

Abstract
Background and purpose:
The neurosteroid, dehydroepiandrosterone sulphate (DHEAS) and its non-sulphated form, DHEA, are considered as crucial endogenous modulators of a number of important physiological events. Evidence suggests that DHEAS and DHEA modulate central nervous system-related functions by activating sigma-1 receptors and/or allosterically inhibiting γ-aminobutyric acic receptor type A (GABAA) receptors. As both the sigma-1 receptor and the GABAA receptor play important roles in spinal pain transmission, the present study was designed to examine whether intrathecally injected DHEAS or DHEA affect nociceptive signalling at the spinal cord level.
Experimental approach:
We first determined whether intrathecal (i.t.) DHEA or DHEAS injection was able to affect nociceptive thresholds to peripheral mechanical stimulation and subsequently examined whether this effect was mediated by sigma-1 or the GABAA receptors.
Key results:
The i.t. DHEAS injection dose-dependently decreased the nociceptive threshold to mechanical stimulation, thus producing mechanical allodynia. Moreover, this DHEAS-induced mechanical allodynia was significantly reduced by administration of the sigma-1 receptor antagonist, BD-1047 or the GABAA receptor agonist, muscimol. Conversely, i.t. DHEA had no effect on mechanical sensitivity. However, when i.t. DHEA was combined with the GABAA receptor antagonist bicuculline, DHEA dose-dependently produced mechanical allodynia similar to that of DHEAS. This effect was blocked by BD-1047 and by muscimol.
Conclusions and implications:
These findings indicate that i.t. injection of DHEAS produces mechanical allodynia and that the development of this mechanical allodynia is mediated by sigma-1 and GABAA receptors. The findings of this study raise several interesting questions for further investigations into the mechanisms underlying neurosteroid modulation of spinal pain transmission.
British Journal of Pharmacology (2009) 157, 666–673; doi:10.1111/j.1476-5381.2009.00197.x; published online 30 April 2009
Keywords: Dehydroepiandrosterone sulphate, dehydroepiandrosterone, sigma-1 receptor, GABAA receptor, mechanical allodynia
Introduction
Neurosteroids are steroid hormones that act as potent endogenous neuromodulators with rapid actions in the central and peripheral nervous systems. They are synthesized either de novo from cholesterol or by in situ metabolism of blood-borne precursors that accumulate in the nervous system independently of classical steroidogenic gland secretion rates (Baulieu, 1998; Compagnone and Mellon, 2000). One of these neurosteroids, dehydroepiandrosterone (DHEA) and its sulphate derivative (DHEAS) are considered as crucial endogenous modulators of numerous physiological functions, including memory, neurogenesis and aging (Baulieu and Robel, 1998; Ren et al., 2004; Dillon, 2005; Maayan et al., 2005; Wojtal et al., 2006). Because of this, exogenous DHEA or DHEAS administration has been tried in an attempt to modulate certain neurobiological mechanisms in animals and in a few clinical trials (Baulieu, 1999; Shirayama et al., 2002). For a number of neuroactive steroids, sulphation at C-3 is an important control point for the activity of endogenous neurosteroids (Park-Chung et al., 1999; Gibbs et al., 2006). While DHEA and DHEAS are interconversion products [DHEAS serving as the major source (60–80%) of DHEA and DHEA serving as a minor source (5–7%) of DHEAS], their actions appear to differ in several systems (Zhang et al., 2002).
Recently, Kibaly et al. (2007) reported that acute intrathecal (i.t.) treatment with DHEA decreases the basal nociceptive thresholds in both neuropathic and control rats, suggesting that i.t. DHEA can affect spinal circuits that are involved in pain signalling. However, the role of the sulphated neurosteroid, DHEAS, on nociception in the spinal cord is unclear. It is important to note, however, that recent work indicates that neurosteroidogenesis is an endogenous mechanism activated in the spinal cord and brainstem for adaptation of the body to chronic peripheral neuropathies (Patte-Mensah and Mensah-Nyagan, 2008).
The possibility that neurosteroids, such as DHEAS and DHEA, could be endogenous activators/inactivators of the sigma-1 receptor and possibly even the ‘endogenous ligands’ for this receptor has generated significant interest in this area (see reviews by Dubrovsky, 2005; Maurice et al., 2006). Recently, Cheng et al. (2008) have shown that DHEAS inhibits persistent sodium currents via the activation of sigma-1 receptors-Gi protein-protein kinase C-coupled signalling pathway, providing the first mechanism by which DHEAS may affect the excitability of neurons via the sigma-1 receptor. With respect to pain, intraplantar injection of DHEAS has been shown to induce nociception via sigma-1 receptors in the peripheral nociceptive flexor test (Ueda et al., 2001). Moreover, accumulating data from our laboratories have shown that spinal sigma-1 receptors are involved with pain facilitatory mechanisms (Kim et al., 2006, 2008). Therefore, it seems likely that DHEA or DHEAS induce pain facilitation in part via activation of spinal sigma-1 receptors. On the other hand, it is well established that DHEAS and DHEA act as allosteric negative modulators of γ-aminobutyric acic receptor type A (GABAA) receptors (Belelli and Lambert, 2005) and that GABAergic inhibitory activity tonically modulates spinal pain transmission (Jasmin et al., 2004). Interestingly, non-sulphated DHEA is less potent than DHEAS in inhibiting GABAA receptors (Imamura and Prasad, 1998; Mehta and Ticku, 2001). Based on this information we hypothesize that there are differences between the effect of DHEAS and DHEA on spinal pain transmission. Moreover, we hypothesized that spinally administered DHEAS would induce a greater facilitatory effect on pain sensation than DHEA and that this effect occurs via activation of sigma-1 receptors and inhibition of GABAA receptors.
To verify this hypothesis, pharmacological approaches were used to study the effects, as well as the mechanism of action, of intrathecally administered DHEAS versus DHEA on mechanical nociceptive thresholds in mice. It is important to point out that the passage of DHEAS into the brain is proportionately much less than DHEA (Baulieu and Robel, 1998), but nonetheless several lines of evidence suggest that peripheral DHEAS can pass through the blood–brain barrier and affect central brain sites (Akwa et al., 1993; Asaba et al., 2000; Chen et al., 2006). On the other hand, it has been reported that intraplantar treatment of DHEAS causes nociceptive flexor responses indicating that DHEAS also has a peripheral site of action (Ueda et al., 2001). Based on these data, it is difficult to discriminate peripheral versus central (spinal/brain) actions of DHEAS or DHEA, as both sites are capable of influencing nociception. More importantly, from a mechanistic point of view, it is currently unclear how spinal DHEAS directly affects pain sensation. Therefore, the present study focused on the role of these neurosteroids in nociception at the spinal level. In order to limit the effect of DHEAS/DHEA to the spinal cord, in this study we used i.t. administration of these neurosteroids. This was accomplished using the following experimental design. First, behavioural tests were performed to determine the effects of i.t. injection of DHEAS or DHEA on mechanical pain threshold. Then the mechanism of action of these neurosteroids was determined by investigating the possible involvement of sigma-1 or GABAA receptors. Finally, this hypothesis was further supported by administration of another neurosteroid, progesterone, which acts as an endogenous blocker of both sigma-1 and GABAA receptors (Tsutsui et al., 2000; Ueda et al., 2001; Bicikova and Hampl, 2007).
Methods
Animals
Male ICR mice (24–30 g) were purchased from the Laboratory Animal Center of Seoul National University (Seoul, South Korea). They were free to access food and water and maintained in temperature- and light-controlled rooms (23 ± 2°C, 12/12 h light/dark cycle with lights on at 08h00min) for at least 1 week prior to beginning an experiment. The experimental protocols for animal usage were reviewed and approved by the SNU Animal Care and Use Committee and conform to NIH guidelines (NIH publication No. 86-23, revised 1985).
Drugs
The following drugs were used: DHEA, DHEAS, muscimol (a GABAA receptor agonist), bicuculline (a GABAA receptor antagonist), BD-1047 {N-[2-(3,4-dichlorophenyl)ethyl]-N-methyl-2-(dimethylamino) ethylamine dihydrobromide; a sigma-1 receptor antagonist} and progesterone. BD-1047 and muscimol were purchased from Tocris (Bristol, UK). The remaining drugs were obtained from Sigma Chemical Company (St. Louis, MO, USA). DHEA and progesterone were diluted in 1% DMSO (dimethyl sulphoxide) in corn oil. The vehicle for DHEAS and bicuculline was 1% DMSO in saline. The remaining drugs were diluted in physiological saline. The drug and molecular target nomenclature used in this article conforms to the British Journal of Pharmacology's ‘Guide to receptors and channels’ (Alexander et al., 2008).
Intrathecal drug administration
Drugs were dissolved in 5 µL of vehicle. We injected a 5 µL volume intrathecally because data suggest that this is likely to be the upper limit that can be reliably injected into a mouse without any appreciable redistribution of the drug through the cerebrospinal fluid to the basal cisterns of the brain (Rieselbach et al., 1962). Intrathecal injections were made into the L5-L6 intervertebral space of unanaesthetized mice using a 10 µL Hamilton syringe connected to a 30-gauge needle as previously described by Hylden and Wilcox (1980). The flick of the tail was considered indicative of a successful i.t. administration. The control group received an i.t. injection of vehicle.
Mechanical allodynia test
The von Frey monofilaments (Stoelting, Wood Dale, IL, USA) were used to quantify mechanical allodynia. On each testing day, mice (n= 7–8) were habituated inside a small plastic chamber (12 cm wide × 11 cm long × 15 cm high) with a floor of soft wire mesh for 1 h. Mechanical sensitivity was tested with von Frey hairs with bending forces from 0.07–6 g. The 50% mechanical withdrawal threshold (the force of the von Frey hair in g to which an animal reacts in 50% of the presentations) was determined on the basis of the Dixon up-and-down method (Dixon, 1980; Chaplan et al., 1994) modified for mice (Sommer and Schafers, 1998). Testing was initiated with the 0.6 g hair. The von Frey filaments were applied from underneath the grid floor perpendicular to the plantar surface until slight buckling occurred. They were held for a period of approximately 3 s before being removed. A positive response was recorded if the paw was withdrawn. Ambulation was considered an ambiguous response, and in such cases the stimulation was repeated. In the event of a positive response, the next weaker filament was then applied and the next measurement recorded. In the absence of a response, then the next stronger filament was presented. This consecutive way of applying filaments was continued until six responses in the immediate vicinity of the 50% threshold were obtained. The resulting sequence of positive and negative responses was used to interpolate the 50% withdrawal threshold. Data are presented as % of the baseline value.
Statistical analysis
All values are expressed as the mean ± SEM. Statistical analysis was performed using Prism 4.0 (Graph Pad Software, San Diego, USA). The area under the curve was analysed using anova followed by a Newman–Keuls post hoc test. For analysis of nociceptive sensitivity to mechanical stimulation at the different time points and under different treatment conditions, the data were analysed using a two-way repeated-measures anova followed by a post hoc Bonferroni analysis. Differences with P < 0.05 were considered significant.
Results
The effect of i.t. DHEAS on nociceptive threshold
The mean value for the 50% nociceptive threshold for mechanical stimulation using the up–down method was 1.32 ± 0.25 g before treatment. The baseline values obtained for each treatment group were not statistically different. However, i.t. injection of DHEAS (150, 300 or 600 pmol) dose-dependently decreased withdrawal threshold to mechanical stimulation compared with that of the vehicle control group (Figure 1A and B). In particular, the mechanical allodynia induced by the 300 and 600 pmol doses of DHEAS peaked during the 10–60 min time period following i.t. injection of DHEAS and then returned to baseline by 180 min post injection (Figure 1A). Conversely, i.t. injection of DHEA had no effect on the mechanical nociceptive threshold, even at the highest dose given (1 µmol, Figure 2).
Figure 1.
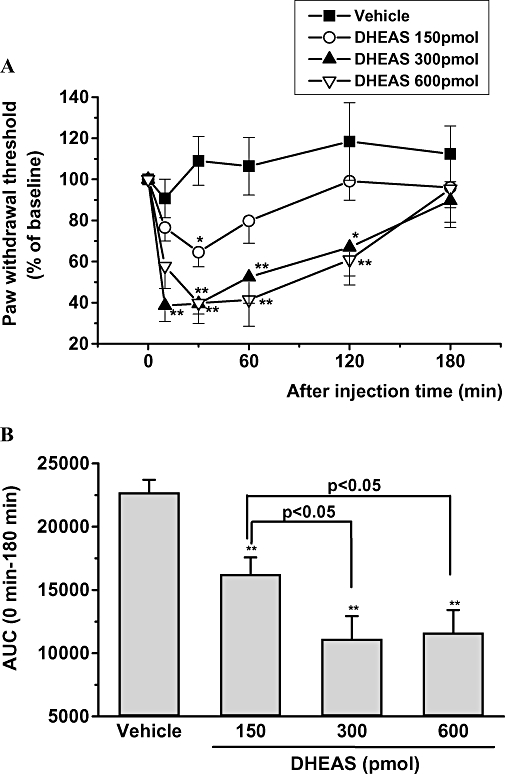
(A) The effect of intrathecal administration of three different doses of dehydroepiandrosterone sulphate (DHEAS) (150, 300 and 600 pmol per mouse) on the nociceptive threshold to mechanical stimulation. Data are presented as the percentage of pre-baseline value. **P < 0.01, *P < 0.05: significantly different from the value of the vehicle group (dimethyl sulphoxide 1% in saline) at same time point. (B) The mean area under the curve for paw withdrawal threshold for the vehicle-treated group and for each of the three groups receiving different doses of DHEAS for the period from 0 to 120 min after injection. **P < 0.01, different from the value of the vehicle group; vertical lines show SEM, n= 8 mice per group. AUC, area under the curve.
Figure 2.
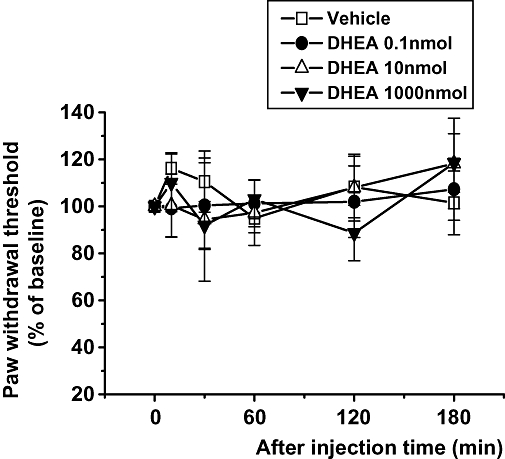
The effect of intrathecal administration of three different doses of dehydroepiandrosterone (DHEA) (0.1, 10 and 1000 nmol per mouse) on the nociceptive threshold to mechanical stimulation. The vehicle control group was treated with 1% dimethyl sulphoxide in corn oil. Data are presented as a percentage of pre-baseline values; n= 8 mice per group.
The role of sigma-1 receptor or GABAA receptor on DHEAS-induced pain facilitation
The 300 pmol dose of DHEAS was selected for these experiments because this dose showed a maximum mechanical allodynia response. The development of DHEAS-induced mechanical allodynia was dose-dependently reversed by pretreatment with the sigma-1 receptor antagonist, BD-1047 (Figure 3A) or by pretreatment with the GABAA receptor agonist, muscimol (Figure 3B). Neither administration of BD-1047 (100 nmol) nor muscimol (0.3 nmol) alone modified the mechanical nociceptive threshold (data not shown).
Figure 3.
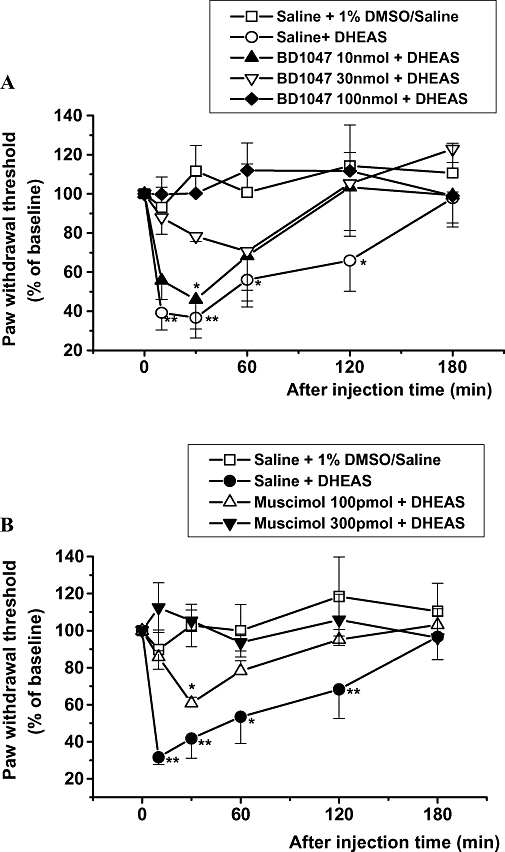
The effect of intrathecal dehydroepiandrosterone sulphate (DHEAS) (300 pmol per mouse) administered 5 min after i.t. pretreatment with BD-1047 (A) or muscimol (B) on the nociceptive threshold to mechanical stimulation. Data are presented as a percentage of the pre-baseline values. **P < 0.01, *P < 0.05: different from the vehicle control group [saline + 1% dimethyl sulphoxide (DMSO) in saline] value; n= 7 mice per group.
The effect of i.t. DHEA or DHEA plus bicuculline on pain sensation
We first examined whether administration of different doses of the GABAA receptor inhibitor, bicuculline, had an effect on the normal nociceptive threshold. We found that i.t. injection of bicuculline induced mechanical allodynia in a dose-dependent fashion as shown in Figure 4A. Interestingly, although the 30 pmol dose of bicuculline alone had no effect on the mechanical nociceptive threshold (Figure 4A and B), when this dose of bicuculline was combined with DHEA (150, 300 or 600 pmol), it dose-dependently produced mechanical allodynia (Figure 4B). Moreover, the mechanical allodynia induced by DHEA (300 pmol) + bicuculline was reversed by pretreatment with BD-1047 or muscimol (Figure 4C).
Figure 4.
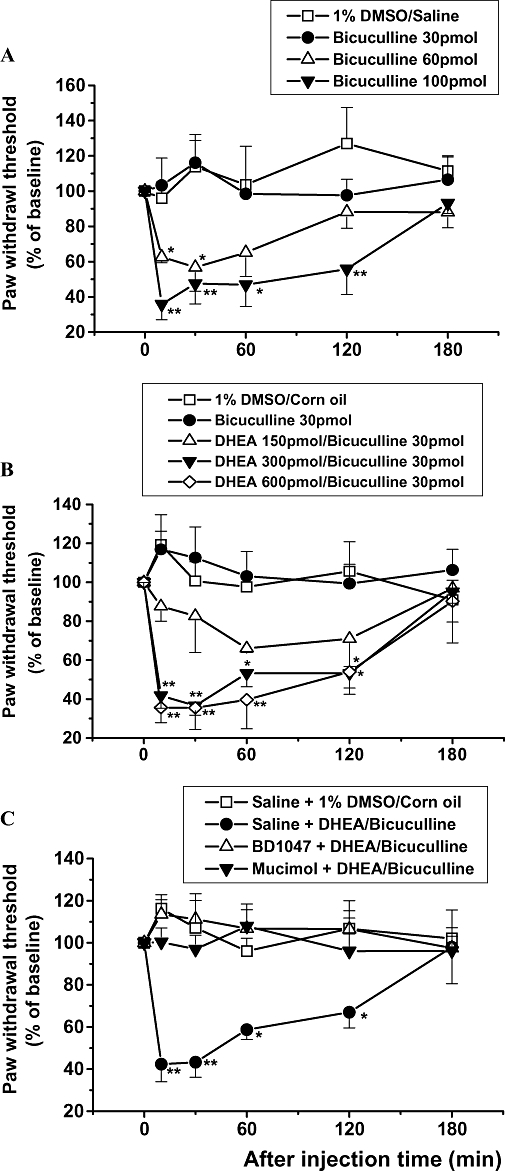
(A) The effect of intrathecal injection of bicuculline on the paw withdrawal threshold to mechanical stimulation. (B) The effect of a combination of dehydroepiandrosterone (DHEA) and bicuculline (DHEA/bicuculline) on the paw withdrawal threshold to mechanical stimulation. (C) The effect of intrathecal pretreatment with BD-1047 (100 nmol) or muscimol (0.3 nmol) on mechanical allodynia induced by DHEA + bicuculline. Data are presented as the percentage of pre-baseline values. **P < 0.01, *P < 0.05: different from vehicle control group [(A) 1% dimethyl sulphoxide (DMSO) in saline; (B) 1% DMSO in corn oil; (C) saline + 1% DMSO in corn oil] value; n= 7 mice per group.
The role of progesterone in the mechanical allodynia induced by DHEAS or DHEA plus bicuculline
Intrathecal pretreatment with progesterone completely blocked DHEAS (300 pmol)-induced mechanical allodynia (Figure 5A). Moreover, i.t. pretreatment with progesterone also blocked the mechanical allodynia produced by DHEA (300 pmol) combined with bicuculline (30 pmol; Figure 5B). Conversely, i.t. progesterone (10 nmol) treatment alone had no effect on nociceptive thresholds to mechanical stimulation.
Figure 5.
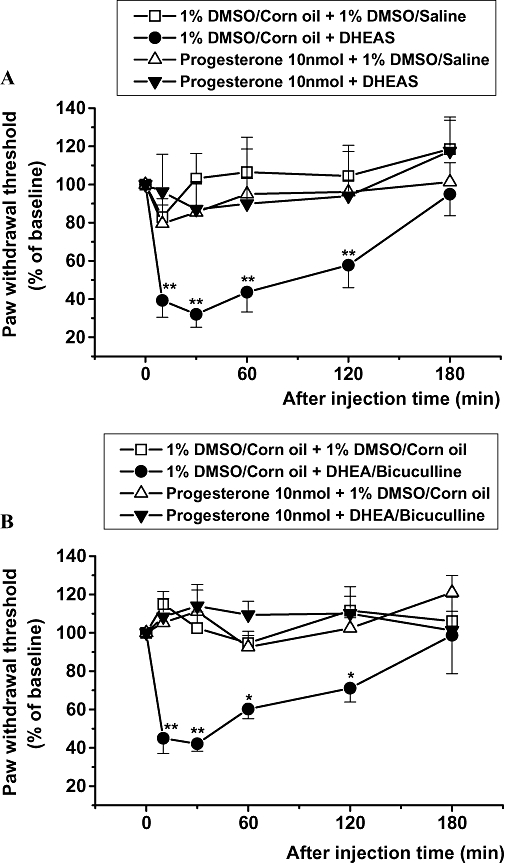
The effect of intrathecal dehydroepiandrosterone sulphate (DHEAS) (300 pmol per mouse) (A) or a combination of DHEA plus bicuculline (B) given 5 min after intrathecal pretreatment with progesterone on the nociceptive threshold for mechanical stimulation. Data are presented as the percentage of pre-baseline values. **P < 0.01, *P < 0.05: different from vehicle control group [(A) 1% dimethyl sulphoxide (DMSO) in corn oil + 1% DMSO in saline, (B) 1% DMSO in corn oil + 1% DMSO in corn oil] value; n= 7 mice per group.
Discussion
The present study demonstrated that i.t. injection of DHEAS in mice enhanced the animal's sensitivity to mechanical stimuli and that this resulting mechanical allodynia was associated with the effect of DHEAS on sigma-1 and GABAA receptors. Previous studies have shown that alterations in the levels of DHEA or DHEAS in the brain can allosterically modulate GABAA, N-methyl-D-aspartate and sigma-1 receptors (McEwen, 1991; Genud et al., 2009). With respect to sigma-1 receptors, recent studies from our laboratories have demonstrated that i.t administration of sigma-1 agonists significantly enhance peripheral nociceptive responses, suggesting that spinal sigma-1 receptors play an important role in nociceptive processing (Kim et al., 2006; 2008; Roh et al., 2008a,b) Moreover, the results of the present study indicate that sigma-1 receptors also play an important role in DHEAS-induced mechanical allodynia. On the other hand, fast inhibitory controls, which act at the spinal cord level and are mediated by GABAA receptors, appear to play an important role in preventing or reducing the development of mechanical allodynia in a number of different pathological pain states (Galluzzi, 2007). Indeed, data obtained in both the present study and previous research studies have demonstrated that effective blockade of spinal GABAA receptors by i.t. bicuculline produces mechanical allodynia (Onaka et al., 1996). The present study extends these findings by indicating that DHEAS-induced mechanical allodynia is also mediated by negative modulation of spinal GABAA receptors.
In contrast to DHEAS, i.t. injection of non-sulphated DHEA failed to produce a pain facilitatory effect on mechanical stimulation, even at the highest dose (1 µmol) tested in this study. One plausible explanation for this discrepancy stems from that fact that there are multiple recognition sites associated with GABAA receptors, including barbiturate, GABA and benzodiazepine-type ligand-binding sites. In this regard, Park-Chung and his colleagues (Park-Chung et al., 1999) demonstrated that sulphated neurosteroids, such as DHEAS and allopregnanolone sulphate, act at distinct sites on the GABAA receptor in comparison with their unsulphated form. In support of this finding, DHEA does not inhibit the binding of DHEAS to brain membranes, suggesting that they may also bind to distinct sites (Demirgoren et al., 1991). DHEAS inhibits the binding of [3H]-GABA to both cerebral cortex and cerebellar membranes, while DHEA (up to 100 µmol·L−1) has no effect on GABA binding to these brain regions (Sousa and Ticku, 1997). Similarly, the affinity of DHEA to GABA-binding sites (using a [3H]-muscimol assay) was too low to detect in rat cerebral cortex, while the affinity of DHEAS was more prominent (IC50= 557 µmol·L−1, Mehta and Ticku, 2001). Taken together, DHEA has very low affinity for the GABA/muscimol binding site on GABAA receptors and this characteristic may produce the pharmacological differences observed when comparing DHEA with DHEAS. In support of this assumption, we have also demonstrated that i.t. administration of a combination of DHEA and a sub-effective dose (30 pmol) of bicuculline, a GABAA receptor antagonist, significantly decreased the nociceptive thresholds to mechanical stimulation.
Interestingly, Kibaly et al. (2007) have shown that i.t. DHEA administration induces mechanical and thermal hyperalgesia in naïve rats and this effect is totally blocked by administration of a sigma-1 receptor antagonist. Although DHEA is less potent at inhibiting GABAA receptors, it is still a sigma-1 activator. Therefore, a high dose of DHEA could induce pain facilitation. In this regard, it is important to note that the dose of DHEA used in the study by Kibaly et al. (2007) was significantly higher (approximately 10 µmol) than that used in the present study. Thus, the discrepancy between the present data and those of Kibaly et al. (2007) may relate to the dose of DHEA used or to possible species differences between mice and rats.
The present results suggest that the allodynia effects of DHEAS or DHEA are mediated through both sigma-1 and GABAA receptors in a possible synergistic, but not additive way. Supporting this assumption, the allodynia effects of DHEAS were completely blocked by administration of either a sigma-1 receptor antagonist or a GABAA agonist. Moreover, while DHEA did not induce allodynia, even at the highest dose (1000 nmol) tested, a very low dose (0.3 nmol) of DHEA induced allodynia when it was combined with a low dose of bicuculline, which by itself did not induce allodynia. Importantly, this pain facilitatory effect of DHEA plus bicuculline was completely blocked by administration of either a sigma-1 antagonist or a GABAA receptor agonist. A possible synergistic interaction between sigma-1 and GABAA receptors has not been convincingly described to date. However, Mtchedlishvili and Kapur (2003) reported that low concentrations of pregnenolone sulphate, which also acts as negative modulator of GABAA receptors, inhibited presynaptic GABA release in cultured hippocampal neurons. This effect was also abolished by the sigma-1 receptor antagonist, suggesting that sigma-1 receptor activation modulates GABA release and neurotransmission. As this observation does not provide a mechanistic picture of how the GABAA and the sigma-1 receptors interact, nor any insight into whether this interaction is direct or indirect, it is clear that further work is required to determine the exact mechanisms by which such synergistic interaction occurs in the spinal cord resulting in decreased mechanical nociceptive sensitivity.
Under chronic pain conditions, such as the neuropathic pain that often develops as a result of peripheral nerve injury, the effect of these neurosteroids on spinal pain transmission may in fact be altered. Our laboratory has recently reported that sigma-1 receptor expression significantly increases in the ipsilateral spinal cord dorsal horn from day 1 to day 3 post surgery in a rat chronic constriction injury model of neuropathic pain (Roh et al., 2008b). Moreover, following peripheral nerve injury, there is an apparent loss of GABAergic inhibitory interneurons in the dorsal horn of the spinal cord (Zeilhofer, 2008). Collectively, these findings raise the possibility that changes in sigma-1 receptor expression and alterations in GABAergic transmission may play important roles in the development of central sensitization of pathological pain states, such as neuropathic pain. In support of this assumption, it has demonstrated that i.t. injection of the sigma-1 receptor antagonist, BD-1047, significantly blocks the induction of mechanical allodynia in rats with neuropathic pain induced by chronic constriction injury (Roh et al., 2008b). Conversely, BD-1047 administered alone does not show any pain-inhibitory effect. Additionally, GABAA receptors appear to play an important role in neuropathic pain, as it has been demonstrated that the GABAA receptor agonist, isoguvacine (Malan et al., 2002), reverses both the tactile allodynia and thermal hyperalgesia produced by spinal nerve ligation. In naïve animals, however, i.t. administration of isoguvacine does not cause any anti-nociceptive effects as measured by the tail-flick test. Moreover, i.t. treatment with another GABAA agonist, muscimol, also resulted in a dose-dependent antagonism of the allodynia associated with a rodent neuropathic pain model (Hwang and Yaksh, 1997). Collectively, these data together with the current findings suggest that co-administration of a sigma-1 agonist with a GABAA receptor antagonist would have a greater therapeutic effect on neuropathic pain than administration of either drug alone. In addition, under pathological pain conditions, the effect of DHEAS or DHEA on spinal pain transmission may be altered. Thus, the combination of an increase in spinal sigma-1 receptor expression and the loss of GABAergic inhibitory interneurons may induce an augmentation in the pain facilitatory effect of DHEAS in pathological pain states compared with its spinal cord effects in normal animals or humans. Further study is required to establish the actual relationship between DHEA and DHEAS and the development or maintenance of chronic pain, as well as the potential interaction of these neurosteroids with GABAA receptors and sigma-1 receptors in patients with chronic pain.
Progesterone and its metabolites have also been proposed to act as an endogenous sigma-1 receptor antagonist as well as a potent positive allosteric modulator of the GABAA receptor (Maurice et al., 2006). Therefore, it is not surprising that we found that pretreatment with progesterone completely blocked the mechanical allodynia induced by DHEAS and DHEA plus bicuculline. This suggests that spinal nociceptive transmission can be positively or negatively regulated by endogenous neurosteroids.
In conclusion, in the present study we observed that i.t. injection of the neurosteroid, DHEAS, produced mechanical allodynia and that this effect was mediated by activation of sigma-1 receptors and inhibition of GABAA receptors. Although i.t. injection of DHEA alone or of a sub-effective dose of the GABAA receptor antagonist, bicuculline, alone did not alter mechanical nociceptive thresholds, injection of a combination of DHEA and bicuculline induced mechanical allodynia similar to that induced by DHEAS alone. This suggests that DHEA is pharmacologically much less potent at inhibiting GABAA receptors than DHEAS. These results also indicate that i.t. DHEAS produces a significantly greater pain facilitatory effect than DHEA. Moreover, i.t. pretreatment with the neurosteroid, progesterone, reversed the DHEAS-induced mechanical allodynia, indicating that endogenous neurosteroids can have both nociceptive and anti-nociceptive effects. Because neurosteroids are synthesized in the dorsal horn of the spinal cord and have limited side effects, this study opens interesting possibilities for further investigations into the mechanisms underlying neurosteroid modulation of spinal pain transmission and more importantly for potential therapeutic use of exogenous neurosteroids to treat pathological pain conditions.
Acknowledgments
This research was supported by a grant (M103KV010015-06K2201-01510) from Brain Research Center of the 21st Century Frontier Research Program funded by the Ministry of Science and Technology, the Republic of Korea. The publication of this manuscript was also supported by a Research Fund from the Research Institute for Veterinary Science in the College of Veterinary Medicine, Seoul National University.
Glossary
Abbreviations:
- BD-1047
N-[2-(3,4-dichlorophenyl)ethyl]-N-methyl-2-(dimethylamino) ethylamine dihydrobromide
- DHEA
dehydroepiandrosterone
- DHEAS
dehydroepiandrosterone sulphate
- GABAA
γ-aminobutyric acic receptor type A
Conflicts of interest
None.
References
- Akwa Y, Sananes N, Gouezou M, Robel P, Baulieu EE, Le Goascogne C. Astrocytes and neurosteroids: metabolism of pregnenolone and dehydroepiandrosterone. Regulation by cell density. J Cell Biol. 1993;121:135–143. doi: 10.1083/jcb.121.1.135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SP, Mathie A, Peters JA. Guide to receptors and channels (GRAC) Br J Pharmacol. (3rd edn.) 2008;153(Suppl. 2):S1–209. doi: 10.1038/sj.bjp.0707746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asaba H, Hosoya K, Takanaga H, Ohtsuki S, Tamura E, Takizawa T, et al. Blood-brain barrier is involved in the efflux transport of a neuroactive steroid, dehydroepiandrosterone sulfate, via organic anion transporting polypeptide 2. J Neurochem. 2000;75:1907–1916. doi: 10.1046/j.1471-4159.2000.0751907.x. [DOI] [PubMed] [Google Scholar]
- Baulieu EE. Neurosteroids: a novel function of the brain. Psychoneuroendocrinology. 1998;23:963–987. doi: 10.1016/s0306-4530(98)00071-7. [DOI] [PubMed] [Google Scholar]
- Baulieu EE. Neuroactive neurosteroids: dehydroepiandrosterone (DHEA) and DHEA sulphate. Acta Paediatr Suppl. 1999;88:78–80. doi: 10.1111/j.1651-2227.1999.tb14408.x. [DOI] [PubMed] [Google Scholar]
- Baulieu EE, Robel P. Dehydroepiandrosterone (DHEA) and dehydroepiandrosterone sulfate (DHEAS) as neuroactive neurosteroids. Proc Natl Acad Sci USA. 1998;95:4089–4091. doi: 10.1073/pnas.95.8.4089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belelli D, Lambert JJ. Neurosteroids: endogenous regulators of the GABA(A) receptor. Nat Rev Neurosci. 2005;6:565–575. doi: 10.1038/nrn1703. [DOI] [PubMed] [Google Scholar]
- Bicikova M, Hampl R. Neurosteroids and their function. Cas Lek Cesk. 2007;146:223–226. [PubMed] [Google Scholar]
- Chaplan SR, Bach FW, Pogrel JW, Chung JM, Yaksh TL. Quantitative assessment of tactile allodynia in the rat paw. J Neurosci Methods. 1994;53:55–63. doi: 10.1016/0165-0270(94)90144-9. [DOI] [PubMed] [Google Scholar]
- Chen L, Dai XN, Sokabe M. Chronic administration of dehydroepiandrosterone sulfate (DHEAS) primes for facilitated induction of long-term potentiation via sigma 1 (sigma1) receptor: optical imaging study in rat hippocampal slices. Neuropharmacology. 2006;50:380–392. doi: 10.1016/j.neuropharm.2005.10.015. [DOI] [PubMed] [Google Scholar]
- Cheng ZX, Lan DM, Wu PY, Zhu YH, Dong Y, Ma L, et al. Neurosteroid dehydroepiandrosterone sulphate inhibits persistent sodium currents in rat medial prefrontal cortex via activation of sigma-1 receptors. Exp Neurol. 2008;210:128–136. doi: 10.1016/j.expneurol.2007.10.004. [DOI] [PubMed] [Google Scholar]
- Compagnone NA, Mellon SH. Neurosteroids: biosynthesis and function of these novel neuromodulators. Front Neuroendocrinol. 2000;21:1–56. doi: 10.1006/frne.1999.0188. [DOI] [PubMed] [Google Scholar]
- Demirgoren S, Majewska MD, Spivak CE, London ED. Receptor binding and electrophysiological effects of dehydroepiandrosterone sulfate, an antagonist of the GABAA receptor. Neuroscience. 1991;45:127–135. doi: 10.1016/0306-4522(91)90109-2. [DOI] [PubMed] [Google Scholar]
- Dillon JS. Dehydroepiandrosterone, dehydroepiandrosterone sulfate and related steroids: their role in inflammatory, allergic and immunological disorders. Curr Drug Targets Inflamm Allergy. 2005;4:377–385. doi: 10.2174/1568010054022079. [DOI] [PubMed] [Google Scholar]
- Dixon WJ. Efficient analysis of experimental observations. Annu Rev Pharmacol Toxicol. 1980;20:441–462. doi: 10.1146/annurev.pa.20.040180.002301. [DOI] [PubMed] [Google Scholar]
- Dubrovsky BO. Steroids, neuroactive steroids and neurosteroids in psychopathology. Prog Neuropsychopharmacol Biol Psychiatry. 2005;29:169–192. doi: 10.1016/j.pnpbp.2004.11.001. [DOI] [PubMed] [Google Scholar]
- Galluzzi KE. Managing neuropathic pain. J Am Osteopath Assoc. 2007;107(10) Suppl 6:ES39–ES48. [PubMed] [Google Scholar]
- Genud R, Merenlender A, Gispan-Herman I, Maayan R, Weizman A, Yadid G. DHEA lessens depressive-like behaviour via GABA-ergic modulation of the mesolimbic system. Neuropsgchopharmacology. 2009;34:577–584. doi: 10.1038/npp.2008.46. [DOI] [PubMed] [Google Scholar]
- Gibbs TT, Russek SJ, Farb DH. Sulfated steroids as endogenous neuromodulators. Pharmacol Biochem Behav. 2006;84:555–567. doi: 10.1016/j.pbb.2006.07.031. [DOI] [PubMed] [Google Scholar]
- Hwang JH, Yaksh TL. The effect of spinal GABA receptor agonists on tactile allodynia in a surgically-induced neuropathic pain model in the rat. Pain. 1997;70:15–22. doi: 10.1016/s0304-3959(96)03249-6. [DOI] [PubMed] [Google Scholar]
- Hylden JL, Wilcox GL. Intrathecal morphine in mice: a new technique. Eur J Pharmacol. 1980;67:313–316. doi: 10.1016/0014-2999(80)90515-4. [DOI] [PubMed] [Google Scholar]
- Imamura M, Prasad C. Modulation of GABA-gated chloride ion influx in the brain by dehydroepiandrosterone and its metabolites. Biochem Biophys Res Commun. 1998;243:771–775. doi: 10.1006/bbrc.1998.8177. [DOI] [PubMed] [Google Scholar]
- Jasmin L, Wu MV, Ohara PT. GABA puts a stop to pain. Curr Drug Targets CNS Neurol Disord. 2004;3:487–505. doi: 10.2174/1568007043336716. [DOI] [PubMed] [Google Scholar]
- Kibaly C, Meyer L, Patte-Mensah C, Mensah-Nyagan AG. Biochemical and functional evidence for the control of pain mechanisms by dehydroepiandrosterone endogenously synthesized in the spinal cord. FASEB J. 2007;22:93–104. doi: 10.1096/fj.07-8930com. [DOI] [PubMed] [Google Scholar]
- Kim HW, Kwon YB, Roh DH, Yoon SY, Han HJ, Kim KW, et al. Intrathecal treatment with sigma1 receptor antagonists reduces formalin-induced phosphorylation of NMDA receptor subunit 1 and the second phase of formalin test in mice. Br J Pharmacol. 2006;148:490–498. doi: 10.1038/sj.bjp.0706764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HW, Roh DH, Yoon SY, Seo HS, Kwon YB, Han HJ, et al. Activation of the spinal sigma-1 receptor enhances NMDA-induced pain via PKC- and PKA-dependent phosphorylation of the NR1 subunit in mice. Br J Pharmacol. 2008;154:1125–1134. doi: 10.1038/bjp.2008.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maayan R, Morad O, Dorfman P, Overstreet DH, Weizman A, Yadid G. The involvement of dehydroepiandrosterone (DHEA) and its sulfate ester (DHEAS) in blocking the therapeutic effect of electroconvulsive shocks in an animal model of depression. Eur Neuropsychopharmacol. 2005;15:253–262. doi: 10.1016/j.euroneuro.2004.10.005. [DOI] [PubMed] [Google Scholar]
- Malan TP, Mata HP, Porreca F. Spinal GABA(A) and GABA(B) receptor pharmacology in a rat model of neuropathic pain. Anesthesiology. 2002;96:1161–1167. doi: 10.1097/00000542-200205000-00020. [DOI] [PubMed] [Google Scholar]
- Maurice T, Gregoire C, Espallergues J. Neuro(active)steroids actions at the neuromodulatory sigma1 (sigma1) receptor: biochemical and physiological evidences, consequences in neuroprotection. Pharmacol Biochem Behav. 2006;84:581–597. doi: 10.1016/j.pbb.2006.07.009. [DOI] [PubMed] [Google Scholar]
- McEwen BS. Non-genomic and genomic effects of steroids on neural activity. Trends Pharmacol Sci. 1991;12:141–147. doi: 10.1016/0165-6147(91)90531-v. [DOI] [PubMed] [Google Scholar]
- Mehta AK, Ticku MK. Unsulfated and sulfated neurosteroids differentially modulate the binding characteristics of various radioligands of GABA(A) receptors following chronic ethanol administration. Neuropharmacology. 2001;40:668–675. doi: 10.1016/s0028-3908(00)00200-8. [DOI] [PubMed] [Google Scholar]
- Mtchedlishvili Z, Kapur J. A presynaptic action of the neurosteroid pregnenolone sulfate on GABAergic synaptic transmission. Mol Pharmacol. 2003;64:857–864. doi: 10.1124/mol.64.4.857. [DOI] [PubMed] [Google Scholar]
- Onaka M, Minami T, Nishihara I, Ito S. Involvement of glutamate receptors in strychnine- and bicuculline-induced allodynia in conscious mice. Anesthesiology. 1996;84:1215–1222. doi: 10.1097/00000542-199605000-00024. [DOI] [PubMed] [Google Scholar]
- Park-Chung M, Malayev A, Purdy RH, Gibbs TT, Farb DH. Sulfated and unsulfated steroids modulate gamma-aminobutyric acidA receptor function through distinct sites. Brain Res. 1999;830:72–87. doi: 10.1016/s0006-8993(99)01381-5. [DOI] [PubMed] [Google Scholar]
- Patte-Mensah C, Mensah-Nyagan AG. Peripheral neuropathy and neurosteroid formation in the central nervous system. Brain Res Rev. 2008;57:454–459. doi: 10.1016/j.brainresrev.2007.05.007. [DOI] [PubMed] [Google Scholar]
- Ren X, Noda Y, Mamiya T, Nagai T, Nabeshima T. A neuroactive steroid, dehydroepiandrosterone sulfate, prevents the development of morphine dependence and tolerance via c-fos expression linked to the extracellular signal-regulated protein kinase. Behav Brain Res. 2004;152:243–250. doi: 10.1016/j.bbr.2003.10.013. [DOI] [PubMed] [Google Scholar]
- Rieselbach RE, Di Chiro G, Freireich EJ, Rall DP. Subarachnoid distribution of drugs after lumbar injection. N Engl J Med. 1962;267:1273–1278. doi: 10.1056/NEJM196212202672502. [DOI] [PubMed] [Google Scholar]
- Roh DH, Kim HW, Yoon SY, Seo HS, Kwon YB, Kim KW, et al. Intrathecal administration of sigma-1 receptor agonists facilitates nociception: involvement of a protein kinase C-dependent pathway. J Neurosci Res. 2008a;86:3644–3654. doi: 10.1002/jnr.21802. [DOI] [PubMed] [Google Scholar]
- Roh DH, Kim HW, Yoon SY, Seo HS, Kwon YB, Kim KW, et al. Intrathecal injection of the sigma(1) receptor antagonist BD1047 blocks both mechanical allodynia and increases in spinal NR1 expression during the induction phase of rodent neuropathic pain. Anesthesiology. 2008b;109:879–889. doi: 10.1097/ALN.0b013e3181895a83. [DOI] [PubMed] [Google Scholar]
- Shirayama Y, Hashimoto K, Suzuki Y, Higuchi T. Correlation of plasma neurosteroid levels to the severity of negative symptoms in male patients with schizophrenia. Schizophr Res. 2002;58:69–74. doi: 10.1016/s0920-9964(01)00367-x. [DOI] [PubMed] [Google Scholar]
- Sommer C, Schafers M. Painful mononeuropathy in C57BL/Wld mice with delayed wallerian degeneration: differential effects of cytokine production and nerve regeneration on thermal and mechanical hypersensitivity. Brain Res. 1998;784:154–162. doi: 10.1016/s0006-8993(97)01327-9. [DOI] [PubMed] [Google Scholar]
- Sousa A, Ticku MK. Interactions of the neurosteroid dehydroepiandrosterone sulfate with the GABA(A) receptor complex reveals that it may act via the picrotoxin site. J Pharmacol Exp Ther. 1997;282:827–833. [PubMed] [Google Scholar]
- Tsutsui K, Ukena K, Usui M, Sakamoto H, Takase M. Novel brain function: biosynthesis and actions of neurosteroids in neurons. Neurosci Res. 2000;36:261–273. doi: 10.1016/s0168-0102(99)00132-7. [DOI] [PubMed] [Google Scholar]
- Ueda H, Inoue M, Yoshida A, Mizuno K, Yamamoto H, Maruo J, et al. Metabotropic neurosteroid/sigma-receptor involved in stimulation of nociceptor endings of mice. J Pharmacol Exp Ther. 2001;298:703–710. [PubMed] [Google Scholar]
- Wojtal K, Trojnar MK, Czuczwar SJ. Endogenous neuroprotective factors: neurosteroids. Pharmacol Rep. 2006;58:335–340. [PubMed] [Google Scholar]
- Zeilhofer HU. Loss of glycinergic and GABAergic inhibition in chronic pain-contributions of inflammation and microglia. Int Immunopharmacol. 2008;8:182–187. doi: 10.1016/j.intimp.2007.07.009. [DOI] [PubMed] [Google Scholar]
- Zhang L, Li B, Ma W, Barker JL, Chang YH, Zhao W, et al. Dehydroepiandrosterone (DHEA) and its sulfated derivative (DHEAS) regulate apoptosis during neurogenesis by triggering the Akt signaling pathway in opposing ways. Brain Res Mol Brain Res. 2002;98:58–66. doi: 10.1016/s0169-328x(01)00315-1. [DOI] [PubMed] [Google Scholar]