

Abstract
Background and purpose:
There are interactions between endothelin-1 (ET-1) and endothelial vascular injury in hyperhomocysteinemia (HHcy), but the underlying mechanisms are poorly understood. Here we evaluated the effects of HHcy on the endothelin system in rat carotid arteries.
Experimental approach:
Vascular reactivity to ET-1 and ETA and ETB receptor antagonists was assessed in rings of carotid arteries from normal rats and those with HHcy. ETA and ETB receptor expression was assessed by mRNA (RT-PCR), immunohistochemistry and binding of [125I]-ET-1.
Key results:
HHcy enhanced ET-1-induced contractions of carotid rings with intact endothelium. Selective antagonism of ETA or ETB receptors produced concentration-dependent rightward displacements of ET-1 concentration response curves. Antagonism of ETA but not of ETB receptors abolished enhancement in HHcy tissues. ETA and ETB receptor gene expressions were not up-regulated. ETA receptor expression in the arterial media was higher in HHcy arteries. Contractions to big ET-1 served as indicators of endothelin-converting enzyme activity, which was decreased by HHcy, without reduction of ET-1 levels. ET-1-induced Rho-kinase activity, calcium release and influx were increased by HHcy. Pre-treatment with indomethacin reversed enhanced responses to ET-1 in HHcy tissues, which were reduced also by a thromboxane A2 receptor antagonist. Induced relaxation was reduced by BQ788, absent in endothelium-denuded arteries and was decreased in HHcy due to reduced bioavailability of NO.
Conclusions and implications:
Increased ETA receptor density plays a fundamental role in endothelial injury induced by HHcy. ET-1 activation of ETA receptors in HHcy changed the balance between endothelium-derived relaxing and contracting factors, favouring enhanced contractility.
British Journal of Pharmacology (2009) 157, 568–580; doi:10.1111/j.1476-5381.2009.00165.x; published online 9 April 2009
This article is part of a themed section on Endothelium in Pharmacology. For a list of all articles in this section see the end of this paper, or visit: http://www3.interscience.wiley.com/journal/121548564/issueyear?year=2009
Keywords: hyperhomocysteinemia; carotid artery; endothelin-1; vasoconstriction; ETA receptors; relaxation, ETB receptors; nitric oxide; calcium
Introduction
Hyperhomocysteinemia (HHcy) is considered to be an independent risk factor for cardiovascular disease (Pisciotta et al., 2005). Many recent clinical studies have observed a positive association between increased plasma homocysteine and atherosclerosis (Weiss et al., 2003; Au-Yeung et al., 2004; Dusitanond et al., 2005). Experiments in animals demonstrated that homocysteine could be a pathogenic factor responsible for arterial damage, such as cell proliferation, increased matrix formation and endothelial injury (Tyagi et al., 1998; Pruefer et al., 1999; Chen et al., 2000). The underlying molecular mechanisms remain conjectural and are likely to be multifactorial.
The adverse vascular effects of homocysteine on endothelial function appear to be an increased oxidant stress with a depletion of biologically active NO (de Andrade et al., 2006). Although decreased NO bioavailability has generally been considered the leading process, recent evidence suggests that raised concentrations of endothelin might have a role in such vascular injury. There is a growing body of evidence suggesting that the endothelin system is affected by homocysteine (Drunat et al., 2001; 2002; Demuth et al., 2002). In particular, the treatment of human endothelial cells with non-cytotoxic concentrations of homocysteine leads to a dose-dependent decrease in both mRNA levels and endothelin-1 (ET-1) secretion (Demuth et al., 1999).
ET-1, the predominant member of the endothelin peptide family, is mainly synthesized by endothelial and vascular cells from a large precursor, prepro-ET-1, which is converted through enzymatic pathways into ET-1, a 21-amino-acid polypeptide (see Masaki, 1995). ET-1 has potent vasoconstrictor, mitogenic and pro-inflammatory properties and is implicated in numerous cardiovascular diseases (Yanagisawa et al., 1988; Tostes and Muscara, 2005). We previously demonstrated the existence of both ETA and ETB vasoconstrictor receptors located on the smooth muscle of rat carotid arteries and endothelial ETB receptors responsible for ET-1-induced vasorelaxation via the NO-cGMP pathway, vasodilator cyclooxygenase product(s) and the activation of voltage-dependent K+ (KV) channels (Tirapelli et al., 2005).
Despite evidence showing an interaction between HHcy and ET-1 on endothelial function, the underlying mechanisms were not fully clarified. Particularly, we aimed to investigate the effects of HHcy on endothelin receptors and the interactions of HHcy with mediators of vascular reactivity.
Methods
Homocysteine diet-induced HHcy
All animal procedures and the experimental protocols were carried out in accordance with the standards and policies of the University of São Paulo Animal Care and Use Committee, Brazil. Male Wistar rats (total 51) were provided by the Central Biotery of University of São Paulo – Campus Ribeirão Preto, and housed at the Biotery of Faculty of Pharmaceutical Sciences of Ribeirão Preto (FCFRP-USP), and kept on a 12 h light/12 h dark lighting schedule, at 23 ± 2°C, with food and water ad libitum. HHcy was induced by feeding rats a diet rich in homocysteine (equivalent to 1 g·kg−1) for 15 days, as previously described (de Andrade et al., 2006). After the 15 days, the HHcy group of rats weighed less (312 ± 5 g) than the age-matched control diet group (338 ± 6 g), although they were the same weights at the start of the diet (HHcy, 301 ± 9 g; control, 298 ± 12 g; n < 15 for both groups).
Determination of plasma homocysteine levels
Blood samples from control and HHcy rats were collected into EDTA and centrifuged at 3000×g for 20 min. In order to minimize the release of homocysteine from blood cells, iced tubes were used to collect blood and centrifugation was carried out at 4°C. Plasma was then stored at −70°C until assayed. Total homocysteine concentration was measured by mass spectrometry using the Q-TRAP system (de Andrade et al., 2006; Haddad et al., 2006).
Functional study of carotid rings
Animals were anaesthetized and killed by aortic exsanguination. The arteries were prepared for organ bath studies as described previously (de Andrade et al., 2006). The composition of the Krebs solution was (mmol·L−1): NaCl, 118.4; KCl, 4.7; KH2PO4, 1.2; MgSO4, 1.2; NaHCO3, 25.0; glucose, 11.6; CaCl2, 1.9.
The rings were stretched to an optimal basal tension of 1 g, which was determined by length–tension relationship experiments. Endothelial integrity was assessed qualitatively by the degree of relaxation caused by acetylcholine (1 µmol·L−1) in the presence of contractile tone induced by phenylephrine (0.1 µmol·L−1), the ring was discarded if relaxation with acetylcholine was less than 80%. For studies using endothelium-denuded vessels, the rings were discarded if there was any degree of relaxation.
Effects of HHcy on ET-1-induced contraction
To delineate the effect of HHcy on the contraction induced by ET-1, concentration–response curves for this peptide (1 pmol·L−1–30 nmol·L−1) were obtained in intact and denuded endothelium carotid rings from control and HHcy groups, in the absence or presence of BQ123 (1 µmol·L−1), a selective ETA antagonist (Ihara et al., 1992; Tirapelli et al., 2005), and BQ788 (100 nmol·L−1), a selective ETB antagonist (Ishikawa et al., 1994; Tirapelli et al., 2005). Contractile curves for the selective ETB receptor agonist {succinyl-[Glu9,Ala11,15]-ET-1(8-210)} (IRL1620) (Takai et al., 1992) were also obtained in intact-endothelium and denuded-endothelium rings.
The measurement of the contraction induced by big ET-1 served as an indicative of functional endothelin-converting enzyme (ECE) activity and concentration–response curves for this peptide (1 pmol·L−1–300 nmol·L−1) were obtained in the absence or in the presence of phosphoramidon (a non-selective ECE inhibitor; 10 mmol·L−1, 30 min).
The curves for ET-1 were performed in endothelium-intact rings, in the absence or after incubation for 30 min with the NO synthase inhibitor, NG-nitro-l-arginine methyl ester (L-NAME, 100 µmol·L−1) or a cyclooxygenase inhibitor, indomethacin (10 µmol·L−1). In addition, endothelium-intact rings were pre-incubated for 30 min with AH6809 (antagonist of PGF2α receptors, 10 µmol·L−1) or SQ29548 [PGH2/thromboxane A2 (TXA2) receptor antagonist, 1 µmol·L−1].
To analyse the relative contribution of intracellular calcium (Ca2+) to the ET-1 contractile response, the normal Krebs solution was replaced by a Ca2+-free solution. The rings were exposed to this solution for 1 min and then were stimulated with ET-1 (1 nmol·L−1). In addition, the role of extracellular Ca2+ mobilization was investigated by performing concentration–response curves to Ca2+ in the presence of ET-1. Endothelium-intact rings were first contracted for approximately 90 min with phenylephrine (0.1 µmol·L−1) to deplete the intracellular Ca2+ stores in Ca2+-free solution containing EGTA (1 mmol·L−1) and then rinsed in Ca2+-free solution (without EGTA) containing ET-1 (1 nmol·L−1).
Rho-kinase participation in the ET-1-induced response was determined in the presence of increased concentrations of Y-27632 (Rho-kinase selective inhibitor) (Chitaley and Webb, 2002). The inhibitor was added 30 min prior to the data being collected for the construction of the curve and the responses were compared with those observed in time-matched control experiments.
Contractions were recorded as changes in the displacement (mN·mg tissue−1) from baseline.
Effects of HHcy on ET-1-induced relaxations
Endothelium-intact and endothelium-denuded rings were pre-contracted with Phe (0.1 and 0.03 µmol·L−1, respectively, to induce contractions of similar magnitude). After a stable and sustainable contraction was obtained, the selective ETB receptor agonist, IRL1620 (100 pmol·L−1–30 nmol·L−1) was added cumulatively to the organ bath.
In the endothelium-intact rings, the same experiments were conducted in the absence and presence of BQ123 (1 µmol·L−1) and BQ788 (100 nmol·L−1). The effects of IRL1620 were also evaluated in the absence or after incubation for 30 min with L-NAME (100 µmol·L−1), indomethacin (10 µmol·L−1) and tetraethylammonium chloride (TEA, non-selective K+ channel blocker 10 mmol·L−1, Nelson and Quayle (1995). Relaxation was expressed as the percentage change from the phenylephrine-contracted levels. As we noted that L-NAME enhanced phenylephrine-induced contraction, the rings with intact endothelium exposed to these compounds were pre-contracted with phenylephrine 0.03 µmol·L−1, to induce a magnitude of contraction similar to that found in the intact endothelium rings not exposed to the inhibitors. Relaxation was expressed as the percentage change from the phenylephrine-contracted levels.
RT-PCR
The RT-PCR was carried out with rat carotid arteries with intact endothelium, as previously described by Tirapelli et al. (2006). Total cell RNA was isolated from carotid arteries using Trizol Reagent. After DNA digestion, total RNA was used for RT in the presence of an RNase inhibitor (RNase OUT®, Gibco BRL), 400 U of Moloney murine leukemia virus RT (Gibco BRL) and 1 µg of oligo (dT)12-18 primer at 21°C for 10 min, 42°C for 60 min and 99°C for 10 min, according to manufacturer's specifications. PCR primers were designed on the basis of published rat cDNA sequences for glyceraldehyde 3-phosphate dehydrogenase (GAPDH), ETA/ETB receptors and are as follows (5′-3′): ETA, antisense primer CTGTGCTGCTCGCCCTTGTA, sense primer GAAGTCGTCCGTGGGCATCA (216 bp fragment) and ETB, antisense primer CACGATGAGGACAATGAGAT, sense primer TTACAAGACAGCCAAAGACT (565 bp fragment); GAPDH, anti-sense primer CACCACCCTGTTGCTGTA, sense primer TATGATGACATCAAGAAGGTGG (219-bp fragment). PCR products (10 µL per lane) were electrophoresed using 1% agarose gel containing ethidium bromide (0.5 µg·mL−1). The gel was subjected to ultraviolet light and photographed. The band intensities were measured using a software package (Kodak Digital Science, Eastman Kodak Company, New Haven, CT, USA), and the signals are reported relative to the intensity of the GAPDH amplification in each co-amplified sample.
Immunohistochemistry
Carotid arteries were fixed in Methacarn (60% methanol, 30% chloroform and 10% acetic acid). The paraffin-embedded longitudinal sections (7 µm) were incubated with 3% H2O2 and a Pierce solution to block endogenous peroxidase and biotin respectively. Then, the sections were incubated (humidified box, 4°C) with primary polyclonal antibodies against rat ETA and ETB receptors (1:10 dilution; Alomone Labs Ltd, Jerusalem, Israel), with a biotin-conjugated secondary anti-rabbit antibody (1:1000, Vector Laboratories Inc, Burlingame, CA, USA) and with streptavidin-conjugated peroxidase (Vectastain ABC kit, Vector Laboratories Inc, Burlingame, CA, USA). Colour was developed by the addition of DAB (Sigma). Sections were lightly stained in haematoxylin, dehydrated with alcohol and xylene. To evaluate the background reaction, the procedures were also performed on sections incubated only with the secondary antibody (indirect technique) or in the absence of antibodies (direct technique).
Autoradiography
Carotid segments were frozen in isopentane at −30°C, and kept frozen at −70°C. Cross sections (16 µm) of the vessels were cut in a cryostat as previously described (De Oliveira et al., 1995; Viswanathan et al., 1997).
[125I]ET-1 (74 TBq·mmol−1, Amersham Biosciences Corp, Piscataway, NJ, USA) was used as the ligand. The binding assay used was identical to that previously described (De Oliveira et al., 1995; Viswanathan et al., 1997). To characterize ET receptor subtypes, consecutive sections were incubated with [125I]ET-1, with or without single 10 µmol·L−1 concentrations of the ETA receptor antagonist BQ123, and the ETB receptor specific ligand BQ788 (RBI, Natick, MA, USA). Sections were dried after washing and exposed to a pellicle sensitive to radiation (Amersham Corp., Arlington Heights, IL, USA), along with 16-mm-thick sections of 14C (ARC-140B, Art Incorporated, St Louis, MO, USA). Films were developed in ice-cold Kodak D-19 developer (Eastman Kodak Co., Rochester, NY, USA) for 4 min, fixed in Kodak rapid fixer (with a hardener) for 4 min at room temperature, and then rinsed in water for 10 min. The films with autoradiographic images of the binding and the standards were used to measure the mean optical densities from different areas of interest on the images. The mean optical density measurements per unit area were obtained from both the standard and the sample images. The mean optical density measurements from images of the plastic standards and their corresponding disintegrations min−1 per mg of plastic values were used to generate standard curves by nonlinear fitting using the computerized NIH Image 1.4 analysis system (Nazarali et al., 1989). These standard curves were used to obtain the disintegrations min−1·mg−1 of plastic values for the samples. After correcting for the specific activity of the ligand, the values of fmol·mg−1 of protein were obtained. The quantification program that we used initially provided the optical density values per unit area, and statistically significant differences were obtained in these values (results not shown) whenever significant differences were obtained using the fmol·mg−1 protein values.
Measurement of ET-1 by ELISA
Rat carotids (5–6 mm in length) were isolated. The tissues were transferred to an organ bath containing 2.5 mL of Krebs solution at 37°C that was continuously bubbled with a mixture of 95% O2 and 5% CO2. The arteries were placed in the Krebs solution for 30 min. Next, the supernatant were systematically collected and stored at −70°C for later determination of ET-1 using commercially available enzyme immunoassay kits (Amersham). For each experimental group, control samples (for the estimation of basal release) were collected.
Levels of nitrogen oxides in vascular homogenates
Nitrite and nitrate levels were measured in supernatants from total carotid artery homogenates prepared under liquid N2. Following a centrifugation at 3000×g for 10 min, supernatants were assayed for nitrite or nitrate through chemiluminescence, using a Nitric Oxide Analyser (Sievers, Model 280), as described (Leite et al., 2003; de Andrade et al., 2006).
Lucigenin-amplified chemiluminescence assays
Lucigenin assays were performed as previously described (Brandes et al., 1997; Li et al., 1998; de Andrade et al., 2006), with a final lucigenin concentration of 5 µmol·L−1. Also as previously described, we used two cell permeable superoxide dismutase (SOD) mimetics, polyethylene glycol (PEG)-SOD and manganese (III) tetrakis (4-benzoic acid) porphyrin chloride (MnTBAP), and a superoxide scavenger, Tiron, to confirm the presence of superoxide in the assays.
Data and statistical analysis
Contractions were recorded as changes in the displacement (mN·mg tissue−1) from baseline. Relaxation was expressed as the percentage change from the phenylephrine-contracted levels. Agonist concentration–response curves were fitted using a nonlinear interactive fitting program (Graph Pad Prism 3.0; GraphPad Software Inc., San Diego, CA, USA). Agonist potencies were expressed as pD2 (negative logarithm of the molar concentration of agonist producing 50% of the maximum response), and maximum response was expressed as Emax (maximum effect elicited by the agonist). For Rho-kinase experiments, using the inhibitor Y-27632, Emax and pD2 were calculated as the % maximal fall in the ET-1 response, induced by Y-27632.
The results were reported as mean ± SEM. Statistically significant differences were calculated by one-way anova or Student's t-test. Post hoc comparisons were performed using Bonferroni's multiple comparison test. P < 0.05 was considered as statistically significant.
Drugs
The following drugs were used: DL-homocysteine-thiolactone (Acrós Organics/Fischer Scientific, WI, USA); phenylephrine hydrochloride, acetylcholine hydrochloride, ET-1, PEG-SOD and Tiron (Sigma, St. Louis, MO, USA); L-NAME, TEA (Sigma/RBI, Natick, MA, USA); big ET-1, indomethacin, MnTBAP (Calbiochem, USA); BQ123, BQ788, IRL1620, (American Peptide Company, Sunnyvale, CA, USA). Indomethacin was dissolved in Tris buffer (pH: 8.4); the other drugs were dissolved in distilled water.
Results
Plasma homocysteine measurements
Treatment for 15 days with the homocysteine-rich diet (1 g·kg body weight−1) induced a 10-fold increase in plasma homocysteine levels (control: 6.2 ± 0.5 vs. HHcy: 62.7 ± 6.9 µmol·L−1; P < 0.01, Student's t-test).
Effect of HHcy on vascular reactivity to ET-1
Initially, we investigated a possible influence of HHcy on the ET-1-induced contraction. This ET-1-induced contraction was significantly increased in arterial rings from HHcy rats (P < 0.05, anova; Figure 1). This difference was endothelium-dependent, as after removal of endothelium, both sets of rings were equally responsive to ET-1. Note that the ET-1-induced contractions in arteries from the HHcy group were unaffected by removal of endothelium (Figure 1). The pD2 values were not altered.
Figure 1.
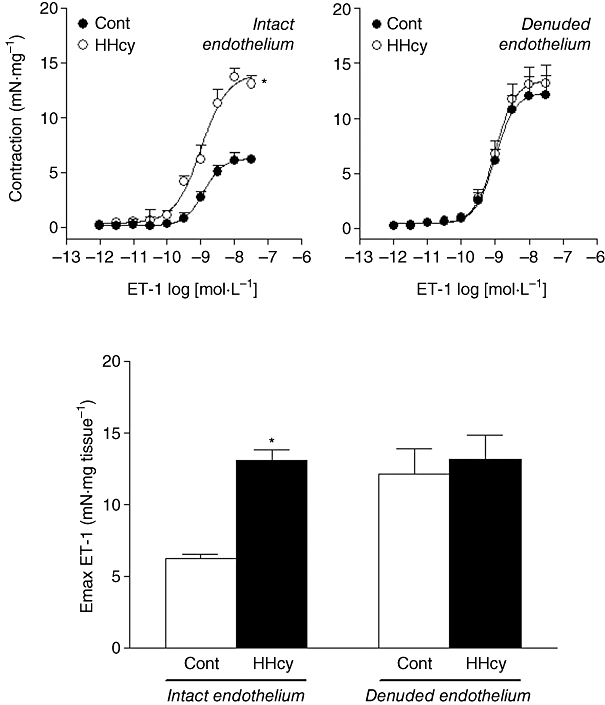
Concentration–response curves for ET-1 obtained in endothelium-intact and endothelium-denuded and isolated carotid rings from control (Cont) and HHcy rats. Values are means ± SEM; n= 8 for intact endothelium and n= 7 for denuded rings. *Compared with respective Cont group (one-way anova followed by Bonferroni's multiple comparison test, P < 0.05). ET-1, endothelin-1; HHcy, hyperhomocysteinemia.
This enhanced contraction was not evoked by alterations in ETB receptors, as IRL1620, a selective agonist for ETB receptors (100 nmol·L−1–300 nmol·L−1), was not able to induce more than a slight contraction in control or HHcy rat arterial rings (Emax values, control: 8.23 ± 0.10 vs. HHcy: 9.48 ± 0.15 mN·mg tissue−1).
Effects of HHcy on ETA and ETB receptors
In carotids from control rats, BQ123 (1 µmol·L−1) produced a concentration-dependent rightward displacement of the ET-1 contractile response with reduction on Emax values. Furthermore, such displacement and Emax reduction were more pronounced in the arteries from the HHcy rats (Figure 2, Table 1). Similarly, BQ788 (0.1 µmol·L−1) right-shifted the ET-1 concentration–response curves and reduced the Emax in arteries from both control and HHcy groups, but without differences between these two groups (Figure 2, Table 1).
Figure 2.
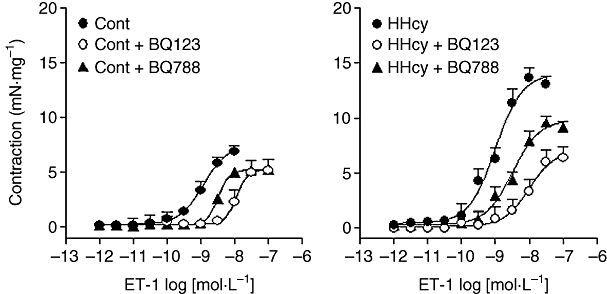
Concentration–response curves for ET-1 obtained in endothelium-intact carotid rings from control (Cont) and HHcy rats in the absence or presence of BQ123 (1 µmol·L−1) or BQ788 (0.1 µmol·L−1), ETA and ETB antagonists respectively. The concentration–response curves in both sets of rings were significantly shifted by either antagonist. Changes in pD2 and Emax values are summarized in Table 1. Data shown here are means ± SEM, from seven preparations in each group. ET-1, endothelin-1; HHcy, hyperhomocysteinemia.
Table 1.
Effect of HHcy on the Emax (mN·mg−1) and pD2 values for endothelin-1 in rat carotid arteries in the absence or presence of BQ123 (1 µmol·L−1) or BQ788 (0.1 µmol·L−1)
|
Control |
HHcy |
|||
|---|---|---|---|---|
| Emax | pD2 | Emax | pD2 | |
| 7.29 ± 0.65 | 8.92 ± 0.09 | 13.82 ± 0.07 | 8.99 ± 0.06 | |
| BQ123 (1 µmol·L−1) | 5.15 ± 0.99* | 7.94 ± 0.10* | 6.35 ± 0.09* | 8.00 ± 0.09* |
| BQ788 (0.1 µmol·L−1) | 5.28 ± 0.33* | 8.48 ± 0.09* | 9.25 ± 0.05* | 8.46 ± 0.08* |
Values are means ± SEM of n= 7 preparations.
Compared with respective control in the absence of antagonists (P < 0.05; one-way anova).
The RT-PCR analysis showed that rat carotid arteries express mRNA for both ETA and ETB receptors and that HHcy did not alter the gene expression for these receptors (Figure 3). The immunohistochemical analysis revealed the presence of ETA and ETB receptors in arterial smooth muscle cells. In endothelial cells, positive immunostaining for ETB, but not for ETA receptors, was detected. The ETA receptor expression on the media layer of arteries from HHcy rats was higher than in arteries from control rats (Figure 4). HHcy also resulted in increased binding of [125I]-ET-1 in the media layer (Figure 5). In addition, ET-1 binding was displaced by unlabelled ET-1 and by the ETA antagonist BQ123, but not by the ETB-specific ligand BQ788. These observations on ET-1 binding indicate that there was a 15% enhancement of ETA receptor density in the arteries obtained from HHcy rats, relative to that in control arteries.
Figure 3.
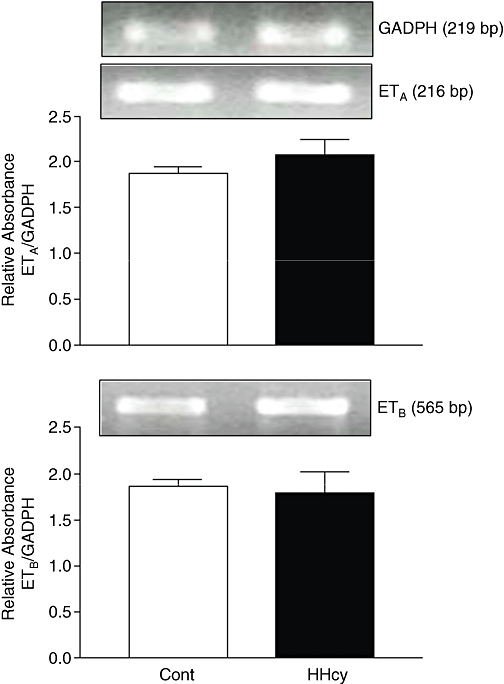
Representative RT-PCR products of 2 µg total RNA extracted from carotid arteries of control (Cont) and HHcy rats. The bar graphs show the relative absorbance values of ETA and ETB receptor bands in endothelium-intact rings. ETA and ETB values were normalized to the corresponding GAPDH values, used as internal standard. Results are reported as means ± SEM from five experiments. GAPDH, glyceraldehyde 3-phosphate dehydrogenase; HHcy, hyperhomocysteinemia.
Figure 4.
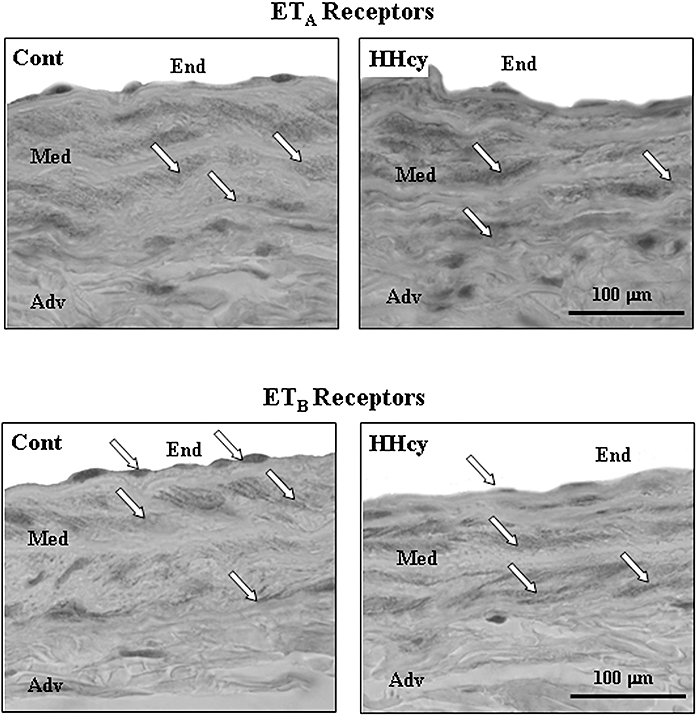
Representative immunohistochemical photomicrographs of ETA and ETB receptors in rat carotid artery sections. Arrows indicate expression of ETA receptor in smooth muscle cells and ETB in both endothelial and smooth muscle cells (magnification: × 400). Cont, control; HHcy, hyperhomocysteinemia.
Figure 5.
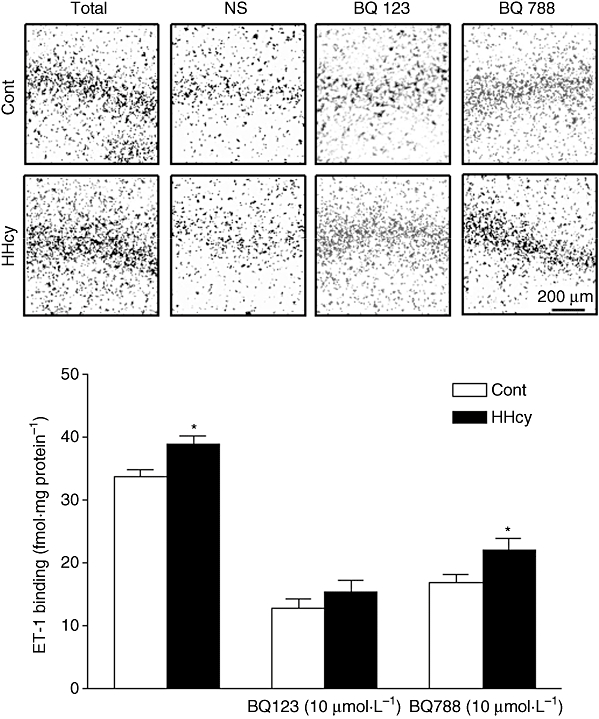
Autoradiography of [H3]-ET-l binding in rat carotid artery. Representative autoradiography of ETA and ETB receptors in rat carotid artery sections: total binding, non-specific binding (NS) and in the presence of BQ123 (10 µmol·L−1) or BQ788 (10 µmol·L−1). Summary data are shown below as means ± SEM from three experiments. *P < 0.05, Student's t-test; different from Cont groups. Cont, control; ET-1, endothelin-1; HHcy, hyperhomocysteinemia.
Effects of HHcy on ET-1 production
The ET-1 precursor peptide, big ET-1, induced contractions of endothelium-intact rat carotid arteries obtained from the control and HHcy group (Figure 6). HHcy induced a decrease in Emax and pD2 values for big ET-1 in arteries with intact endothelium. To confirm that the contraction to big ET-1 was because it was converted to ET-1, the ECE activity was being assayed, the ECE inhibitor, phosphoramidon, was used. As expected, phosphoramidon reduced the contractile response to big ET-1 in arteries from control rats (Figure 6).
Figure 6.
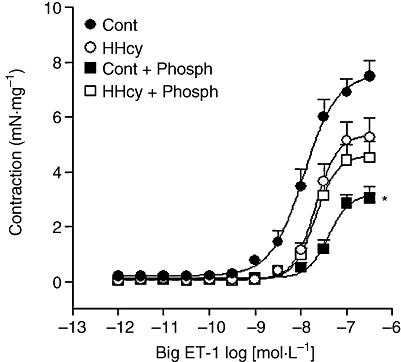
Concentration–response curves for big ET-1 obtained in endothelium-intact carotid rings from control (Cont) and HHcy rats, in the absence or presence of phosphoramidon (Phosph; 10 mmol·L−1). Values are means ± SEM from five preparations in each group. *Compared with respective Cont group in the absence of inhibitor (one-way anova followed by Bonferroni's multiple comparison test, P < 0.05). HHcy, hyperhomocysteinemia.
In addition, HHcy did not alter prepro-ET-1 gene expression in rat carotid arteries (control: 1.09 ± 0.09 vs. HHcy: 0.96 ± 0.13 relative absorbance). Indeed, the basal release of ET-1 did not differ in carotid rings from both groups (control: 136 ± 36 vs. HHcy 140 ± 50 pg mL·mg tissue−1).
Role of Rho-kinase activity on ET-1-induced responses
The ET-1-induced Emax was reduced in a concentration-dependent manner by the selective Rho-kinase inhibitor, Y-27632, in carotid arteries obtained from the control and HHcy rats. The maximal fall in the ET-1 response was 12 ± 1% in control arteries and 13 ± 3% in HHcy arteries. The pD2 values for Y-27632 were 6.74 ± 0.11 in control and 6.15 ± 0.21 in HHcy arteries. These pD2 values were significantly different (P < 0.05, Student's t-test) between control and HHcy tissues.
Contribution of intracellular and extracellular Ca2+ to the changes in ET-1 vascular reactivity
In Ca2+-free medium, ET-1-induced contractions reached a peak and then returned to baseline levels. The magnitude of the peak response to ET-1 (1 nmol·L−1) was enhanced in the arteries with intact endothelium obtained from HHcy rats (1.34 ± 0.23 mN·mg tissue−1) when compared with the control rats (0.38 ± 0.11 mN·mg tissue−1, P < 0.01, Student's t-test). Similarly, CaCl2-induced contraction was greater in carotid rings obtained from HHcy rats (6.86 ± 0.14 mN·mg tissue−1) than in those from control rats (2.82 ± 0.34 mN·mg tissue−1, P < 0.001, Student's t-test).
Contribution of NO and endothelial cyclooxygenase metabolites to the ET-1 response
Incubation with L-NAME significantly enhanced the Emax induced by ET-1 in arterial segments obtained from control, but not in those obtained from HHcy rats (Figure 7). Indomethacin did not alter Emax values for ET-1 in the endothelium-intact carotid rings obtained from control, whereas in the arteries obtained from HHcy rats, such Emax values were reduced, compared with the values obtained when the inhibitor was absent (Figure 7).
Figure 7.
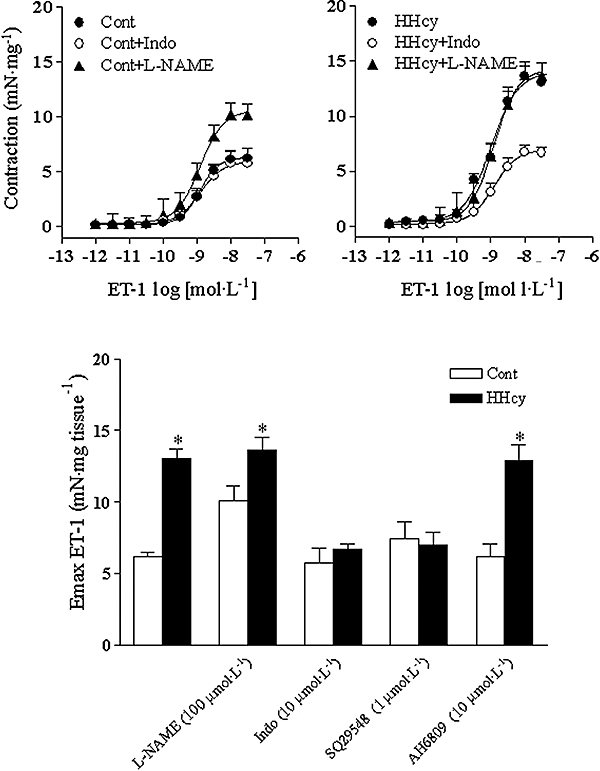
Concentration–response curves for ET-1 obtained in endothelium-intact carotid rings from control (Cont) (left-hand graph) and HHcy (right-hand graph) rats in the absence or presence of L-NAME (100 µmol·L−1), indomethacin (Indo) (10 µmol·L−1), inhibitors of nitric oxide synthase and cyclooxygenase respectively. In the lower graph are shown the effects on Emax for ET-1 of L-NAME (100 µmol·L−1), Indo (10 µmol·L−1), SQ29548 (TXA2 receptor antagonist; 1 µmol·L−1) or AH6809 (PGF2α receptor antagonist, 10 µmol·L−1). Values are means ± SEM of five to seven independent preparations. *Compared with respective Cont group (one-way anova followed by Bonferroni's multiple comparison test, P < 0.05). ET-1, endothelin-1; HHcy, hyperhomocysteinemia; L-NAME, NG-nitro-L-arginine methyl ester; SQ29548.
The PGH2/TXA2 receptor antagonist, SQ29548, reduced the ET-1 Emax in arteries from HHcy group (Figure 7). No changes in pD2 values were detected (data not shown). On the other hand, pre-incubation with AH6809 did not produce changes in the concentration–response curves for ET-1 (data not shown).
IRL1620-induced relaxation
The selective agonist at ETB receptors, IRL1620, induced relaxation in endothelium-intact rings from both groups. This relaxation was significantly reduced by BQ788, but not by BQ123. Moreover, HHcy impaired this IRL1620-induced relaxation (Figure 8 top). There were no differences in the pD2 values of IRL1620 in endothelium-intact rings in the absence or presence of antagonists. Removal of the endothelium abolished this relaxation response in arteries from both groups.
Figure 8.
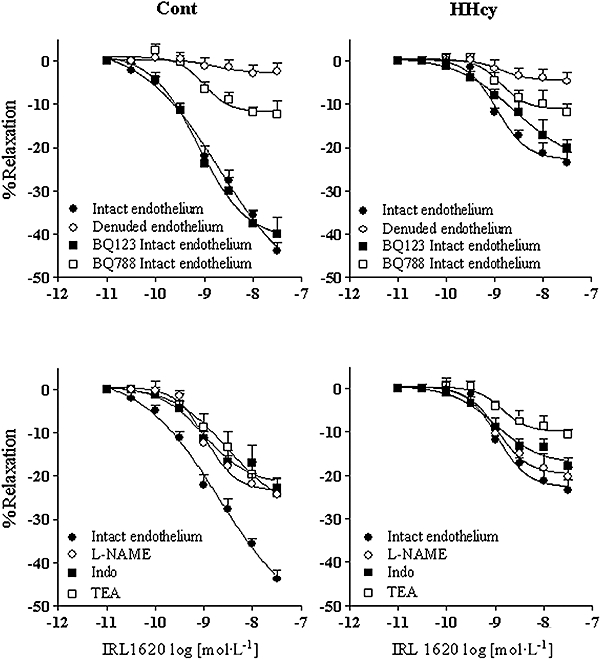
Relaxation responses induced by IRL1620 (ETB agonist) on rat carotid rings pre-contracted with phenylephrine. Top: the concentration–response curves for IRL1620 were obtained in endothelium-denuded and endothelium-intact rings, in the absence or presence of BQ123 and BQ788. Bottom: the concentration–response curves were obtained in the absence or in the presence of L-NAME (100 µmol·L−1), indomethacin (10 µmol·L−1) and TEA (10 mmol·L−1), from Cont and HHcy rats. Values are means ± SEM of five to seven independent preparations. BQ123; BQ788, [N-cis-2,6-dimethyl-piperidinocarbonyl-L-J-methylleucyl1-D-1methoxycarbonyl tryptophanyl-D-norleucine]; Cont, control; ET-1, endothelin-1; HHcy, hyperhomocysteinemia; IRL1620, TEA, tetraethylammonium.
In order to verify the mechanisms underlying ETB-induced relaxation, endothelium-intact rings were exposed to L-NAME or indomethacin. Both inhibitors reduced IRL1620-induced relaxation in the arteries obtained from control, but not in the arteries from the HHcy rats, and abolished the differences between the groups in the IRL1620 responses. TEA also reduced the relaxation induced by IRL1620 (Figure 8 bottom). The mean pD2 of the relaxant IRL1620 concentration–response curves, as well as the Emax values, in the presence or absence of the previously mentioned inhibitors, are given in Table 2.
Table 2.
Relaxation (%) induced by IRL1620 in the presence of BQ123 (1 µmol·L−1), BQ788 (0.1 µmol·L−1), L-NAME (100 µmol·L−1), indomethacin (10 µmol·L−1) or TEA (10 mmol·L−1)
|
Control |
HHcy |
|
|---|---|---|
| IRL1620 Emax (%) | ||
| Intact endothelium | 44 ± 2 | 24 ± 2* |
| Denuded endothelium | 2.5 ± 2.1 | 4.7 ± 2.1 |
| BQ123 (1 µmol·L−1) | 40 ± 4 | 20 ± 2† |
| BQ788 (0.1 µmol·L−1) | 12 ± 3 | 12 ± 2 |
| L-NAME (100 µmol·L−1) | 24 ± 1 | 20 ± 2 |
| Indomethacin (10 µmol·L−1) | 23 ± 2 | 18 ± 2 |
| TEA (10 mmol·L−1). | 24 ± 3 | 11 ± 1 |
| pD2 (−log EC50) | ||
|---|---|---|
| Intact endothelium | 8.68 ± 0.22 | 8.95 ± 0.08 |
| Denuded endothelium | 8.81 ± 0.26 | 8.86 ± 0.09 |
| BQ123 (1 µmol·L−1) | 9.11 ± 0.06 | 8.59 ± 0.08 |
| BQ788 (0.1 µmol·L−1) | 9.01 ± 0.12 | 8.87 ± 0.07 |
| L-NAME (100 µmol·L−1) | 8.95 ± 0.08 | 8.90 ± 0.07 |
| Indomethacin (10 µmol·L−1) | 8.98 ± 0.15 | 8.89 ± 0.12 |
| TEA (10 mmol·L−1). | 8.49 ± 0.07 | 8.86 ± 0.10 |
Values are means ± SEM of n= 5–7 preparations.
Compared with arteries from control (P < 0.05; one-way anova).
Compared with respective control in the absence of inhibitors (P < 0.01; one-way anova).
Carotid arteries obtained from HHcy rats also had reduced nitrite levels (control: 3.42 ± 0.08 vs. HHcy: 2.86 ± 0.21 µmol·L−1, protein−1P < 0.05, Student's t-test), but not in the nitrate levels (control: 10.41 ± 5.74 vs. HHcy: 11.71 ± 4.28 µmol·L−1 protein−1). In parallel, our data indicated an increased output of superoxide from carotid segments, as assessed by chemiluminescence, in HHcy rats (control: 1375 ± 199 vs. HHcy: 2468 ± 305 cpm mg tissue). MnTBAP (5 µmol·L−1) and PEG-SOD (200 U·mL−1), cell permeable SOD mimetics and Tiron, a superoxide scavenger (1 mmol·L−1), abolished superoxide generation.
Discussion and conclusions
The present findings demonstrate that a diet rich in homocysteine thiolactone caused a 10-fold increase in plasma homocysteine in rats and, although this model does not exactly mimic the human condition, it provides important information about the vascular consequences of HHcy in rats (Refsum et al., 1998; Hill et al., 2002; Bonaventura et al., 2004, de Andrade et al., 2006).
Our present results also showed that vascular ET-1 receptor density was enhanced in rat arteries exposed to high concentrations of homocysteine and this increased density played an active role in endothelium-dependent vascular contraction. However, thus far there has been little information regarding the interactions of ET-1 in HHcy. We therefore carried out experiments on isolated carotid arteries from control and HHcy rats. Interestingly, we observed that high plasma levels of homocysteine led to a significant enhancement of vascular reactivity to ET-1.
ET-1 is produced by a variety of cells but predominantly by endothelial cells (Luscher and Barton, 2000). Endothelial dysfunction is accepted as one of the earliest process in the development of vascular diseases such as atherosclerosis (Traupe et al., 2003) and two mechanisms have been described as underlying the endothelial dysfunction in HHcy: endothelial injury and the reduced bioavailability of NO (Powers et al., 2003; de Andrade et al., 2006). The present report addressed changes in ETA/ETB receptor density, intracellular transduction signalling (Ca2+ and Rho-kinase pathways), ET-1 synthesis and/or bioavailability, participation of prostanoids and the relaxing factors released after activation of ETB receptors.
We first evaluated whether endothelial cells modulated ET-1 responses in arteries from HHcy rats. HHcy enhanced ET-1 responses only in arteries with intact endothelium, which reinforces the role of the endothelium in the enhanced ET-1 vascular responsiveness.
Although HHcy had no effects on either ETA or ETB receptor gene expression, autoradiographical or immunohistochemical assays showed that ETA receptor expression is significantly enhanced in arteries obtained from HHcy rats, suggesting that the number of ETA receptors is one of the factors involved in the enhanced reactivity to ET-1 in HHcy rats. This differs from in vitro experiments where high homocysteine levels increased ET-1 mRNA (Drunat et al., 2002), and changes in ET-1 release have previously been associated with an increase in vascular resistance.
It should be pointed out that the effects we observed also involved a reduction of ECE activity. Altered ECE activity might lead to changes in ET-1 production, which could alter the density of endothelin receptors, thus promoting differences in the vascular responsiveness to exogenous ET-1. Exposure of the ECE to increased superoxide, which can occur in specific microenvironments, could decrease ECE activity (Lopez-Ongil et al., 2000). We have previously observed increased generation of O2− in arteries from HHcy rats (de Andrade et al., 2006), which emphasizes the role that this reactive oxygen species has in the observed changes. The precise nature of such interaction requires further study.
Enhanced contractility to ET-1, triggered by HHcy, also involves the activation of intracellular signalling pathways. Basal peripheral and systemic vascular tone depends on Rho-associated kinase pathways (Büssemaker et al., 2007), which negatively regulate the vascular bioavailability of NO by increasing superoxide concentrations. In this study, the effect of the Rho-kinase inhibitor (Y-27632) on HHcy-treated rats clearly indicated that the contraction evoked by ET-1 required the activation of Rho-kinase pathway. Rho/Rho-kinase signalling, as well as Ca2+ mobilization, is thought to play important roles in vasoconstriction and may contribute to the aetiology of cardiovascular diseases both in experimental animals and humans (Jin et al., 2004; Nakakuki et al., 2005). In line with these previous observations, increased O2− levels can modify the contraction of vascular smooth muscle through activation of the Rho-kinase pathway (Jin et al., 2004).
Changes in phosphoinositide turnover and transmembrane Ca2+ influx in vascular smooth muscle cells have been described (Okatani et al., 2001). The present study showed a significant increase in vascular sensitivity to extracellular Ca2+ in HHcy rats, which is in agreement with the suggestion that HHcy increases ET-1-stimulated influx of Ca2+. Furthermore, we found that the potentiating effect of HHcy on ET-1-induced contraction may also involve increased release of intracellular Ca2+. Interestingly, there is a previous report that HHcy induced mobilization of intracellular Ca2+ pools and activated a variety of kinases in the mouse hippocampus (Robert et al., 2005). This suggests a major role for Rho-kinase in HHcy-induced changes in vascular reactivity, in addition to its role in changing intracellular and extracellular Ca2+.
Additionally, L-NAME significantly enhanced the ET-1-induced response in carotid arteries from control, but not in those from HHcy rats, in the presence of endothelium, which suggests that the enhanced responsiveness of HHcy carotid arteries to ET-1 was partially triggered by impaired NO release. Indomethacin, by contrast, significantly inhibited the Emax induced by ET-1 in arteries from HHcy rats. The contribution of endothelial prostanoids derived from the cyclooxygenase pathway on the modulation of the contraction triggered in HHcy rats has been previously reported (Ungvari et al., 2000; Bagi et al., 2001). On analysing the possible prostanoid(s) involved in the enhanced reactivity to ET-1, we found that SQ29548, a competitive antagonist of PGH2/TXA2 receptors, reduced the Emax evoked by ET-1 in carotid rings obtained from HHcy rats, but not in carotid rings obtained from control rats. Thus, our data corroborate previous findings, namely that HHcy enhances the production of TXA2 (Ungvari et al., 2000; Bagi et al., 2002). We discounted the contribution of PGF2α, as AH6809, an antagonist of PGF2α receptors, had no effect on such contractions.
Finally, increased generation of reactive oxygen species in the arteries obtained from HHcy rats may lead to NO inactivation, thus reducing NO bioavailability, and increasing the formation of peroxynitrite and secondary nitro-oxidative species (de Andrade et al., 2006). Indeed, IRL1620-induced relaxation was decreased in the arteries obtained from HHcy rats, possibly due to changes in the NO and cyclooxygenase pathways. The decreased bioavailability of NO could provide an additional mechanism by which HHcy induces enhanced ET-1 vascular reactivity.
In summary, this is the first study using rat carotid rings to investigate the molecular mechanisms of ET-1-induced vascular dysfunction in rats with HHcy. Observations made in HHcy arteries provide direct evidence that ETA receptors not only modulate the contractile response to ET-1, but also increase the generation of potent constrictor factors such as TXA2 and the Rho-kinase pathway. Staining for ETA receptors in the media layer reinforces the premise that ETA is a major player in the vascular system in rats with HHcy. This increase in ETA receptors, combined with redox imbalance, appear to play an important role in ET-1-induced vascular contraction, and are probably key mediators of the long-term vascular structural adaptation to raised homocysteine concentrations.
 |
Acknowledgments
We are most grateful to Juliana A. Vercesi, Miriam C. C. de Melo, Mayara Santos Gomes (FCFRP-USP), Laura Ventura (Incor, FMUSP) and Eleni L. T. Gomes (FMRP-USP), for their technical assistance. This work was supported by FAPESP-Brazil (01/05549-3 and 02/8045-9) and by FAPESP Grant 04/13683-0 to FRML.
Glossary
Abbreviations:
- BQ123
c(DTrp – Dasp-Pro-Dval-Leu)
- BQ788
[N-cis-2,6-dimethyl-piperidinocarbonyl-L-J-methylleucyl1-D-1methoxycarbonyl tryptophanyl-D-norleucine]
- ECE
endothelin-converting enzyme
- ET-1
endothelin-1
- HHcy
hyperhomocysteinemia
- IRL1620
{succinyl-[Glu9,Ala11,15]-ET-1(8-210)}
- L-NAME
NG-nitro-L-arginine methyl ester
- SQ29548
([1S-[1alpha-2alpha(Z)3alpha,4alpha]]-7-[3-[(phenylamino)carbonyl]hydrazino]methyl]-7-oxabicyclo[2.2.1]hept-2-yl]-5-heptenoic acid)
- TEA
tetraethylammonium
- TXA2
thromboxane A2
Conflict of interest
The authors state no conflict of interest.
References
- de Andrade CR, Fukada SY, Olivon VC, de Godoy MA, Haddad R, Eberlin MN, et al. Alpha1D-adrenoceptor-induced relaxation on rat carotid artery is impaired during the endothelial dysfunction evoked in the early stages of hyperhomocysteinemia. Eur J Pharmacol. 2006;543:83–91. doi: 10.1016/j.ejphar.2006.06.003. [DOI] [PubMed] [Google Scholar]
- Au-Yeung KK, Woo CW, Sung FL, Yip JC, Siow YL, Karmin O. Hyperhomocysteinemia activates nuclear factor-kappaB in endothelial cells via oxidative stress. Circ Res. 2004;94:28–36. doi: 10.1161/01.RES.0000108264.67601.2C. [DOI] [PubMed] [Google Scholar]
- Bagi Z, Ungvari Z, Szollar L, Koller A. Flow-induced constriction in arterioles of hyperhomocysteinemic rats is due to impaired nitric oxide and enhanced thromboxane A(2) mediation. Arterioscler Thromb Vasc Biol. 2001;21:233–237. doi: 10.1161/01.atv.21.2.233. [DOI] [PubMed] [Google Scholar]
- Bagi Z, Ungvari Z, Koller A. Xanthine oxidase-derived reactive oxygen species convert flow-induced arteriolar dilation to constriction in hyperhomocysteinemia: possible role of peroxynitrite. Arterioscler Thromb Vasc Biol. 2002;22:28–33. doi: 10.1161/hq0102.101127. [DOI] [PubMed] [Google Scholar]
- Bonaventura D, Tirapelli CR, Haddad R, Hoehr NF, Eberlin MN, de Oliveira AM. Chronic methionine load-induced hyperhomocysteinemia enhances rat carotid responsiveness for angiotensin II. Pharmacology. 2004;70:91–99. doi: 10.1159/000074673. [DOI] [PubMed] [Google Scholar]
- Brandes RP, Barton M, Philippens KM, Schweitzer G, Mugge A. Endothelial-derived superoxide anion in pig coronary arteries; evidence from lucigenin chemiluminescence and histochemical techniques. J Physiol. 1997;500:331–342. doi: 10.1113/jphysiol.1997.sp022024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Büssemaker E, Pistrosch F, Förster S, Herbrig K, Gross P, Passauer J, et al. Rho kinase contributes to basal vascular tone in humans: role of endothelium-derived nitric oxide. Am J Physiol Heart Circ Physiol. 2007;293:H541–H547. doi: 10.1152/ajpheart.00770.2006. [DOI] [PubMed] [Google Scholar]
- Chen C, Halkos ME, Surowiec SM, Conklin BS, Lin PH, Lumsden AB. Effects of homocysteine on smooth muscle cell proliferation in both cell culture and artery perfusion culture models. J Surg Res. 2000;88:26–33. doi: 10.1006/jsre.1999.5756. [DOI] [PubMed] [Google Scholar]
- Chitaley K, Webb RC. Nitric oxide induces dilation of rat aorta via inhibition of rho-kinase signaling. Hypertension. 2002;39:438–442. doi: 10.1161/hy02t2.102960. [DOI] [PubMed] [Google Scholar]
- De Oliveira AM, Viswanathan M, Capsoni S, Heemskerk FM, Correa FM, Saavedra JM. Characterization of endothelinA receptors in cerebral and peripheral arteries of the rat. Peptides. 1995;16:139–144. doi: 10.1016/0196-9781(94)00169-7. [DOI] [PubMed] [Google Scholar]
- Demuth K, Atger V, Borderie D, Benoit MO, Sauvaget D, Lotersztajn S, et al. Homocysteine decreases endothelin-1 production by cultured human endothelial cells. Eur J Biochem. 1999;263:367–376. doi: 10.1046/j.1432-1327.1999.00496.x. [DOI] [PubMed] [Google Scholar]
- Demuth K, Drunat S, Girerd X, Moatti N, Paul JL, Safar M, et al. Homocysteine is the only plasma thiol associated with carotid artery remodeling. Atherosclerosis. 2002;165:167–174. doi: 10.1016/s0021-9150(02)00205-8. [DOI] [PubMed] [Google Scholar]
- Drunat S, Moatti N, Paul JL, Cogny A, Benoit MO, Demuth K. Homocysteine-induced decrease in endothelin-1 production is initiated at the extracellular level and involves oxidative products. Eur J Biochem. 2001;268:5287–5294. doi: 10.1046/j.0014-2956.2001.02460.x. [DOI] [PubMed] [Google Scholar]
- Drunat S, Moatti N, Demuth K. Homocysteine decreases endothelin-1 expression by interfering with the AP-1 signaling pathway. Free Radic Biol Med. 2002;33:659–668. doi: 10.1016/s0891-5849(02)00957-7. [DOI] [PubMed] [Google Scholar]
- Dusitanond P, Eikelboom JW, Hankey GJ, Thom J, Gilmore G, Loh K, et al. Homocysteine-lowering treatment with folic acid, cobalamin, and pyridoxine does not reduce blood markers of inflammation, endothelial dysfunction, or hypercoagulability in patients with previous transient ischemic attack or stroke: a randomized substudy of the VITATOPS trial. Stroke. 2005;36:144–146. doi: 10.1161/01.STR.0000150494.91762.70. [DOI] [PubMed] [Google Scholar]
- Haddad R, Sparrapan R, Eberlin MN. Desorption sonic spray ionization for (high) voltage-free ambient mass spectrometry. Rapid Commun Mass Spectrom. 2006;20:2901–2905. doi: 10.1002/rcm.2680. [DOI] [PubMed] [Google Scholar]
- Hill CH, Mecham R, Starcher B. Fibrillin-2 defects impair elastic fiber assembly in a homocysteinemic chick model. J Nutr. 2002;132:2143–2150. doi: 10.1093/jn/132.8.2143. [DOI] [PubMed] [Google Scholar]
- Ihara M, Noguchi K, Saeki T, Fukuruda T, Tsuchida S, Kimura S, et al. Biological profiles of highly potent novel endothelin antagonists selective for ETA receptor. Life Sci. 1992;50:247–255. doi: 10.1016/0024-3205(92)90331-i. [DOI] [PubMed] [Google Scholar]
- Ishikawa K, Ihara M, Noguchi K, Mase T, Mino N, Saeki T, et al. Biochemical and pharmacological profile of a newly-developed potent and selective endothelin B receptor antagonist, BQ-788. Proc Natl Acad Sci USA. 1994;20:4892–4896. doi: 10.1073/pnas.91.11.4892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin L, Ying Z, Webb RC. Activation of Rho/Rho kinase signaling pathway by reactive oxygen species in rat aorta. Am J Physiol Heart Circ Physiol. 2004;287:H1495–H500. doi: 10.1152/ajpheart.01006.2003. [DOI] [PubMed] [Google Scholar]
- Leite PF, Danilovic A, Moriel P, Dantas K, Marklund S, Dantas AP, et al. Sustained decrease in superoxide dismutase activity underlies constrictive remodeling after balloon injury in rabbits. Arterioscler Thromb Vasc Biol. 2003;23:2197–2202. doi: 10.1161/01.ATV.0000093980.46838.41. [DOI] [PubMed] [Google Scholar]
- Li Y, Zhu H, Kuppusamy P, Roubaud V, Zweier JL, Trush MA. Validation of lucigenin (bis-N- methylacridinium) as a chemiluminescent probe for detecting superoxide anion radical for detecting superoxide anion radical production by enzymatic and cellular sources. J Biol Chem. 1998;273:2015–2023. doi: 10.1074/jbc.273.4.2015. [DOI] [PubMed] [Google Scholar]
- Lopez-Ongil S, Senchak V, Saura M, Zaragoza C, Ames M, Ballermann B, et al. Superoxide regulation of endothelin-converting enzyme. J Biol Chem. 2000;275:26423–26427. doi: 10.1074/jbc.M000767200. [DOI] [PubMed] [Google Scholar]
- Luscher TF, Barton M. Endothelins and endothelin receptor antagonists: therapeutic considerations for a novel class of cardiovascular drugs. Circulation. 2000;102:2434–2440. doi: 10.1161/01.cir.102.19.2434. [DOI] [PubMed] [Google Scholar]
- Masaki T. Possible role of endothelin in endothelial regulation of vascular tone. Annu Rev Pharmacol Toxicol. 1995;35:235–255. doi: 10.1146/annurev.pa.35.040195.001315. [DOI] [PubMed] [Google Scholar]
- Nakakuki T, Ito M, Iwasaki H, Kureishi Y, Okamoto R, Moriki N, et al. Rho/Rho-kinase pathway contributes to C-reactive protein-induced plasminogen activator inhibitor-1 expression in endothelial cells. Arterioscler Thromb Vasc Biol. 2005;25:2088–2093. doi: 10.1161/01.ATV.0000183607.50230.9f. [DOI] [PubMed] [Google Scholar]
- Nazarali AJ, Gutkind JS, Saavedra JM. Calibration of 125I-polymer standards with 125I-brain paste standards for use in quantitative receptor autoradiography. J Neurosci Methods. 1989;30:247–253. doi: 10.1016/0165-0270(89)90135-0. [DOI] [PubMed] [Google Scholar]
- Nelson MT, Quayle JM. Physiological roles and properties of potassium channels in arterial smooth muscle. Am J Physiol. 1995;268:C799–C822. doi: 10.1152/ajpcell.1995.268.4.C799. [DOI] [PubMed] [Google Scholar]
- Okatani Y, Wakatsuki A, Reiter RJ. Melatonin counteracts potentiation by homocysteine of KCL-induced vasoconstriction in human umbilical artery: relation to calcium influx. Biochem Biophys Res Commun. 2001;280:940–944. doi: 10.1006/bbrc.2000.4211. [DOI] [PubMed] [Google Scholar]
- Pisciotta L, Cortese C, Gnasso A, Liberatoscioli L, Pastore A, Mannucci L, et al. Serum homocysteine, methylenetetrahydrofolate reductase gene polymorphism and cardiovascular disease in heterozygous familial hypercholesterolemia. Atherosclerosis. 2005;179:333–338. doi: 10.1016/j.atherosclerosis.2004.10.016. [DOI] [PubMed] [Google Scholar]
- Powers RW, Majors AK, Cerula SL, Huber HA, Schmidt BP, Roberts JM. Changes in markers of vascular injury in response to transient hyperhomocysteinemia. Metabolism. 2003;52:501–507. doi: 10.1053/meta.2003.50081. [DOI] [PubMed] [Google Scholar]
- Pruefer D, Scalia R, Lefer AM. Homocysteine provokes leukocyte-endothelium interaction by downregulation of nitric oxide. Gen Pharmacol. 1999;33:487–498. doi: 10.1016/s0306-3623(99)00045-2. [DOI] [PubMed] [Google Scholar]
- Refsum H, Ueland PM, Nygard O, Vollset SE. Homocysteine and cardiovascular disease. Annu Rev Med. 1998;49:31–62. doi: 10.1146/annurev.med.49.1.31. [DOI] [PubMed] [Google Scholar]
- Robert K, Pages C, Ledru A, Delabar J, Caboche J, Janel N. Regulation of extracellular signal-regulated kinase by homocysteine in hippocampus. Neuroscience. 2005;133:925–935. doi: 10.1016/j.neuroscience.2005.03.034. [DOI] [PubMed] [Google Scholar]
- Takai M, Umemura K, Yamasaki K, Watanabe T, Fujitani Y, Oda K, et al. A potent and specific agonist, Suc-[Glu9 Ala11,15]-endothelin-1 (8-21), IRL 1620, for ETB receptor. Biochem Biophys Res Commun. 1992;184:953–959. doi: 10.1016/0006-291x(92)90683-c. [DOI] [PubMed] [Google Scholar]
- Tirapelli CR, Casolari DA, Yogi A, Montezano AC, Tostes RC, Legros E, et al. Functional characterization and expression of endothelin receptors in rat carotid artery: involvement of nitric oxide, a vasodilator prostanoid and the opening of K(+) channels in ET(B)-induced relaxation. Br J Pharmacol. 2005;146:903–912. doi: 10.1038/sj.bjp.0706388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tirapelli CR, Casolari DA, Montezano AC, Yogi A, Tostes RC, Legros E, et al. Ethanol consumption enhances endothelin-1-induced contraction in the isolated rat carotid. J Pharmacol Exp Ther. 2006;318:819–827. doi: 10.1124/jpet.106.103010. [DOI] [PubMed] [Google Scholar]
- Tostes RC, Muscara MN. Endothelin receptor antagonists: another potential alternative for cardiovascular diseases. Curr Drug Targets Cardiovasc Haematol Disord. 2005;5:287–301. doi: 10.2174/1568006054553390. [DOI] [PubMed] [Google Scholar]
- Traupe T, Ortmann J, Munter K, Barton M. Endothelial therapy of atherosclerosis and its risk factors. Curr Vasc Pharmacol. 2003;1:111–121. doi: 10.2174/1570161033476763. [DOI] [PubMed] [Google Scholar]
- Tyagi SC, Smiley LM, Mujumdar VS, Clonts B, Parker JL. Reduction-oxidation (Redox) and vascular tissue level of homocyst(e)ine in human coronary atherosclerotic lesions and role in extracellular matrix remodeling and vascular tone. Mol Cell Biochem. 1998;181:107–116. doi: 10.1023/a:1006882014593. [DOI] [PubMed] [Google Scholar]
- Ungvari Z, Sarkadi-Nagy E, Bagi Z, Szollar L, Koller A. Simultaneously increased TxA(2) activity in isolated arterioles and platelets of rats with hyperhomocysteinemia. Arterioscler Thromb Vasc Biol. 2000;20:1203–1208. doi: 10.1161/01.atv.20.5.1203. [DOI] [PubMed] [Google Scholar]
- Viswanathan M, De Oliveira AM, Johren O, Saavedra JM. Increased endothelin ET(A) receptor expression in rat carotid arteries after balloon injury. Peptides. 1997;18:247–255. doi: 10.1016/s0196-9781(96)00285-9. [DOI] [PubMed] [Google Scholar]
- Yanagisawa M, Kurihara H, Kimura S, Tomobe Y, Kobayashi M, Mitsui Y, et al. A novel potent vasoconstrictor peptide produced by vascular endothelial cells. Nature. 1988;332:411–415. doi: 10.1038/332411a0. [DOI] [PubMed] [Google Scholar]
- Weiss N, Heydrick SJ, Postea O, Keller C, Keaney JF, Jr, Loscalzo J. Influence of hyperhomocysteinemia on the cellular redox state – impact on homocysteine-induced endothelial dysfunction. Clin Chem Lab Med. 2003;41:1455–1461. doi: 10.1515/CCLM.2003.223. [DOI] [PubMed] [Google Scholar]