

Abstract
Background and purpose:
We investigated the effects of a synthetic flavonol, 3′,4′-dihydroxyflavonol (DiOHF) on the expression of monocyte chemoattractant protein-1 (MCP-1) in rat vascular smooth muscle cells.
Experimental approach:
MCP-1 expression was assessed by quantitative real-time PCR and protein phosphorylation by immunoprecipitation and Western blots.
Key results:
DiOHF (1–30 µmol·L−1) concentration-dependently reduced MCP-1 expression in both quiescent cells and cells stimulated with platelet-derived growth factor (PDGF) or interleukin 1-β. The effect of DiOHF was associated with a suppression of focal adhesion kinase (FAK)-mediated signalling. In vitro kinase assays demonstrated that DiOHF is a potent inhibitor of FAK kinase activity (EC50= 2.4 µmol·L−1). Expression of FAK-related non-kinase reduced basal MCP-1 expression, but not that induced by PDGF or interleukin 1-β. DiOHF also inhibited autophosphorylation of PDGF receptors. The PDGF receptor inhibitor AG-1296 potently suppressed basal and PDGF-induced MCP-1 expression. Inhibition of extracellular signal-regulated kinase activation by DiOHF, either directly or indirectly, may also be involved in its effects on MCP-1 expression. DiOHF had no inhibitory effect on either p38 or nuclear factor-κB activation. Moreover, DiOHF inhibited smooth muscle cell spreading (a FAK-mediated response) and proliferation.
Conclusions and implications:
This is the first report on a flavonoid compound (DiOHF) that is a potent FAK inhibitor. DiOHF also inhibits PDGF receptor autophosphorylation. These effects underlie the inhibitory action of DiOHF on MCP-1 expression in smooth muscle cells. Our results suggest that DiOHF might be a useful tool for dissection of the (patho)physiological roles of FAK signalling.
British Journal of Pharmacology (2009) 157, 597–606; doi:10.1111/j.1476-5381.2009.00199.x; published online 9 April 2009
Keywords: flavonoid, focal adhesion kinase, monocyte chemoattractant protein-1, PDGF receptor, protein tyrosine kinase, smooth muscle cell
Introduction
Monocyte chemoattractant protein-1 (MCP-1), initially identified as the product of the platelet-derived growth factor (PDGF)-inducible gene JE in the mouse, is a C-C type of chemokine that is released by various vascular cells, including endothelial cells, smooth muscle cells and fibroblasts (Rollins et al., 1988; Charo and Taubman, 2004). Via interactions with its receptor CCR2, MCP-1 is the most potent chemoattractant for monocyte migration across the endothelium and their recruitment in the arterial wall (Charo and Taubman, 2004). MCP-1/CCR2 signalling has a crucial role in the development of vascular inflammatory diseases, such as atherosclerosis (Aiello et al., 1999) and intimal hyperplasia (Mori et al., 2002). Moreover, MCP-1 has been implicated in the inflammatory response following ischaemia–reperfusion injury. For example, there is evidence that MCP-1 is involved in mediating the recruitment of monocytes into the ischemic myocardium after reperfusion (Birdsall et al., 1997). Genetic deletion of CCR2 significantly reduced the infarct size after cardiac ischaemia–reperfusion in mice (Hayasaki et al., 2006). In addition, in a stroke model induced by middle cerebral artery occlusion and reperfusion, it was reported that elevated MCP-1 expression in the brain increased monocyte infiltration and the infarct size (Chen et al., 2003). These results indicate that treatments targeting the MCP-1/CCR2 pathway may be useful approaches to cytoprotection after ischaemia and reperfusion injury.
In our previous studies, we found that 3′,4′-dihydroxyflavonol (DiOHF), a synthetic flavonoid, had potent antioxidant activities (Jiang et al., 2007). In vivo treatment with DiOHF significantly reduced ischaemia–reperfusion-induced infarction in the heart (Wang et al., 2004). Numerous studies have shown that these compounds may also have significant anti-inflammatory effects, including inhibition of the expression of adhesion molecules, cytokines and chemokines (Jiang and Dusting, 2003). Given that MCP-1 is involved in mediating inflammatory responses to ischaemia–reperfusion (Frangogiannis, 2004), in this study we examined the pharmacological effects of DiOHF on MCP-1 expression in vascular smooth muscle cells and explored the underlying mechanisms.
Methods
Cell culture
All animal studies were carried out in accordance with the guidelines of the St Vincent's Hospital Animal Ethics Committee and National Health and Medical Research Council of Australia. Rats were killed by pentobarbital sodium overdose (i.p., 150 mg per rat) and the aorta removed. Rat aortic smooth muscle cells (RASMCs) were isolated from thoracic aorta of male Sprague-Dawley rats by enzymatic digestion with collagenase I (1 mg·mL−1), elastase (0.5 mg·mL−1) and trypsin (1.25 mg·mL−1). Cells were maintained in Dulbecco's modification of Eagle's medium (Gibco) supplemented with 5% foetal calf serum in a humidified incubator with 5% CO2 at 37°C. Cells between passages 10–20 were used. The purity of the cells was determined as >95% by anti-smooth muscle α-actin immunocytochemistry.
Plasmids and cell transfection
The control plasmid pRK that encoding vesicular stomatitis virus glycoprotein (VSV-G)-tagged focal adhesion kinase (FAK)-related non-kinase (FRNK), an endogenous inhibitor of FAK (Richardson and Parsons, 1996), was a kind gift from Dr K.M. Yamada (National Institutes of Health, Bethesda, MD, USA) (Gu et al., 1999). For gene transfection, RASMCs were subcultured into 24-well plates in antibiotic-free medium containing 10% serum 24 h before transfection, and then incubated with 2 µg of plasmid per well mixed with 5 µL of Lipofectamine 2000 (Invitrogen) for 24 h.
Real-time PCR
Cells grown in culture plates were collected in 0.5 mL per well TRI Reagent (Ambion) at 4°C for 1 h and the total RNA was extracted according to the manufacture's instruction. Total RNA was reverse-transcribed to cDNA using random hexamer and TaqMan reverse transcription reagents (Applied Biosystems). Quantitative real-time PCR reactions were performed in an ABI Prism 7700 system (Applied Biosystems) using the TaqMan Universal PCR master mix. The 18s RNA was used as housekeeping gene. The sequences for rat MCP-1 were: forward CAGATCTCTCTTCCTCCACCACTAT; reverse CAGGCAGCAACTGTGAACAAC; and probe CAGGTCTCTGTCACGCTTCTGGGCC. The sequences for 18s were: forward CGGCTACCACATCCAAGGAA; reverse GCTGGAATTACCGCGGCT; and probe TGCTGGCACCAGACTTGCCCTC. The thermal cycler parameters were 2 min at 50°C, 10 min at 95°C and 40 cycles of 95°C × 30 s and 60°C × 1 min. The relative amount of target genes normalized to the housekeeping gene in treated samples was converted to fold of control (ΔΔCT method, see the User's Bulletin of Applied Biosystems).
Immunoprecipitation and Western blots
Total protein was extracted in a lysis buffer containing 25 mmol·L−1 Tris (pH 7.5), 100 mmol·L−1 NaCl, 2 mmol·L−1 EDTA, 1 mmol·L−1 dithiothreitol, 1% Triton-X 100, 0.25% deoxycholate, 1 mmol·L−1 Na3VO4, 50 mmol·L−1 NaF and the Complete proteinase inhibitor cocktail (Roche), and ∼200 µg of the protein from each sample was diluted with the same lysis buffer to a final concentration of 1 mg·mL−1. The samples were pre-cleared with 5 µg normal IgG and 20 µL of 50% slurry of protein G agarose beads (P-4691, Sigma) and then incubated with 5 µg of antibody for overnight. The immunocomplex was precipitated with 20 µL of 50% protein G agarose by continuous mixing for 1 h. The beads were collected by centrifugation, washed with phosphate-buffered saline and resuspended in 30 µL (2×) Laemmli SDS-PAGE sample buffer and boiled for 5 min. All other procedures were performed at 4°C.
Total protein or immunoprecipitated samples were separated by 10% SDS-PAGE and transferred to Hybond nitrocellulose membrane (Amersham). The membrane was blocked with 5% non-fat milk powder in Tris-buffered saline (pH 7.5) and hybridized overnight with primary antibodies, which were then detected with horseradish peroxidase-conjugated anti-IgG and visualized with an ECL kit (Amersham).
Kinase activity assay
In vitro tyrosine kinase activity was measured with a non-radioactive tyrosine kinase activity assay kit (Chemicon, Cat. #SGT410). Purified active human FAK and Src kinases were purchased from Cell Signalling Technology (MA, USA). Biotin conjugated poly(Glu4-Tyr) peptide (Upstate) was used as the substrate. The reaction was carried out at 30°C in a mixture of a total volume of 50 µL, containing 60 mmol·L−1 HEPES (pH 7.5), 3 mmol·L−1 MgCl2, 3 mmol·L−1 MnCl2, 3 µmol·L−1 Na3VO4, 1.2 mmol·L−1 dithiothreitol, 0.2% bovine serum albumin, 0.5 µg substrate and either 400 ng of FAK or 5 U of Src.
Cell spreading and proliferation
Cell spreading response was assessed by trypsinization and replating the cells into a 12-well plate in normal medium. After 3 h, the attached cells were washed and photos taken using an inverted phase-contrast microscope (Olympus CK40) equipped with a digital camera (Olympus DP10). The percentage of cells that showed spreading (vs. those remained round) was counted in at least five randomly selected fields. Cell proliferation was assessed by counting the total number of cells maintained in normal culture medium.
Statistical analysis
Data are expressed as mean ± SEM. The mean data were analysed with Student's t-test or one-way anova (analysis of the variance) followed by Newman–Keul's t-test as appropriate. A value of P < 0.05 was regarded as statistically significant.
Materials
DiOHF was obtained from Indofine Chemical Company Inc. (Hillsborough, NJ, USA). Genistein, recombinant human interleukin 1-β (IL-1β), human PDGF-BB and Tempo (4-hydroxy-2,2,6,6-tetramethylpiperidinyloxy) were all purchased from Sigma. PP2, AG-1296 and SB-202190 were from BIOMOL (PA, USA). Quercetin was from ICN Biomedicals (OH, USA).
Results
DiOHF inhibits MCP-1 expression
In RASMCs arrested by serum starvation, treatment with DiOHF (1–30 µmol·L−1) for 6 h suppressed MCP-1 expression by a maximum of 93% as measured by quantitative real-time PCR. This effect is concentration-dependent (Figure 1A). As compared with DiOHF, quercetin (a naturally occurring analogue of DiOHF with equal antioxidant activity) (Jiang et al., 2007) did not significantly reduce MCP-1 expression at 10 and 30 µmol·L−1 (Figure 1B, P= 0.34, one-way anova, n= 3). We then treated the cells with the superoxide scavenger Tempo (1 mmol·L−1), which suppressed MCP-1 expression by 40% (Figure 1C). The much greater effect of DiOHF, compared with those of the other antioxidants, suggests that the antioxidant activity of DiOHF is unlikely to have a major role in inhibiting MCP-1 expression. To further clarify the role of redox mechanisms in MCP-1 expression in smooth muscle cells, we treated the cells with exogenous H2O2 (100 µmol·L−1) or the catalase inhibitor 3-amino-1, 2, 4-triazole (10 mmol·L−1). Both agents reduced the expression of MCP-1 (Figure 1D).
Figure 1.
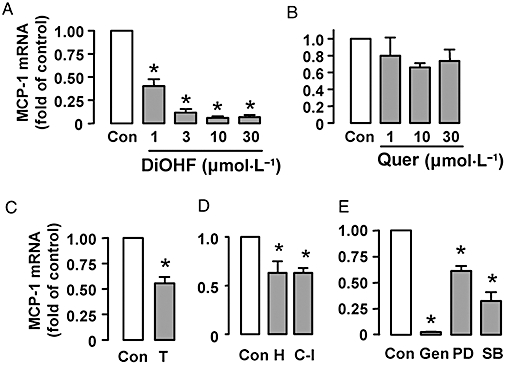
Quantitative real-time PCR analysis of monocyte chemoattractant protein-1 (MCP-1) mRNA expression in growth arrested rat aortic smooth muscle cells treated with 3′,4′-dihydroxyflavonol (DiOHF), quercetin (Quer), Tempo (T, 1 mmol·L−1), H2O2 (H, 100 µmol·L−1), the catalase inhibitor (C-I) 3-amino-1, 2, 4-triazole (10 mmol·L−1), genistein (Gen, 100 µmol·L−1), PD-98059 (PD, 25 µmol·L−1) and SB-202190 (SB, 10 µmol·L−1). The duration of treatment for all agents was 6 h. The results are expressed as fold of control (Con). *P < 0.05 versus control, n= 3–6.
To characterize the signalling pathways that may be involved in mediating the effects of DiOHF, we treated the cells with genistein (100 µmol·L−1) (a non-selective tyrosine kinase inhibitor), PD-98059 (25 µmol·L−1) [mitogen-activated protein kinase (MAPK)/extracellular signal-regulated kinase (ERK) kinase inhibitor] and SB-202190 (10 µmol·L−1) (p38 MAPK inhibitor). As shown in Figure 1E, both PD-98059 and SB-202190 significantly reduced MCP-1 expression, while it was nearly abolished by genistein.
DiOHF inhibits MCP-1 expression under pro-inflammatory conditions
We next examined whether DiOHF has similar effects on MCP-1 expression under pro-inflammatory conditions. Treatment of the cells with IL-1β (10 ng·mL−1) or PDGF-BB (20 ng·mL−1) for 6 h significantly increased MCP-1 expression (Figure 2); these responses to cytokines were significantly suppressed by DiOHF at 10 µmol·L−1 and nearly abolished at 30 µmol·L−1 (Figure 2). Treatment with Tempo had a minor effect on IL-1β-induced MCP-1 expression (data not shown), suggesting that oxidant-independent mechanisms were involved in mediating MCP-1 expression under inflammatory conditions (see Figure 8 below).
Figure 2.
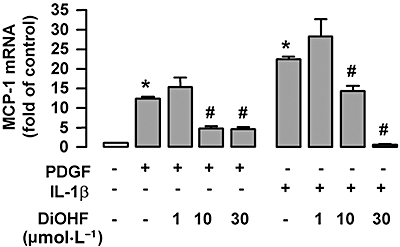
The effects of 3′,4′-dihydroxyflavonol (DiOHF) on monocyte chemoattractant protein-1 (MCP-1) mRNA expression in the presence of interleukin 1-β (IL-1β) (10 ng·mL−1) or platelet-derived growth factor (PDGF)-BB (20 ng·mL−1). The duration of treatment for all agents was 6 h. The results are expressed as fold of control (Con). *P < 0.05 versus control; #P < 0.05 versus cells treated with IL-1β or PDGF respectively; n= 3–6.
Figure 8.
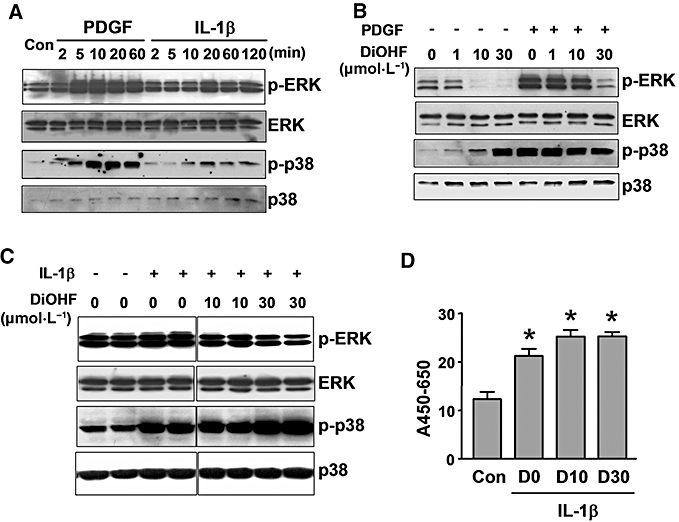
Effects of 3′,4′-dihydroxyflavonol (DiOHF) on extracellular signal-regulated kinase (ERK)/p38 phosphorylation and nuclear factor-κB (NF-κB) activation. (A) Time course of ERK and p38 phosphorylation induced by platelet-derived growth factor (PDGF) (20 ng·mL−1) and interleukin 1-β (IL-1β) (10 ng·mL−1). Example from two independent experiments. (B) Concentration-dependent effects of DiOHF on basal and PDGF-induced ERK1/2 and p38 phosphorylation. Cells were pretreated with DiOHF for 1 h followed by PDGF stimulation (for 20 min). (C) Effects of DiOHF on IL-1β-induced ERK1/2 and p38 phosphorylation. Cells were pretreated with DiOHF for 1 h followed by IL-1β (for 20 min). All Western blot experiments in (B) and (C) were repeated three times. (D) Effects of DiOHF on IL-1β-induced p50 NF-κB activation. Cells were pretreated with DiOHF for 1 h followed by IL-1β for another hour. The DNA binding activity of p50 was assessed with a colorimetric transcription factor assay kit from Millipore (Billerica, MA, USA). The results were expressed as the difference of absorbance at 450 and 650 nm normalized to protein content. *P < 0.05 versus control, one-way anova, n= 3.
DiOHF suppressed tyrosine phosphorylation of paxillin
Given that DiOHF contains a polyphenol structure, similar to that of genistein, we then examined whether the effects of DiOHF were related to modulation of tyrosine kinase(s) activities. We first performed Western blots using a monoclonal anti-phosphotyrosine antibody to detect total tyrosine-phosphorylated proteins. As shown in Figure 3A, DiOHF treatment altered the profile of protein tyrosine phosphorylation in smooth muscle cells. Specifically, we observed that DiOHF consistently diminished a band at ∼70 kDa. Moreover, we found that DiOHF markedly suppressed PDGF-induced phosphorylation of a protein at ∼130 kDa (Figure 3A). To clarify whether the phosphorylated 70 kDa protein is paxillin (Hassid et al., 1999), we performed immunoprecipitation experiments using the anti-phosphotyrosine antibody, followed by Western blotting using anti-paxillin. As shown in Figure 3B, DiOHF significantly reduced paxillin phosphorylation in a concentration-dependent manner, whereas quercetin was much less effective. Next we examined the effects of DiOHF on FAK, which is the upstream kinase of paxillin. As shown in Figure 3B, DiOHF only had a marginal inhibitory effect on FAK tyrosine phosphorylation. These results suggest that DiOHF-induced inhibition of paxillin phosphorylation is not explained by reduced FAK phosphorylation and activation, but may be possibly because of a direct inhibition of FAK kinase activity (see below). As the non-receptor tyrosine kinase Src forms a complex with FAK at the focal adhesions and also can phosphorylate paxillin, we then examined how DiOHF modulated Src-mediated effects. We found (Figure 3B) that treatment of the cells with a specific Src inhibitor PP2 did not significantly reduce paxillin phosphorylation, suggesting that Src is not involved in paxillin phosphorylation at basal conditions. We further demonstrated that treatment with DiOHF significantly increased the phosphorylation of another Src substrate, pp120. The increase in pp120 phosphorylation induced by DiOHF was not caused by altered Src expression (Figure 3B), but was blocked by PP2 (Figure 3B).
Figure 3.
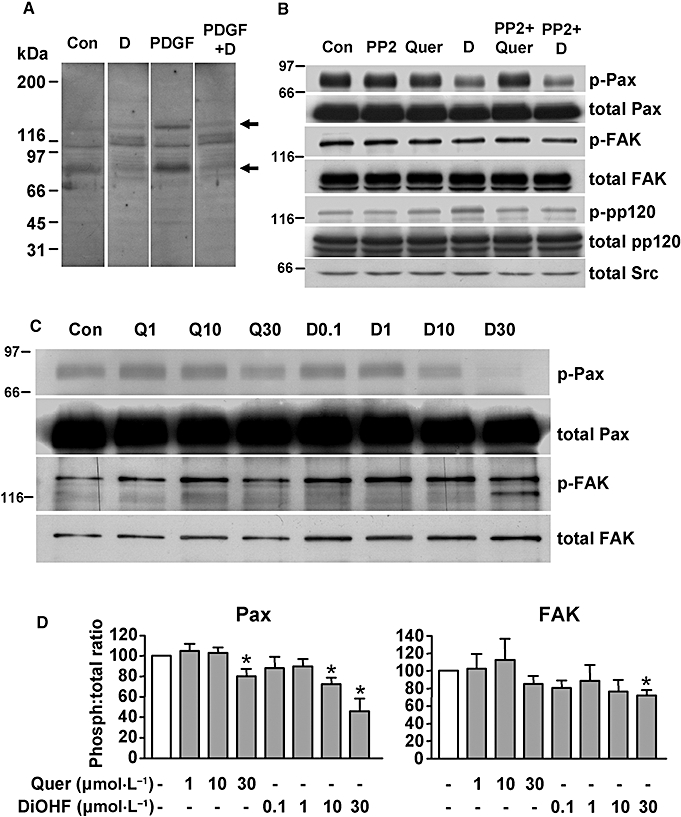
Effects of 3′,4′-dihydroxyflavonol (DiOHF), quercetin (Quer) and PP2 on protein tyrosine phosphorylation. (A) Effects of DiOHF (D; 30 µmol·L−1 for 1 h) on total tyrosine-phosphorylated proteins detected by a monoclonal anti-phosphotyrosine antibody in rat aortic smooth muscle cells with or without platelet-derived growth factor (PDGF) (20 ng·mL−1) treatment. The arrows indicate the 130 and 70 kDa bands that were significantly modified by DiOHF treatment (n= 3). Con, vehicle control. The blot strips were from the same membrane, in which equal amounts of protein were loaded for all lanes. (B) Cells were treated with DiOHF (D; 30 µmol·L−1), quercetin (Quer; 30 µmol·L−1) or PP2 (1 µmol·L−1) for 1 h. Total cell lysates were immunoprecipitated with the anti-phosphotyrosine antibody followed by Western blotting with antibodies against paxillin (Pax), pp120 or focal adhesion kinase (FAK). Total paxillin, pp120, FAK and Src were determined by western blot with the cell lysates without immunoprecipitation. n= 3. The lower thin band in total FAK is likely to be an artefact because of the relatively high protein load; in experiments with a lower protein load, this band disappeared. (C) Concentration-dependent effects of DiOHF (D, concentration shown in µmol·L−1) and quercetin (Q) treatment for 1 h on tyrosine phosphorylation of paxillin (Pax) and FAK. n= 3. (D) Summary data of the Western blots in (C) measured by densitometry. *P < 0.05 versus control (normalized to 100%), n= 3.
We further analysed the concentration–response relationship of the effects of DiOHF on paxillin phosphorylation. We performed immunoprecipitation with anti-paxillin followed by Western blotting with anti-phosphotyrosine. DiOHF reduced paxillin phosphorylation in a concentration-dependent manner (Figure 3C and D), while the effect of quercetin was much less potent (Figure 3C and D). The phosphorylation of FAK was only slightly reduced by DiOHF at highest concentration tested, supporting that DiOHF may have a direct inhibition effect on activated (phosphorylated) FAK kinase.
To examine the effects of Src on MCP-1 expression, we treated the cells with the specific Src inhibitor, PP2. As shown in Figure 4, treatment with PP2 significantly suppressed MCP-1 expression under basal conditions and after PDGF or IL-1β stimulation, suggesting that Src is also involved in mediating MCP-1 expression.
Figure 4.
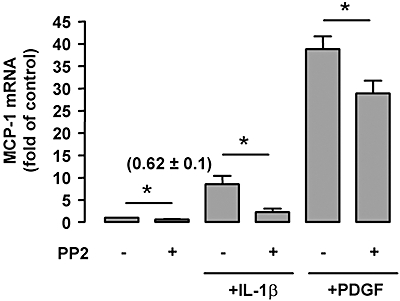
Effects of the Src inhibitor PP2 (1 µmol·L−1) on monocyte chemoattractant protein-1 (MCP-1) mRNA expression under basal conditions and after platelet-derived growth factor (PDGF) (20 ng·mL−1 for 6 h) or interleukin 1-β (IL-1β) (10 ng·mL−1) stimulation. Data shown as mean ± SEM, *P < 0.05, n= 4–6. The numbers in parenthesis are the results for PP2 alone and show that the low basal expression of MCP-1 mRNA was significantly inhibited by PP2.
DiOHF inhibits FAK kinase activity in vitro
Because our previous experiments suggest that DiOHF may have a direct inhibitory action on FAK kinase activity, we performed in vitro kinase assays using recombinant human FAK and Src. As shown in Figure 5, DiOHF inhibited the FAK kinase activity in a concentration-dependent manner, with an EC50 value of 2.4 µmol·L−1. In contrast, DiOHF only slightly suppressed Src activity at 30 µmol·L−1.
Figure 5.
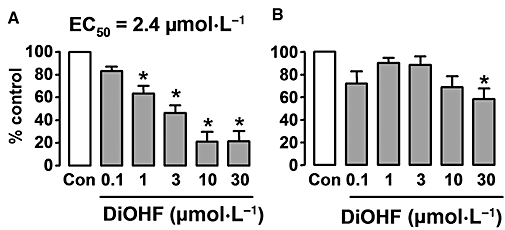
Concentration-dependent effects of 3′,4′-dihydroxyflavonol (DiOHF) on the kinase activity of (A) focal adhesion kinase (FAK) and (B) Src measured with an in vitro kinase assay using recombinant human FAK or Src. *P < 0.05 versus control, n= 8 assays.
Inhibition of FAK signalling reduced MCP-1 expression
To elucidate the role of FAK-mediated signalling in MCP-1 expression, we transiently expressed VSV-G-tagged FRNK, the carboxy-terminal domain of FAK that acts as an endogenous inhibitor of FAK activity (Richardson and Parsons, 1996). Using an anti-VSV-G antibody, we found that FRNK expression was maximal 24 h after transfection, and the expression diminished after 48 h (Figure 6A). Using this transfection condition, we demonstrated that expression of FRNK did not modify the expression levels of FAK or paxillin (Figure 6B). FRNK expression slightly but significantly (P= 0.0027, t-test, n= 7) reduced MCP-1 expression at basal conditions, but had no effect on PDGF- or IL-1β-induced expression of MCP-1 (Figure 6C–E).
Figure 6.

Effects of focal adhesion kinase-related non-kinase (FRNK) on monocyte chemoattractant protein-1 (MCP-1) expression. (A) Time course of FRNK expression. Rat aortic smooth muscle cells transfected with no vector (N), the control pRK vector (P) or pRK-vesicular stomatitis virus (VSV)-FRNK (F) for different times as indicated were analysed for FRNK expression with Western blot using an anti-VSV-glycoprotein (VSV-G) antibody. (B) Western blots showing that FRNK expression did not affect the total expression levels of focal adhesion kinase (FAK) or paxillin (Pax). (C,D) Effects of FRNK expression on MCP-1 mRNA levels under basal condition and after platelet-derived growth factor (PDGF) or interleukin 1-β (IL-1β) stimulation measured by real-time PCR. *P < 0.05 versus control, n= 6–7.
DiOHF inhibited PDGF receptor autophosphorylation
In Figure 3A, we demonstrated that DiOHF markedly suppressed PDGF-induced tyrosine phosphorylation of a protein of ∼130 kDa. We hypothesized that this protein could be the PDGF receptor (PDGFR). To further analyse this, we treated the cells with PDGF-BB (20 ng·mL−1) in the absence or presence of DiOHF. Using immunoprecipitation with anti-phosphotyrosine followed by Western blot with anti-PDGFR-α, we showed that the PDGF-responsive 130 kDa band appeared to be PDGFR (Figure 7A). Moreover, we found that PDGFR autophosphorylation was indeed inhibited by DiOHF. To investigate the concentration–response relation of DiOHF effects on PDGFR autophosphorylation, we treated the cells with PDGF in the presence of increasing concentrations of DiOHF. The blot of total protein was probed with anti-phosphotyrosine, and the same blot was then stripped and reprobed with anti-PDGFR-α to determine the total PDGFR-α. The identity of PDGFR of the 130 kDa band was further confirmed by the anti-PDGFR-α antibody that detected the bands at the same position as did anti-phosphotyrosine. As shown in Figure 7B, PDGF-induced autophosphorylation was concentration-dependently reduced by DiOHF (1–100 µmol·L−1).
Figure 7.
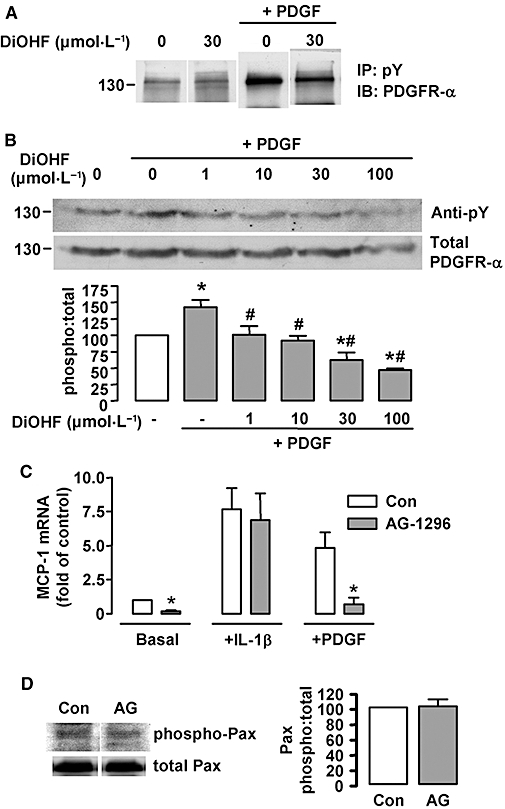
Role of platelet-derived growth factor receptor (PDGFR) activation in 3′,4′-dihydroxyflavonol (DiOHF)-induced inhibition on monocyte chemoattractant protein-1 (MCP-1) expression. (A) Effects of DiOHF on ligand-induced PDGFR autophosphorylation analysed by immunoprecipitation (IP) with anti-phosphotyrosine (pY) followed by Western blotting (IB) with anti-PDGFR-α. DiOHF alone had little effects on PDGFR phosphorylation but inhibited PDGF-induced PDGFR phosphorylation (n= 3 experiments). (B) Concentration–response effects of DiOHF on PDGFR autophosphorylation induced by PDGF (20 ng·mL−1 for 10 min). The phosphorylation of PDGFR was detected by Western blot with anti-phosphotyrosine (pY) followed by membrane stripping and reprobing with anti-PDGFR-α. *P < 0.05 versus control (open bar); #P < 0.05 versus PDGF alone, n= 3. (C) Effects of the PDGFR inhibitor AG-1296 (10 µmol·L−1 for 6 h) on MCP-1 expression (*P < 0.05 versus control, n= 3). (D) Effects of AG-1296 (10 µmol·L−1) on paxillin tyrosine phosphorylation in resting RASMCs. Paxillin phosphorylation was measured with an anti-phospho-paxillin (phospho Pax) antibody. The same membrane was then stripped and reprobed with anti-paxillin. The summary data are shown on the right (n= 3).
Then we examined whether inhibition of PDGFR activation can mimic the effects of DiOHF on MCP-1 expression. As shown in Figure 7C, (co)treatment with the specific PDGFR inhibitor AG-1296 drastically reduced both basal and PDGF-induced MCP-1 expression. These effects are similar to those observed with DiOHF. In contrast to DiOHF, AG-1296 did not change IL-1β-stimulated MCP-1 expression. Because there is evidence that FAK may be activated downstream of PDGFR activation (Hauck et al., 2000), we next examined whether AG-1296 had any effect on paxillin phosphorylation. As shown in Figure 7D, AG-1296 did not modify paxillin phosphorylation in resting cells.
Effects of DiOHF on ERK, p38 and nuclear factor-κB
Because we found that inhibition of ERK or p38 MAPKs suppressed MCP-1 expression, we then examined whether DiOHF had any effects on ERK or p38 activation. As shown in Figure 8A, both PDGF and IL-1β induced time-dependent phosphorylation of ERK1/2 and p38, which peaked at 20 min. In resting cells, treatment with DiOHF concentration-dependently reduced the basal level of phospho-ERK, but conversely induced p38 phosphorylation in a concentration-dependent manner (Figure 8B). Moreover, DiOHF at 30 µmol·L−1 inhibited PDGF-induced ERK phosphorylation, whereas phosphorylation of p38 was unchanged. Similar effects of DiOHF on ERK and p38 phosphorylation were also observed in IL-1β-stimulated cells (Figure 8C). Furthermore, we examined whether DiOHF could inhibit nuclear factor-κB (NF-κB) activation. As shown in Figure 8D, DiOHF at 10 or 30 µmol·L−1 had no effect on IL-1β-induced NF-κB activity.
DiOHF suppressed spreading and proliferation of RASMCs
Because the in vitro kinase assay demonstrated that DiOHF had a direct inhibitory action on kinase activity of FAK, we next examined whether or not DiOHF modulated cell spreading. This response of vascular smooth muscle cells depends on FAK-mediated mechanisms (Owen et al., 1999). As shown in Figure 9A and B, DiOHF 10 and 30 µmol·L−1 significantly delayed attachment-induced spreading of cells in the culture dish. Because FAK-mediated signalling is also involved in smooth muscle cell proliferation (Taylor et al., 2001), we then examined the effects of DiOHF on RASMC proliferation in the absence of exogenous PDGF, by counting the cell number over a period of 14 days. As shown in Figure 9C, the rate of proliferation of RASMCs was significantly reduced in the presence of DiOHF (6 µmol·L−1) in the culture medium. This concentration was selected because at higher concentrations DiOHF exhibited some cell toxicity after a chronic (>24 h) exposure (Figure 9D).
Figure 9.
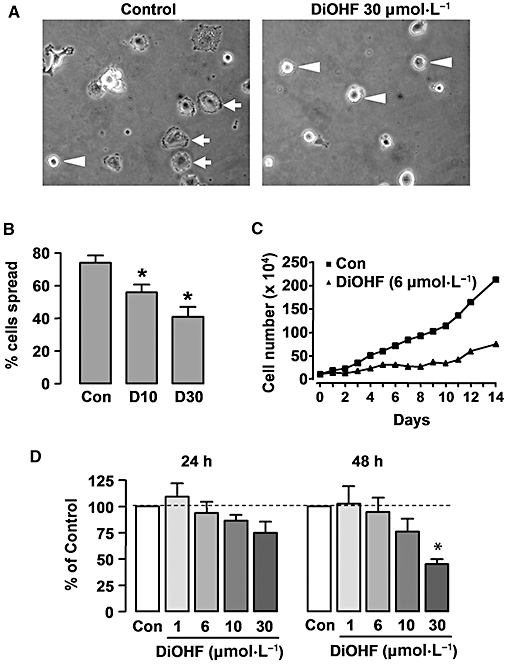
Effects of 3′,4′-dihydroxyflavonol (DiOHF) on cell spreading and proliferation. (A) Phase contrast microscopy showing the morphology of rat aortic smooth muscle cells (RASMCs) 3 h after plating in culture dish without (control) or with DiOHF treatment. Cells that showed spreading were indicated by arrows, while the cells remained round were indicated by arrowheads. (B) Percentage of spread cells at 3 h after plating without (Con) or with DiOHF (D, concentrations in µmol·L−1) treatment. *P < 0.05 versus Con, n= 3. (C) Effects of DiOHF on RASMC proliferation. Cells were cultured in medium containing 10% serum. DiOHF remained in the culture medium throughout the culture period. The data are averaged numbers from triplicate wells. (D) Effects of DiOHF on RASMC viability. Cells were growth arrested by incubating in serum-free medium for 48 h. Then DMSO (control) or DiOHF was added and cultured for another 24 or 48 h period. Cell proliferation was assessed with the CellTiter-96 AQueous One Solution kit (Promega) and measured as absorbance at 490 nm. *P < 0.05 versus control, one-way anova, n= 4.
Discussion
In the present study, we extended our previous studies on the synthetic flavonoid compound DiOHF (Wang et al., 2004; Jiang et al., 2007). We examined the pharmacological effects and underlying mechanisms of DiOHF action on MCP-1 expression in vascular smooth muscle cells. We demonstrated that DiOHF markedly suppressed MCP-1 expression in cultured RASMCs under basal conditions. We further demonstrated that this inhibitory effect of DiOHF is unlikely to be attributable to its antioxidant activities, because the naturally occurring analogue of DiOHF, quercetin, had minimal effects on MCP-1 expression, although our previous studies revealed that these two compounds have comparable antioxidant activities (Jiang et al., 2007). In addition, we found that MCP-1 expression was indeed reduced by exogenous H2O2 or by inhibition of catalase, as had been previously observed in human mesangial cells (Rovin et al., 1997). The mechanism of the difference between DiOHF and quercetin on MCP-1 expression is not totally clear and this is likely to be explained by the difference in chemical structures between the two flavonoids (Jiang et al., 2007). Using inhibitors of various kinases, we found that inhibition of protein tyrosine phosphorylation with genistein had a comparable inhibitory effect on MCP-1 expression as DiOHF, supporting the importance of protein tyrosine kinase-mediated mechanisms in maintaining MCP-1 expression in both vascular (Shyy et al., 1993; Okada et al., 1998; Rong et al., 2002) and non-vascular cells (Zen et al., 1995; Grandaliano et al., 2000; Lee et al., 2001). These results prompted us to hypothesize that the inhibition of MCP-1 expression by DiOHF might be related to its interactions with tyrosine kinase(s).
Interestingly, using an in vitro kinase assay, we have identified that DiOHF is an inhibitor of the non-receptor protein tyrosine kinase FAK, with an EC50 of 2.4 µmol·L−1, consistent with the observation that treatment of RASMCs with DiOHF also significantly reduced tyrosine phosphorylation of the FAK substrate paxillin. The direct inhibition of FAK activity is also consistent with the observation that DiOHF has only a marginal inhibitory effect on FAK tyrosine phosphorylation (and activation), supporting that DiOHF-induced inhibition of paxillin is a result of direct inhibition of activated FAK kinase. As the non-receptor tyrosine kinase Src forms a complex with FAK at the focal adhesions and also can phosphorylate paxillin, we also examined how DiOHF modulated Src-mediated effects. We found that treatment of the cells with a specific Src inhibitor PP2 did not significantly reduce paxillin phosphorylation, suggesting that Src is not involved in paxillin phosphorylation at basal conditions. We further demonstrated that treatment with DiOHF significantly increased the phosphorylation of another Src substrate, pp120, suggesting that Src-mediated actions were indeed increased after DiOHF treatment. The increase in pp120 phosphorylation induced by DiOHF was not caused by altered Src expression (Figure 2), but was blocked by PP2, supporting that this effect was mediated by increased Src activation. Although we observed that PP2 also significantly suppressed MCP-1 expression, suggesting that Src is indeed involved in mediating MCP-1 expression, taken the above data together, our results indicate that the inhibitory effect of DiOHF on MCP-1 expression is unlikely to be due to an inhibition of Src activity.
Activation of FAK is primarily induced by engagement of integrins at the cell surface, resulting in phosphorylation of multiple tyrosine residues and recruitment of a number of Src homology 2- and Src homology 3-domain-containing proteins (Parsons, 2003). Experimental evidence indicates that FAK may act as a signalling scaffold via its connection to multiple signalling pathways, including the MAPK cascade, phosphatidylinositol 3-kinase and small GTPases such as Rac and Rho (Hanks et al., 2003; Mitra et al., 2005). Activation of FAK is involved in modulating inflammatory responses by promoting expression of cytokines (Funakoshi-Tago et al., 2003), adhesion molecules (Nakayamada et al., 2003) and chemokines such as MCP-1 (Watanabe et al., 2003; Kanegae et al., 2005). In the present study, we demonstrated that expression of FRNK, an endogenous inhibitor of FAK (Richardson and Parsons, 1996), suppressed MCP-1 expression in resting RASMCs, indicating that FAK-mediated signalling is indeed involved in modulating MCP-1 expression in vascular smooth muscle cells, and this is consistent with the observations made in mesangial cells (Watanabe et al., 2003; Kanegae et al., 2005). Within focal adhesions, Src is closely associated with FAK, and the two kinases have many overlapping functions (Mitra et al., 2005). Thus we examined whether suppression of Src activity can also modulate MCP-1 expression. Consistent with the findings in previous studies (Jalali et al., 1998), we demonstrated that the Src inhibitor PP2 significantly reduced basal and cytokine-induced MCP-1 expression, suggesting that Src also has an important role in mediating MCP-1 expression in smooth muscle cells. However, our results suggest that the inhibition of MCP-1 expression by DiOHF was not likely to be mediated by suppression of Src activity. First, DiOHF at 30 µmol·L−1 had only a small inhibitory action on Src activity as revealed by the kinase assay, and second, DiOHF treatment resulted in an increase of tyrosine phosphorylation of pp120 in a PP2-sensitive manner, suggesting that in intact cells, there was indeed an increased Src activation after DiOHF treatment. The mechanism of increased Src activity after DiOHF treatment is unclear, but could be related to the reduced paxillin phosphorylation in DiOHF-treated cells. There is evidence that tyrosine phosphorylation of paxillin creates high-affinity binding sites for C-terminal Src kinase, a negative regulator of Src activity (Sabe et al., 1994; Schaller and Parsons, 1995). In both fibroblasts and some tumour cell lines, it has been shown that dephosphorylation of paxillin was accompanied by increased Src activation, suggesting that phosphorylated paxillin may participate in a feedback inhibition of Src by recruiting C-terminal Src kinase. This mechanism may provide an explanation of the enhanced pp120 phosphorylation after FAK inhibition induced by DiOHF.
Our study clearly demonstrated that inhibition of FAK activity only has a partial role in the reduced MCP-1 expression in resting cells, whereas FAK inhibition had little effects on PDGF- or IL-1β-induced expression of MCP-1. Furthermore, we showed that DiOHF also potently inhibited PDGFR autophosphorylation in RASMCs. Notably, MCP-1 was initially identified as a PDGF-inducible protein in murine cells (Rollins et al., 1988). Treatment with the PDGFR inhibitor AG-1296 abolished both basal and PDGF-induced expression of MCP-1. These results indicate that suppression of PDGFR-meditated events by DiOHF may have a major role in its effects on MCP-1 expression. From our results, we could not exclude the possibility that inhibition of FAK has a redundant role in mediating the inhibitory effects of DiOHF, because FAK is a downstream kinase in PDGFR-induced signalling (Hauck et al., 2000). However, we found that AG-1296 did not modify paxillin phosphorylation in resting cells, suggesting that the basal FAK activity in these cells is independent of PDGFR activation. Therefore, at least in resting cells, suppression of FAK activity may contribute additionally to the reduced MCP-1 expression induced by DiOHF.
Taken together, our results indicate that inhibition of FAK-mediated signalling may have a role in DiOHF-induced inhibition of MCP-1 in unstimulated cells, whereas inhibition of PDGF receptor activation may be involved in MCP-1 inhibition at both basal and cytokine-stimulated conditions. Because both FAK and PDGF receptors are upstream of MAPK activation, we also examined the effects of DiOHF on ERK and p38 phosphorylation. We found that DiOHF increased p38 phosphorylation in a concentration-dependent manner, suggesting that the p38 pathway is not involved in the inhibitory effects of DiOHF on MCP-1 expression. In contrast, we showed that treatment with DiOHF concentration-dependently inhibited phosphorylation of ERK, suggesting that inhibition of ERK activation may have a role in the inhibition of MCP-1 expression by DiOHF. However, it should be noted that DiOHF-induced inhibition on ERK phosphorylation may be an indirect effect based on its inhibitory actions on PDGFR and FAK. Moreover, the stronger activation of p38 by PDGF than IL-1β may provide an explanation to the lower effectiveness of 30 µmol·L−1 DiOHF on PDGF-induced MCP-1 expression (Figure 2B). The ERK phosphorylation induced by PDGF in the presence of DiOHF at lower concentrations may be explained by the PDGFR-independent activation of ERK by the ligand (Cartel et al., 2001). In kidney glomerular cells, quercetin may inhibit IL-1β-triggered MCP-1 expression by suppressing NF-κB activity (Ishikawa et al., 1999). Given the similar structures of DiOHF and quercetin, we examined whether DiOHF could inhibit NF-κB activation. However, we found that DiOHF had no effect on IL-1β-induced NF-κB activity, suggesting that NF-κB pathway is unlikely to be involved in DiOHF effects. Therefore, a limitation of the present study is that we are still not clear about how DiOHF suppresses the up-regulation of MCP-1 by IL-1β in smooth muscle cells.
It has previously been suggested that FAK may have important roles in mediating the pathological neointimal hyperplasia, which is a result of accumulation of activated smooth muscle cells in the arterial intima. For example, FAK is overexpressed in neointimal smooth muscle cells in human arteries (Owens et al., 2001). Inhibition of FAK function with FRNK reduced PDGF-induced vascular smooth muscle cell migration and proliferation (Hauck et al., 2000; Taylor et al., 2001). However, the role of FAK in neointimal hyperplasia in vivo has not been proven, probably because of the lack of a specific inhibitor and the lethal phenotype of the FAK knockout mouse. In our experiments, we demonstrated that DiOHF significantly inhibited attachment-induced cell spreading in vascular smooth muscle cells, a process involving FAK-mediated mechanisms (Owen et al., 1999). Our observation that DiOHF suppressed cell spreading is consistent with its inhibitory effects on the kinase activity of FAK. We also demonstrated that DiOHF markedly suppressed proliferation of RASMCs. However, because DiOHF also inhibited PDGFR activation, it is difficult to dissect FAK- and PDGFR-mediated pathways in relation to the cytostatic effect of DiOHF in smooth muscle cells.
Taken together, our results show that DiOHF is a potent inhibitor of FAK, a kinase for which a small molecule inhibitor is still not available. DiOHF also inhibits PDGFR-mediated signalling. Although both effects on FAK and PDGFR may be responsible for the inhibitory action of DiOHF on MCP-1 expression in vascular smooth muscle cells, our data indicate that DiOHF might be a useful tool for dissection of the (patho)physiological roles of FAK signalling in the future.
Acknowledgments
The authors thank Dr K.M. Yamada (National Institutes of Health, Bethesda, MD, USA) for providing us the pRK-VSV-FRNK plasmids. This work was supported by Project Grants from the National Health and Medical Research Council of Australia and Grants-in-Aid from the National Heart Foundation of Australia. G.J.D. receives an National Health and Medical Research Council Principal Research Fellowship.
Glossary
Abbreviations:
- DiOHF
3′,4′-dihydroxyflavonol
- ERK
extracellular signal-regulated kinase
- FAK
focal adhesion kinase
- FRNK
FAK-related non-kinase
- IL-1β
interleukin 1-β
- MAPK
mitogen-activated protein kinase
- MCP-1
monocyte chemoattractant protein-1
- NF-κB
nuclear factor-κB
- PDGF
platelet-derived growth factor
- PDGFR
PDGF receptor
- RASMCs
rat aortic smooth muscle cells
- VSV-G
vesicular stomatitis virus glycoprotein
Conflicts of interest
None.
References
- Aiello RJ, Bourassa PA, Lindsey S, Weng W, Natoli E, Rollins BJ, et al. Monocyte chemoattractant protein-1 accelerates atherosclerosis in apolipoprotein E-deficient mice. Arterioscler Thromb Vasc Biol. 1999;19:1518–1525. doi: 10.1161/01.atv.19.6.1518. [DOI] [PubMed] [Google Scholar]
- Birdsall HH, Green DM, Trial J, Youker KA, Burns AR, MacKay CR, et al. Complement C5a, TGF-beta 1, and MCP-1, in sequence, induce migration of monocytes into ischemic canine myocardium within the first one to five hours after reperfusion. Circulation. 1997;95:684–692. doi: 10.1161/01.cir.95.3.684. [DOI] [PubMed] [Google Scholar]
- Cartel NJ, Liu J, Wang J, Post M. PDGF-BB-mediated activation of p42(MAPK) is independent of PDGF beta-receptor tyrosine phosphorylation. Am J Physiol Lung Cell Mol Physiol. 2001;281:L786–L798. doi: 10.1152/ajplung.2001.281.4.L786. [DOI] [PubMed] [Google Scholar]
- Charo IF, Taubman MB. Chemokines in the pathogenesis of vascular disease. Circ Res. 2004;95:858–866. doi: 10.1161/01.RES.0000146672.10582.17. [DOI] [PubMed] [Google Scholar]
- Chen Y, Hallenbeck JM, Ruetzler C, Bol D, Thomas K, Berman NE, et al. Overexpression of monocyte chemoattractant protein 1 in the brain exacerbates ischemic brain injury and is associated with recruitment of inflammatory cells. J Cereb Blood Flow Metab. 2003;23:748–755. doi: 10.1097/01.WCB.0000071885.63724.20. [DOI] [PubMed] [Google Scholar]
- Frangogiannis NG. The role of the chemokines in myocardial ischemia and reperfusion. Curr Vasc Pharmacol. 2004;2:163–174. doi: 10.2174/1570161043476375. [DOI] [PubMed] [Google Scholar]
- Funakoshi-Tago M, Sonoda Y, Tanaka S, Hashimoto K, Tago K, Tominaga S, et al. Tumor necrosis factor-induced nuclear factor kappaB activation is impaired in focal adhesion kinase-deficient fibroblasts. J Biol Chem. 2003;278:29359–29365. doi: 10.1074/jbc.M213115200. [DOI] [PubMed] [Google Scholar]
- Grandaliano G, Monno R, Ranieri E, Gesualdo L, Schena FP, Martino C, et al. Regenerative and proinflammatory effects of thrombin on human proximal tubular cells. J Am Soc Nephrol. 2000;11:1016–1025. doi: 10.1681/ASN.V1161016. [DOI] [PubMed] [Google Scholar]
- Gu J, Tamura M, Pankov R, Danen EH, Takino T, Matsumoto K, et al. Shc and FAK differentially regulate cell motility and directionality modulated by PTEN. J Cell Biol. 1999;146:389–403. doi: 10.1083/jcb.146.2.389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanks SK, Ryzhova L, Shin NY, Brabek J. Focal adhesion kinase signaling activities and their implications in the control of cell survival and motility. Front Biosci. 2003;8:d982–d996. doi: 10.2741/1114. [DOI] [PubMed] [Google Scholar]
- Hassid A, Huang S, Yao J. Role of PTP-1B in aortic smooth muscle cell motility and tyrosine phosphorylation of focal adhesion proteins. Am J Physiol. 1999;277:H192–H198. doi: 10.1152/ajpheart.1999.277.1.H192. [DOI] [PubMed] [Google Scholar]
- Hauck CR, Hsia DA, Schlaepfer DD. Focal adhesion kinase facilitates platelet-derived growth factor-BB-stimulated ERK2 activation required for chemotaxis migration of vascular smooth muscle cells. J Biol Chem. 2000;275:41092–41099. doi: 10.1074/jbc.M005450200. [DOI] [PubMed] [Google Scholar]
- Hayasaki T, Kaikita K, Okuma T, Yamamoto E, Kuziel WA, Ogawa H, et al. CC chemokine receptor-2 deficiency attenuates oxidative stress and infarct size caused by myocardial ischemia-reperfusion in mice. Circ J. 2006;70:342–351. doi: 10.1253/circj.70.342. [DOI] [PubMed] [Google Scholar]
- Ishikawa Y, Sugiyama H, Stylianou E, Kitamura M. Bioflavonoid quercetin inhibits interleukin-1-induced transcriptional expression of monocyte chemoattractant protein-1 in glomerular cells via suppression of nuclear factor-kappaB. J Am Soc Nephrol. 1999;10:2290–2296. doi: 10.1681/ASN.V10112290. [DOI] [PubMed] [Google Scholar]
- Jalali S, Li YS, Sotoudeh M, Yuan S, Li S, Chien S, et al. Shear stress activates p60src-Ras-MAPK signaling pathways in vascular endothelial cells. Arterioscler Thromb Vasc Biol. 1998;18:227–234. doi: 10.1161/01.atv.18.2.227. [DOI] [PubMed] [Google Scholar]
- Jiang F, Dusting GJ. Natural phenolic compounds as cardiovascular therapeutics: potential role of their antiinflammatory effects. Curr Vasc Pharmacol. 2003;1:135–156. doi: 10.2174/1570161033476736. [DOI] [PubMed] [Google Scholar]
- Jiang F, Guo N, Dusting GJ. Modulation of NADPH oxidase expression and function by 3′,4′-dihydroxyflavonol in phagocytic and vascular cells. J Pharmacol Exp Ther. 2007;324:261–269. doi: 10.1124/jpet.107.131433. [DOI] [PubMed] [Google Scholar]
- Kanegae K, Tamura M, Kabashima N, Serino R, Tokunaga M, Oikawa S, et al. Synergistic induction of monocyte chemoattractant protein-1 by integrins and platelet-derived growth factor via focal adhesion kinase in mesangial cells. Nephrol Dial Transplant. 2005;20:2080–2088. doi: 10.1093/ndt/gfh998. [DOI] [PubMed] [Google Scholar]
- Lee SK, Kim BS, Yang WS, Kim SB, Park SK, Park JS. High glucose induces MCP-1 expression partly via tyrosine kinase-AP-1 pathway in peritoneal mesothelial cells. Kidney Int. 2001;60:55–64. doi: 10.1046/j.1523-1755.2001.00770.x. [DOI] [PubMed] [Google Scholar]
- Mitra SK, Hanson DA, Schlaepfer DD. Focal adhesion kinase: in command and control of cell motility. Nat Rev Mol Cell Biol. 2005;6:56–68. doi: 10.1038/nrm1549. [DOI] [PubMed] [Google Scholar]
- Mori E, Komori K, Yamaoka T, Tanii M, Kataoka C, Takeshita A, et al. Essential role of monocyte chemoattractant protein-1 in development of restenotic changes (neointimal hyperplasia and constrictive remodeling) after balloon angioplasty in hypercholesterolemic rabbits. Circulation. 2002;105:2905–2910. doi: 10.1161/01.cir.0000018603.67989.71. [DOI] [PubMed] [Google Scholar]
- Nakayamada S, Saito K, Fujii K, Yasuda M, Tamura M, Tanaka Y. beta1 integrin-mediated signaling induces intercellular adhesion molecule 1 and Fas on rheumatoid synovial cells and Fas-mediated apoptosis. Arthritis Rheum. 2003;48:1239–1248. doi: 10.1002/art.10941. [DOI] [PubMed] [Google Scholar]
- Okada M, Matsumori A, Ono K, Furukawa Y, Shioi T, Iwasaki A, et al. Cyclic stretch upregulates production of interleukin-8 and monocyte chemotactic and activating factor/monocyte chemoattractant protein-1 in human endothelial cells. Arterioscler Thromb Vasc Biol. 1998;18:894–901. doi: 10.1161/01.atv.18.6.894. [DOI] [PubMed] [Google Scholar]
- Owen JD, Ruest PJ, Fry DW, Hanks SK. Induced focal adhesion kinase (FAK) expression in FAK-null cells enhances cell spreading and migration requiring both auto- and activation loop phosphorylation sites and inhibits adhesion-dependent tyrosine phosphorylation of Pyk2. Mol Cell Biol. 1999;19:4806–4818. doi: 10.1128/mcb.19.7.4806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owens LV, Xu L, Marston WA, Yang X, Farber MA, Iacocca MV, et al. Overexpression of the focal adhesion kinase (p125FAK) in the vascular smooth muscle cells of intimal hyperplasia. J Vasc Surg. 2001;34:344–349. doi: 10.1067/mva.2001.114814. [DOI] [PubMed] [Google Scholar]
- Parsons JT. Focal adhesion kinase: the first ten years. J Cell Sci. 2003;116:1409–1416. doi: 10.1242/jcs.00373. [DOI] [PubMed] [Google Scholar]
- Richardson A, Parsons T. A mechanism for regulation of the adhesion-associated proteintyrosine kinase pp125FAK. Nature. 1996;380:538–540. doi: 10.1038/380538a0. [DOI] [PubMed] [Google Scholar]
- Rollins BJ, Morrison ED, Stiles CD. Cloning and expression of JE, a gene inducible by platelet-derived growth factor and whose product has cytokine-like properties. Proc Natl Acad Sci USA. 1988;85:3738–3742. doi: 10.1073/pnas.85.11.3738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rong JX, Berman JW, Taubman MB, Fisher EA. Lysophosphatidylcholine stimulates monocyte chemoattractant protein-1 gene expression in rat aortic smooth muscle cells. Arterioscler Thromb Vasc Biol. 2002;22:1617–1623. doi: 10.1161/01.atv.0000035408.93749.71. [DOI] [PubMed] [Google Scholar]
- Rovin BH, Dickerson JA, Tan LC, Fassler J. Modulation of IL-1-induced chemokine expression in human mesangial cells through alterations in redox status. Cytokine. 1997;9:178–186. doi: 10.1006/cyto.1996.0152. [DOI] [PubMed] [Google Scholar]
- Sabe H, Hata A, Okada M, Nakagawa H, Hanafusa H. Analysis of the binding of the Src homology 2 domain of Csk to tyrosine-phosphorylated proteins in the suppression and mitotic activation of c-Src. Proc Natl Acad Sci USA. 1994;91:3984–3988. doi: 10.1073/pnas.91.9.3984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaller MD, Parsons JT. pp125FAK-dependent tyrosine phosphorylation of paxillin creates a high-affinity binding site for Crk. Mol Cell Biol. 1995;15:2635–2645. doi: 10.1128/mcb.15.5.2635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shyy YJ, Li YS, Kolattukudy PE. Activation of MCP-1 gene expression is mediated through multiple signaling pathways. Biochem Biophys Res Commun. 1993;192:693–699. doi: 10.1006/bbrc.1993.1470. [DOI] [PubMed] [Google Scholar]
- Taylor JM, Mack CP, Nolan K, Regan CP, Owens GK, Parsons JT. Selective expression of an endogenous inhibitor of FAK regulates proliferation and migration of vascular smooth muscle cells. Mol Cell Biol. 2001;21:1565–1572. doi: 10.1128/MCB.21.5.1565-1572.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S, Dusting GJ, May CN, Woodman OL. 3′,4′-Dihydroxyflavonol reduces infarct size and injury associated with myocardial ischaemia and reperfusion in sheep. Br J Pharmacol. 2004;142:443–452. doi: 10.1038/sj.bjp.0705815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe Y, Tamura M, Osajima A, Anai H, Kabashima N, Serino R, et al. Integrins induce expression of monocyte chemoattractant protein-1 via focal adhesion kinase in mesangial cells. Kidney Int. 2003;64:431–440. doi: 10.1046/j.1523-1755.2003.00122.x. [DOI] [PubMed] [Google Scholar]
- Zen K, Masuda J, Sasaguri T, Kosaka C, Shimokado K, Ogata J. Protein tyrosine kinases and protein kinase C are required for gene expression of monocyte chemoattractant protein-1 in human monocytes. Ann N Y Acad Sci. 1995;748:508–509. doi: 10.1111/j.1749-6632.1994.tb17349.x. [DOI] [PubMed] [Google Scholar]