

Abstract
Background and purpose:
Vascular ATP-sensitive potassium (KATP) channels are activated by cyclic AMP elevating vasodilators through protein kinase A (PKA). Direct channel phosphorylation is a critical mechanism, though the phosphatase opposing these effects is unknown. Previously, we reported that calcineurin, a Ca2+-dependent phosphatase, inhibits KATP channels, though neither the site nor the calcineurin isoform involved is established. Given that the type-2 regulatory (RII) subunit of PKA is a substrate for calcineurin we considered whether calcineurin regulates channel activity through interacting with PKA.
Experimental approach:
Whole-cell recordings were made in HEK-293 cells stably expressing the vascular KATP channel (KIR6.1/SUR2B). The effect of intracellular Ca2+ and modulators of the calcineurin and PKA pathway on glibenclamide-sensitive currents were examined.
Key results:
Constitutively active calcineurin Aα but not Aβ significantly attenuated KATP currents activated by low intracellular Ca2+, whereas calcineurin inhibitors had the opposite effect. PKA inhibitors reduced basal KATP currents and responses to calcineurin inhibitors, consistent with the notion that some calcineurin action involves inhibition of PKA. However, raising intracellular Ca2+ (equivalent to increasing calcineurin activity), almost completely inhibited KATP channel activation induced by the catalytic subunit of PKA, whose enzymatic activity is independent of the RII subunit. In vitro phosphorylation experiments showed calcineurin could directly dephosphorylate a site in Kir6.1 that was previously phosphorylated by PKA.
Conclusions and implications:
Calcineurin Aα regulates KIR6.1/SUR2B by inhibiting PKA-dependent phosphorylation of the channel as well as PKA itself. Such a mechanism is likely to directly oppose the action of vasodilators on the KATP channel.
British Journal of Pharmacology (2009) 157, 554–564; doi:10.1111/j.1476-5381.2009.00221.x; published online 7 May 2009
This article is commented on by Tammaro, pp. 551–553 of this issue and is part of a themed section on Endothelium in Pharmacology. For a list of all articles in this section see the end of this paper, or visit: http://www3.interscience.wiley.com/journal/121548564/issueyear?year=2009
Keywords: Calcineurin, ATP-sensitive potassium channels, KIR6.1/SUR2B, vascular, protein kinase A, whole-cell patch clamp
Introduction
Vascular ATP-sensitive K+ (KATP) channels contribute to the maintenance of resting membrane potential and local blood flow. They modulate vascular tone because of the steep relationship between membrane potential and Ca2+ influx through voltage-dependent Ca2+ channels (Quayle et al., 1997). Vasoconstrictor and vasodilator hormones will influence this relationship through opposing effects on the channel (Quayle et al., 1997; Buckley et al., 2006). The structure of the KATP channel is an octomeric complex composed of a pore-forming subunit (KIR6.x) to which ATP binds and a sulphonylurea receptor (SUR), the primary target for sulphonylureas, K+ channel opening drugs (KCOs) and nucleotide diphosphates (NDPs) (Seino and Miki, 2003). Based on subunit expression, channel characteristics and gene deletion experiments, KIR6.1/SUR2B almost certainly constitutes the vascular KATP channel (termed KNDP) that is relatively insensitive to ATP, activated by NDPs and inhibited by glibenclamide (Chutkow et al., 2002; Cui et al., 2002; Miki et al., 2002; Li et al., 2003). The consequence of deleting either the SUR2 or KIR6.1 gene, is to produce coronary vasospasm, sudden death and a markedly reduced vasodilatory response to KCOs (Chutkow et al., 2002; Miki et al., 2002; Kane et al., 2006). In addition, SUR2−/− mice are hypertensive while KIR6.1−/− mice have increased mortality towards endotoxin, and do not display the classic hypotensive response to this bacterial toxin (Kane et al., 2006).
KATP channels represent an important target for vasodilators that elevate cyclic AMP. These include hormones such as calcitonin-gene related peptide, adenosine, prostacyclin and vasoactive intestinal peptide, whose effects of relaxation and hypotension are sensitive to glibenclamide (Quayle et al., 1997; Buckley et al., 2006; Yang et al., 2008). Recently, the mechanism of channel activation was shown to involve direct phosphorylation by protein kinase A (PKA) of Ser and Thr residues located on SUR2B and KIR6.1 (Quinn et al., 2004; Shi et al., 2007; 2008a,b). While, the nature of the phosphatase opposing such phosphorylation is unknown, we have shown that the Ca2+-dependent phosphatase, calcineurin can regulate KATP channels both in vitro (Wilson et al., 2000) and in vivo (Singer et al., 2005). Such a mechanism allows the channel to sense changes in intracellular Ca2+ ([Ca2+]i), being activated at resting levels and inhibited as [Ca2+]i approaches micromolar levels (Wilson et al., 2000). Thus, hormonal regulation of KATP channels is likely to be influenced by calcineurin, though this has yet to be demonstrated. Moreover, the mechanism by which this phosphatase inhibits the channel is unknown.
Calcineurin is a highly conserved, Ca2+/calmodulin-dependent Ser/Thr phosphatase with a catalytic subunit A (calcineurin A or CnA) that binds calmodulin and a regulatory subunit B (calcineurin B or CnB) that binds Ca2+ (Rusnak and Mertz, 2000). The CnA subunit is encoded by three separate genes, which give rise to CnAα, CnAβ and CnAγ isoforms (Herzig and Neumann, 2000; Rusnak and Mertz, 2000). While CnAα and CnAβ are co-expressed in most tissues, expression of the γ isoform is restricted to the testis and discrete regions of the brain (Herzig and Neumann, 2000; Eastwood et al., 2005). Calcineurin is activated in response to a sustained elevation of cytoplasmic Ca2+ and is best known for its role in the Ca2+-dependent regulation of nuclear factor of activated T-cells (NFAT) which controls T-cell activation (Crabtree, 2001). In addition, this phosphatase is a major regulator of ion channel function, inhibiting or activating voltage-dependent Ca2+ channels (Schuhmann et al., 1997), Ca2+-activated Cl− channels (Greenwood et al., 2004) and a variety of K+ channels (Czirjak and Enyedi, 2006; Loane et al., 2006; Park et al., 2006). While several different mechanisms may underlie the effects of calcineurin on ion channels, the type-2 regulatory (RII) subunit of PKA is a well-known cellular substrate for calcineurin (Perrino et al., 2002) and has been shown to co-localize with calcineurin in cardiac cells (Santana et al., 2002). This raises the possibility that calcineurin may oppose PKA activation of the KATP channel through inhibiting PKA activity. Alternatively, it may directly dephosphorylate the channel itself. We therefore investigated Ca2+/calcineurin and PKA regulation of KIR6.1/SUR2B channels stably expressed in HEK-293 cells.
Methods
Stable lines were generated in human embryonic kidney 293 (HEK-293) cells containing KIR6.1 with SUR2B as described previously (Cui et al., 2001).
Whole-cell recording
Membrane currents were recorded under voltage-clamp in the whole-cell recording configuration of the patch-clamp technique using an Axopatch 200B amplifier (Axon Instruments, Foster City, CA). Currents were filtered at 1 kHz and sampled at 2 kHz via a Digidata 1322A (Axon Instruments) interface. Data were acquired and analysed using pClamp8 computer software (Axon Instruments). Patch pipettes were made from thin walled (OD 1.5 mm) borosilicate glass capillaries (Harvard Apparatus, Edenbridge, Kent), which were pulled and fire-polished using a DMZ-Universal puller (Zeitz-Instruments, Müchen, Germany) to give resistances of 2–4 MΩ. Electrode capacitance was reduced by coating tips with a parafilm/mineral oil suspension and was compensated electronically. Series resistance was compensated to 70% using the amplifier. Whole-cell bath solutions (pH 7.4) contained the following (in mmol·L−1): 140 KCl, 5 HEPES, 1.2 MgCl2 and 2.6 CaCl2. Pipette solutions (pH 7.2) contained the following (in mmol·L−1): 140 KCl, 5 HEPES, 1.2 MgCl2, 10 EGTA, 3 MgATP, 0.5 Na2GDP and 0–1.78 CaCl2. CaCl2 was added in an amount to yield 0, 18 and 36 nmol·L−1 intracellular free Ca2+, as calculated with the CaBuf computer program (G Droogmans, Physiology Laboratory, KU Leuven, Belgium), which takes into account the buffering capacity of ATP, NDPs and Mg2+. All measurements of basal or drug-activated currents were made at least 10–15 min after ‘break-in’. The magnitude of KATP current was assessed by sensitivity to 10 µmol·L−1 glibenclamide (IGlib). Drugs were applied either through the pipette or in the bath solutions.
Synthesis of constitutively active calcineurin
Constitutively active calcineurin isoforms were created by introducing stop codons into the cDNA for CnA, causing the translated CnA subunits to truncate immediate to the C-terminal of the calmodulin-binding domain and delete the auto-inhibitory domain. Methodologies for cDNA manipulation, baculovirus screening and purification of CnA constructs using cultures of Sf21 cells have been described previously (Perrino et al., 2002).
In vitro phosphorylation assay
Cloning, expression and purification of the maltose-binding protein (MBP) and the C-terminus of KIR6.1 (MBP-KIR6.1C) were carried out as previously described (Quinn et al., 2003). In vitro phosphorylation with the catalytic subunit of PKA was carried out as previously described (Quinn et al., 2004) except that 2 h after phosphorylation, samples were washed 3 times with 1 mL of HEPES buffer followed by a single wash with 1 mL of calcineurin reaction buffer (50 mmol·L−1 HEPES, pH 7.4, 18 nmol·L−1 CaCl2, 100 mmol·L−1 NaCl, 6 mmol·L−1 MgCl2, 1 mmol·L−1 DTT, 1% Triton X-100, EDTA-free complete protease inhibitor cocktail). Samples were then resuspended in 50 µL of calcineurin reaction buffer and a 10 µL aliquot removed for SDS analysis. Samples were subsequently centrifuged and all of the supernatant removed before addition of 9 µmol·L−1 calmodulin ± 100 nmol·L−1 of human recombinant CnAα subunit in calcineurin reaction buffer. Samples were incubated for 1 h at 37°C and then washed 4 times with 1 mL of calcineurin reaction buffer. The protein was subsequently eluted with 2× Laemmli gel loading buffer, run on a 10% SDS-PAGE gel and subjected to autoradiography.
Statistical analysis
Data were analysed using Clampfit 8.2 programme (Axon Instruments) and Graphpad Prism 4 (San Diego, CA). Values are given as means ± standard error of the mean (SEM) of glibenclamide-sensitive current (Iglib) densities (pA/pF), and n indicates the number of cells. Statistical significance was assessed using a paired or unpaired Student's t-test or one-way analysis of variance (anova) with correction for multiple comparisons between different groups of cells. P-values < 0.05 were considered to be statistically significant.
Materials
Glibenclamide, Mg2+-adenosine triphosphate, Na2 guanosine 5′ diphosphate (GDP), forskolin, levcromakalim and the catalytic subunit of PKA (PKAcat) were obtained from Sigma Chemical Co. (Poole, Dorset, UK). Cyclosporin A (CsA), calcineurin auto inhibitory peptide (CAP), okadaic acid, N-[2-(p-Bromocinnamylamino)ethyl]-5-isoquinolinesulphonamide·2HCl (H-89) Rp-2′-O-monobutyryl-cAMPS (Rp-cAMPS) and human recombinant CnAα subunit were all from Biomol (Exeter, UK). Gö6976 and calmodulin were bought from Calbiochem (San Diego, USA). Forskolin, H-89, Gö6976, levcromakalim and glibenclamide were dissolved in dimethyl sulphoxide but final concentrations after dilution were less than 0.1%, which had no effect on whole-cell currents. PKAcat was prepared as a stock of 1 or 10 U·µL−1 of a solution containing 6 mg·mL−1 of DTT. All other stock solutions were made up in distilled water.
Nomenclature of molecular targets, including receptors and ion channels, is in accordance with the Guide to Receptor and Channels (Alexander et al., 2008).
Results
Effect of Ca2+ on whole-cell KATP currents
We have previously demonstrated that increasing [Ca2+]i inhibited KATP currents in freshly isolated vascular smooth muscle cells (Wilson et al., 2000). We therefore wished to establish if KIR6.1/SUR2B channels stably expressed in HEK-293 cells were similarly regulated by [Ca2+]i. When cells were dialysed with solutions containing 0, 18 or 36 nmol·L−1 free Ca2+ concentrations, basal currents evoked at all potentials were reduced as [Ca2+]i was increased (Figure 1A). Likewise, the magnitude of glibenclamide-sensitive current (Iglib) was largest in the 0 Ca2+ pipette solution and smallest in cells dialysed with 36 nmol·L−1 Ca2+, as shown by the average current-voltage (I–V) curves in Figure 1B. Thus, the magnitude of current measured at −80 mV dropped by 33% with 18 nmol·L−1 Ca2+ (P < 0.05) and by 71% with 36 nmol·L−1 Ca2+ (P < 0.001) compared with 0 nmol·L−1 Ca2+ (Figure 1C). These results confirm that Ca2+ regulates KIR6.1/SUR2B.
Figure 1.
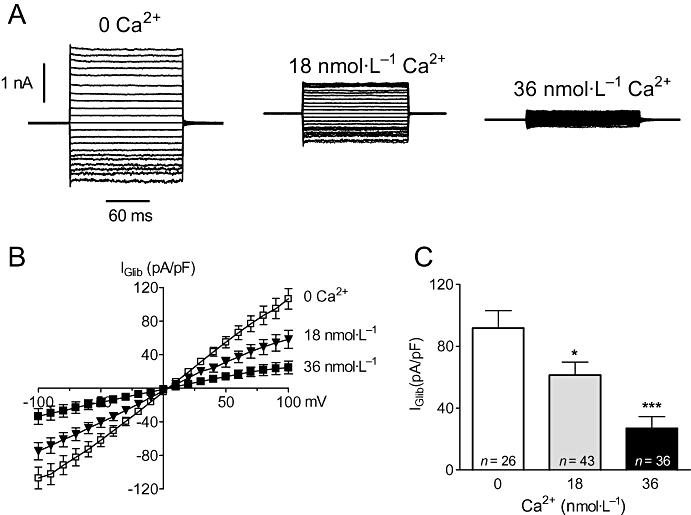
Intracellular Ca2+ inhibits whole-cell KATP currents in HEK-293 cells stably expressing KIR6.1/SUR2B. (A) Recordings of membrane currents from three separate cells dialysed with a pipette solution containing 0, 18 or 36 nmol·L−1 free Ca2+. Currents were evoked from a holding potential of 0 mV by stepping the voltage for 150 ms in 10 mV increments from −100 mV to +100 mV. (B) Mean current-voltage (I–V) relationships of steady-state current recorded under the three different [Ca2+]i conditions shown in A. Data have been plotted as glibenclamide-sensitive (Iglib) current (control current minus that in the presence of 10 µmol·L−1 glibenclamide) and currents normalized to cell capacitance. (C) Mean IGlib evoked at −80 mV taken from data in B. *P < 0.05, ***P < 0.001 when compared with 0 Ca2+.
Role of calcineurin
Having confirmed that Ca2+ regulates the channel, we investigated whether this involved signalling through calcineurin. We used two chemically unrelated inhibitors, calcineurin auto-inhibitory peptide (CAP; 100 µmol·L−1) and the immunophilin, CsA (Cyclo A; 10 µmol·L−1). Representative time-dependent plots comparing the magnitude of currents at −80 mV with and without 100 µmol·L−1 CAP in the pipette solution is shown in Figure 2A. In the presence of CAP, currents were noticeably larger, and gave rise to bigger glibenclamide-sensitive currents (IGlib). In a series of experiments, CAP completely reversed the Ca2+-dependent inhibition of the channel, doubling the magnitude of IGlib seen with 18 nmol·L−1 intracellular free Ca2+ (Figure 2B,C) and increasing it ∼3.5 fold in cells dialysed with 36 nmol·L−1 Ca2+ (Figure 2E,F). Likewise, IGlib in the presence of 10 µmol·L−1 Cyclo A was significantly greater (P < 0.05; Figure 2C) than that observed in control cells.
Figure 2.
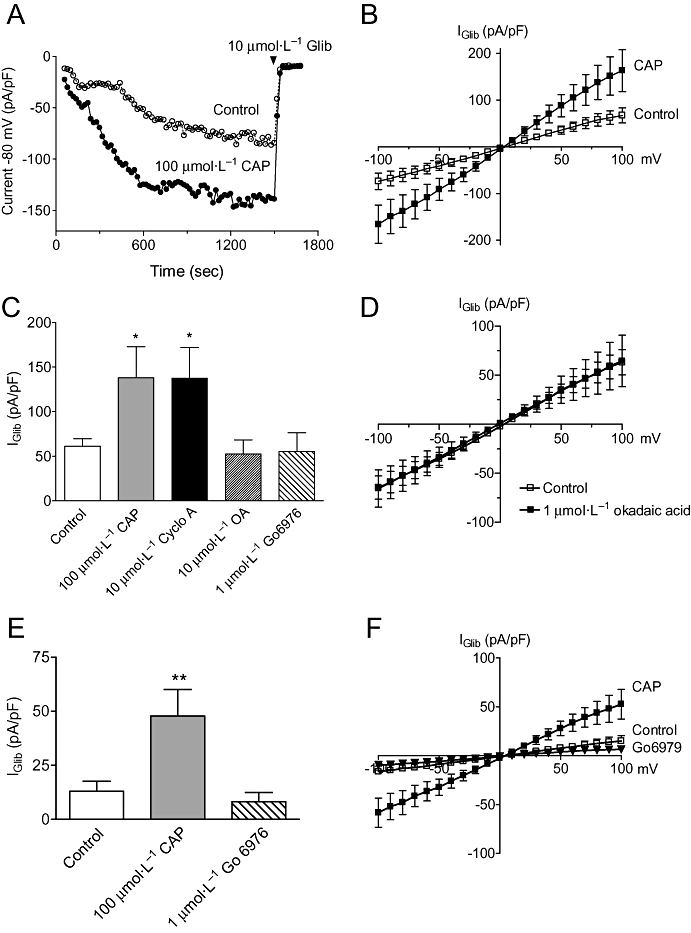
Calcineurin but not PP1 or PKC inhibitors increase KIR6.1/SUR2B currents. (A) Time-course of currents recorded from two cells dialysed in the absence (control) and presence of calcineurin auto-inhibitory peptide (CAP; 100 µmol·L−1) in the pipette. Currents were evoked by voltage steps (150 ms duration) applied from a holding potential of 0 mV to −80 mV and repeated every 15 s. Time 0 represents the onset of recording, and glibenclamide (10 µmol·L−1) was given at 25 min to assess the size of basal KATP current. (B) Mean IGlib plotted at different potentials in the absence (control, n= 19) or presence of 100 µmol·L−1 CAP (n= 8). Currents were elicited by 150 ms voltage steps from −100 to +100 mV. (C) Mean IGlib evoked at −80 mV in control cells (n= 43) compared with cells dialysed with CAP (n= 8), cyclosporin A (Cyclo A, n= 10), Gö6976 (n= 17) or okadaic acid (OA, n= 4). (D) I–V plots generated as in B in the absence (control) or presence of OA. (E) Mean IGlib evoked at −80 mV in cells dialysed with 36 nmol·L−1 under control conditions (n= 16) or in the presence of CAP (n= 8) or Gö6976 (n= 5) in the pipette. (F) I–V plots generated using the protocol described in B in the absence (control) or presence of CAP or Gö6976 in pipette solutions containing 36 nmol·L−1 Ca2+. In all other cases (A–D), the pipette solution contained 18 nmol·L−1 Ca2+. *P < 0.05, **P < 0.01 when compared with control.
Although our data indicated the involvement of calcineurin in the Ca2+-dependent regulation of KIR6.1/SUR2B, we wished to know whether regulation involved protein phosphatase type 1 (PP1), whose activity can be enhanced by calcineurin-dependent dephosphorylation of the PP1 inhibitory peptide inhibitor-1 (Perrino and Soderling, 1998; Herzig and Neumann, 2000). Therefore, the observed effects of CAP and CsA could in part reflect a decrease in PP1 activation. To examine this possibility, we internally applied the PP1 inhibitor, okadaic acid (1 µmol·L−1) in cells dialysed with 18 nmol·L−1 free Ca2+. This failed to change the magnitude of Iglib recorded at any potential (Figure 2C,D) leading us to conclude that PP1 is neither involved in the Ca2+-dependent regulation of KIR6.1/SUR2B nor has direct effects on the KATP channel itself. We also investigated the possibility that Ca2+ inhibits KATP current through activation of protein kinase C (PKC), which is known to inhibit both native and cloned vascular KATP channels (Quayle et al., 1997; Thorneloe et al., 2002; Quinn et al., 2003). We found that 1 µmol·L−1 Gö6976, an inhibitor of Ca2+-dependent isoforms of PKC, had no effect on KATP current magnitude at any potential whether cells were dialysed with 18 or 36 nmol·L−1 Ca2+ (Figure 2C–E).
To further confirm that calcineurin could indeed inhibit KATP channels, cells were dialysed with constitutively active recombinant isoforms of the catalytic subunit, CNAα or CNAβ (Perrino et al., 2002). These were added to the patch pipette at a concentration of 100 nmol·L−1 and experiments carried out in 0 nmol·L−1 Ca2+ to ensure minimal contribution from endogenous calcineurin. Figure 3 shows that CNAα but not CNAβ inhibited KATP current, giving rise to basal currents (35 ± 9.6 pA/pF, n= 6) of similar magnitude to that observed in 36 nmol·L−1 Ca2+ (27.0 ± 7.5 pA/pF, n= 16). These results therefore suggest that CNAα is the isoform likely to be involved in channel regulation.
Figure 3.
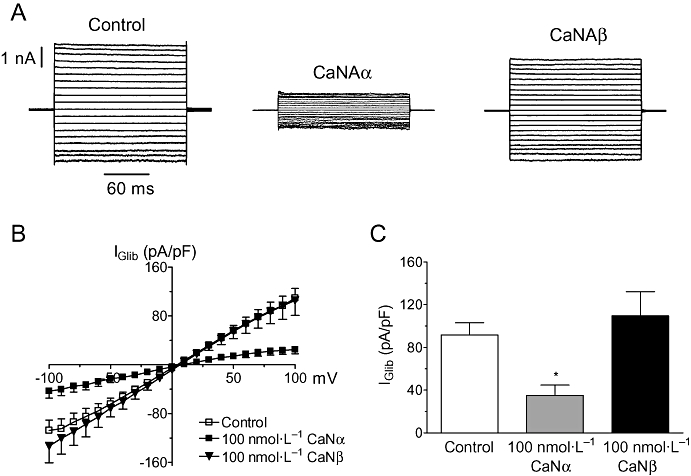
CnAα but not CnAβ inhibits whole-cell KATP currents. (A) Recordings of membrane currents from three separate cells dialysed with a pipette solution containing 0 added Ca2+ in the absence (left panel) or presence of constitutively active CnAα (middle panel) and CnAβ (right panel). Currents were evoked from a holding potential of 0 mV by stepping the voltage in 10 mV increments from −100 mV to +100 mV. (B) Mean IGlib plotted at different potentials in the absence (control, n= 19) or presence of either 100 nmol·L−1 CnAα (n= 6) or CnAβ (n= 7) in the pipette. (C) Mean IGlib evoked at −80 mV in control cells compared with cells dialysed with constitutively active CnAβ or CnAα. *P < 0.05 when compared with CnAβ.
Modulation by PKA
As PKA phosphorylation of KIR6.1/SUR2B can lead to channel opening (Quinn et al., 2004) and calcineurin is known to dephosphorylate and inhibit the RII subunit of PKA (Perrino et al., 2002), we investigated whether calcineurin/Ca2+ effects on the channel involved inhibition of the PKA pathway. As a first step, we sought to confirm that PKA regulated basal channel activity by using two inhibitors, H-89 (10 µmol·L−1) and Rp-cAMPs (100 µmol·L−1). We found that both these agents significantly attenuated the magnitude of KATP current measured in the presence of 18 nmol·L−1 Ca2+ (Figure 4A,B) by 56% and 70% respectively. Next, we examined the combined effect of H-89 (10 µmol·L−1) and CAP (100 µmol·L−1) included in the same patch pipette. Using this approach, the magnitude of the IGlib, while greater than that observed with H-89 alone (Figure 4B), was still significantly less (P < 0.05) than that observed if cells were dialysed with CAP alone under the same experimental conditions (Figure 2C). These results are consistent with the notion that a portion of calcineurin action involved inhibition of PKA.
Figure 4.
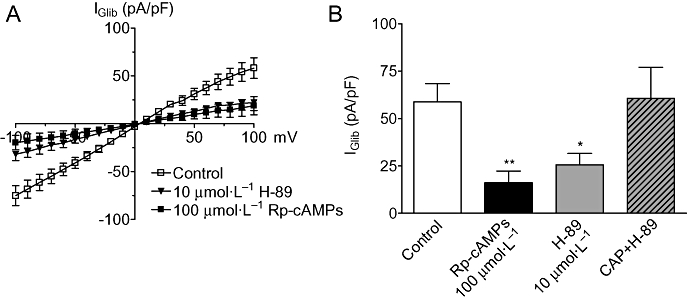
Ca2+/calcineurin regulation of KATP channels is intrinsically linked to PKA. (A) Mean IGlib plotted at different potentials in the absence (control, n= 15) or presence of either H-89 (n= 8) or Rp-cAMPs (n= 9) in the pipette. (B) Mean IGlib evoked at −80 mV in control cells compared with cells dialysed with either Rp-cAMPs, H-89 or H-89 with 100 µmol·L−1 CAP (n= 8). In both A & B the pipette contained 18 nmol·L−1 Ca2+. *P < 0.05, **P < 0.01 when compared with contol.
Likewise, the effects of the adenylate cyclase activator, forskolin, whose stimulatory action on KATP channels in HEK-293 cells is completely dependent on PKA (Quinn et al., 2004), were essentially abolished when Ca2+ was raised from 18 to 36 nmol·L−1 (Figure 5A,B). Again, this could be partially restored (P < 0.001, n= 8) when the pipette also contained 100 µmol·L−1 CAP (Figure 5B), suggesting that suppression of forskolin activation of the channel by Ca2+ involved calcineurin either acting on PKA or on the channel itself. To distinguish between these possibilities, two experimental approaches were used. First, we examined channel regulation using the constitutively active catalytic subunit of PKA (PKAcat), whose activity can no longer be inhibited by calcineurin because it lacks the RII subunit. Dialysis of cells with PKAcat (10 U·mL−1) produced KATP currents that were 3 times larger than in control cells (Figure 5C,D). However, on raising the intracellular free Ca2+ to 36 nmol·L−1 (equivalent to increasing calcineurin activity), the same concentration of PKAcat failed to have any effect on whole-cell currents. Some activation was achieved when the concentration was raised to 100 U·mL−1, though this failed to reach significance. These results strongly suggest that PKA and calcineurin can compete for the same phosphorylation sites on the KATP channel. This contrasts with responses to the classical KATP channel opener, levcromakalim (10 µmol·L−1) which activated currents of similar magnitude in both 18 and 36 nmol·L−1 free Ca2+ (Figure 5E). The other experimental approach was to examine the ability of calcineurin to dephosphorylate MBP-KIR6.1C, a C-terminal domain of Kir6.1 that contains a serine site (S385) capable of being phosphorylated by PKA (Quinn et al., 2004). Figure 6A shows a Coomassie-stained SDS-PAGE gel loaded with either MBP or MBP-KIR6.1C in the absence or presence of PKAcat and CnAα. In essence, the gel shows roughly equivalent loading of MBP-KIR6.1C (doublet between 60 and 75 kD) in the phosphorylation assay shown in Figure 6B. As described previously, MBP-KIR6.1C was a substrate for PKA-mediated phosphorylation, an effect that was subsequently reduced in the presence of CnAα (Figure 6B). In contrast, PKAcat does not phosphorylate the MBP control protein nor does CnAα have any effect. Therefore, these results strongly suggest that the pore of KIR6.1 can act as a direct substrate for CnAα mediated dephosphorylation.
Figure 5.
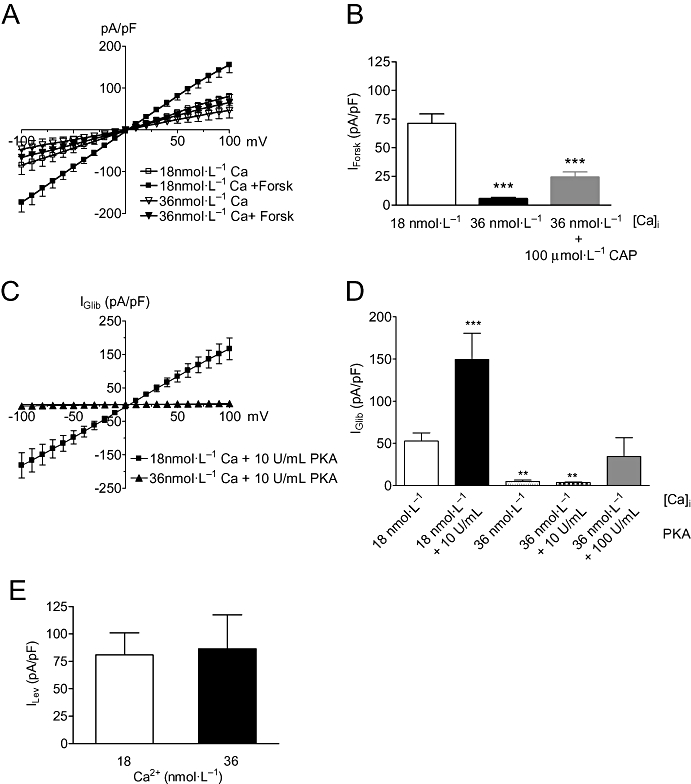
Ca2+/calcineurin disrupts PKA but not levcromakalim activation of KATP currents. (A) Mean current (pA/pF) plotted at different potentials before and 5 min after the application of 10 µmol·L−1 forskolin at two different internal Ca2+ concentrations of 18 (n= 12) and 36 nmol·L−1 (n= 9). (B) Average forskolin-induced current (IForsk) at −80 mV in cells in the absence or presence of 100 µmol·L−1 CAP (n= 8). (C) Mean IGlib induced by the catalytic subunit of PKA in the presence of either 18 (n= 11) or 36 (n= 17) nmol·L−1 free Ca2+ in the pipette. (D) Column graph of currents evoked at −80 mV in the absence and presence of activated PKA in the patch pipette. (E) Mean levcromakalim current (ILev) induced at −80 mV with either 18 (n= 5) or 36 nmol·L−1 (n= 7) free Ca2+ in the pipette. Currents in the presence of 10 µmol·L−1 levcromakalim were subtracted from those obtained before application. **P < 0.01, ***P < 0.001 when compared with control.
Figure 6.
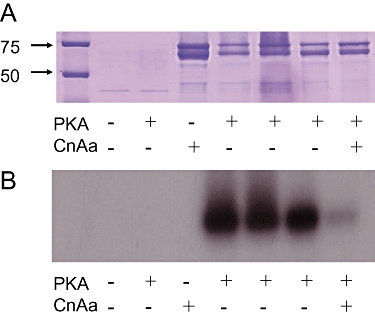
(A) 10% SDS-PAGE stained with Coomassie showing 2 µg of either MBP (lanes 2–3) or MBPKir6.1C (lanes 4–8) ± protein kinase A (PKA) and ±calcineurin Aα (CnAα). Marker sizes in kDa are indicated. B. Autoradiograph of the gel in A showing phosphorylation via PKA following a 2 h incubation and subsequent dephosphorylation in the presence of CnAα after 1 h. Presence or absence of PKA and CnAα are indicated.
Discussion
Previously, we demonstrated that Ca2+ regulates basal KATP channel activity in vascular smooth muscle cells isolated from rat aorta (Wilson et al., 2000). In the present study, we investigated the mechanism underlying this regulation in HEK-293 cells stably expressing KIR6.1/SUR2B, a model system where many of the properties of native vascular KATP channels are faithfully reconstituted (Thorneloe et al., 2002; Quinn et al., 2003; 2004). Our data revealed that calcineurin is likely to be the main mediator of Ca2+-dependent inhibition of KIR6.1/SUR2B and that this phosphatase inhibited channel function by opposing PKA-dependent phosphorylation. This conclusion is based on a number of observations. First, there was selective reversal of Ca2+-induced channel inhibition by two chemically unrelated inhibitors of calcineurin, CsA and CAP. The latter corresponds to a C-terminal domain (residues 457–482) of the calmodulin-binding domain of calcineurin making it a highly specific inhibitor with little effect on PP1, PP2A or CaM kinase II activity. Likewise, it was concluded that all Ca2+-dependent regulation of KV2.1 was attributable to calcineurin activity in HEK-293 cells (Park et al., 2006). Second, constitutively active CnAα was able to inhibit KATP currents activated under conditions of low endogenous calcineurin activity (no added Ca2+ in the pipette), confirming our cyclosporin and CAP data. Third, PKA inhibitors substantially reduced basal KATP currents and responses to calcineurin inhibitors, suggesting crosstalk between these two signalling pathways (Figure 7). Fourthly, raising [Ca2+]i to 36 nmol·L−1 essentially abolished the activating effects of both PKAcat and forskolin. As PKAcat is constitutively active and therefore not subject to deactivation by calcineurin, this failure could only result from dephosphorylation of sites normally phosphorylated by PKA. Lastly, consistent with direct channel effects, CnAα dephosphorylated MBP-KIR6.1C, a C-terminal domain of KIR6.1 which contains a serine site (S385) directly phosphorylated by PKA and critical for channel activation by either forskolin or PKAcat (Quinn et al., 2004). In other studies, direct binding of calcineurin to K+ channels has been demonstrated, though the interaction sites are different, involving CnB subunit for the Ca2+-activated K+ channel (Loane et al., 2006) or the NFAT binding site on CnAα for TRESK (Czirjak and Enyedi, 2006).
Figure 7.
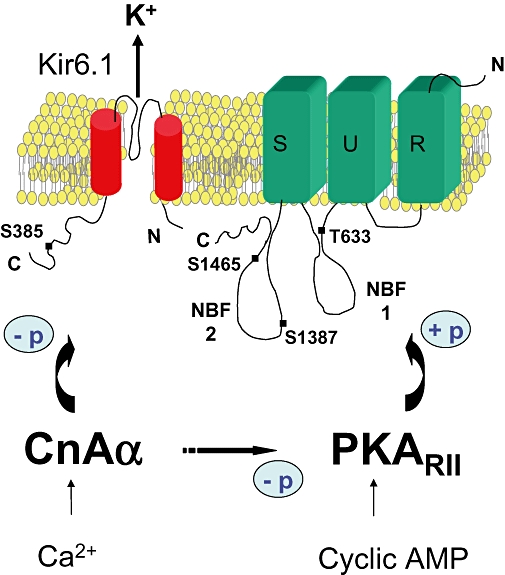
Diagram of possible mechanisms of calcineurin regulation of the vascular KATP channel. The putative sites involved in PKA and calcineurin regulation are shown on KIR6.1 and the nucleotide binding domains 1 (NB1) and 2 (NB2) in SUR2B.
One of the significant findings in the present study was the demonstration that CnAα but not CnAβ regulated KIR6.1/SUR2B, with only the former significantly inhibiting whole-cell currents. Such an isoform-selective effect of CnAα has previously been described for Ca2+-activated Cl− channels in pulmonary artery (Greenwood et al., 2004). So far this is the only isoform reported to regulate either Ca2+-activated K+ channels (Loane et al., 2006) or TRESK, a member of the twin pore family of K+ channels (Czirjak and Enyedi, 2006). Moreover, CnAα has the appropriate cytosolic cellular distribution to regulate plasmalemmal ion channels in rat aortic smooth muscle whereas CnAβ does not, being largely confined to the perinuclear region, where it is differentially translocated into the nucleus in response to smooth muscle mitogens (Jabr et al., 2007). In pulmonary artery, the cellular distribution of the two isoforms was found to be more homogeneous, though translocation to the membrane was only observed with CnAα, but not CnAβ when [Ca2+]i was elevated to 500 nmol·L−1 (Greenwood et al., 2004). Despite an 85% homology between CnAα and CnAβ, different substrate affinities and catalytic activities are displayed by these two isoforms (Perrino et al., 2002), which may contribute to their selective cellular function.
In our study we found no evidence that either PP2A or PP1 regulated SUR2B/KIR6.1 under basal conditions. This conclusion is based on no discernible effect of okadaic acid, which blocks either phosphatase in the low to high nanomolar range respectively (Herzig and Neumann, 2000). Lack of involvement from PP1 was contrary to our expectation because calcineurin is known to activate PP1 through dephosphorylating either PP1 and/or its endogenous inhibitors (DARPP32, inhibitor-1), thus making its activity potentially dependent on Ca2+/calcineurin in the intact cell (Hubbard and Cohen, 1989; Halpain et al., 1990; Mulkey et al., 1994). Given that HEK-293 cells do express endogenous PP1 (Morimoto et al., 2004; Huang et al., 2005), this might suggest that either the cellular localization of this phosphatase is not in the vicinity of the KATP channel or that it cannot oppose PKA phosphorylation of functional channel sites. Little is known about the role of phosphatases in vascular smooth muscle, though in rat mesenteric artery, PP1 and PP2A inhibitors either had no effect or slightly increased basal KATP currents while actually decreasing them in guinea-pig mesenteric artery (Firth et al., 2000; Hayabuchi et al., 2001). In contrast, PKC-mediated inhibition of KATP channels was significantly enhanced by okadaic acid in both vascular and non-vascular smooth muscle (Firth et al., 2000) suggesting Ca2+-independent phosphatases may play a more prominent role in opposing PKC signalling. In tissues expressing other KATP channel subtypes, PP2A inhibits channel activity, promotes rundown and opposes the effects of PKC (Kubokawa et al., 1995; Kwak et al., 1996; Light et al., 1996), while calcineurin appears a major regulator of skeletal muscle channels in humans (Singer et al., 2005), but not in rat renal tubule cells (Kubokawa et al., 1995).
We also considered the possibility that Ca2+ might inhibit the KATP channel through activation of Ca2+-dependent isoforms of PKC. We think this unlikely based on lack of effect of Gö6976, an inhibitor of conventional PKC isoforms (α,β,γ) which are activated by both Ca2+ and diacylglycerol. This finding is also consistent with previous observations showing angiotensin II inhibition of vascular KATP channels involves PKCepsilon, a Ca2+-insenstive isoform of PKC (Hayabuchi et al., 2001; Sampson et al., 2007). Moreover, the same isoform is responsible for PKC-mediated inhibition of KIR6.1/SUR2B induced by phorbol esters (Quinn et al., 2003).
We sought to investigate whether calcineurin could act by modulating the degree of phosphorylation of residues located on either the channel itself or a regulatory protein such as PKA. Our results support a mechanism whereby calcineurin and PKA can directly compete for the same residues on the KATP channel, with the degree of channel activation depending on the relative activities of PKA and calcineurin (Figure 7). Based on our phosphorylation data, we would surmise that S385 in Kir6.1 is one of the sites involved in this reciprocal regulation. This is consistent with the corresponding site in Kir6.2 (S372) being the major site of phosphorylation promoted by Gs-coupled receptors or direct stimulators of PKA (Beguin et al., 1999). We believe however that additional PKA sites (Quinn et al., 2004; Shi et al., 2007; 2008a) contribute to the action of calcineurin. We have previously shown two other sites in SUR2B (T633, S1465) to be functionally involved in the activation of KIR6.1/SUR2B by cyclic AMP elevating agents (Quinn et al., 2004; Shi et al., 2007), while in other studies S1387, a site in the second nucleotide-binding domain of SUR2B, is reported to be the main site (Shi et al., 2007; 2008a). It is also possible other serine sites (S1571 in SUR1) might contribute to basal channel activation (Beguin et al., 1999). Taken together, it is likely that multiple sites are dephosphorylated by calcineurin. Indeed KV2.1 channels expressed in HEK cells are extensively phosphorylated at several serine sites under basal conditions (Park et al., 2006). These sites can be variably dephosphorylated by calcineurin to give graded regulation of channel activity, a mechanism which allows for the fine tuning of neuronal action potential firing (Park et al., 2006).
A multisite phosphorylation mechanism for PKA activation of the vascular KATP channel is not strongly supported by the data of Shi and colleagues. Based on systematic mutational analysis of all putative PKA sites, it was concluded that the main phosphorylation site responsible for the functional effects of forskolin or the β2-receptor agonist isoprenaline is S1387 (Shi et al., 2007; 2008a). Moreover, on making the exact mutations they were unable to repeat the observations in our original paper (Quinn et al., 2004). How, therefore, do we reconcile these seemingly conflicting data? The most notable difference in their studies is that experiments were performed in the presence of EGTA in the bath or pipette solutions with no added Ca2+ (Shi et al., 2007; 2008a). Thus, based on our results presented here, we would predict that in 0 Ca2+, more PKA sites (or the relative proportion of an individual site) would become phosphorylated due to the low calcineurin activity. The latter would also promote higher basal PKA activity, due to increased phosphorylation of the RII subunit (Figure 7). This may explain why forskolin-induced currents relative to activation by the KATP channel opener were smaller than we observed in the present study in 18 nmol·L−1 Ca2+ (40% vs. 100% respectively). Thus, it is likely that both groups are investigating contributions from different PKA sites under the different calcium levels. Furthermore, Shi and colleagues could not completely rule out a role for S385, as mutating this site to glutamate instead of alanine, did cause about a 30% reduction in the forskolin-induced activation of KIR6.1/SUR2B (Shi et al., 2007). Lastly, contributions from other unidentified sites could not be ruled out given the moderate activation by PKAcat observed in their S1387 mutant (Shi et al., 2007).
What might be the functional significance of calcineurin regulation of the KATP channel? We postulate that under conditions where Ca2+ levels are elevated, for example, in the presence of a vasoconstrictor, this would promote channel closure. Indeed angiotensin II inhibits KATP channels in part through inhibiting basal PKA activity (Hayabuchi et al., 2001), which we would argue results from calcineurin opposing PKA phosphorylation. The other consequence might be to limit and/or impair channel opening by vasodilator agents known to activate PKA. Interestingly, KATP channels do not contribute to forskolin-induced relaxation of phenylephrine contractions unless tissues are first incubated with endotoxin (Wilson and Clapp, 2002). In this context, opening may result from prolonged NO production and/or superoxide generation inhibiting calcineurin activity (Sommer et al., 2002). The net effect would be to promote hyperphosphorylation of the KATP channel even at relatively high Ca2+ levels. A parallel might be drawn in patients on cyclosporin therapy who develop life-threatening hyperkalemia (raised plasma K+ levels), which can successfully be treated with glibenclamide (Singer et al., 2005). Thus low calcineurin activity is likely to drive channel opening under physiological and pathophysiological conditions.
In conclusion, this study reveals that calcineurin Aα is the main mediator of the inhibitory action of Ca2+ on KIR6.1/SUR2B and that the mechanism involves dephosphorylation of PKA-phosphorylated sites on the channel. Such a mechanism may oppose the action of vasodilator hormones known to signal though PKA.
 |
Acknowledgments
This work was supported by the Medical Research Council, UK (G117/440) and the British Heart Foundation (PG/07/078/2355).
Glossary
Abbreviations:
- [Ca2+]i
intracellular Ca2+
- CAP
calcineurin auto inhibitory peptide
- CnA
catalytic subunit A of calcineurin
- CnB
regulatory subunit B of calcineurin
- CsA
Cyclosporin A
- H-89
N-[2-(p-Bromocinnamylamino)ethyl]-5-isoquinolinesulphonamide·2HCl
- HEK-293
human embryonic kidney 293
- KATP
ATP-sensitive potassium
- KCOs
K+ channel opening drugs
- MBP
maltose-binding protein
- NFAT
nuclear factor of activated T-cells
- NDPs
nucleotide diphosphates
- PKA
protein kinase A
- PKAcat
catalytic subunit of protein kinase A
- Rp-cAMPS
Rp-2′-O-monobutyryl-cAMPS
- SUR
sulphonylurea receptor
- RII
type-2 regulatory subunit
Conflict of interest
None.
References
- Alexander SP, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC) Br J Pharmacol. (3rd edn.) 2008;153(Suppl. 2):S1–209. doi: 10.1038/sj.bjp.0707746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beguin P, Nagashima K, Nishimura M, Gonoi T, Seino S. PKA-mediated phosphorylation of the human KATP channel: separate roles of Kir6.2 and SUR subunit phosphorylation. EMBO J. 1999;18:4722–4732. doi: 10.1093/emboj/18.17.4722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckley JF, Singer M, Clapp LH. The role of KATP channels in sepsis. Cardiovasc Res. 2006;72:220–230. doi: 10.1016/j.cardiores.2006.07.011. [DOI] [PubMed] [Google Scholar]
- Chutkow WA, Pu J, Wheeler MT, Wada T, Makielski JC, Burant CF, et al. Episodic coronary artery vasospasm and hypertension develop in the absence of Sur2 KATP channels. J Clin Invest. 2002;110:203–208. doi: 10.1172/JCI15672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crabtree GR. Calcium, calcineurin, and the control of transcription. J Biol Chem. 2001;276:2313–2316. doi: 10.1074/jbc.R000024200. [DOI] [PubMed] [Google Scholar]
- Cui Y, Giblin JP, Clapp LH, Tinker A. A mechanism for ATP-sensitive potassium channel diversity: functional coassembly of two pore forming subunits. Proc Natl Acad Sci USA. 2001;98:729–734. doi: 10.1073/pnas.011370498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui Y, Tran S, Tinker A, Clapp LH. The molecular composition of KATP channels in human pulmonary artery smooth muscle cells and their modulation by growth. Am J Respir Cell Mol Biol. 2002;26:135–143. doi: 10.1165/ajrcmb.26.1.4622. [DOI] [PubMed] [Google Scholar]
- Czirjak G, Enyedi P. Targeting of calcineurin to an NFAT-like docking site is required for the calcium-dependent activation of the background K+ channel, TRESK. J Biol Chem. 2006;281:14677–14682. doi: 10.1074/jbc.M602495200. [DOI] [PubMed] [Google Scholar]
- Eastwood SL, Burnet PW, Harrison PJ. Decreased hippocampal expression of the susceptibility gene PPP3CC and other calcineurin subunits in schizophrenia. Biol Psychiatry. 2005;57:702–710. doi: 10.1016/j.biopsych.2004.12.029. [DOI] [PubMed] [Google Scholar]
- Firth TA, Mawe GM, Nelson MT. Pharmacology and modulation of KATP channels by protein kinase C and phosphatases in gallbladder smooth muscle. Am J Physiol. 2000;278:C1031–C1037. doi: 10.1152/ajpcell.2000.278.5.C1031. [DOI] [PubMed] [Google Scholar]
- Greenwood IA, Ledoux J, Sanguinetti A, Perrino BA, Leblanc N. Calcineurin Aα but not Aβ augments ICl(Ca) in rabbit pulmonary artery smooth muscle cells. J Biol Chem. 2004;279:38830–38837. doi: 10.1074/jbc.M406234200. [DOI] [PubMed] [Google Scholar]
- Halpain S, Girault JA, Greengard P. Activation of NMDA receptors induces dephosphorylation of DARPP-32 in rat striatal slices. Nature. 1990;343:369–372. doi: 10.1038/343369a0. [DOI] [PubMed] [Google Scholar]
- Hayabuchi Y, Davies NW, Standen NB. Angiotensin II inhibits rat arterial KATP channels by inhibiting steady-state protein kinase A activity and activating protein kinase Ce. J Physiol. 2001;530:193–205. doi: 10.1111/j.1469-7793.2001.0193l.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herzig S, Neumann J. Effects of serine/threonine protein phosphatases on ion channels in excitable membranes. Physiol Rev. 2000;80:173–210. doi: 10.1152/physrev.2000.80.1.173. [DOI] [PubMed] [Google Scholar]
- Huang HS, Pozarowski P, Gao Y, Darzynkiewicz Z, Lee EY. Protein phosphatase-1 inhibitor-3 is co-localized to the nucleoli and centrosomes with PP1gamma1 and PP1alpha, respectively. Arch Biochem Biophys. 2005;443:33–44. doi: 10.1016/j.abb.2005.08.021. [DOI] [PubMed] [Google Scholar]
- Hubbard MJ, Cohen P. Regulation of protein phosphatase-1G from rabbit skeletal muscle. 1. Phosphorylation by cAMP-dependent protein kinase at site 2 releases catalytic subunit from the glycogen-bound holoenzyme. Eur J Biochem. 1989;186:701–709. doi: 10.1111/j.1432-1033.1989.tb15263.x. [DOI] [PubMed] [Google Scholar]
- Jabr RI, Wilson AJ, Riddervold MH, Jenkins AH, Perrino BA, Clapp LH. Nuclear translocation of calcineurin Aβ but not calcineurin Aα by platelet-derived growth factor in rat aortic smooth muscle. Am J Physiol. 2007;292:C2213–C2225. doi: 10.1152/ajpcell.00139.2005. [DOI] [PubMed] [Google Scholar]
- Kane GC, Lam CF, O'Cochlain F, Hodgson DM, Reyes S, Liu XK, et al. Gene knockout of the KCNJ8-encoded Kir6.1 KATP channel imparts fatal susceptibility to endotoxemia. FASEB J. 2006;20:2271–2280. doi: 10.1096/fj.06-6349com. [DOI] [PubMed] [Google Scholar]
- Kubokawa M, Mcnicholas CM, Higgins MA, Wang W, Giebisch G. Regulation of ATP-sensitive K+ channel by membrane-bound protein phosphatases in rat principal tubule cell. Am J Physiol. 1995;269:F355–F362. doi: 10.1152/ajprenal.1995.269.3.F355. [DOI] [PubMed] [Google Scholar]
- Kwak YG, Park SK, Cho KP, Chae SW. Reciprocal modulation of ATP-sensitive K+ channel activity in rat ventricular myocytes by phosphorylation of tyrosine and serine threonine residues. Life Sci. 1996;58:897–904. doi: 10.1016/0024-3205(96)00032-x. [DOI] [PubMed] [Google Scholar]
- Li L, Wu J, Jiang C. Differential expression of Kir6.1 and SUR2B mRNAs in the vasculature of various tissues in rats. J Membr Biol. 2003;196:61–69. doi: 10.1007/s00232-003-0625-z. [DOI] [PubMed] [Google Scholar]
- Light PE, Sabir AA, Allen BG, Walsh MP, French RJ. Protein kinase C-induced changes in the stoichiometry of ATP binding activate cardiac ATP-sensitive K+ channels. A possible mechanistic link to ischemic preconditioning. Circ Res. 1996;79:399–406. doi: 10.1161/01.res.79.3.399. [DOI] [PubMed] [Google Scholar]
- Loane DJ, Hicks GA, Perrino BA, Marrion NV. Inhibition of BK channel activity by association with calcineurin in rat brain. Eur J Neurosci. 2006;24:433–441. doi: 10.1111/j.1460-9568.2006.04931.x. [DOI] [PubMed] [Google Scholar]
- Miki T, Suzuki M, Shibasaki T, Uemura H, Sato T, Yamaguchi K, et al. Mouse model of Prinzmetal angina by disruption of the inward rectifier Kir6.1. Nat Med. 2002;8:466–472. doi: 10.1038/nm0502-466. [DOI] [PubMed] [Google Scholar]
- Morimoto H, Okamura H, Yoshida K, Kitamura S, Haneji T. Double-stranded RNA mediates selective gene silencing of protein phosphatase type 1 delta isoform in HEK-293 cells. J Enzyme Inhib Med Chem. 2004;19:327–331. doi: 10.1080/14756360409162445. [DOI] [PubMed] [Google Scholar]
- Mulkey RM, Endo S, Shenolikar S, Malenka RC. Involvement of a calcineurin/inhibitor-1 phosphatase cascade in hippocampal long-term depression. Nature. 1994;369:486–488. doi: 10.1038/369486a0. [DOI] [PubMed] [Google Scholar]
- Park KS, Mohapatra DP, Misonou H, Trimmer JS. Graded regulation of the Kv2.1 potassium channel by variable phosphorylation. Science. 2006;313:976–979. doi: 10.1126/science.1124254. [DOI] [PubMed] [Google Scholar]
- Perrino BA, Soderling TR. Biochemistry and pharmacology of calmodulin-regulated phosphatase calcinuerin. In: van Eldik L, Watterson DM, editors. Calmodulin and Signal Transduction. New York: Academic Press; 1998. pp. 169–236. [Google Scholar]
- Perrino BA, Wilson AJ, Ellison P, Clapp LH. Substrate selectivity and sensitivity to inhibition by FK506 and cyclosporin A of calcineurin heterodimers composed of the alpha or beta catalytic subunit. Eur J Biochem. 2002;269:3540–3548. doi: 10.1046/j.1432-1033.2002.03040.x. [DOI] [PubMed] [Google Scholar]
- Quayle JM, Nelson MT, Standen NB. ATP-sensitive and inwardly rectifying potassium channels in smooth muscle. Physiol Rev. 1997;77:1166–1232. doi: 10.1152/physrev.1997.77.4.1165. [DOI] [PubMed] [Google Scholar]
- Quinn K, Cui Y, Giblin JP, Clapp LH, Tinker A. Do anionic phospholipids serve as cofactors or second messengers for the regulation of activity of cloned ATP-sensitive K+ channels? Circ Res. 2003;93:646–655. doi: 10.1161/01.RES.0000095247.81449.8E. [DOI] [PubMed] [Google Scholar]
- Quinn KV, Giblin JP, Tinker A. Multisite phosphorylation mechanism for protein kinase A activation of the smooth muscle ATP-sensitive K+ channel. Circ Res. 2004;94:1359–1366. doi: 10.1161/01.RES.0000128513.34817.c4. [DOI] [PubMed] [Google Scholar]
- Rusnak F, Mertz P. Calcineurin: form and function. Physiol Rev. 2000;80:1483–1521. doi: 10.1152/physrev.2000.80.4.1483. [DOI] [PubMed] [Google Scholar]
- Sampson LJ, Davies LM, Barrett-Jolley R, Standen NB, Dart C. Angiotensin II-activated protein kinase C targets caveolae to inhibit aortic ATP-sensitive potassium channels. Cardiovasc Res. 2007;76:61–70. doi: 10.1016/j.cardiores.2007.05.020. [DOI] [PubMed] [Google Scholar]
- Santana LF, Chase EG, Votaw VS, Nelson MT, Greven R. Functional coupling of calcineurin and protein kinase A in mouse ventricular myocytes. J Physiol. 2002;544:57–69. doi: 10.1113/jphysiol.2002.020552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuhmann K, Romanin C, Baumgartner W, Groschner K. Intracellular Ca2+ inhibits smooth muscle L-type Ca2+ channels by activation of protein phosphatase type 2B and by direct interaction with the channel. J Gen Physiol. 1997;110:503–513. doi: 10.1085/jgp.110.5.503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seino S, Miki T. Physiological and pathophysiological roles of ATP-sensitive K+ channels. Prog Biophys Mol Biol. 2003;81:133–176. doi: 10.1016/s0079-6107(02)00053-6. [DOI] [PubMed] [Google Scholar]
- Shi Y, Wu Z, Cui N, Shi W, Yang Y, Zhang X, et al. PKA phosphorylation of SUR2B subunit underscores vascular KATP channel activation by beta-adrenergic receptors. Am J Physiol Regul Integr Comp Physiol. 2007;293:R1205–R1214. doi: 10.1152/ajpregu.00337.2007.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, Chen X, Wu Z, Shi W, Yang Y, Cui N, et al. cAMP-dependent protein kinase phosphorylation produces interdomain movement in SUR2B leading to activation of the vascular KATP channel. J Biol Chem. 2008a;283:7523–7530. doi: 10.1074/jbc.M709941200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, Cui N, Shi W, Jiang C. A short motif in Kir6.1 consisting of four phosphorylation repeats underlies the vascular KATP channel inhibition by protein kinase C. J Biol Chem. 2008b;283:2488–2494. doi: 10.1074/jbc.M708769200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singer M, Coluzzi F, O'Brien AJ, Clapp LH. Reversal of life-threatening, drug-related potassium-channel syndrome by glibenclamide. Lancet. 2005;365:1873–1875. doi: 10.1016/S0140-6736(05)66619-6. [DOI] [PubMed] [Google Scholar]
- Sommer D, Coleman S, Swanson SA, Stemmer PM. Differential susceptibilities of serine/threonine phosphatases to oxidative and nitrosative stress. Arch Biochem Biophys. 2002;404:271–278. doi: 10.1016/s0003-9861(02)00242-4. [DOI] [PubMed] [Google Scholar]
- Thorneloe KS, Maruyama Y, Malcolm AT, Light PE, Walsh MP, Cole WC. Protein kinase C modulation of recombinant ATP-sensitive K+ channels composed of Kir6.1 and/or Kir6.2 expressed with SUR2B. J Physiol. 2002;541:65–80. doi: 10.1113/jphysiol.2002.018101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson AJ, Clapp LH. The molecular site of actions determines the ability of KATP channel inhibitors to reverse iNOS-mediated vasorelaxation. Cardiovasc Res. 2002;56:154–163. doi: 10.1016/s0008-6363(02)00504-7. [DOI] [PubMed] [Google Scholar]
- Wilson AJ, Jabr RI, Clapp LH. Calcium modulation of vascular smooth muscle ATP-sensitive K+ channels: role of protein phosphatase-2B. Circ Res. 2000;87:1019–1025. doi: 10.1161/01.res.87.11.1019. [DOI] [PubMed] [Google Scholar]
- Yang Y, Shi Y, Guo S, Zhang S, Cui N, Shi W, et al. PKA-dependent activation of the vascular smooth muscle isoform of KATP channels by vasoactive intestinal polypeptide and its effect on relaxation of the mesenteric resistance artery. Biochim Biophys Acta. 2008;1778:88–96. doi: 10.1016/j.bbamem.2007.08.030. [DOI] [PMC free article] [PubMed] [Google Scholar]