

Abstract
We examine the contribution of residues at the dimer interface of the transcriptional regulator OxyR to oligomerization. Residues in contact across the dimer interface of OxyR were identified using the program Quaternary Contacts (QContacts). Site-directed mutagenesis was performed on the non-alanine or glycine residues identified in the resultant contact profile and the oligomerization ability of the mutant proteins was tested using the λcI repressor system to identify residues that are hot spots in OxyR. We compared the properties of these hot spots to those described in the literature from other systems. The hot spots identified in this study are not especially conserved amongst a set of OxyR orthologs.
Keywords: protein–protein interactions, LysR-type transcriptional regulators, OxyR, oligomerization
Introduction
Many biological processes rely on the assembly of oligomeric proteins from conserved fold families. Although different members of these families share structural similarity, they only assemble with specific partners. The LysR-type transcriptional regulators are one such family of related proteins whose assembly must be specific within the same cell; in E. coli, there are 46 (LTTRs), whereas P. aeruginosa encodes 123.1 Understanding the specificity of assembly first requires understanding the contributions of individual residues in the oligomer interfaces of specific family members to oligomerization. Here, we focus on one E. coli LTTR, OxyR.
OxyR is a global response regulator to oxidative stress.2 In E. coli, OxyR controls a regulon including katG, dps, ahpCF, sufA, grxA, gorA, and oxyS. OxyR has been extensively studied to understand the molecular basis of the oxidative stress response in E. coli. The active form of OxyR is homoteterameric, consisting of a dimer of dimers where each dimer is made up of 34 kDa monomers.3 Mutational studies identified Cys199 and Cys208 as necessary for the response to oxidative stress.4 The crystal structures of the reduced and oxidized forms of the regulatory domain (residues 80–305), solved by Choi et al.5 demonstrated a large rearrangement of the subunits, creating two distinct structural forms of the protein characterized by a 30° rotation between the subunits. This rearrangement was brought about by the formation of an intrasubunit disulfide bond between Cys199 and Cys208, which are 17 Å apart in the reduced form.
Alanine scanning has been used to investigate individual residues' contributions to the oligomerization of many oligomers.6 In this report, we examine the roles of residues in the subunit interfaces of OxyR in stabilizing the oligomeric forms. We identified residues with potential for contributing to the oligomerization of OxyR utilizing QContacts.7 The resultant list of residues in contact, or contact profile, was used as a guide for site-directed mutagenesis, and the ability to form oligomers was assessed using the lambda repressor fusion assay.8 We find a small subset of the candidate residues have strong oligomerization phenotypes in the tetramer, consistent with these residues being hot spots in one or both oligomeric forms of OxyR. Other residues in the interface have weaker but significant energetic contributions to oligomerization, as determined from repressor fusion assays using the regulatory domain instead of the full-length OxyR.
Results
Identification of interface contact residues
Residues in contact in both the reduced and oxidized forms of OxyR were identified from the two regulatory domain crystal structures as targets for alanine mutagenesis. Choi et al.5 described several residues that comprise the interfaces of the two forms of OxyR, including I110, H114, E121, L124, D172, H218, F219, and A233, but they do not present a comprehensive analysis of the interfaces. To provide such an analysis, we used the program QContacts to identify residues in Voronoi contact across the interfaces of the reduced and oxidized forms.7 The nonredundant contact profiles generated by QContacts are shown in Supplementary Tables SI and SII. The contact profile yields two types of information: the residues involved at the interface and the atomic-level interaction across the interface. QContacts identifies the specific residues described in the paper, as well as additional residues. As shown in Figure 1, we mapped these contacts on the reduced form of OxyR.
Figure 1.

Comparison of identified structure residues with QContacts. The interface of one monomer of the reduced form of OxyR is shown. Residues detected by QContacts are colored green. Residues described by both Choi et al.5 and identified by QContacts are colored purple. There were no residues described by Choi et al. that were not detected by QContacts.
In the crystal structures solved by Choi et al.,5 the transition from the reduced to the oxidized form is mediated by a large rotation between the two subunits. To examine whether this involves formation of contacts between different sets of residues or changes in the contacts made by a common set of residues, we compared the contact profiles of the reduced and oxidized forms. A total of 32 and 34 residues were identified in the contact profiles of the reduced and oxidized forms, respectively. A total of 24 residues were found in the contact profiles for both forms, whereas 18 are specific to only one of the two forms. When we evaluated the contact pairs between the two contact profiles, a total of 43 and 49 contacts were identified for the reduced and oxidized contact profiles, respectively. However, when we examined the pairwise contacts between the two profiles, only four are shared between the two forms. Additionally, though some atom–atom interactions are in common, many are different. For example, in the reduced form, P103 Cα is making contacts with E225 Cβ and Oɛ1, whereas in the oxidized form this Cα is not making any contacts (not shown).
Individual residues' contributions to oligomerization
To systematically determine the contribution to oligomerization of the residues identified by QContacts, non-alanine or glycine residues were mutated to alanine. To assay for assembly in vivo, the mutant oxyR genes were subcloned into the λ repressor fusion vectors pLM1000, pAZ299, and pGK751 (see Methods).8 These vectors contain a combination of two features that allow for increasing amounts of fusion protein to be expressed. First, the promoter in plasmids pLM1000 and pAZ299, P7107, is a weak, constitutive promoter derived from an operatorless PlacUV5. Plasmid pGK751 contains the PlacUV5 promoter. Plasmids pGK751 and pLM1000 contain an amber mutation at codon 103 of the cI segment. There is roughly a twofold expression level difference between fusions that contain this mutation and fusions that do not. Thus, we expect expression of fusions from pLM1000 to be approximately twofold lower than those expressed from pAZ299. Furthermore, because of the promoter differences, we expect expression of fusions from pGK751 to be substantially higher than those from pLM1000 and pGK751.
Each fusion's ability to oligomerize was assessed by its ability to confer immunity to phage λ infection, as described in the Methods. In all three vectors, full-length wild-type OxyR is immune. We sorted the immunity results of the full-length OxyR mutations into three classes: (1) those that were not able to confer immunity in any of the vectors, (2) those that were unable to oligomerize in pLM1000, but able to oligomerize in pAZ299 or pGK751, and (3) those that behaved as wild-type, being immune in all three vectors. These results are listed in Table I. There are 7 residues in Class I, 17 in Class II, and 3 in Class III.
Table I.
Immunity Result Classes
| Position | Full-length | Regulatory domain | Sub-class |
|---|---|---|---|
| E126 | I | I | 1A |
| R228 | I | I | 1A |
| E248 | I | I | 1A |
| H125 | I | II | 1B |
| H218 | I | II | 1B |
| M230 | I | II | 1B |
| S235 | I | III | 1C |
| Y104 | II | I | 2A |
| L106 | II | I | 2A |
| I110 | II | I | 2A |
| P111 | II | I | 2A |
| H114 | II | I | 2A |
| L120 | II | I | 2A |
| Q131 | II | I | 2A |
| T217 | II | I | 2A |
| S223 | II | I | 2A |
| E225 | II | I | 2A |
| D252 | II | I | 2A |
| Q128 | II | II | 2B |
| D172 | II | II | 2B |
| R220 | II | II | 2B |
| E121 | II | III | 2C |
| T226 | II | III | 2C |
| P107 | III | I | 3A |
| P103 | III | I | 3A |
| M122 | III | I | 3A |
| F219 | III | I | 3A |
| N229 | III | I | 3A |
| Y170 | III | II | 3B |
| T222 | III | III | 3C |
| H94 | ND | II | |
| L97 | ND | I | |
| L124 | ND | I |
Though the full-length protein is tetrameric, the crystal structure of the OxyR regulatory domain is dimeric.3,5 Crystal structures of other LTTRs such as CbnR from Ralstonia eutropha NH99 and DntR from Burkholderia sp. strain DNT10 show the tetramer as a dimer of dimers. To determine whether the oligomerization interactions in the tetramer and dimer behaved differently, we constructed λcI fusions to mutant and wild-type regulatory domains in all three vectors and assayed for immunity as described in the Methods. The wild-type OxyR regulatory domain was sensitive in pLM1000, being unable to oligomerize, whereas in pAZ299 and pGK751, the OxyR regulatory domain was able to oligomerize, as indicated by the immune phenotype. The regulatory domain results were grouped into three classes: (1) those that were unable to confer immunity in any of the vectors, (2) those that were unable to oligomerize in pLM1000 and pAZ299, but able to oligomerize in pGK751, and (3) those that behaved as wild-type, being sensitive in pLM1000 and immune in pAZ299 and pGK751. These results are also listed in Table I. There are 20 Class I, 8 Class II, and 4 Class III residues. Comparing the full-length results to the regulatory results allowed for subcategorization of the residues and they are also listed in Table I, generating a range of classes for both the full-length and regulatory immunity results combined. For example, Class IA positions show that both the full-length and regulatory domain phenotype is sensitive, while Class 3C positions show the wild-type phenotype in the full-length and regulatory domain. As expected, the regulatory domain fusions are generally weaker repressors in the absence of tetramer-specific contacts. However, the pattern observed is not simply a shift of all the alleles to lower activity.
Conservation of residues
To determine if there was a correlation between residue conservation amongst other OxyR orthologs and oligomerization, 97 OxyR orthologs were identified (see Methods) and the residue composition at each position in the sequence alignment was calculated. Conservation was calculated based on four categories of amino acid types: hydrophobic, polar uncharged, positively charged, and negatively charged (see Methods). Important residues for regulatory domain assembly [see Fig. 2A] range in identity from 20 to 100% (red bars). The same range of values is seen for residues that do not significantly contribute to oligomerization of OxyR (blue bars).
Figure 2.
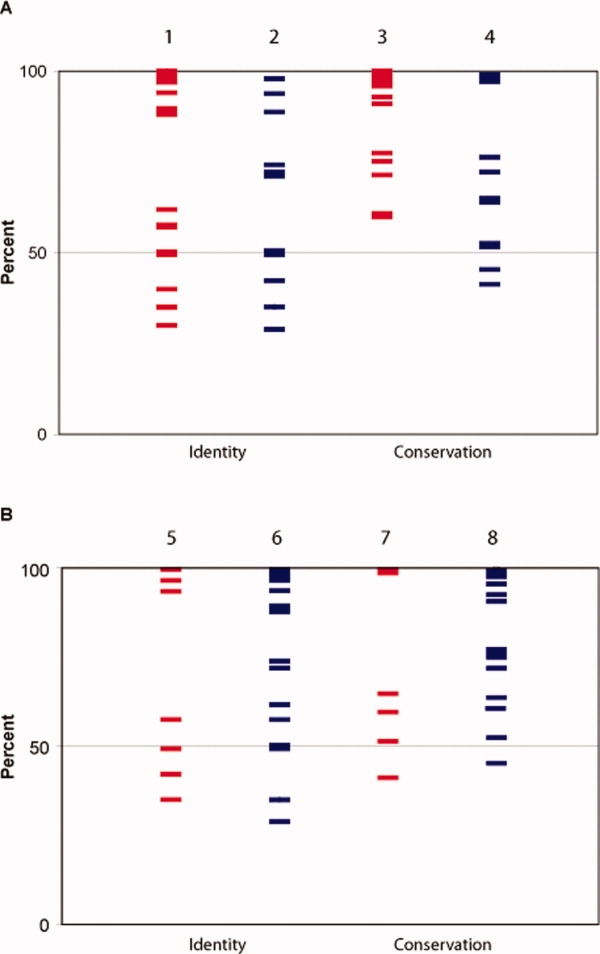
Comparison of identity and conservation of residues based on oligomerization results. Each line represents a residue mutated to alanine. Residues important for oligomerization (red) and those that did not show a sensitive phenotype (blue) in the regulatory domain (Panel A) or full-length OxyR fusions (Panel B) are plotted based on the identity (columns 1, 2, 5, and 6) or conservation (columns 3, 4, 7, and 8) at that particular position within a family of determined OxyR orthologs, as described in the Methods. Overall, the seven hot spots had conservation scores as follows: H125, 51%, E126, 100%, H218, 65%, R228, 97%, M230, 99%, S235, 41%, and E248, 60%.
The same analysis was applied to the full-length protein. Considering identity only, the same range of percent identity found in the regulatory-domain results is found in both residues important and not important for oligomerization in the full-length [Fig. 2(B)]. Taken together, the residues important for oligomerization of OxyR are not more conserved than other interface residues.
Discussion
In this study, we examined the contribution of residues in the OxyR interface, as identified by QContacts, to oligomerization of this LTTR. Of the 38 positions identified by QContacts, only seven, E126, R228, E248, H125, H218, M230, and S235, were identified as making major contributions to the ability of cI-OxyR fusions to assemble. These positions could affect oligomerization directly or indirectly through effects on monomer stability. We were unable to express the wild-type fusions in soluble form, and thus cannot rule out effects on monomer folding. However, the seven putative hot spot positions are occupied by mostly hydrophilic residues and several are not especially conserved among OxyR orthologs. None of the residues is completely buried in the OxyR monomer, and one residue, H218, is hyperexposed when the other monomer in the regulatory domain dimer is removed in silico. Thus, we discuss these seven residues as hot spots in the oligomerization interface of OxyR below.
Hot spots in proteins have been catalogued by several previous studies and several general trends are found.11–13 Although the hot spots in this study appear to largely contribute to OxyR oligomerization, they are atypical with respect to most of these trends.
Keskin et al.,14 as did Bogan and Thorn,12 found that the hot spots tended to be clustered together, terming these clusters hot regions. These regions tended to be protected from bulk solvent and located toward the center of the interface. We find that this is quite the opposite of our relevant OxyR positions. As seen in Figure 3(A, B), the hot spots in OxyR are scattered over the surface of the interface.
Figure 3.
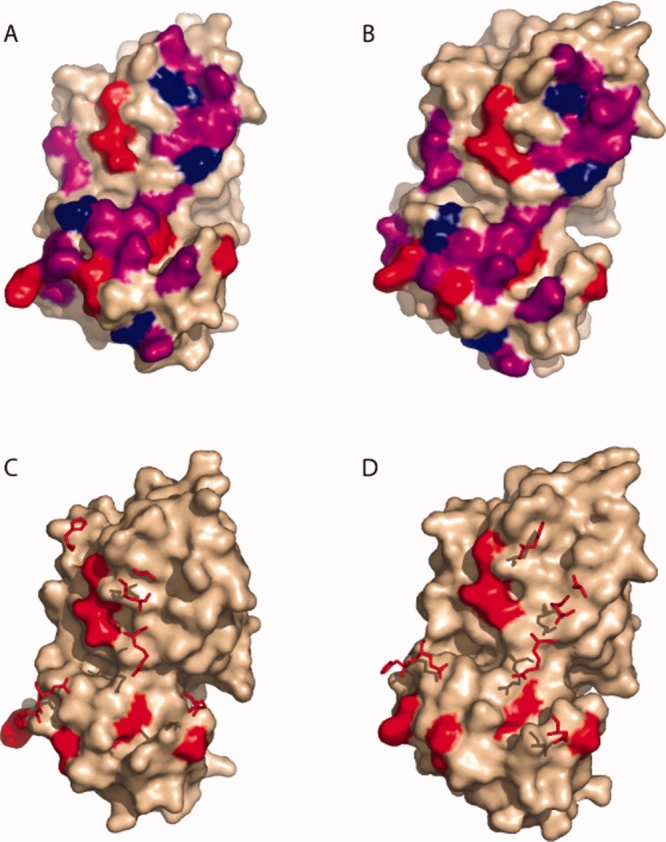
Positions of mutated residues. Mutated residues are colored on the dimer interface of the monomer structures. In panels (A) and (B), Class I (hot spot) residues are colored red. Class II residues are purple and Class III residues are blue in the reduced and oxidized forms, respectively. Panels (C) and (D) show how the hot spot residues on one monomer are positioned relative to hot spots across the interface. The surface of one monomer is shown (wheat colored), with the seven Class I positions highlighted red. The seven Class I positions from the other monomer are shown as stick structures. (C) Reduced Form. (D) Oxidized form.
As noted by Keskin et al.,14 hot spot residues on one side of a protein–protein interface tend to interact with other hot spot residues on the other side of the interface. By contrast, there are no direct contacts between OxyR hot spots identified here and hot spots on the opposite subunit. Figure 3(C, D) shows the hot spot residues of one monomer superimposed on the surface of the other monomer in the reduced [Fig. 3(B)] and oxidized [Fig. 3(C)] forms. All seven identified hot spots interact with residues that can be mutated to alanine without dramatically disrupting the oligomerization of OxyR. We note that, in the absence of a full-length OxyR structure, we cannot formally rule out the possibility that the subunit interface in the full-length protein involves different residue pairs than are seen in the two regulatory domain structures we use to interpret our results (see below).
By contrast, several positions that tolerate mutation to alanine contact more than one hot spot. T222 is the position that is least sensitive to mutation; T222A is a Class III mutant in both the full-length and regulatory domain contexts. Located in the middle of the interface, T222 interacts with atoms from residues V97, L106, L124, H125, and E126 in the oxidized form and only E126 in the reduced form. H125 and E126 are hot spots, which interact indirectly through T222 in the reduced form.
Despite using different criteria for what constitutes conservation, Guharoy and Chakrabarti15 and Keskin et al.14 all find that hot spots conserved among orthologs. By contrast, we did not observe a correlation between conservation among OxyR orthologs and sensitivity to mutation to alanine. This lack of correlation (see Fig. 2) was not significantly altered by using different classifications of amino residues, such as those described by Keskin et al.14 and Guharoy and Chakrabarti15 (data not shown). The hot spots in this study are enriched in polar amino acids, consistent with the observations of Hu et al.13 However, only one of the seven, R228, is one of the preferred hot spot residue types identified by Bogan and Thorn.12
Why are the OxyR hot spots so atypical? The large rotation between the subunits observed in the transition from the reduced to oxidized form may provide an explanation for the uncharacteristic properties of the hot spots observed in this study. With such a large rotation, having hot spots in the center of the interface may inhibit the molecule's ability to undergo this conformational change. Closer examination of the hot spots' contacts in each crystal form illustrates this point. In the reduced form, M230 makes interactions with M122, Y123, and L124. In the oxidized form, these interactions are replaced with interacting residues L106, P107, I110, and P111. Additional examples are found in other hot spots' contacts.
Our results also suggest that both the oxidized and reduced forms must be stable for a protein to score as oligomeric in our assay. Two residues, E248 and R228, are unique to the oxidized and reduced contact profiles, respectively. QContacts identifies only one interatomic contact across the interface for H218 in the reduced structure, but the imidazole group of H218 makes 23 contacts in the oxidized form. This was surprising, as we did not expect the oxidized conformation to be populated because E. coli is not thought to be under significant oxidative stress under our assay conditions. However, the assay requires exposure to phage, which may stress the cells in ways that induce the oxidized form of OxyR.
Testing the contribution of each residue in both a full-length and regulatory domain repressor fusion suggests that the assembly of the full-length tetramer is not simply the additive contribution of the dimer contacts found in the regulatory domain plus additional interactions. As would be expected from losing tetramer contacts, the regulatory domain fusions are more sensitive to disruption than the full-length fusions, and in an additive model we would expect that mutations would disrupt assembly of both the full-length and regulatory domain fusions, neither the full-length nor the regulatory domain, or the regulatory domain only. Although most positions tested fit this pattern (see Classes 2A, 3A, 2B, and 3B), four hot spots in subclasses 1B and 1C disrupt assembly of the full-length fusions while retaining immunity as cI-regulatory domain fusions.
Methods
Generation of the contact profile
The crystal structures used in these studies were PDB files 1I69 (reduced) and 1I6A (oxidized).5 The dimeric form of 1I6A was constructed using the PQS Server.16 QContacts7 was used to calculate the residues in contact across each chain. The output of QContacts was filtered for redundant pairwise interactions and the subsequent culled residue contact profiles are shown in Supplementary Tables SI and SII.
Site-directed mutagenesis
Using the contact profiles as a guide, the identified non-alanine and glycine residues of OxyR in the λcI OxyR repressor fusion were mutated to alanine. PrimerX (http://www.bioinformatics.org/primerx) was used to generate the primer sequence and primers were ordered from IDT (Iowa). Pfu Turbo (Stratagene) or Pfx Platinum (Invitrogen) was used for the polymerase. pGK702 was the DNA template. Reactions were treated with 2U of DpnI for 2 h and transformed into Mach1-T1R, Top1-T1R (Invitrogen), or XL-1Blue Supercompetent (Stratagene) cells. Transformants were recovered and sequenced (LPGT, TAMU).
Identification of candidates
Gateway Cloning Technology (Invitrogen) was used to facilitate moving the mutated gene to other vectors. pGK702 contains the λcI OxyR repressor fusion. oxyR is flanked by the att sites for the Gateway system, effectively making this vector a Gateway expression vector. The mutated oxyR gene was moved into pDONR201 via the back reaction to generate a Gateway Entry Clone.17 The entry clones were used to move the gene into several destination vectors: pLM1000,18 pAZ299, and pGK751 (this study) via the LR reaction (see Table II for descriptions). The reactions were transformed into Mach1-T1R cells. Candidate DNA was transformed into JH78719 or AG1688,20 streak purified, and freezer stocks made.
Table II.
Strains and Plasmids
| AG1688 | araD139 Δ(ara, leu)7697 ΔlacX74 galU galK hsdR strA F'128 lacIq lacZ::Tn5 |
| JH787 | AG1688 Φ80 su3 |
| Mach1-T1R | F− ΔlacX74 hsdR(rk− mk+) Δrec1398 endA1 tonA |
| pAZ299 | P7107-cI-Gateway cassette, CmR, AmpR |
| pDONR201 | Gateway entry clone vector; contains ccdB, CmR, KmR |
| pGK702 | P7107-cI-attB1-OxyR(2-305)-attB2, AmpR |
| pGK751 | PlacUV5-cI-amber-Gateway cassette CmR, AmpR |
| pLM1000 | P7107-cI-amber-Gateway cassette CmR, AmpR |
Immunity assay
To test for the repressor fusion's ability to oligomerize, cross-streak assays were done. Strains were struck out for single colonies and incubated for 16 h at 37°C. Three individual colonies were challenged against lines of phage λKH54,8 λvir, and control phage λi21c8 on tryptone agar plates and incubated for 7 h at 37°C. Those that were able to grow across the λKH54 line were called immune, and those that were not able to grow were sensitive. All colonies died at the control λi21c phage. Strains immune to λKH4 were given a score of one, whereas sensitive phenotypes received a score of zero. Each strain was tested three times with three individual colonies and the average calculated.
Generation of λcI regulatory domain repressor fusions
Entry clones were generated of the 233 amino acids that comprise the regulatory domain of OxyR. Residues 80–305 of the regulatory domain of the full-length oxyR mutants were amplified with the primers OxyR RD GW-f and attB2 to attach in-frame attB sites. Using the PCR product, entry clones were generated in pDONR201 and the mutated genes subsequently moved to pLM1000, pAZ299, and pGK751 to generate λcI-regulatory domain repressor fusions.
Conservation calculations
Ninety-seven OxyR orthologs were identified by performing a BLAST search. We required the sequences have a cysteine at the equivalent E. coli positions of 199 and 208. A multiple sequence alignment was generated using the default settings on CLUSTAL-W. The different residues present at each position identified in QContacts were counted using the program QConAAtally.pl and the identity and conservation at the identified positions compared. Identical residues were scored for an exact match to the E. coli K-12 OxyR sequence. Conservation of residues was based on the following categories: hydrophobic AIFLMPVW, polar non-charged CGNQSTY, positively charged HKR, and negatively charged ED. A complete list of the orthologs is shown in Supplemental Table S3.
Supplemental material
References
- 1.Tatusov RL, Galperin MY, Natale DA, Koonin EV. The COG database: a tool for genome-scale analysis of protein functions and evolution. Nucleic Acids Res. 2000;28:33–36. doi: 10.1093/nar/28.1.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Christman MF, Storz G, Ames BN. OxyR, a positive regulator of hydrogen peroxide-inducible genes in Escherichia coli and Salmonella typhimurium, is homologous to a family of bacterial regulatory proteins. Proc Natl Acad Sci USA. 1989;86:3484–3488. doi: 10.1073/pnas.86.10.3484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kullik I, Stevens J, Toledano MB, Storz G. Mutational analysis of the redox-sensitive transcriptional regulator OxyR: regions important for DNA binding and multimerization. J Bacteriol. 1995;177:1285–1291. doi: 10.1128/jb.177.5.1285-1291.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zheng M, Aslund F, Storz G. Activation of the OxyR transcription factor by reversible disulfide bond formation. Science. 1998;279:1718–1721. doi: 10.1126/science.279.5357.1718. [DOI] [PubMed] [Google Scholar]
- 5.Choi H, Kim S, Mukhopadhyay P, Cho S, Woo J, Storz G, Ryu S. Structural basis of the redox switch in the OxyR transcription factor. Cell. 2001;105:103–113. doi: 10.1016/s0092-8674(01)00300-2. [DOI] [PubMed] [Google Scholar]
- 6.Clackson T, Wells JA. A hot spot of binding energy in a hormone-receptor interface. Science. 1995;267:383–386. doi: 10.1126/science.7529940. [DOI] [PubMed] [Google Scholar]
- 7.Fischer TB, Holmes JB, Miller IR, Parsons JR, Tung L, Hu JC, Tsai J. Assessing methods for identifying pair-wise atomic contacts across binding interfaces. J Struct Biol. 2006;153:103–112. doi: 10.1016/j.jsb.2005.11.005. [DOI] [PubMed] [Google Scholar]
- 8.Hu JC, O'Shea EK, Kim PS, Sauer RT. Sequence requirements for coiled-coils: analysis with lambda repressor-GCN4 leucine zipper fusions. Science. 1990;250:1400–1403. doi: 10.1126/science.2147779. [DOI] [PubMed] [Google Scholar]
- 9.Muraoka S, Okumura R, Ogawa N, Nonaka T, Miyashita K, Senda T. Crystal structure of a full-length LysR-type transcriptional regulator, CbnR: unusual combination of two subunit forms and molecular bases for causing and changing DNA bend. J Mol Biol. 2003;328:555–566. doi: 10.1016/s0022-2836(03)00312-7. [DOI] [PubMed] [Google Scholar]
- 10.Smirnova IA, Dian C, Leonard GA, McSweeney S, Birse D, Brzezinski P. Development of a bacterial biosensor for nitrotoluenes: the crystal structure of the transcriptional regulator DntR. J Mol Biol. 2004;340:405–418. doi: 10.1016/j.jmb.2004.04.071. [DOI] [PubMed] [Google Scholar]
- 11.Tsai CJ, Lin SL, Wolfson HJ, Nussinov R. A dataset of protein-protein interfaces generated with a sequence-order-independent comparison technique. J Mol Biol. 1996;260:604–620. doi: 10.1006/jmbi.1996.0424. [DOI] [PubMed] [Google Scholar]
- 12.Bogan AA, Thorn KS. Anatomy of hot spots in protein interfaces. J Mol Biol. 1998;280:1–9. doi: 10.1006/jmbi.1998.1843. [DOI] [PubMed] [Google Scholar]
- 13.Hu Z, Ma B, Wolfson H, Nussinov R. Conservation of polar residues as hot spots at protein interfaces. Proteins. 2000;39:331–342. [PubMed] [Google Scholar]
- 14.Keskin O, Ma B, Nussinov R. Hot regions in protein–protein interactions: the organization and contribution of structurally conserved hot spot residues. J Mol Biol. 2005;345:1281–1294. doi: 10.1016/j.jmb.2004.10.077. [DOI] [PubMed] [Google Scholar]
- 15.Guharoy M, Chakrabarti P. Conservation and relative importance of residues across protein-protein interfaces. Proc Natl Acad Sci USA. 2005;102:15447–15452. doi: 10.1073/pnas.0505425102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Henrick K, Thornton JM. PQS: a protein quaternary structure file server. Trends Biochem Sci. 1998;23:358–361. doi: 10.1016/s0968-0004(98)01253-5. [DOI] [PubMed] [Google Scholar]
- 17.Walhout AJ, Temple GF, Brasch MA, Hartley JL, Lorson MA, van den Heuvel S, Vidal M. GATEWAY recombinational cloning: application to the cloning of large numbers of open reading frames or ORFeomes. Methods Enzymol. 2000;328:575–592. doi: 10.1016/s0076-6879(00)28419-x. [DOI] [PubMed] [Google Scholar]
- 18.Mariño-Ramírez L, Minor JL, Reading N, Hu JC. Identification and mapping of self-assembling protein domains encoded by the Escherichia coli K-12 genome by use of lambda repressor fusions. J Bacteriol. 2004;186:1311–1319. doi: 10.1128/JB.186.5.1311-1319.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mariño-Ramírez L, Hu JC. Isolation and mapping of self-assembling protein domains encoded by the Saccharomyces cerevisiae genome using lambda repressor fusions. Yeast. 2002;19:641–650. doi: 10.1002/yea.867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hu JC, Newell NE, Tidor B, Sauer RT. Probing the roles of residues at the e and g positions of the GCN4 leucine zipper by combinatorial mutagenesis. Protein Sci. 1993;2:1072–1084. doi: 10.1002/pro.5560020701. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.