DEA/NO was more resistant to the inhibitory effects of ODQ as compared with AS, a finding we have observed previously (Irvine et al., 2007). Thus DEA/NO may activate cGMP-independent mechanisms and indeed, in RMA, NO• mediates vasorelaxation and smooth muscle hyperpolarization in part by activation of BKCa channels (Mistry and Garland, 1998; Sampson et al., 2001). Importantly however, DEA/NO-induced relaxation was unaffected by KV channel blockade (with 4-AP), thus suggesting a different mechanism of action for HNO and NO•. These findings also highlight that 4-AP can serve as an additional tool to distinguish between HNO and NO•.
Having demonstrated clear differences between the vasorelaxant actions of HNO and NO•, and shown that we can distinguish between these redox siblings using known pharmacological tools, we then sought to determine the relative contribution of each to endothelium-dependent relaxation in small resistance-like arteries. Both MMA and RMA were utilized in order to elucidate the possible contribution of HNO to endothelium-dependent relaxation in vessels where NO and EDHF predominate respectively. Although previous studies have reported a contribution of EDHF (ChTx- and apamin-sensitive component) to endothelium-dependent relaxation in MMA (Chataigneau et al., 1999; Brandes et al., 2000; Ding et al., 2000; Fitzgerald et al., 2007), we did not observe a role for EDHF under the level of resting tension employed in this study. We have previously found that under low levels of resting tension (1 mN), as utilized in the present study, NO predominates, whereas at higher levels of resting tone (3 mN and above) such as that generated following normalization to D100, a major contribution of EDHF is observed (K. L. Andrews, unpubl. obs.). Furthermore, there is evidence for contributions from hydrogen peroxide (Matoba et al., 2000; Takaki et al., 2008) and cytochrome P450 metabolites (Pannirselvam et al., 2006) to endothelium-dependent relaxations in MMA. Indeed, we observed a vasorelaxation, which was resistant to inhibitors of NO, prostacyclin and EDHF (see Figure 1A) and these mediators may prove to play a minor role under the conditions used in this study. However, in our studies, in RMA, we found EDHF was a major mediator of endothelium-derived relaxation with a contribution of NO only apparent when the effects of EDHF were blocked.
The effects of the NO• scavenger HXC clearly suggest a significant contribution of NO• to endothelium-dependent relaxation in both the MMA and RMA, although HXC only impaired the response to ACh in RMA following inhibition of EDHF. Interestingly, in MMA HXC did not attenuate ACh-mediated vasorelaxation to the same extent as l-NAME alone, suggesting either an incomplete scavenging of NO• by HXC, or a possible NOS-derived HNO component to endothelium-derived relaxation. Indeed, the latter theory is supported by previous findings that HNO can be derived directly through NOS (Hobbs et al., 1994; Schmidt et al., 1996; Adak et al., 2000). In RMA however, in the absence of EDHF, HXC attenuates vasorelaxation to a greater extent than l-NAME, indicative of incomplete blockade of NO-mediated responses by this NOS inhibitor. Indeed, a NOS-independent source of NO• in the RMA has previously been reported (Chauhan et al., 2003). Such findings do not preclude a contribution of HNO in RMA as vasorelaxation to ACh is not abolished following inhibition of NO• (l-NAME and HXC) and of EDHF (ChTx and apamin). Moreover, the residual K+-sensitive component may be mediated by HNO given its potential ability to activate KV channels independently of sGC/cGMP in this vessel (Irvine et al., 2003).
Interestingly, we clearly demonstrated a role for HNO as an EDRF in both MMA and RMA. Thus negating the effects of HNO with l-cysteine or the KV channel inhibitor 4-AP significantly shifted the concentration–response curve to ACh to the right in MMA, and in RMA when in the presence of ChTx and apamin. Furthermore, with the additional scavenging of NO• by HXC, there was almost complete loss of vasorelaxation to ACh. Thus it appears that in both species, the NO component to endothelium-derived relaxation is mediated equally by HNO and NO•. This is in agreement with previous findings in conduit arteries that suggest that HNO contributes to endothelium-derived relaxation (Ellis et al., 2000; Wanstall et al., 2001).
Exogenous HNO opens Kv channels and coupled with the inhibitory effects of 4-AP upon ACh-mediated vasorelaxation, suggests that endogenously generated HNO may also contribute to endothelium-dependent hyperpolarization evoked by ACh. Excitingly, we have provided the first evidence that HNO serves as an EDHF in resistance-like arteries. Specifically, ACh-mediated, endothelium-dependent smooth muscle hyperpolarization of both MMA and RMA (in presence of ChTx and apamin) was sensitive to 4-AP. Interestingly, in MMA, NOS inhibition in combination with 4-AP impaired the hyperpolarization to ACh to a greater extent than 4-AP alone suggesting that endogenously generated NO• may also contribute to vascular smooth muscle hyperpolarization. These findings require further investigation but support the recent concept that endothelium-dependent hyperpolarization in mice is dependent upon the NOS system (Takaki et al., 2008). Strikingly, in RMA a substantial KCa channel-insensitive hyperpolarization to ACh exists and appears to be mediated predominantly by HNO.
The identification of HNO as an endothelium-derived relaxing and hyperpolarizing factor raises the question as to its endogenous source. HNO may be generated from both NOS (Hobbs et al., 1994; Schmidt et al., 1996; Adak et al., 2000) and non-NOS sources, including cytochrome c (Sharpe and Cooper, 1998), haemoglobin (Gow and Stamler, 1998), xanthine oxidase (Saleem and Ohshima, 2004) and S-nitrosothiols (Arnelle and Stamler, 1995; Wong et al., 1998). As speculated earlier, in MMA HNO appears to be derived from NOS as neither HXC nor l-cysteine had any further inhibitory effect beyond that of l-NAME alone. In contrast in RMA, HNO appears to be, at least in part, non-NOS-derived as 4-AP attenuated and l-cysteine abolished ACh-mediated relaxation following NOS and BKCa channel inhibition. Similarly, a NOS inhibitor-resistant source of NO, which is sensitive to haemoglobin and high extracellular K+, has been previously identified by ourselves (Kemp and Cocks, 1997) and others (Chauhan et al., 2003) in human coronary arteries and RMA respectively. Given HNO is scavenged by haemoglobin (J. C. Irvine, unpubl. obs.) and can target K+ channels (Irvine et al., 2003), it may account for these previous observations. Although the source of HNO in RMA remains to be elucidated, it is known that HNO can be generated from S-nitrosothiols (Arnelle and Stamler, 1995; Wong et al., 1998) and we and others have previously identified a S-nitrosothiol store that can be activated by UV light and ACh to release an as yet undetermined redox species of NO (Chauhan et al., 2003; Ng et al., 2007). Thus, HNO in RMA may be derived from a pre-formed thiol store. New experiments are required to explore this concept further.
The endogenous production of HNO will not be proven conclusively until direct detection methods for HNO are developed (Irvine et al., 2008). Although detection of HNO has been recently achieved by using xerogel sensing films (Dobmeier et al., 2008), unfortunately, specific measurement of HNO from either cultured endothelial cells or the intact blood vessel has not been possible thus far. As such, in the present study we have utilized several pharmacological tools that are currently available and that clearly distinguish between the redox species of NO. Future studies to elucidate the contribution of HNO to endothelium-derived relaxation and hyperpolarization under pathophysiological conditions are necessary, particularly given that vascular disease is associated with an uncoupling of NOS and increased oxidative stress, leading to decreased NO• bioavailability (Kojda et al., 1994; Kojda and Harrison, 1999) and in some instances a loss of classical EDHF (ChTx- and apamin-sensitive) responses (Wigg et al., 2001; Mori et al., 2006). Under such conditions HNO may serve as a compensatory vasorelaxant (Irvine et al., 2008).
In conclusion, NO is the predominant endothelium-dependent vasodilator in MMA, the rank orders of contributors to vasorelaxation being NO• = HNO > EDHF. In the Wistar Kyoto RMA, EDHF is the predominant endothelium-dependent vasodilator, the order of contributors to vasorelaxation, in this tissue, being EDHF > NO• = HNO. This study identifies HNO as an endothelium-derived hyperpolarizing and relaxing factor in resistance vessels.
 |
Acknowledgments
Dr Karen L Andrews and Dr Joanne L Favaloro are Peter Doherty Fellows of the National Health and Medical Research Council (Australia). Dr Barbara K Kemp-Harper is a Foundation for High Blood Pressure Research Postdoctoral Fellow (Australia) and supported by grants from the National Health and Medical Research Council (NHMRC, Australia). Professor Chris Triggle is supported by grants from the Heart & Stroke Foundation of Alberta, Northwest Territories & Nunavut (Canada) and the Australian Research Council. Dr Marianne Tare is supported by grants from the NHMRC (Australia) and the Diabetes Australia Research Trust.
Glossary
Abbreviations:
- 4-AP
4-aminopyridine
- AS
Angeli's salt
- ChTx
charybdotoxin
- EDHF
endothelium-derived hyperpolarizing factor
- EDRF
endothelium-derived relaxing factor
- KCa
calcium-activated K+ channel
- Kv
voltage-dependent K+ channel
- MMA
mouse mesenteric arteries
- RMA
rat mesenteric arteries
Conflict of interest
None.
References
- Alexander SPH, Mathie A, Peters JA. Guide to receptors and channels. Br J Pharmacol. (3rd edn.) 2008;153(Suppl. 2):S1–S209. doi: 10.1038/sj.bjp.0707746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adak S, Wang Q, Stuehr DJ. Arginine conversion of nitroxide by tetrahydrobiopterin-free neuronal nitric-oxide synthase. J Biol Chem. 2000;275:33554–33561. doi: 10.1074/jbc.M004337200. [DOI] [PubMed] [Google Scholar]
- Arnelle DR, Stamler JS. NO+, NO•, NO− donation by S-nitrosothiols: implications for regulation of physiological functions by S-nitrosylation and acceleration of disulfide formation. Arch Biochem Biophys. 1995;318:279–285. doi: 10.1006/abbi.1995.1231. [DOI] [PubMed] [Google Scholar]
- Brandes RP, Schmitz-Winnenthal FH, Feletou M, Godecke A, Huang PL, Vanhoutte PM, et al. An endothelium-derived hyperpolarizing factor distinct from NO and prostacyclin is a major endothelium-dependent vasodilator in resistance vessels of wild-type and endothelial NO synthase knockout mice. Proc Natl Acad Sci USA. 2000;97:9747–9752. doi: 10.1073/pnas.97.17.9747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chataigneau T, Feletou M, Huang PL, Fishman MC, Duhault J, Vanhoutte PM. Acetylcholine-induced relaxation in blood vessels from endothelial nitric oxide synthase knockout mice. Br J Pharmacol. 1999;126:219–226. doi: 10.1038/sj.bjp.0702300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chauhan S, Rahman A, Nilsson H, Clapp L, MacAllister R, Ahluwalia A. NO contributes to EDHF-like responses in rat small arteries: a role for NO stores. Cardiovasc Res. 2003;57:207–216. doi: 10.1016/s0008-6363(02)00611-9. [DOI] [PubMed] [Google Scholar]
- Ding H, Kubes P, Triggle C. Potassium- and acetylcholine-induced vasorelaxation in mice lacking endothelial nitric oxide synthase. Br J Pharmacol. 2000;129:1194–1200. doi: 10.1038/sj.bjp.0703144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobmeier KP, Riccio DA, Schoenfisch MH. Xerogel optical sensor films for quantitative detection of nitroxyl. Anal Chem. 2008;80:1247–1254. doi: 10.1021/ac702024t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donzelli S, Espey MG, Flores-Santana W, Switzer CH, Yeh GC, Huang J, et al. Generation of nitroxyl by heme protein-mediated peroxidation of hydroxylamine but not N-hydroxy-L-arginine. Free Radic Biol Med. 2008;45:578–584. doi: 10.1016/j.freeradbiomed.2008.04.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellis A, Li CG, Rand MJ. Differential actions of l-cysteine on responses to nitric oxide, nitroxyl anions and EDRF in the rat aorta. Br J Pharmacol. 2000;129:315–322. doi: 10.1038/sj.bjp.0703058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Favaloro JL, Kemp-Harper BK. The nitroxyl anion (HNO) is a potent dilator of rat coronary vasculature. Cardiovasc Res. 2007;73:587–596. doi: 10.1016/j.cardiores.2006.11.018. [DOI] [PubMed] [Google Scholar]
- Fitzgerald SM, Kemp-Harper BK, Parkington HC, Head GA, Evans RG. Endothelial dysfunction and arterial pressure regulation during early diabetes in mice: roles for nitric oxide and endothelium-derived hyperpolarizing factor. Am J Physiol. 2007;293:R707–R713. doi: 10.1152/ajpregu.00807.2006. [DOI] [PubMed] [Google Scholar]
- Fukuto JM, Chiang K, Hszieh R, Wong PSY, Chaudhurri G. The pharmacological activity of nitroxyl: a potent vasodilator with activity similar to nitric oxide and/or endothelium-derived relaxing factor. J Pharmacol Exp Ther. 1992a;263:546–551. [PubMed] [Google Scholar]
- Fukuto JM, Wallace GC, Hszieh R, Chaudhuri G. Chemical oxidation of N-hydroxyguanidine compounds. Release of nitric oxide, nitroxyl and possible relationship to the mechanism of biological nitric oxide generation. Biochem Pharmacol. 1992b;43:607–613. doi: 10.1016/0006-2952(92)90584-6. [DOI] [PubMed] [Google Scholar]
- Gow AJ, Stamler JS. Reactions between nitric oxide and haemoglobin under physiological conditions. Nature. 1998;391:169–173. doi: 10.1038/34402. [DOI] [PubMed] [Google Scholar]
- Hobbs AJ. Soluble guanylate cyclase: the forgotten sibling. Trends Pharmacol Sci. 1997;18:484–491. doi: 10.1016/s0165-6147(97)01137-1. [DOI] [PubMed] [Google Scholar]
- Hobbs AJ, Fukuto JM, Ignarro LJ. Formation of free nitric oxide from L-arginine by nitric oxide synthase: direct enhancement of generation by superoxide dismutase. Proc Natl Acad Sci USA. 1994;91:10992–10996. doi: 10.1073/pnas.91.23.10992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irvine JC, Favaloro JL, Kemp-Harper BK. NO− activates soluble guanylate cyclase and Kv channels to vasodilate resistance arteries. Hypertension. 2003;41:1301–1307. doi: 10.1161/01.HYP.0000072010.54901.DE. [DOI] [PubMed] [Google Scholar]
- Irvine JC, Favaloro JL, Widdop RE, Kemp-Harper BK. Nitroxyl anion donor, Angeli's salt, does not develop tolerance in rat isolated aortae. Hypertension. 2007;49:885–892. doi: 10.1161/01.HYP.0000259328.04159.90. [DOI] [PubMed] [Google Scholar]
- Irvine JC, Ritchie RH, Favaloro JL, Andrews KL, Widdop RE, Kemp-Harper BK. Nitroxyl (HNO): the Cinderella of the nitric oxide story. Trends Pharmacol Sci. 2008;29:601–608. doi: 10.1016/j.tips.2008.08.005. [DOI] [PubMed] [Google Scholar]
- Kemp BK, Cocks TM. Evidence that mechanisms dependent and independent of nitric oxide mediate endothelium-dependent relaxation to bradykinin in human small resistance-like coronary arteries. Br J Pharmacol. 1997;120:757–762. doi: 10.1038/sj.bjp.0700928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kojda G, Harrison D. Interactions between NO and reactive oxygen species: pathophysiological importance in atherosclerosis, hypertension, diabetes and heart failure. Cardiovasc Res. 1999;43:562–571. doi: 10.1016/s0008-6363(99)00169-8. [DOI] [PubMed] [Google Scholar]
- Kojda G, Beck JK, Meyer W, Noack E. Nitrovasodilator-induced relaxation and tolerance development in porcine vena cordis magna: dependence on intact endothelium. Br J Pharmacol. 1994;112:533–540. doi: 10.1111/j.1476-5381.1994.tb13106.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li CG, Rand MJ. Effects of hydroxocobalamin and haemoglobin on NO-mediated relaxations in the rat anococcygeus muscle. Clin Exp Pharmacol Physiol. 1993;20:633–640. doi: 10.1111/j.1440-1681.1993.tb01645.x. [DOI] [PubMed] [Google Scholar]
- McPherson GA. Optimal conditions for assessing vascular reactivity in small resistance arteries in the small vessel myograph. Clin Exp Pharmacol Physiol. 1992;19:815–825. doi: 10.1111/j.1440-1681.1992.tb00420.x. [DOI] [PubMed] [Google Scholar]
- Matoba T, Shimokawa H, Nakashima M, Hirakawa Y, Mukai Y, Hirano K, et al. Hydrogen peroxide is an endothelium-derived hyperpolarizing factor in mice. J Clin Invest. 2000;106:1521–1530. doi: 10.1172/JCI10506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mistry DK, Garland CJ. Nitric oxide (NO)-induced activation of large conductance Ca2+-dependent K+ channels (BKCa) in smooth muscle cells isolated from the rat mesenteric artery. Br J Pharmacol. 1998;124:1131–1140. doi: 10.1038/sj.bjp.0701940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moncada S, Palmer RM, Higgs EA. Nitric oxide: physiology, pathophysiology, and pharmacology. Pharmacol Rev. 1991;43:109–140. [PubMed] [Google Scholar]
- Mori Y, Ohyanagi M, Koida S, Ueda A, Ishiko K, Iwasaki T. Effects of endothelium-derived hyperpolarizing factor and nitric oxide on endothelial function in femoral resistance arteries of spontaneously hypertensive rats. Hypertens Res. 2006;29:187–195. doi: 10.1291/hypres.29.187. [DOI] [PubMed] [Google Scholar]
- Murphy ME, Sies H. Reversible conversion of nitroxyl anion to nitric oxide by superoxide dismutase. Proc Natl Acad Sci USA. 1991;88:10860–10864. doi: 10.1073/pnas.88.23.10860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelli S, Hillen M, Buyukafsar K, Martin W. Oxidation of nitroxyl anion to nitric oxide by copper ions. Br J Pharmacol. 2000;131:356–362. doi: 10.1038/sj.bjp.0703550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng ESM, Cheng Z-J, Ellis A, Ding H, Jiang Y, Li Y, et al. Nitrosothiol stores in vascular tissue: modulation by ultraviolet light, acetylcholine and ionomycin. Eur J Pharmacol. 2007;560:183–192. doi: 10.1016/j.ejphar.2007.01.016. [DOI] [PubMed] [Google Scholar]
- Pannirselvam M, Ding H, Anderson TJ, Triggle CR. Pharmacological characteristics of endothelium-derived hyperpolarizing factor-mediated relaxation of small mesenteric arteries from db/db mice. Eur J Pharmacol. 2006;551:98–107. doi: 10.1016/j.ejphar.2006.08.086. [DOI] [PubMed] [Google Scholar]
- Pino RZ, Feelisch M. Bioassay discrimination between nitric oxide (NO•) and nitroxyl (NO−) using l-cysteine. Biochem Biophys Res Commun. 1994;201:54–62. doi: 10.1006/bbrc.1994.1668. [DOI] [PubMed] [Google Scholar]
- Pufahl RA, Wishnok JS, Marletta MA. Hydrogen peroxide-supported oxidation of NG-hydroxyl-L-arginine by nitric oxide synthase. Biochemistry. 1995;34:1930–1941. doi: 10.1021/bi00006a014. [DOI] [PubMed] [Google Scholar]
- Rusche KM, Spiering MM, Marletta MA. Reactions catalyzed by tetrahydrobiopterin-free nitric oxide synthase. Biochemistry. 1998;37:15503–15512. doi: 10.1021/bi9813936. [DOI] [PubMed] [Google Scholar]
- Saleem M, Ohshima H. Xanthine oxidase converts nitric oxide to nitroxyl that inactivates the enzyme. Biochem Biophys Res Commun. 2004;315:455–462. doi: 10.1016/j.bbrc.2004.01.081. [DOI] [PubMed] [Google Scholar]
- Sampson LJ, Plane F, Garland CJ. Involvement of cyclic GMP and potassium channels in relaxation evoked by the nitric oxide donor, diethylamine NONOate, in the rat small isolated mesenteric artery. Naunyn Schmiedebergs Arch Pharmakol. 2001;364:220–225. doi: 10.1007/s002100100453. [DOI] [PubMed] [Google Scholar]
- Schmidt HHW, Hofmann H, Schindler U, Shutenko ZS, Cunningham DD, Feelisch M. No NO• from NO synthase. Proc Natl Acad Sci USA. 1996;93:14492–14497. doi: 10.1073/pnas.93.25.14492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharpe MA, Cooper CE. Reactions of nitric oxide with mitochondrial cytochrome c: a novel mechanism for the formation of nitroxyl anion and peroxynitrite. Biochem J. 1998;332:9–19. doi: 10.1042/bj3320009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takaki A, Morikawa K, Tsutsui M, Murayama Y, Tekes E, Yamagishi H, et al. Crucial role of nitric oxide synthases system in endothelium-dependent hyperpolarization in mice. J Exp Med. 2008;205:2053–2063. doi: 10.1084/jem.20080106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tare M, Parkington HC, Coleman HA. EDHF, NO and a prostanoid: hyperpolarization-dependent and independent relaxation in guinea-pig arteries. Br J Pharmacol. 2000;130:605–618. doi: 10.1038/sj.bjp.0703332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wanstall JC, Homer KL, Doggrell SA. Evidence for, and importance of, cGMP-independent mechanisms with NO and NO donors on blood vessels and platelets. Curr Vasc Pharmacol. 2005;3:41–53. doi: 10.2174/1570161052773933. [DOI] [PubMed] [Google Scholar]
- Wanstall JC, Jeffery TK, Gambino A, Lovren F, Triggle CR. Vascular smooth muscle relaxation mediated by nitric oxide donors: a comparison with acetylcholine, nitric oxide and nitroxyl ion. Br J Pharmacol. 2001;134:463–472. doi: 10.1038/sj.bjp.0704269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wigg SJ, Tare M, Tonta MA, O'Brien RC, Meredith IT, Parkington HC. Comparison of effects of diabetes mellitus on an EDHF-dependent and an EDHF-independent artery. Am J Physiol. 2001;281:H232–H240. doi: 10.1152/ajpheart.2001.281.1.H232. [DOI] [PubMed] [Google Scholar]
- Wong PSY, Hyun J, Fukuto JM, Shirota FN, DeMaster EG, Shoeman DW, et al. Reaction between S-nitrosothiols and thiols: generation of nitroxyl (HNO) and subsequent chemistry. Biochemistry. 1998;37:5362–5371. doi: 10.1021/bi973153g. [DOI] [PubMed] [Google Scholar]