

Abstract
Background and purpose:
Bepridil is an anti-arrhythmic agent with anti-electrical remodelling effects that target many cardiac ion channels, including the voltage-gated Na+ channel. However, long-term effects of bepridil on the Na+ channel remain unclear. We explored the long-term effect of bepridil on the Na+ channel in isolated neonatal rat cardiomyocytes and in a heterologous expression system of human Nav1.5 channel.
Experimental approach:
Na+ currents were recorded by whole-cell voltage-clamp technique. Na+ channel message and protein were evaluated by real-time RT-PCR and Western blot analysis.
Key results:
Treatment of cardiomyocytes with 10 µmol·L−1 bepridil for 24 h augmented Na+ channel current (INa) in a dose- and time-dependent manner. This long-term effect of bepridil was mimicked or masked by application of W-7, a calmodulin inhibitor, but not KN93 [2-[N-(2-hydroxyethyl)-N-(4-methoxy benzenesulphonyl)]-amino-N-(4-chlorocinnamyl)-N-methylbenzylamine], a Ca2+/calmodulin-dependent kinase inhibitor. During inhibition of protein synthesis by cycloheximide, the INa increase due to bepridil was larger than the increase without cycloheximide. Bepridil and W-7 significantly slowed the time course of Nav1.5 protein degradation in neonatal cardiomyocytes, although the mRNA levels of Nav1.5 were not modified. Bepridil and W-7 did not increase INa any further in the presence of the proteasome inhibitor MG132 [N-[(phenylmethoxy)carbonyl]-L-leucyl-N-[(1S)-1-formyl-3-methylbutyl]-L-leucinamide]. Bepridil, W-7 and MG132 but not KN93 significantly decreased 20S proteasome activity in a concentration-dependent manner.
Conclusions and implications:
We conclude that long-term exposure of cardiomyocytes to bepridil at therapeutic concentrations inhibits calmodulin action, which decreased degradation of the Nav1.5 α-subunit, which in turn increased Na+ current.
Keywords: bepridil, Na+ channel, anti-electrical remodelling, calmodulin, post-transcriptional, proteasome
Introduction
Bepridil is known as a powerful anti-arrhythmic agent with anti-anginal properties (Hollingshead et al., 1992; Prystowsky, 1992). Although bepridil is primarily classified as a Ca2+ channel antagonist, it is reported to block many cardiac ion channels including the slow (IKs), rapid (IKr), and ultrarapid (IKur) delayed rectifier K+ channels (Wang et al., 1999; Kobayashi et al., 2001; Kamiya et al., 2006), the ATP-sensitive K+ (IKATP) channel (Sato et al., 2006), the Na+-activated K+ (IKNa) channel (Li et al., 1999; Sato et al., 2006), the L- and T-type Ca2+ channels (Yatani et al., 1986; Uchino et al., 2005) and the Na+ channel (Nawada et al., 1995; Sato et al., 1996). Probably because of its multi-channel blocking properties, bepridil is effective for the treatment of intractable cardiac arrhythmias including ventricular tachycardia (Levy et al., 1984; Brembilla-Perrot et al., 1992; Izumi et al., 2007) and persistent atrial fibrillation (AF) (Nakazato et al., 2005;Miyaji et al., 2007). Furthermore, several recent reports have demonstrated that bepridil exhibits anti-electrical remodelling effects in the heart (Fujiki et al., 2003; Nishida et al., 2007), similar to those of amiodarone in clinical and experimental models of AF (Shinagawa et al., 2003; Ashikaga et al., 2006; Yamashita et al., 2006). Moreover, after conversion or maintenance of sinus rhythm (SR) in patients with persistent AF, contractile functions as well as electrophysiological features of atrial muscles were highly preserved by long-term treatment with bepridil (Fujiki et al., 2004; Nishida et al., 2007). Importantly, Fujiki et al. (2003) demonstrated that the addition of aprindine, a class I anti-arrhythmic drug, enhanced the rate of AF termination, although intravenous aprindine had failed to terminate AF before bepridil treatment. Based on these findings, we hypothesized that bepridil, in the long-term, preserves Na+ channel function in cardiac myocytes apart from its acute blocking effects on ion channels.
In the present study, we explored the long-term effects of bepridil on the Na+ channel in neonatal rat isolated cardiomyocytes and in a heterologous expression system for the human Nav1.5 channel. Our results indicate that the short- and long-term applications of bepridil have different effects on Na+ channel current (INa) in cardiomyocytes; long-term application of bepridil up-regulates INa by blunting proteasome signals through the inhibition of calmodulin (CaM) action.
Methods
Neonatal rat cardiomyocytes: preparations and culture
Animal care and the experimental protocols were approved by the Ethics Review Committee for Animal Experimentation of Oita University School of Medicine. Neonatal cardiomyocytes were prepared from 1–3-day-old Wistar rats as described previously (Wang et al., 2007). The cardiomyocytes were plated onto 35 mm culture dishes and cultured in Dulbecco's modified Eagle's medium, supplemented with 5% fetal bovine serum at 37°C under 5% CO2. The cells were seeded onto glass-bottom dishes and incubated in a culture medium for 24–48 h before electrophysiological measurements.
Expression of Na+ channel proteins and cell culture
The Na+ channel α-subunit (Nav1.5) derived from human hearts and forming cardiac Na+ channels, was stably expressed in human embryonic kidney HEK-293 (HEK-Nav1.5) cells (Hartmann et al., 1994; Nagatomo et al., 1998). The HEK-Nav1.5 cells were maintained in Dulbecco's modified Eagle's medium, supplemented with 10% fetal bovine serum, 100 U·mL−1 penicillin and 100 µg·mL−1 streptomycin in an air atmosphere plus 5% CO2 at 37°C. This medium was supplemented with 300 µg·mL−1 G418 (neomycin analogue) for the selection of recombinant HEK-293 cells.
Electrophysiological measurements
Macroscopic INa were recorded in whole-cell configuration by using an EPC-9 amplifier (HEKA Electronik, Lambrecht, Germany) at room temperature (20–23°C). Patch pipettes were pulled from 75 mm plain capillary tubes (Drummond Scientific Co., Broomall, PA, USA) with a micropipette puller, Model P-97 (Sutter Instrument, Co., Novato, CA, USA), and were fire-polished subsequently. The electrode had a resistance of 1.5–2.5 MΩ when the pipette was filled with the pipette solution (see below). Series resistance was compensated electrically as much as possible without oscillation (60–75%). Capacitive artefacts were minimized by using the built-in circuitry of the amplifier. The current signals were filtered at 3.3 kHz and digitized at 10 kHz under the control of a data acquisition programme, Pulse/Pulsefit (V.8.11, HEKA Electronik). To investigate the channel availability (steady-state inactivation), a conventional double-pulse protocol was applied every 5 s: 30 ms of test pulses at −25 mV following 500 ms of prepulses from −120 to −20 mV (increment = 5 mV) were applied. The reversal potential and the chord conductance were calculated by fitting the current–voltage (I–V) relationship to a Boltzmann distribution function: I = Gmax(Vm – Vrev)/(1 + exp[(Vm – Va, 1/2)/k]), where I is the peak INa at the given test potential Vm, Vrev is the reversal potential, Gmax is the maximal chord conductance, Va, 1/2 is the mid-point of the relationship, and k is the slope factor. The voltage-dependent inactivation was similarly determined with a Boltzmann equation: I/Imax = 1/(1 + exp[(Vm – Vi, 1/2)/k]), where Vm is the membrane potential, Vi, 1/2 is the half-point of the relationship, and k is the slope factor. Only a single patch was obtained from each cell. For evaluation of a short-term effect of bepridil, INa from a single cell was compared in many experiments. For evaluation of a long-term effect of bepridil, INa from cells in distinct culture conditions were compared.
The recording chamber was filled with the bath solution of the following composition (mmol·L−1): NaCl 20, MgCl2 0.5, TEA-Cl 125, CsCl 5, 4-AP 5, DIDS 0.1, HEPES 10, glucose 10, CaCl2 1.8 (pH of 7.4 adjusted with 1 N TEA-OH). The patch-clamp electrode was filled with the pipette solution of the following composition (mmol·L−1): CsF 20, CsCl 120, EGTA 2, HEPES 5, (pH of 7.2 adjusted with 1 N CsOH). The data were acquired by using computer software (Pulse/Pulsefit, V.8.11), and all curve fittings were made on SigmaPlot (V9.01, SPSS Inc., Chicago, IL, USA).
Western blot analysis
Cells were lysed in cold cell lysis buffer containing (in mmol·L−1) NaCl 150, Tris-HCl 50, EDTA 1, phenylmethyl sulphonyl fluoride 0.02 with 1% deoxycholic acid sodium salt monohydrate, 0.1% sodium dodecyl sulphate (SDS) and 10% Triton X-100 (v/v) followed by centrifugation at 12 000×g for 15 min at 4°C to remove cell debris, nuclei and large particulates. The supernatant portion that contains membrane proteins and cytosolic proteins was used for Western blot analysis. In all, 40 µg of protein was denatured by boiling for 5 min in the loading buffer containing 250 mmol·L−1 Tris-HCl (pH 6.8), 4% SDS, 1% β-mercaptoethanol, 1% bromophenol blue and 20% glycerol. The proteins were electrophoresed to SDS-polyacrylamide gel and transferred to a nitrocellulose membrane (PROTRAN, S&S, Bioscience, Germany). The membrane was blocked by using 5% skim milk in Tris-buffered saline containing 0.05% Tween 20 for 1 h and incubated with a rabbit anti-Nav1.5 antibody (1:200, Alomone Labs Ltd., Jerusalem, Israel), rabbit anti-Navβ1 antibody (1:200, Cell Application Inc., San Diego, CA, USA), rabbit anti-Navβ2 antibody (1:200, Alomone Labs Ltd.). The blot was visualized with anti-rabbit IgG horseradish peroxidase-conjugate secondary antibody (1:2000, Biosource International, Camarillo, CA, USA) and an ECL detection system (Amersham Pharmacia Biotech, Aylesbury, UK). Blots were stripped in stripping buffer containing 62.5 mmol·L−1 Tris-HCl, 100 mmol·L−1 2-mercaptoethanol and 2% SDS, at 50°C and pH of 6.8 for 40 min for a second-round immunoblotting.
Quantitative real-time RT-PCR
Total RNA was extracted from rat neonatal cardiomyocytes and recombinant HEK-293 cells by using Isogen (Nippongene, Tokyo, Japan). The cDNA was synthesized from 1 µg of total RNA by using Transcriptor First Strand cDNA Synthesis Kit (Roche Molecular System Inc., Alameda, CA, USA). The real-time PCR was performed on Light Cycler (Roche) by using the FastStart DNA Master SYBR Green I (Roche) as a detection reagent. The sequences of the specific primers are shown in Table 1. Data were calculated by 2−ΔΔCT and presented as fold change of transcripts for Nav1.5, Navβ1, Navβ2, Navβ3 and Navβ4 genes in cardiomyocytes normalized to GAPDH. Size of PCR products were confirmed by 2% agarose electrophoresis.
Table 1.
Sequence of oligonucleotides used as real-time PCR primers
| Subunit | Gene | Sequence |
|---|---|---|
| Nav1.5 | SCN5A | F: 5′-CTGCGGCGGCGACCTAAGAAGC-3′ |
| R: 5′-GTCCTCAGGGGTCTCACCGCAC-3′ | ||
| Navβ1 | SCN1B | F: 5′-CCAGAAGGGCACAGAGGAAT-3′ |
| R: 5′-TCGCCAGAGTGGTTGTAGGT-3′ | ||
| Navβ2 | SCN2B | F: 5′-CTGCTACATCACCAACCCTC-3′ |
| R: 5′-GTCATCCGTGCTCAGCTTCT-3′ | ||
| Navβ3 | SCN3B | F: 5′-TGCCTTCAACAGATTGCTTC-3′ |
| R: 5′-GGCCATTCCGATACTCATAT-3′ | ||
| Navβ4 | SCN4B | F: 5′-GCCACCACCATCTACGCTAT-3′ |
| R: 5′-ATCTGCCCGTGTCACTGAAC-3′ |
Measurement of 20S proteasome activity
The 20S proteasome activity was assessed by fluorescence of free 7-amino-4-methylcoumarin (AMC; excitation: 380 nm, emission: 460 nm) liberated from a substrate peptide (Suc-Leu-Leu-Val-Tyr-AMC), by using an assay kit (Calbiochem, San Diego, CA, USA). The reaction mixture contained 20S proteasome (500 µg·mL−1), the substrate peptide (10 µmol·L−1) and indicated drugs in a buffer (25 mmol·L−1 HEPES, 0.5 mmol·L−1 EDTA and 0.03% SDS, pH 7.6). The mixture was incubated at 37°C for 1 h. AMC fluorescence liberated in the absence of drugs was taken as the basal value (1.0). Because the proteasome in this reaction is activated by SDS, the fluorescence liberated in the absence of SDS was taken as the background value (0.0).
Data analysis
The group data show means ± SD. Between groups and among groups comparisons were conducted with one-way anova with Scheffé test. IC50 and EC50 values were estimated by using non-linear least square curve-fitting programmes in SigmaPlot (V9.01, SPSS Inc.). P < 0.05 was considered significant.
Materials
Bepridil hydrochloride was a kind gift from Daiichi-Sankyo Pharmaceutical Co., Tokyo, Japan. All other chemicals were purchased from Wako Chemical Co., Osaka, Japan. All drug and molecular target nomenclature conforms to the British Journal of Pharmacology's Guide to Receptors and Channels (Alexander et al., 2008).
Results
Long-term effects of bepridil on INa in cardiomyocytes
In conventional whole-cell patch-clamp experiments using neonatal rat cardiomyocytes, the voltage-gated INa was recorded. Figure 1A shows representative INa families in the control condition, during the acute action of 10 µmol·L−1 bepridil, after the long-term treatment (24 h) with vehicle, 10 µmol·L−1 bepridil, 20 µmol·L−1 W-7, 20 µmol·L−1 W-7 with 10 µmol·L−1 bepridil, 10 µmol·L−1 KN93 [2-[N-(2-hydroxyethyl)-N-(4-methoxy benzenesulphonyl)]-amino-N-(4-chlorocinnamyl)-N-methylbenzylamine] and 10 µmol·L−1 KN93 with 10 µmol·L−1 bepridil. Consistent with previous studies, bepridil, as a short-term effect, decreased INa by 21% in this example and by 23 ± 1% (n = 6) on average (Figure 1B). Conversely, long-term treatment of cardiomyocytes with 10 µmol·L−1 bepridil for 24 h augmented INa by 36 ± 2% (n = 13) (Figure 1B). While the chord conductance of the channel was decreased by short-term application of 10 µmol·L−1 bepridil by −21 ± 1% (n = 6), it was increased by long-term application of 10 µmol·L−1 bepridil (31 ± 2%) (n = 13) (Figure 1D).
Figure 1.
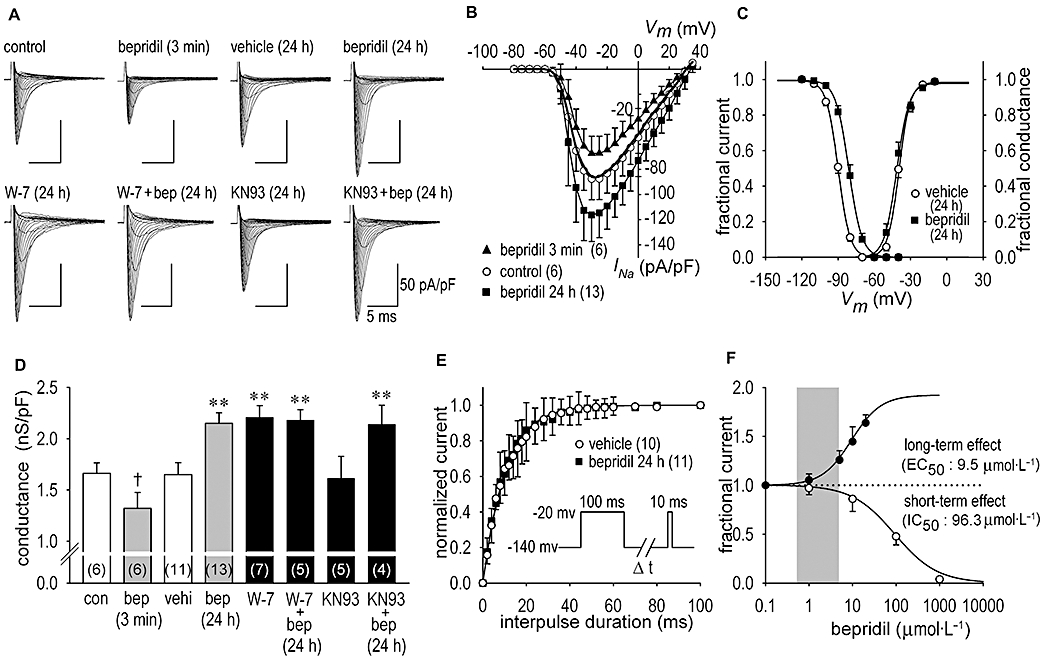
Short- and long-term effects of bepridil on Na+ channel current (INa) in neonatal rat cardiomyocytes. (A) Representative INa families in the control condition and during the acute (3 min) application of 10 µmol·L−1 bepridil given to the same patch are shown. Also, representative INa families after the long-term (24 h) treatment with vehicle (saline), 10 µmol·L−1 bepridil, 20 µmol·L−1 W-7, 20 µmol·L−1 W-7 with 10 µmol·L−1 bepridil (bep), 10 µmol·L−1 KN93 [2-[N-(2-hydroxyethyl)-N-(4-methoxy benzenesulphonyl)]-amino-N-(4-chlorocinnamyl)-N-methylbenzylamine, a Ca2+/calmodulin -dependent kinase inhibitor] and 10 µmol·L−1 KN93 with 10 µmol·L−1 bepridil are shown respectively. INa was elicited by a depolarization pulse of 35 ms duration, ranging from −80 to 30 mV in 5 mV steps, applied from the holding potential of −140 mV. During INa recordings, bepridil was excluded from the bath solution, except for the short-term application of bepridil. (B) I–V relationships constructed by using group data in control during the application of 10 µmol·L−1 bepridil in 3 min and after long-term treatment of 10 µmol·L−1 bepridil for 24 h. I–V relationship in the vehicle treatment for 24 h is shown in bold solid line without symbol for clarity. (C) The steady-state activation and inactivation curves of the Na+ channel obtained from cardiomyocytes treated with bepridil for 24 h. Mid-points of the voltage relation for the activation (Va, 1/2) and slope factors (k) were −39.9 ± 0.8 mV and −4.5 ± 0.6 (n = 11) in vehicle and −41.7 ± 0.6 mV and −4.9 ± 0.6 (n = 13) after treatment with bepridil for 24 h respectively. For inactivation, the Vi, 1/2 values and slope factors (k) were −89.8 ± 2.0 mV and 4.8 ± 0.3 (n = 10) in vehicle and −81.2 ± 2.5 mV and 5.4 ± 0.4 (n = 11) after treatment with bepridil for 24 h respectively. Curves were fitted by the Boltzmann equation (see Methods section). (D) The maximum chord conductance in the control condition (con), during acute (3 min) application of bepridil (bep), in vehicle (vehi) for 24 h, after the long-term (24 h) action of 10 µmol·L−1 bepridil, after the long-term action of 20 µmol·L−1 W-7 in the absence and presence of 10 µmol·L−1 bepridil and after the long-term action of 10 µmol·L−1 KN93 in the absence and presence of 10 µmol·L−1 bepridil. Numbers of experiments are shown in parentheses. (E) Time course of recovery from inactivation of INa. Curves were fitted to the data by using a single exponential equation yielding time constant (τ) of 10.5 ± 0.2 ms (n = 10) in vehicle and 10.8 ± 0.2 ms (n = 11) after treatment with bepridil for 24 h. Pulse protocols are shown in inset. (F) Concentration-dependent short-term (3–20 min) and long-term (24 h) effects of bepridil on INa elicited at the frequency of 0.1 Hz at −30 mV. The short-term inhibition and long-term increase of INa was plotted against the bepridil concentration. The short-term IC50 value for INa was estimated to be 96.3 µmol·L−1, whereas the long-term EC50 value for INa was estimated to be 9.5 µmol·L−1. The grey area indicates effective plasma concentration (0.5–5 µmol·L−1). †P < 0.05 compared with control, **P < 0.01 compared with vehicle.
Because bepridil is known to inhibit CaM action apart from its pharmacological effects on various ion channels (Itoh et al., 1984; Zimmer and Hofmann, 1987; Schaeffer et al., 1991), long-term actions of bepridil were postulated to mediate CaM activity in myocytes. To this end, neonatal rat cardiomyocytes were incubated with 20 µmol·L−1 W-7, a CaM inhibitor, for 24 h in the presence or absence of 10 µmol·L−1 bepridil. The application of 20 µmol·L−1 W-7 increased the maximum INa by 39 ± 2% (n = 7) (Figure 1A) and increased maximum chord conductance by 33 ± 2% (n = 7) (Figure 1D), similar to the effects of bepridil. Importantly, no further increase of INa by bepridil was observed in the presence of W-7 (Figure 1D). To further examine whether Ca2+/CaM-dependent kinases (CaM-K) were involved in long-term actions of bepridil, cardiomyocytes were incubated with 10 µmol·L−1 KN93, a CaM-K inhibitor, for 24 h in the presence or absence of 10 µmol·L−1 bepridil. The application of 10 µmol·L−1 KN93 did not modify the change of INa (Figure 1A) and the maximum chord conductance caused by bepridil (Figure 1D), suggesting CaM-K-independent long-term actions of bepridil on the Na+ channel. To determine the mechanisms of the effect of bepridil on the activation and steady-state inactivation kinetics of the Na+ channel, the fractional INa and fractional Na+ channel conductance were compared in myocytes with or without 24 h bepridil treatment (Figure 1C). Treatment of myocytes with 10 µmol·L−1 bepridil for 24 h had no significant effect on the voltage dependency of the activation curve. However, the steady-state inactivation curve was significantly shifted in the direction of depolarization by 8.6 ± 2.1 mV (n = 10) by long-term treatment with bepridil (Figure 1C). To further study long-term actions of bepridil on the channel kinetics, we determined whether the time course of recovery from inactivation of INa was modified by long-term treatment with bepridil. As illustrated in Figure 1E, the time constant for the recovery from inactivation was unaltered after long-term (24 h) treatment with bepridil: τ was 10.8 ± 0.2 ms with bepridil (n = 11) and 10.3 ± 0.2 ms without bepridil (vehicle) (n = 10). Short-term (3–20 min) application of bepridil inhibited INa as a tonic block in a dose-dependent manner with IC50 of 96.3 µmol·L−1, whereas long-term (24 h) application of bepridil increased INa in a dose-dependent manner with EC50 of 9.5 µmol·L−1 (Figure 1F).
Long-term effects of bepridil on INa in a heterologous system
To determine whether long-term effects of bepridil on the Na+ channel are restricted to cardiomyocytes, we used a heterologous system involving non-cardiac cells. Because changes in intracellular Ca2+ concentration have a significant impact on ion channel remodelling under pathological conditions of the heart, and bepridil has high affinity for both L- and T-type Ca2+ channels, we used HEK-293 cells, a stable heterologous expression system transfected with the Na+ channel α1 subunit (HEK-Nav1.5), the major Na+ channel pore-forming subunit identified in the human heart. As HEK-293 cells have no endogenous voltage-activated Ca2+ channels, a possible interaction of bepridil with the Ca2+ channels would be avoided in this context. We tested the long-term effect of bepridil (10 µmol·L−1, 24 h) on HEK-Nav1.5 in the same fashion as we did for the experiments in Figure 1A. Not only the peak current but also the Na+ channel conductance in HEK-Nav1.5 containing cells were augmented by bepridil treatment (Figure 2A–C), identical to results for neonatal myocytes. Consistent with results observed in neonatal myocytes, the peak INa was decreased by the short-term application of 10 µmol·L−1 bepridil (−26 ± 2%) (n = 6) and was increased by the long-term application of 10 µmol·L−1 bepridil (37 ± 2%) (n = 12) (Figure 2B). Dose- and time-dependent actions of bepridil on INa in HEK-Nav1.5 are shown in Figure 2D and E. Treatment of HEK-Nav1.5 cells with 5 µmol·L−1 or higher concentrations of bepridil for 24 h, and or with exposure times of 12 h or longer for 10 µmol·L−1 bepridil augmented INa significantly.
Figure 2.
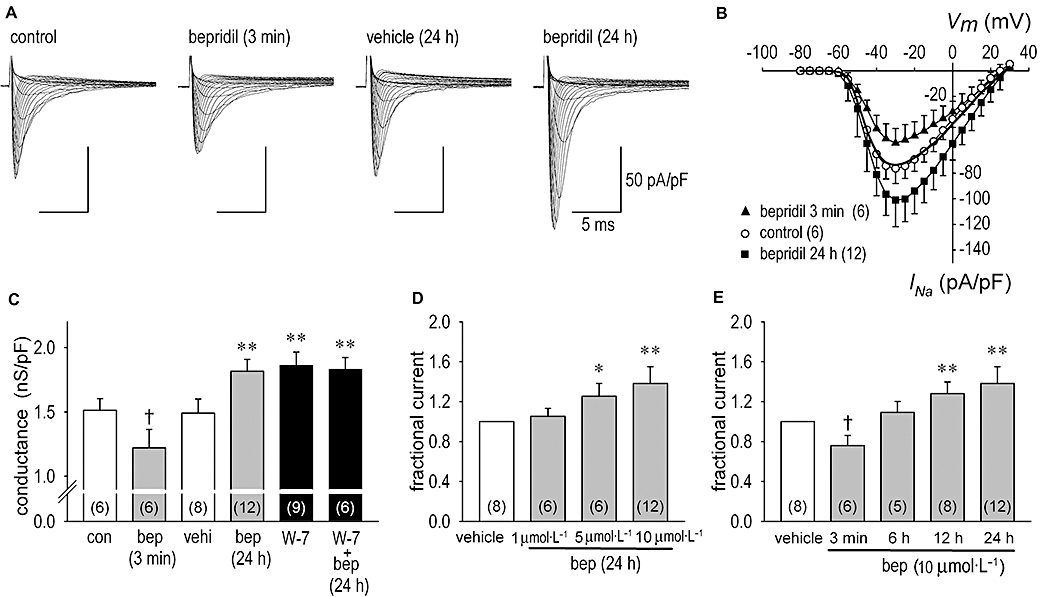
Short- and long-term effects of bepridil on Na+ channel current (INa) in human embryonic kidney (HEK)-Nav1.5 cells. (A) Representative INa families in the control condition, during the acute action of 10 µmol·L−1 bepridil given to the same patch shown in control condition in 3 min, in vehicle for 24 h and after the long-term action of 10 µmol·L−1 bepridil for 24 h. INa was elicited by a depolarization pulse of 35 ms duration, ranging from −80 to 30 mV in 5 mV steps, applied from the holding potential of −140 mV. (B) I–V relationships constructed by using group data in control, during the application of 10 µmol·L−1 bepridil in 3 min and after long-term treatment of 10 µmol·L−1 bepridil for 24 h. I–V relationship in the vehicle treatment for 24 h is shown in bold solid line without symbol for clarity. (C) The maximum chord conductance in the control condition (con), during acute (3 min) application of bepridil (bep), in vehicle (vehi) for 24 h, after treatment with 10 µmol·L−1 bepridil for 24 h, after treatment with 20 µmol·L−1 W-7 for 24 h and after treatment with W-7 (20 µmol·L−1) plus bepridil (10 µmol·L−1) for 24 h. (D) Dose-dependent long-term actions of bepridil on INa. HEK-Nav1.5 cells were incubated in vehicle, 1, 5 and 10 µmol·L−1 bepridil for 24 h. INa at each condition was obtained at the test potential of −30 mV and normalized to the value in vehicle. (E) Time-dependent actions of bepridil on INa. HEK-Nav1.5 cells were incubated (bathed) for 3 min, 6 h, 12 h and 24 h in the presence of 10 µmol·L−1 bepridil. INa was obtained by the same fashion as shown in panel (D). During INa recordings bepridil was excluded from the bath solution, except for the 3 min applications of bepridil. Numbers of experiments are shown in parentheses. †P < 0.05 compared with control, *P < 0.05 compared with vehicle, **P < 0.01 compared with vehicle.
Na+ channel messages were not altered by bepridil
Bepridil modulation of cardiac INa may be caused by activation of the channel synthesis, which includes activation of mRNA transcription and protein expression. Therefore we did real-time RT-PCR quantification of the cardiac Na+ channel genes, SCN5A, SCN1B, SCN2B, SCN3B and SCN4B, using RNA samples extracted from myocytes treated with or without bepridil for 24–48 h. As shown in Figure 3A–D, the mRNA levels were not modified by bepridil.
Figure 3.
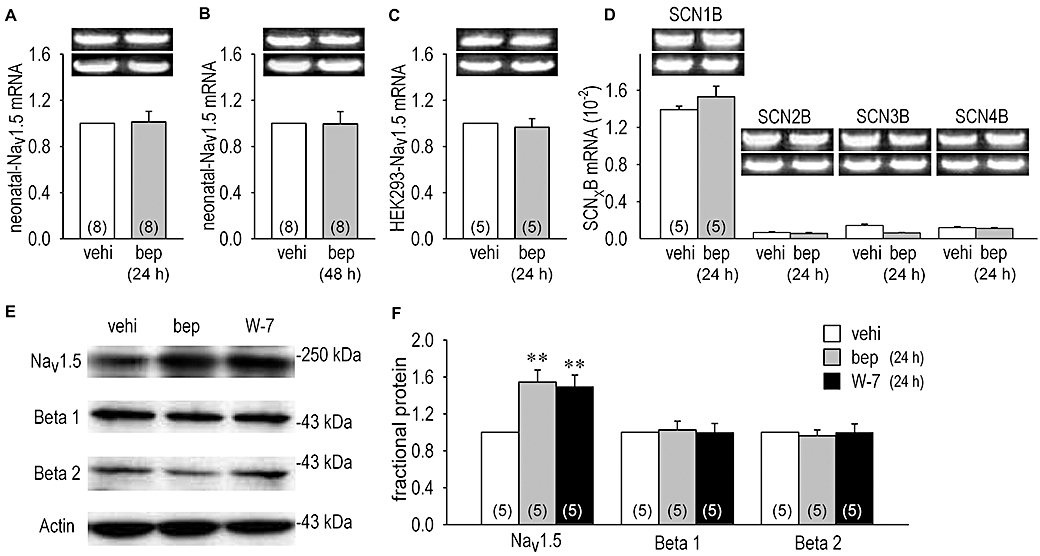
Quantitative real-time RT-PCR experiments and expression of cardiac Na+ channel proteins (Nav1.5, Navβ1 and Navβ2) assessed by Western blots analysis. Bar graphs represent amount of Nav1.5 mRNA or β-subunits mRNA. (A and B) mRNA for Nav1.5 extracted from cardiomyocytes with or without 10 µmol·L−1 bepridil (bep) treatment for 24 h (A) and 48 h (B). (C) mRNA for Nav1.5 from human embryonic kidney (HEK)-Nav1.5 cells with or without 10 µmol·L−1 bepridil treatment for 24 h. (D) Comparison of four different β-subunits genes (SCN1B, SCN2B, SCN3B and SCN4B) level in cardiomyocytes treated with or without 10 µmol·L−1 bepridil for 24 h. Representative PCR products are shown in inset with the reference gene GAPDH (below). Each mRNA product was normalized to that of GAPDH. Numbers of experiment are indicated in parentheses. (E) Examples of Nav1.5, Navβ1, Navβ2 and actin protein expression in neonatal rat cardiomyocytes. (F) Protein expression levels of Nav1.5, Navβ1 and Navβ2 determined from the density of blotted bands in panel (E). Na+ channel proteins extracted from cardiomyocytes treated with vehicle (vehi), 10 µmol·L−1 bepridil and 20 µmol·L−1 W-7 for 24 h. **P < 0.01 compared with vehicle.
Nav1.5 proteins were increased by bepridil
Then, we carried out Western blot analysis of the Na+ channel proteins isolated from neonatal rat cardiomyocytes. Figure 3E and F show that both 10 µmol·L−1 bepridil and 20 µmol·L−1 W-7 increased Nav1.5 protein expression, the former by 54 ± 1% (n = 5) and the latter by 49 ± 1% (n = 5). No significant effect of bepridil on Navβ proteins expression was observed. Thus, increased Nav1.5 protein levels may be due to the interruption of the Nav1.5 protein degradation. To investigate the rate of breakdown of Nav1.5 proteins during inhibition of protein synthesis, we monitored effects of bepridil on INa and Nav1.5 protein expression in the presence of cycloheximide. The neonatal rat cardiomyocytes were incubated with cycloheximide, a protein synthesis inhibitor, for 1, 3, 6, 12 and 24 h in the presence or absence of bepridil. Although cycloheximide had no significant short-term effect on INa (INa was decreased by 3.4 ± 0.8%, n = 5, data not shown), it decreased the peak INa as a long-term effect by 63 ± 3% (n = 6) (Figure 4A and B). When protein synthesis was intact, bepridil increased INa by 36 ± 2% (n = 13) (Figure 4B which is from the same data set as shown in Figure 1B). In the presence of cycloheximide, however, bepridil augmented INa by 64.6 ± 3.5% (n = 5), a greater increase than without cycloheximide. The time course for Nav1.5 protein degradation was significantly slowed in the presence of bepridil (Figure 4C), which is consistent with INa changes shown in Figure 4B in the presence of cycloheximide, although this analysis evaluated Nav1.5 proteins in plasma membrane as well as those in the cytoplasm. The application of 20 µmol·L−1 W-7 also slowed Nav1.5 protein degradation, mimicking the effects of bepridil (Figure 4C).
Figure 4.
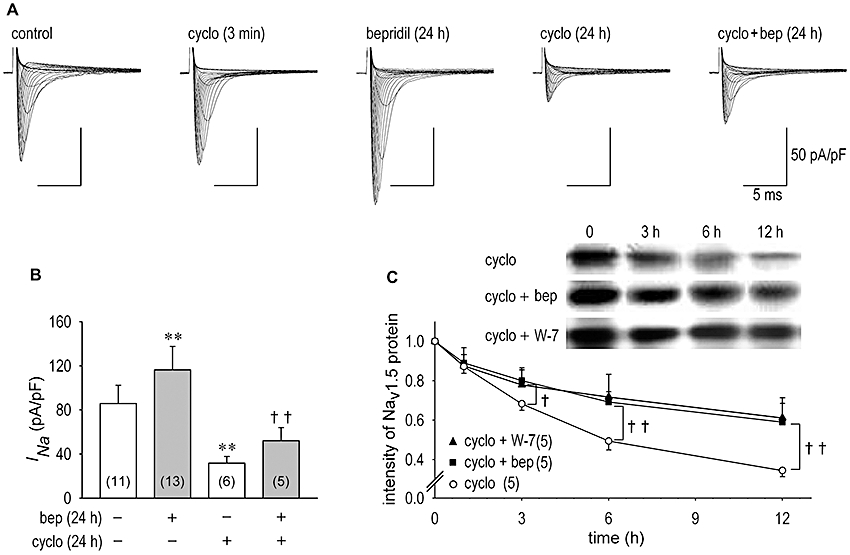
Na+ channel current (INa) and Nav1.5 channel expression during protein synthesis inhibition. (A) Representative INa families in the control condition, during the acute application of 10 µg·mL−1 cycloheximide (cyclo) given to the same patch shown in control condition in 3 min, after the long-term action of 10 µmol·L−1 bepridil for 24 h, after the long-term action of 10 µg·mL−1 cycloheximide for 24 h and after the long-term action of 10 µmol·L−1 bepridil in the presence of 10 µg·mL−1 cycloheximide for 24 h. (B) INa was evaluated as the maximum INa recorded from cardiomyocytes in vehicle or treated with 10 µmol·L−1 bepridil, 10 µg·mL−1 cycloheximide and concomitant application of 10 µg·mL−1 cycloheximide plus 10 µmol·L−1 bepridil for 24 h. Bepridil augmented INa by 65 ± 4% (n = 5) with a higher increase rate in the presence of cycloheximide than that without cycloheximide by 36 ± 2% (n = 13). (C) Time-dependent changes in Nav1.5 protein during inhibition of protein synthesis by 10 µg·mL−1 cycloheximide with or without 10 µmol·L−1 bepridil and 20 µmol·L−1 W-7 up to 12 h. **P < 0.01 compared with vehicle, ††P < 0.01 compared with cycloheximide alone.
The bepridil and ubiquitin–proteasome pathway
Recent studies indicate that some ion channel proteins are degraded through the ubiquitin–proteasome pathway. Therefore we evaluated the action of a proteasome activator and a proteasome inhibitor on INa and Nav1.5 protein expression to specify the cellular mechanism underlying the effect of bepridil on Na+ channel degradation. Figure 5A and C show representative INa families as we explored the short-term (Figure 5A) and long-term (Figure 5C) effects of SDS (a proteasome activator) and MG132 [N-[(phenylmethoxy)carbonyl]-L-leucyl-N-[(1S)-1-formyl-3-methylbutyl]-L-leucinamide, a proteasome inhibitor] with or without bepridil or W-7. As a short-term effect (3–5 min), W-7 (20 µmol·L−1), SDS (0.06%) or MG132 (50 µmol·L−1) had negligibly small actions on INa (Figure 5A and B). Meanwhile, treatment of cardiomyocytes with SDS decreased INa as the long-term effect (24 h) by 54% in this example (Figure 5C) and by 54 ± 3% (n = 4) on average (Figure 5D). Importantly, up-regulation of INa by bepridil was halted in the presence of SDS (Figure 5C and D). On the other hand, treatment of cardiomyocytes with MG132 for 24 h augmented INa by 39% in this example (Figure 5C) and by 41 ± 3% (n = 6) on average (Figure 5D). The steady-state inactivation curve but not the activation curve was shifted in the direction of depolarization by 8.6 ± 1.3 mV (n = 6) by long-term treatment of cardiomyocytes with MG132 for 24 h (data not shown). This result is identical to the long-term effect of bepridil shown in Figure 1C. However, both bepridil and W-7 failed to increase INa any further in the presence of MG132 for 24 h (Figure 5D). When cells were exposed to long-term treatment with MG132, chord conductance of the Na+ channel and Nav1.5 protein expression were significantly increased, the former by 38 ± 2% (n = 6) and the latter by 58 ± 4% (n = 4). Additional application of bepridil or W-7 did not modify any further the change in chord conductance or in Nav1.5 protein expression caused by MG132 (Figure 5E and F).
Figure 5.
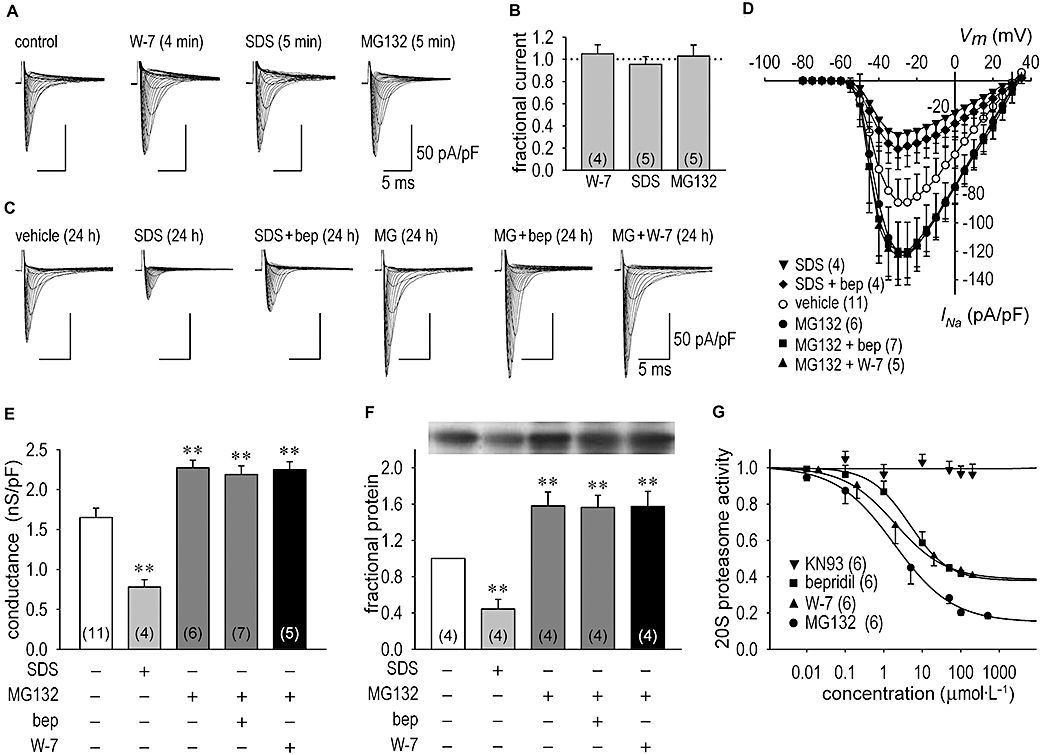
Na+ channel current (INa) and Nav1.5 channel expression through modulation of the ubiquitin–proteasome pathway. (A) Representative INa families in the control condition, during the acute application of 20 µmol·L−1 W-7, 0.06% SDS (sodium dodecyl sulphate, a proteasome activator) and 50 µmol·L−1 MG132 [N-[(phenylmethoxy)carbonyl]-L-leucyl-N-[(1S)-1-formyl-3-methylbutyl]-L-leucinamide] in 4–5 min. (B) A summary of short-term (3–20 min) actions of W-7, SDS and MG132 on INa. INa at each condition was obtained at the test potential of −30 mV and normalized to the control value. (C) Representative INa families in vehicle (24 h), after the long-term action of 0.06% SDS (a proteasome activator), after the long-term action of SDS plus 10 µmol·L−1 bepridil for 24 h, after the long-term action of 50 µmol·L−1 MG132 for 24 h, and after the long-term action of 50 µmol·L−1 MG132 plus 10 µmol·L−1 bepridil (bep) or 20 µmol·L−1 W-7 for 24 h. (D) I–V relationships constructed by using group data in vehicle, after the long-term action of 0.06% SDS for 24 h, after 0.06% SDS treatment for 24 h in the presence of 10 µmol·L−1 bepridil, after long-term treatment of 50 µmol·L−1 MG132 for 24 h, after bepridil (10 µmol·L−1) treatment for 24 h in the presence of 50 µmol·L−1 MG132 and after W-7 (20 µmol·L−1) treatment for 24 h in the presence of 50 µmol·L−1 MG132. (E) The maximum chord conductance in vehicle (24 h), after treatment with 0.06% SDS for 24 h, after treatment with 50 µmol·L−1 MG132 for 24 h, after bepridil (10 µmol·L−1) treatment for 24 h in the presence of 50 µmol·L−1 MG132 and after W-7 (20 µmol·L−1) treatment for 24 h in the presence of 50 µmol·L−1 MG132. Numbers of experiments are shown in parentheses. (F) Expression of cardiac Nav1.5 proteins assessed by Western blots analysis. Na+ channel proteins extracted from cardiomyocytes treated with vehicle, after treatment with 0.06% SDS for 24 h, after the treatment with 50 µmol·L−1 MG132 for 24 h, after bepridil (10 µmol·L−1) treatment for 24 h in the presence of 50 µmol·L−1 MG132 and after W-7 (20 µmol·L−1) treatment for 24 h in the presence of 50 µmol·L−1 MG132. Representative protein expression levels of Nav1.5 are shown in inset. (G) Effect of bepridil, W-7 and KN93 [2-[N-(2-hydroxyethyl)-N-(4-methoxy benzenesulphonyl)]-amino-N-(4-chlorocinnamyl)-N-methylbenzylamine] on 20S proteasome activity in vitro in comparison with that of MG132. 20S proteasome activities were assessed when cardiomyocytes were exposed to 10 µmol·L−1 bepridil, 20 µmol·L−1 W-7, 10 µmol·L−1 KN93 and 50 µmol·L−1 MG132 for 24 h, as relative to the values in the absence of drugs (1.0). **P < 0.01 compared with vehicle.
CaM-kinases (CaM-K) potentially phosphorylate a site in the proteasome, and the Ca2+/CaM signalling pathway is known as a major positive regulator of proteasome activity in neuronal cells (Kawahara and Yokosawa, 1994; Boutillier et al., 1999). To obtain direct evidence that bepridil modulates proteasome activity, we tested whether bepridil and W-7 inhibited in vitro activity of the 20S proteasome, which acts as a catalytic core of the 26S proteasome complex. As expected, MG132 (at 100 µmol·L−1 or higher concentrations) almost completely inhibited the 20S proteasome activity. Importantly, bepridil and W-7 reduced 20S proteasome activity to a similar extent (Figure 5G), while KN93 had no effect on 20S proteasome activity. These results strongly support our hypothesis that bepridil up-regulates cardiac INaas a long-term effect by slowing the degradation of Na+ channel proteins.
Discussion
In the present study, we investigated the long-term effects of bepridil on the Na+ channel in isolated neonatal rat cardiomyocytes and in a heterologous system expressing human Nav1.5. Our three major findings regarding the long-term effects of bepridil are as follows: (i) up-regulation of INa in a dose-dependent manner, apart from its acute blocking effect; (ii) a shift of the steady-state inactivation curve of INa in the direction of depolarization; and (iii) slowing of proteasomal degradation of the Nav1.5 α-subunit through the inhibition of CaM action.
Short- and long-term effects of bepridil on the Na+ channel
Bepridil is a diarylaminopropylamine derivative and is a potent blocker of various cardiac ion channels. Acute applications of bepridil block ICa.L and ICa.T with IC50 values of 0.5–1.6 µmol·L−1 and 0.4–10.6 µmol·L−1 respectively (Yatani et al., 1986; Hara and Nakaya, 1995; Uchino et al., 2005). Wang et al. (1999) reported that bepridil blocks IKs and IKr in guinea pig with IC50 values of 6.2 µmol·L−1 and 13.2 µmol·L−1 respectively. Bepridil inhibits other cardiac ion channels or transporters (IC50values): the Na+-activated K+ channel (IKNa) in guinea pig (2.2 µmol·L−1) (Li et al., 1999), transient outward current (Ito) in sheep Purkinjie fibre (∼3 µmol·L−1) (Berger et al., 1989), the IKATP in guinea pig (6.6–10.0 µmol·L−1) (Li et al., 1999), the Na+-Ca+ exchanger current (8.1 µmol·L−1) (Watanabe and Kimura, 2001) and the Kv1.5 channel (IKur) (6.6 µmol·L−1) (Kobayashi et al., 2001). Bepridil blocks INa in guinea pig cardiomyocytes in a dose-dependent manner with IC50 values of 30–300 µmol·L−1 (Yatani et al., 1986; Sato et al., 1996), shifts the steady-state inactivation curve of INa in the negative direction (Nawada et al., 1995; Sato et al., 1996) and prolongs the recovery from Na+ channel inactivation (Yatani et al., 1986). Moreover, comparing the effects on ICa.L, the half-blocking concentration for INa is 100 times greater than that for ICa.L (Yatani et al., 1986). Thus, the effect of bepridil on INa blocking is less potent compared with its effect on the Ca2+ channels or the K+ channels. Therapeutic plasma concentrations of bepridil reportedly range from 0.5 to 5 µmol·L−1 (Benet, 1985). In the present study, short-term application of bepridil inhibited INa with an IC50 of 96.3 µmol·L−1 in neonatal rat cardiomyocytes and an IC50 of 82.0 µmol·L−1 in HEK-Nav1.5 cells (data not shown). However, as far as we know, the long-term effects of bepridil on ion channels have not been clearly clarified. Our study indicates that long-term application of bepridil significantly increased INa in neonatal rat cardiomyocytes by 25% with 5 µmol·L−1 and 38% with 10 µmol·L−1. Based on these observations, the short-time inhibitory effect of bepridil on INa is considered to play a minor role in comparison with its long-term stimulatory effect on INa.
Atrial fibrillation is the most common sustained cardiac arrhythmia found in clinical practice. However, pharmacological treatment of AF remains challenging. Pharmacological therapy to reverse the remodelling is desirable, as most patients are given treatment only after the onset of AF. A number of studies have investigated various pharmacological approaches to the prevention of atrial electrical and structural remodelling. In most studies pharmacological cardioversion has been attempted by intravenous administration of class I anti-arrhythmic drugs. Interestingly, it was reported that, in patients who are resistant to class I anti-arrhythmic drugs, an application of bepridil effectively suppressed AF attacks and maintained SR (Miyaji et al., 2007). Moreover, several recent reports demonstrated that bepridil in combination with Na+channel blockers exhibits anti-arrhythmic effects on AF and anti-electrical remodelling effects in the hearts of AF patients (Fujiki et al., 2003; Nishida et al., 2007), which is similar to the effects of amiodarone (Shinagawa et al., 2003; Ashikaga et al., 2006; Yamashita et al., 2006). In addition, after conversion to, or maintenance of, SR in patients with persistent AF, contractile functions as well as electrophysiological features of atrial muscles were well preserved by long-term treatment with bepridil (Fujiki et al., 2004). In this context, our results demonstrating an increase in INa and a shift of the steady-state inactivation curve in the direction of depolarization by bepridil in cardiomyocytes suggest the effectiveness of bepridil in long-term application for AF patients. Although the molecular mechanism for the positive shift of the steady-state inactivation curve by bepridil was unknown, it could account for the increase of INa particularly in cardiomyocytes at depolarized potentials. An increase in the INa by bepridil in heart tissue during the therapeutic application may result in a prolongation of both the action potential duration and the effective refractory period. By reducing outward current by a short-term effect on K+ channels and by increasing inward INa as a long-term effect on the Na+channel, bepridil may contribute to modulate fibrillation wave characteristics as, for instance, in the recovery of the conduction delay at pivotal points of the re-entry.
Cellular mechanisms of bepridil-dependent regulation of cardiac ion channels
One may speculate that bepridil modulation of cardiac INa was caused by an increase in channel density through activation of mRNA transcription and consequent protein expression. In the present study, however, the levels of Nav1.5 mRNAs and Navβ mRNAs were not increased by long-term treatment of cardiomyocytes with bepridil. These findings strongly suggested that the observed increase in INa was due to the interruption of channel protein degradation or a redistribution of the channels located in an intracellular pool towards the cell membrane compartment. Because of pharmacological cardioversion, bepridil reverses atrial remodelling gradually and finally terminates AF usually after a treatment period of 3–10 weeks (Fujiki et al., 2003; Nakazato et al., 2005; Nishida et al., 2007). This time course of the action of bepridil suggests an additional effect on channel protein expression as well. Actually, little is known about the molecular determinants of trafficking and membrane turnover of Nav1.5. When protein synthesis was inhibited, we found that the breakdown rate of Nav1.5 protein was significantly decreased by bepridil. The Navβ subunit, SCN1B, has been reported to increase Nav1.5-mediated current in cardiac myocytes and in heterologous expression system (Qu et al., 1995; Fahmi et al., 2001). However, no Navβ mRNAs or Navβ proteins were up-regulated by bepridil and these proteins are, therefore, not likely to be involved in the effects of bepridil that we observed.
The Nav1.5 channels are degraded through the ubiquitin–proteasome pathway in heterologous expression systems (Abriel et al., 2000; van Bemmelen et al., 2004). Therefore, we analysed the effect of SDS, a proteasome activator, and MG132, a proteasome inhibitor, on INa and Nav1.5 protein expression. Inhibition of the ubiquitin–proteasome system accounted for an increase in the number of the Na+ channels in the plasma membrane. Indeed, we found that MG132 increased cell-surface expression of Nav1.5 as shown by an increase in INa, and that INa was not further increased by bepridil. Moreover, up-regulation of INa by bepridil was halted in the presence of SDS. These results indicate that bepridil regulates the degradation process of Na+ channel protein, presumably by modulating the ubiquitin–proteasome pathway.
The number of the Na+ channels in the cardiomyocyte surface membrane depends on the balance between protein insertion and protein breakdown and internalization. Recent studies have highlighted the importance of signal transduction in modulation of cell surface Na+ channel expression through the Ca2+/CaM pathways. Actually, among nearly 300 sequences known to bind CaM, a possible binding motif is postulated in the β-subunit of the 20S proteasome (-RNKERISVAAA-) in rat and human according to the CaM Target Database Web site (http://calcium.uhnres.utoronto.ca/ctdb). Bepridil is widely known to inhibit CaM action apart from its pharmacological effects on various ion channels (Itoh et al., 1984; Zimmer and Hofmann, 1987; Schaeffer et al., 1991). In this context, the present study revealed that W-7, a CaM inhibitor, increased INa as a long-term effect, which is similar to the effect of bepridil, and that bepridil lost its effects on INa in the presence of W-7. Importantly, KN93, a CaM-K inhibitor, had no effect on the bepridil-induced increase of INa (Figure 1) and 20S proteasome activity (Figure 5G). Therefore, it is likely that bepridil increases INa as a long-term effect via CaM inhibition, independently of CaM-K action, by decreasing 20S proteasome activity. A recent study identified a functional relationship between Ca2+/CaM and ubiquitin-specific protease activity (Shen et al., 2005). As we only provide results supporting an inhibitory action of bepridil on CaM that accounts for the inactivation of proteasomal molecules, a systematic investigation such as a comprehensive post-translational analysis of the Na+ channel in myocytes treated with bepridil will be needed to understand more thoroughly the molecular and cellular mechanisms underlying the possible interaction of CaM and the ubiquitin–proteasome system.
In conclusion, the short- and long-term applications of bepridil have different effects on INa in cardiomyocytes. Long-term application of bepridil up-regulates INa by blunting proteasome signals through the inhibition of CaM action, which suggests that the clinical use of bepridil for the treatment of arrhythmias, particularly in combination with Na+ channel blockers, would have a pharmacologically complex outcome. Also caution may be needed in evaluating anti-arrhythmic effects of bepridil in pathological conditions of the heart, particularly when intracellular Ca2+ overload is shown to activate CaM actions.
Acknowledgments
We thank Dr Makielski (University of Wisconsin, Madison, WI) for the generous gift of Nav1.5 vector constructions. This work was supported in part by Grants-in-aid for Scientific Research from the Ministry of Education, Culture, Sports, Science and Technology of Japan (No. 15590759, 17590775) to KO.
Glossary
Abbreviations:
- AF
atrial fibrillation
- CaM
calmodulin
- CaM-K
Ca2+/CaM-dependent kinase
- HEK
human embryonic kidney
- I–V
current–voltage
- INa
Na+ channel current
- KN93
2-[N-(2-hydroxyethyl)-N-(4-methoxy benzenesulphonyl)]-amino-N-(4-chlorocinnamyl)-N-methylbenzylamine
- MG132
N-[(phenylmethoxy)carbonyl]-L-leucyl-N-[(1S)-1-formyl-3-methylbutyl]-L-leucinamide
- SDS
sodium dodecyl sulphate
- SR
sinus rhythm
Conflict of interest
The authors state no conflict of interest.
References
- Abriel H, Kamynina E, Horisberger JD, Staub O. Regulation of the cardiac voltage-gated Na+ channel (H1) by the ubiquitin-protein ligase Nedd4. FEBS Lett. 2000;466:377–380. doi: 10.1016/s0014-5793(00)01098-x. [DOI] [PubMed] [Google Scholar]
- Alexander SPH, Mathie A, Peters JA. Guide to receptors and channels (GRAC), 3rd edn. Br J Pharmacol. 2008;153(Suppl.)(2):S1–S209. doi: 10.1038/sj.bjp.0707746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashikaga K, Kobayashi T, Kimura M, Owada S, Sasaki S, Iwasa A, et al. Effects of amiodarone on electrical and structural remodeling induced in a canine rapid pacing-induced persistent atrial fibrillation model. Eur J Pharmacol. 2006;536:148–153. doi: 10.1016/j.ejphar.2006.02.023. [DOI] [PubMed] [Google Scholar]
- van Bemmelen MX, Rougier JS, Gavillet B, Apotheloz F, Daidie D, Tateyama M, et al. Cardiac voltage-gated sodium channel Nav1.5 is regulated by Nedd4-2 mediated ubiquitination. Circ Res. 2004;95:284–291. doi: 10.1161/01.RES.0000136816.05109.89. [DOI] [PubMed] [Google Scholar]
- Benet LZ. Pharmacokinetics and metabolism of bepridil. Am J Cardiol. 1985;55:8C–13C. doi: 10.1016/0002-9149(85)90799-4. [DOI] [PubMed] [Google Scholar]
- Berger F, Borchard U, Hafner D. Effects of the calcium entry blocker bepridil on repolarizing and pacemaker currents in sheep cardiac Purkinje fibres. Naunyn Schmiedebergs Arch Pharmacol. 1989;339:638–646. doi: 10.1007/BF00168656. [DOI] [PubMed] [Google Scholar]
- Boutillier AL, Kienlen-Campard P, Loeffler JP. Depolarization regulates cyclin D1 degradation and neuronal apoptosis: a hypothesis about the role of the ubiquitin/proteasome signalling pathway. Eur J Neurosci. 1999;11:441–448. doi: 10.1046/j.1460-9568.1999.00451.x. [DOI] [PubMed] [Google Scholar]
- Brembilla-Perrot B, Aliot E, Clementy J, Cosnay P, Djiane P, Fauchier JP, et al. Evaluation of bepridil efficacy by electrophysiologic testing in patients with recurrent ventricular tachycardia: comparison of two regimens. Cardiovasc Drugs Ther. 1992;6:187–193. doi: 10.1007/BF00054570. [DOI] [PubMed] [Google Scholar]
- Fahmi AI, Patel M, Stevens EB, Fowden AL, John JE, III, Lee K, et al. The sodium channel β-subunit SCN3b modulates the kinetics of SCN5a and is expressed heterogeneously in sheep heart. J Physiol. 2001;537:693–700. doi: 10.1111/j.1469-7793.2001.00693.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujiki A, Tsuneda T, Sugao M, Mizumaki K, Inoue H. Usefulness and safety of bepridil in converting persistent atrial fibrillation to sinus rhythm. Am J Cardiol. 2003;92:472–475. doi: 10.1016/s0002-9149(03)00672-6. [DOI] [PubMed] [Google Scholar]
- Fujiki A, Tsuneda T, Sakabe M, Nakagawa K, Mizumaki K, Hirai T, et al. Maintenance of sinus rhythm and recovery of atrial mechanical function after cardioversion with bepridil or in combination with aprindine in long-lasting persistent atrial fibrillation. Circ J. 2004;68:834–839. doi: 10.1253/circj.68.834. [DOI] [PubMed] [Google Scholar]
- Hara Y, Nakaya H. SD-3212, a new class I and IV antiarrhythmic drug: a potent inhibitor of the muscarinic acetylcholine-receptor-operated potassium current in guinea-pig atrial cells. Br J Pharmacol. 1995;116:2750–2756. doi: 10.1111/j.1476-5381.1995.tb17237.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartmann HA, Tiedeman AA, Chen SF, Brown AM, Kirsch GE. Effects of III-IV linker mutations on human heart Na+ channel inactivation gating. Circ Res. 1994;75:114–122. doi: 10.1161/01.res.75.1.114. [DOI] [PubMed] [Google Scholar]
- Hollingshead LM, Faulds D, Fitton A. Bepridil. A review of its pharmacological properties and therapeutic use in stable angina pectoris. Drugs. 1992;44:835–857. doi: 10.2165/00003495-199244050-00009. [DOI] [PubMed] [Google Scholar]
- Itoh H, Ishikawa T, Hidaka H. Effects on calmodulin of bepridil, an antianginal agent. J Pharmacol Exp Ther. 1984;230:737–741. [PubMed] [Google Scholar]
- Izumi D, Chinushi M, Watanabe H, Washizuka T, Okamura K, Komura S, et al. Bepridil for drug-refractory ventricular tachyarrhythmias. Intern Med. 2007;46:119–124. doi: 10.2169/internalmedicine.46.6024. [DOI] [PubMed] [Google Scholar]
- Kamiya K, Niwa R, Mitcheson JS, Sanguinetti MC. Molecular determinants of hERG channel block. Mol Pharmacol. 2006;69:1709–1716. doi: 10.1124/mol.105.020990. [DOI] [PubMed] [Google Scholar]
- Kawahara H, Yokosawa H. Intracellular calcium mobilization regulates the activity of 26 S proteasome during the metaphase-anaphase transition in the ascidian meiotic cell cycle. Dev Biol. 1994;166:623–633. doi: 10.1006/dbio.1994.1342. [DOI] [PubMed] [Google Scholar]
- Kobayashi S, Reien Y, Ogura T, Saito T, Masuda Y, Nakaya H. Inhibitory effect of bepridil on hKv1.5 channel current: comparison with amiodarone and E-4031. Eur J Pharmacol. 2001;430:149–157. doi: 10.1016/s0014-2999(01)01381-4. [DOI] [PubMed] [Google Scholar]
- Levy S, Cointe R, Metge M, Faugere G, Valeix B, Gerard R. Bepridil for recurrent sustained ventricular tachycardias: assessment using electrophysiologic testing. Am J Cardiol. 1984;54:579–581. doi: 10.1016/0002-9149(84)90252-2. [DOI] [PubMed] [Google Scholar]
- Li Y, Sato T, Arita M. Bepridil blunts the shortening of action potential duration caused by metabolic inhibition via blockade of ATP-sensitive K+ channels and Na+-activated K+ channels. J Pharmacol Exp Ther. 1999;291:562–568. [PubMed] [Google Scholar]
- Miyaji K, Tada H, Fukushima Kusano K, Hashimoto T, Kaseno K, Hiramatsu S, et al. Efficacy and safety of the additional bepridil treatment in patients with atrial fibrillation refractory to class I antiarrhythmic drugs. Circ J. 2007;71:1250–1257. doi: 10.1253/circj.71.1250. [DOI] [PubMed] [Google Scholar]
- Nagatomo T, Fan Z, Ye B, Tonkovich GS, January CT, Kyle JW, et al. Temperature dependence of early and late currents in human cardiac wild-type and long Q-T ΔKPQ Na+ channels. Am J Physiol. 1998;275:H2016–H2024. doi: 10.1152/ajpheart.1998.275.6.H2016. [DOI] [PubMed] [Google Scholar]
- Nakazato Y, Yasuda M, Sasaki A, Iida Y, Kawano Y, Nakazato K, et al. Conversion and maintenance of sinus rhythm by bepridil in patients with persistent atrial fibrillation. Circ J. 2005;69:44–48. doi: 10.1253/circj.69.44. [DOI] [PubMed] [Google Scholar]
- Nawada T, Tanaka Y, Hisatome I, Sasaki N, Ohtahara A, Kotake H, et al. Mechanism of inhibition of the sodium current by bepridil in guinea-pig isolated ventricular cells. Br J Pharmacol. 1995;116:1775–1780. doi: 10.1111/j.1476-5381.1995.tb16662.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishida K, Fujiki A, Sakamoto T, Iwamoto J, Mizumaki K, Hashimoto N, et al. Bepridil reverses atrial electrical remodeling and L-type calcium channel downregulation in a canine model of persistent atrial tachycardia. J Cardiovasc Electrophysiol. 2007;18:765–772. doi: 10.1111/j.1540-8167.2007.00833.x. [DOI] [PubMed] [Google Scholar]
- Prystowsky EN. Effects of bepridil on cardiac electrophysiologic properties. Am J Cardiol. 1992;69:63D–67D. doi: 10.1016/0002-9149(92)90961-w. [DOI] [PubMed] [Google Scholar]
- Qu Y, Isom LL, Westenbroek RE, Rogers JC, Tanada TN, McCormick KA, et al. Modulation of cardiac Na+ channel expression in Xenopus oocytes by β1 subunits. J Biol Chem. 1995;270:25696–25701. doi: 10.1074/jbc.270.43.25696. [DOI] [PubMed] [Google Scholar]
- Sato N, Nishimura M, Kawamura Y, Ward CA, Kikuchi K. Block of Na+ channel by bepridil in isolated guinea-pig ventricular myocytes. Eur J Pharmacol. 1996;314:373–379. doi: 10.1016/s0014-2999(96)00567-5. [DOI] [PubMed] [Google Scholar]
- Sato T, Costa AD, Saito T, Ogura T, Ishida H, Garlid KD, et al. Bepridil, an antiarrhythmic drug, opens mitochondrial KATP channels, blocks sarcolemmal KATP channels, and confers cardioprotection. J Pharmacol Exp Ther. 2006;316:182–188. doi: 10.1124/jpet.105.094029. [DOI] [PubMed] [Google Scholar]
- Schaeffer P, Lugnier C, Stoclet JC. Interactions of calmodulin antagonists with calcium antagonists binding sites. Eur J Pharmacol. 1991;206:325–332. doi: 10.1016/0922-4106(91)90117-z. [DOI] [PubMed] [Google Scholar]
- Shen C, Ye Y, Robertson SE, Lau AW, Mak DO, Chou MM. Calcium/calmodulin regulates ubiquitination of the ubiquitin-specific protease TRE17/USP6. J Biol Chem. 2005;280:35967–35973. doi: 10.1074/jbc.M505220200. [DOI] [PubMed] [Google Scholar]
- Shinagawa K, Shiroshita-Takeshita A, Schram G, Nattel S. Effects of antiarrhythmic drugs on fibrillation in the remodeled atrium: insights into the mechanism of the superior efficacy of amiodarone. Circulation. 2003;107:1440–1446. doi: 10.1161/01.cir.0000055316.35552.74. [DOI] [PubMed] [Google Scholar]
- Uchino T, Lee TS, Kaku T, Yamashita N, Noguchi T, Ono K. Voltage-dependent and frequency-independent inhibition of recombinant Cav3.2 T-type Ca2+ channel by bepridil. Pharmacology. 2005;74:174–181. doi: 10.1159/000085329. [DOI] [PubMed] [Google Scholar]
- Wang JC, Kiyosue T, Kiriyama K, Arita M. Bepridil differentially inhibits two delayed rectifier K+ currents, IKr and IKs, in guinea-pig ventricular myocytes. Br J Pharmacol. 1999;128:1733–1738. doi: 10.1038/sj.bjp.0702959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Morishima M, Zheng M, Uchino T, Mannen K, Takahashi A, et al. Transcription factors Csx/Nkx2.5 and GATA4 distinctly regulate expression of Ca2+ channels in neonatal rat heart. J Mol Cell Cardiol. 2007;42:1045–1053. doi: 10.1016/j.yjmcc.2007.03.905. [DOI] [PubMed] [Google Scholar]
- Watanabe Y, Kimura J. Blocking effect of bepridil on Na+/Ca2+ exchange current in guinea pig cardiac ventricular myocytes. Jpn J Pharmacol. 2001;85:370–375. doi: 10.1254/jjp.85.370. [DOI] [PubMed] [Google Scholar]
- Yamashita N, Kaku T, Uchino T, Isomoto S, Yoshimatsu H, Ono K. Short- and long-term amiodarone treatments regulate Cav3.2 low-voltage-activated T-type Ca2+ channel through distinct mechanisms. Mol Pharmacol. 2006;69:1684–1691. doi: 10.1124/mol.105.021253. [DOI] [PubMed] [Google Scholar]
- Yatani A, Brown AM, Schwartz A. Bepridil block of cardiac calcium and sodium channels. J Pharmacol Exp Ther. 1986;237:9–17. [PubMed] [Google Scholar]
- Zimmer M, Hofmann F. Differentiation of the drug-binding sites of calmodulin. Eur J Biochem. 1987;164:411–420. doi: 10.1111/j.1432-1033.1987.tb11073.x. [DOI] [PubMed] [Google Scholar]