

Abstract
The binding of the Bacillus anthracis protective antigen (PA) to the host cell receptor is the first step toward the formation of the anthrax toxin, a tripartite set of proteins that include the enzymatic moieties edema factor (EF), and lethal factor (LF). PA is cleaved by a furin-like protease on the cell surface followed by the formation of a donut-shaped heptameric prepore. The prepore undergoes a major structural transition at acidic pH that results in the formation of a membrane spanning pore, an event which is dictated by interactions with the receptor and necessary for entry of EF and LF into the cell. We provide direct evidence using 1-dimensional 13C-edited 1H NMR that low pH induces dissociation of the Von-Willebrand factor A domain of the receptor capillary morphogenesis protein 2 (CMG2) from the prepore, but not the monomeric full length PA. Receptor dissociation is also observed using a carbon-13 labeled, 2-fluorohistidine labeled CMG2, consistent with studies showing that protonation of His-121 in CMG2 is not a mechanism for receptor release. Dissociation is likely caused by the structural transition upon formation of a pore from the prepore state rather than protonation of residues at the receptor PA or prepore interface.
Keywords: anthrax, protective antigen, histidine, pH, membrane, pore, fluorine
Introduction
The protective antigen (PA),1 an 83 kDa protein secreted by Bacillus anthracis, is a component of the anthrax toxin, a tripartite set of proteins that also includes the enzymatic moieties lethal factor (LF)2 and edema factor (EF).3 PA recognizes on host cells the von-Willebrand factor A (VWA) domain of one of two possible cell surface receptors, anthrax toxin receptor/tumor endothelial marker 8 (ATR/TEM8) or capillary morphogenesis protein 2 (CMG2).4,5 Binding to the receptor is followed by proteolysis on the cell surface by a furin-like protease, an event which cleaves PA into two separate fragments of 20 and 63 kDa. The 20 kDa fragment diffuses away whereas the 63 kDa fragment remains bound to the receptor. This fragment subsequently undergoes oligomerization into a seven-member ring-shaped structure called the prepore [(PA63)7].6
Formation of the prepore creates binding sites for LF and/or EF, with a stoichiometry of three molecules of LF and/or EF per prepore.7,8 The toxin is then endocytosed into an early endosome and subsequently trafficked to an acidified late endosome.9 In this low pH environment (pH ˜5–6), residues 285–340 (β2-β3 strands) within Domain 2 of each of the seven monomers of [(PA63)7] insert into the vesicle membrane bilayer, forming a 14-stranded extended β-barrel called the pore.10–12 LF and EF then enter through the narrow channel of the pore in an unfolded state and are translocated through the pore into the cytosol.13–15 Upon entry, these proteins must refold and through manifestation of their enzymatic activities, disrupt cell signaling mechanisms required for maintenance of host immunity.
Early in vitro studies16,17 have shown that the purified prepore can form a pore by lowering the pH from 8 to ˜7, leading to the hypothesis that pore formation involves the protonation of histidine residues (which have a pKa of ˜6). However, more recent studies have shown that when the prepore is first bound to the isolated VWA domain of the receptor, the pH required for pore formation is lowered to ˜5–6.6,18 In addition, the pH threshold necessary to induce pore formation is dependent on the type of receptor VWA domain bound to the prepore, which also exhibit differences in binding affinity for PA. For instance, the dissociation constant for the VWA domain of ATR/TEM8 is ˜130 nM and the pH required for pore formation is ˜6, whereas the dissociation constant for VWA domain of CMG2 is ˜0.15 nM and the pH is lowered to ˜5.18,19 Also, mutagenesis of residues in CMG2 that are at the PA-receptor interface raise the pH required for pore formation from ˜5 to ˜6 and more closely approach that of ATR/TEM8.20
Although the receptor lowers the pH threshold for pore formation, experiments based upon immunoprecipitation of PA-CMG2 or PA-ATR/TEM8 complexes as a function of pH have shown that the receptor VWA domain dissociates from the prepore at the same acidic pH which induces pore formation.18 Whether dissociation is a prerequisite to pore formation, or happens as a consequence of pore formation, is not understood. The crystal structures of the PA21 and prepore-CMG27 complexes are virtually identical with identical interfaces (see Fig. Fig 1), indicating that little change in the interface occurs after the initial binding event of PA to CMG2. On the basis, we hypothesized that if protonation of receptor interface residues was the cause of receptor dissociation, that release of the VWA domain of CMG2 (referred hereafter as just CMG2) should occur when low pH binding is monitored between either monomeric PA or prepore. In this study, we present the use of 1-dimensional (1D) 13C-edited 1H NMR to monitor binding of a carbon-13 labeled CMG2 to PA and prepore as a function of pH. Our results are consistent with a model whereby the large structural change that occurs in the prepore to pore transition at low pH induces dissociation of the receptor.
Figure 1.
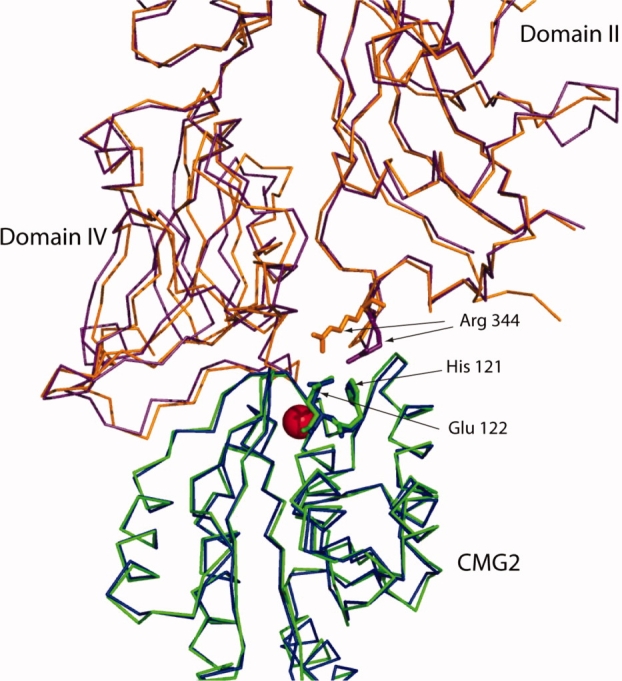
Overlay of structures of PA(purple) bound to CMG2 (green) and of prepore (orange) bound to CMG2 (blue) (pdb 1T6B and 1TZN, respectively). The gray region comprises the β3-β4 loop and the β2-β3 strands from Domain 2. The red sphere is manganese. Overlay was carried out using combinatorial extension.22 The figure was generated using Pymol v.99.23
Results
To monitor binding of CMG2 to either PA (PA83, the full-length monomeric protein) or the prepore, we used a method termed strongest methyl resonance of carbon-13 (SMRC).24 This technique utilizes the first dimension of a 1H-13C-heteronuclear single quantum coherence experiment (HSQC), focusing on the 1H-methyl resonance (˜0.8 ppm) of a 13C-labeled protein (CMG2), in the presence and absence of an unlabeled protein (PA or prepore). SMRC is highly sensitive, allowing nanomolar to micromolar concentrations to be used in binding assays. A protein binding event causes line broadening and a subsequent decrease in peak height, due to an increase in molecular mass.24–26
As seen in Figure Fig. 2, a significant decrease in SMRC intensity of 13C-CMG2 (4 μM) is observed at pH 8 upon addition of PA (4 μM), indicating formation of the 13C-CMG2-PA complex. A similar decrease in SMRC intensity is observed upon binding of 13C-CMG2 (6.25 μM) to the prepore (3.1 μM PA63) at pH 8. In the latter case, we used a twofold higher stoichiometry of 13C-CMG2 to prepore. This was done in order to be more quantitative in terms of degree of binding, because binding of 13C-CMG2 (˜20 kDa) to prepore (444 kDa) results in a complete loss of SMRC intensity. At pH 5, however, although we observed very little change in the intensity of the resonance of 13C-CMG2 bound to PA, a significant increase in the SMRC peak intensity is observed for the prepore complex, indicating that a large proportion of the receptor present in solution is no longer associated with the prepore. This is the first direct evidence that the receptor dissociates from the prepore at low pH and is consistent with a model in which the prepore-to-pore structural transition induces dissociation.
Figure 2.
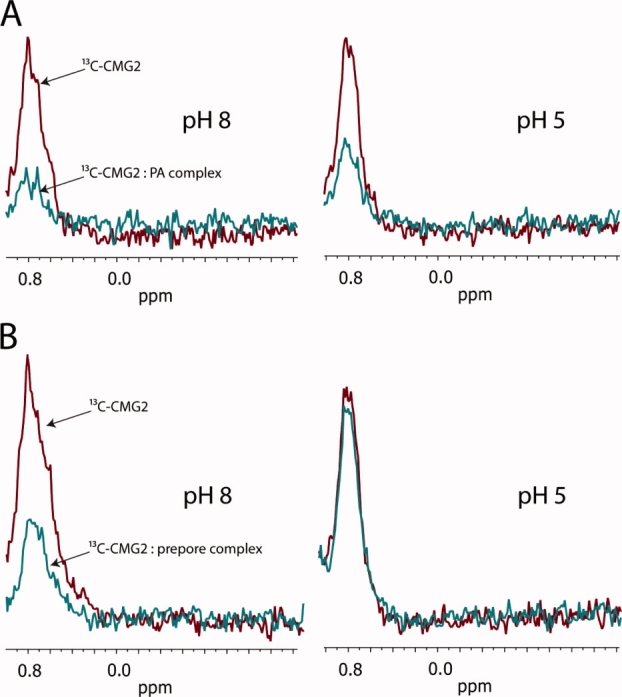
(A) SMRC of 13C-labeled CMG2 (4 μM), either in the absence (red) or presence (blue) of PA (4 μM) at pH 8.0 (left spectra) or pH 5 (right spectra). (B) SMRC of 13C-labeled CMG2 (6.25 μM), either in the absence (red) or presence (blue) of prepore (3.1 μM PA63) at pH 8.0 (left panel) or pH 5 (right panel). Spectra represent 512 transients 20°C.
Role of CMG2 His-121 in pore formation
An early hypothesis for low pH mediated release of CMG2, based on the crystal structures of PA-CMG2 and prepore-CMG2 complexes, focused on His-121 in CMG2 as a likely candidate for the pH sensor that governs pore formation.6,21 His-121 is within 5 Å of Arg-344 in PA, a residue adjacent to the Domain 2 β2-β3 strands that form the transmembrane pore (see Fig. Fig. 1). Thus, it was thought that protonation of His-121 in CMG2 at low pH results in charge repulsion of Arg-344, triggering the release of the β3-β4 loop from the CMG2 surface and allowing insertion of the β2-β3 strands into the membrane. Experimentally, mutagenesis of His-121 (to Ala, Asn and Glu) only slightly shifts the pH required for pore formation to higher values (0.2–0.6 pH units), and a His-121 to Gln mutant behaved in a manner similar to WT CMG2 in cytotoxicity assays.20,27
We also investigated the role of His-121 by labeling this residue with 2-fluorohistidine (2-FHis), which not only would allow us to probe the role of this residue in pH-dependent pore formation, but also allow us to probe pH-dependent interactions with PA or prepore by 19F NMR.28,29 2-FHis is isosteric for histidine and is perhaps the most conservative change one could make for this residue. However, 2-FHis possesses a side chain pKa of ˜1, and thus prevents protonation at low pH.28 The fluorine nucleus is small and in general structurally nonperturbing, but is highly sensitive to local environmental changes and should allow the detection of CMG2 binding to PA or the prepore via 19F NMR.30,31
We labeled CMG2 with 2-FHis, and the degree of labeling was confirmed to be >90% by mass spectrometry, as had been observed previously when the periplasmic chaperone PapD was labeled with this residue.28 The 2-FHis-CMG2 was also shown to form a 1 : 1 complex with PA using a gel-shift assay, and the stability to pH was similar to that of WT CMG2 (see Fig. Fig. 3). 19F NMR experiments were carried out in a manner similar to that used to monitor binding by the SMRC technique. As seen if Figure Figure. 4(A), 19F NMR experiments show that PA (40 μM) bound to 2-FHis-CMG2 (40 μM) at pH 8, as indicated by a large chemical shift change from δ = −35 ppm (unbound) to δ = −30.9 ppm (bound). However, when the pH was lowered to 5, a visible precipitate was observed in the NMR tube, preventing observation of the spectral changes at this pH.
Figure 3.
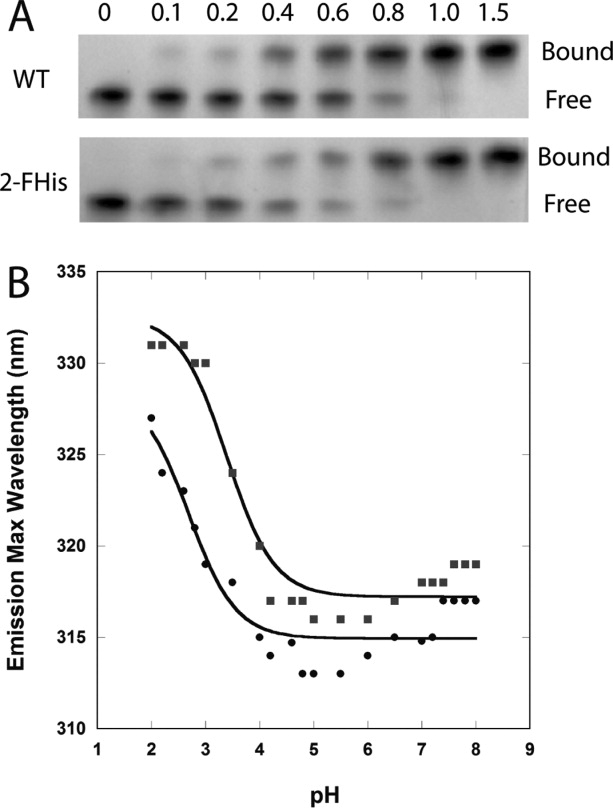
(A) Binding assay of WT PA with varying molar ratios of WT-CMG2 (top) and 2-FHis-CMG2 (bottom). Free refers to free PA and bound refers to the PA—CMG2 complex. (B) WT CMG2 (▪) and 2-FHis-CMG2 (•) pH dependence on fluorescence (Ex. 280 nm) was monitored by following the peak maximum wavelength. Data were acquired on a Cary Eclipse fluorescence instrument, and samples maintained at 20°C with a Peltier cooling system. Protein concentration was 0.8 μM, and the buffer consisted of 10 mM each of BisTris/Hepes/cacodylic acid/citric acid. The data were fit according to the Henderson-Hasselbalch equation assuming a two-state protonation equilbrium: Fl(obs) = (Fl(N) + Fl(I) 10pH-pKapp)/(1 + 10pH-pKap) where pKapp represents an apparent pKa encompassing all classes of titratable sites. The fits (solid lines) gave a pKapp of 3.4 ± 0.1 for the WT protein and 2.7 ± 0.2 for the 2-FHis labeled protein.
Figure 4.
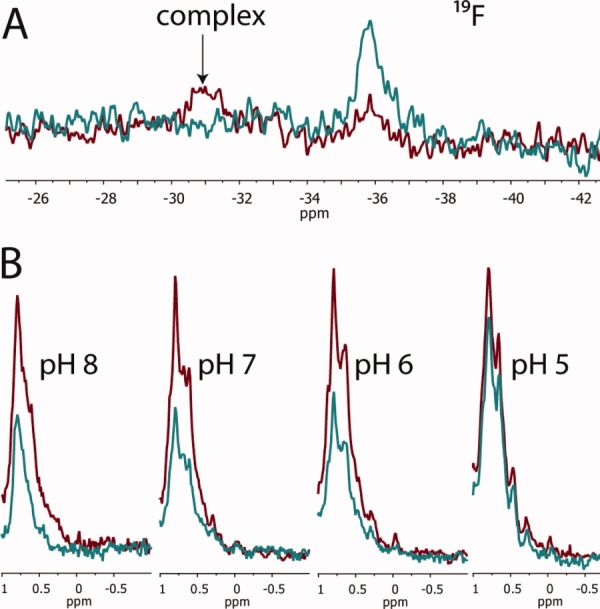
(A) Binding 2-FHis-CMG2 (40 μM) in the absence (blue) and presence (red) of PA (40 μM) monitored by 19F NMR. Spectra were acquired at 20°C, pH 8.0 and represent 4096 transients. (B) SMRC of 13C-2-FHis-CMG2 (12.5 μM), either in the absence (red) or presence (blue) of prepore (6.25 μM PA63), as a function of pH. Spectra represent 512 transients acquired at 20°C.
Given that dissociation of CMG2 from PA could not be detected using 19F NMR, CMG2 was colabeled with carbon-13 and 2-FHis, and receptor binding to the prepore was monitored as a function of pH using the SMRC technique. As shown in Figure Figure. 4(B), binding was observed for 13C-2-FHis-CMG2 (12.5 μM) to prepore (6.25 μM PA63) from pH 8 to 6, but at pH 5 there is a ˜30% increase in SMRC intensity, indicative of receptor dissociation. Our results are consistent with studies showing that protonation of His-121 is not a mechanism for receptor dissociation and provide further support for a model in which the structural transition from a prepore to a pore induces receptor dissociation.
Discussion
In this study, we provide direct evidence using NMR for low pH induced dissociation of CMG2 from the prepore form of PA. Because dissociation is not observed at low pH when monitoring association of CMG2 to full-length PA, this suggests that the structural change in the prepore to pore conversion, rather than protonation of residues at the receptor-PA or prepore interface, is the cause of receptor dissociation. Indeed, recent cryo-EM studies of the pore bound to GroEL, induced in 1M urea, indicate a large-scale global conformational change that is not just specific to Domain 2, which contains the transmembrane segment but Domain 4 as well.12 In fact, Domain 4 is not observed in the cryo-EM images. Therefore, receptor-prepore interactions may be lost through the global unfolding of the prepore to a pore, but rather than being initiated by protonation of residues in the receptor, is likely initiated by protonation of residues within the prepore.
Recent studies from our laboratory have shown that labeling PA with 2-FHis prevents unfolding from pH 8 to 5, but does not inhibit the prepore to pore conversion at pH 5.29 However, if the 2-FHis-labeled prepore is first bound to CMG2, we observe that both low pH induced receptor release and pore formation is blocked (manuscript in preparation). This supports the model proposed here, whereby protonation of histidines residues within the prepore (bound to CMG2) causes a structural transition from a prepore to a pore, and either in a concomitant or subsequent timeframe, dissociation of CMG2. By using the SMRC method, it should be possible to probe the timing of release of the receptor in relation to pore formation, thereby possibly determining which residues in the PA initiate pore formation.
Materials and Methods
Protein production and labeling
Production and purification of the B. anthracis PA was carried out as described previously.29 A human cDNA corresponding to full-length CMG2 from Origene Technologies (Rockville, MD) was used to clone the VWA domain of CMG2 (residues 38–218) into the expression plasmid pGEX-4T1, using BamH1-Not1 restriction sites as described previously.19 2-FHis labeling: cultures of the E.coli strain UTH780, harboring pGEX-4T1-CMG238-218, in minimal media31 were grown to an A600 of 2.0. The cells were harvested, washed three times with 0.9% NaCl, and resuspended in minimal media supplemented with 0.2 mM 2-FHis. The cells were incubated for 10 min, and then induced for 3 h at 37°C by addition of IPTG to 1 mM. 13C-labeling: cultures of the E. coli strain BL21, harboring pGEX-4T1-CMG238-218, were grown in M9 minimal media supplemented with 0.2% U-13C-glucose at 37°C. Cultures were grown to an A600 of 2.0 and induced with 1 mM IPTG, grown for an additional 3 h at 37°C, and then harvested. For the combined (13C and 2-FHis) labeling, we used strain UTH780 and grew cultures in M9 minimal media supplemented with 0.2% U-13C-glucose and 0.2 mM histidine. Cultures were grown to an A600 of 2, harvested, washed twice with 0.9% NaCl, and resuspended in fresh M9 media containing 0.2% U-13C-glucose, supplemented with 0.2 mM 2-FHis. Cultures were grown for 10 min, induced with 1.0 mM IPTG for 3 h at 37°C, and then harvested. Cells were frozen at −20°C prior to purification.
Purification of CMG2
Frozen cells (13C, 2-FHis labeled) were resuspended in phosphate buffered saline and lysed using a sonicator (Branson). After centrifugation (15,000g, 30 min), the supernatant was applied to a 5 × 5 mL glutathione–sepharose HP column (GE HealthCare; equilibrated in phosphate-buffered saline, pH 7.4). Elution of the protein was achieved using thrombin (80 Units/mL; GE Healthcare), which cleaves between CMG2 and glutathione, and passage through a benzamidine column (5 × 5 mL) afforded pure CMG2.
SMRC experiments
All experiments were performed at 20°C. 13C-labeled CMG2 or 13C-labeled, 2-FHis-CMG2 in PBS pH 7.4 was incubated with 20 mM Tris d11, pH 8.5, 0.4M NaCl alone or with either PA or PA63 (in the prepore) in 0.4M NaCl, 20 mM Tris-d11, pH 8.5 in a volume of 100 μL. The binding reaction was incubated overnight at room temperature, to which 400 μL of a universal pH buffer (200 mM each of cacodylic acid-d7, Bis-Tris-d19 and Tris-d11, 1.0% β-d-octyl-glucoside-d24, and 12.5% D2O) at pH 8, 7, 6, or 5 was added. 1D 1H-13C-HSQC spectra were acquired on a Varian INOVA 700 MHz spectrometer and represent 512 transients.
19F NMR spectroscopy
Spectra were acquired at 20°C on a Varian INOVA 500 MHz spectrometer using a dedicated Varian 19F-CryoQ probe operating at 20 K. Spectra represent 4096 transients with a recycle delay of 4 s and were referenced to an external standard of 5 mM trifluoroacetic acid. 2-FHis-CMG2 (47.4 μL) in PBS was incubated with 52.6 μL of either 20 mM Tris d11 pH 8.0, 0.4M NaCl, or with PA (same buffer) to give final concentrations of 200 μM 2-FHis-CMG2 and PA. The binding reaction was incubated overnight, to which 400 μL of a universal pH buffer (200 mM each of cacodylic acid-d7, Bis-Tris-d19 and Tris-d11, 1.0% β-d-octyl-glucoside-d24, and 12.5% D2O) at pH 8 or 5 was added. Final concentration of PA and 2-FHis-CMG2 was 40 μM. No spectra were recorded at pH 5 due to aggregation.
Stability of CMG2, 2-FHis-CMG2 to pH
Data were acquired on a Cary Eclipse spectrofluorimeter, using an excitation wavelength of 280 nm, and samples maintained at 20°C with a Peltier cooling system. Protein concentration was 0.8 μM, and the buffer consisted of 10 mM each of BisTris/Hepes(sodium salt)/cacodylic acid(sodium salt)/citric acid. The data were fit according to the Henderson-Hasselbalch equation assuming a two-state protonation equilibrium: Fl(obs) = (Fl(N) + Fl(I) 10pH-pKapp)/(1 + 10pH-pKap) where pKapp represents an apparent pKa encompassing all classes of titratable sites. The fits gave a pKapp of 3.4 ± 0.1 for the WT protein and 2.7 ± 0.2 for the 2-FHis labeled protein. We note that very little change occurs in the peak max of either protein down to ˜pH 4.
Mass spectrometry
Mass for CMG2 (residues 38–218, plus an N-terminal Gly from the thrombin cleavage) is 19,876 (expected 19875.8) and 2-FHis-CMG2 is 19,894 (expected 19893.8). For LC/MS measurements of whole protein mass, an aliquot of protein (˜20 picomoles) in 5% acetic acid was applied to a reversed phase column (zorbax 300SB-C3 2.1 × 150 mm, Agilent), the column was washed for 25 min (0.2 mL/minute 5% acetic acid in water), and then eluted with a steep gradient (3.6%/minute to 95%). Starting solvent was 5% acetic acid in water and the elution solvent was neat acetonitrile. The HPLC system was an HP1100 (Agilent) stack with degasser and column heater (the column was heated at 40°C). The protein eluted into an HP MSD single quad mass spectrometer (Agilent), which was set up to scan between 500 and 1700 M/Z beginning after the first 25 min. LC/MS data were analyzed using the Chemstation software package. A prominent peak appearing only in the sample run was summed together and subjected to the transform program present in the software package.
Native PAGE binding assay
WT PA (1.6 μL of 31 μM) was placed into eppendorf tubes and either WT CMG2 (15 μM) or 2-FHis-CMG2 (52 μM) was added in increments to give final concentrations of 2.5 μM PA and 0, 0.25, 0.5, 1.0, 1.5, 2.0, 2.5, and 3.75 μM CMG2 proteins. The volumes were brought up to 20 μL by adding appropriate amounts of 10 mM Tris-HCl pH 8. Glycerol (80%) was then added to each sample to a final concentration of 5% and incubated at room temperature for 1 h. The protein samples were then loaded (20 μL) onto a 4–20% native PAGE gel and run at constant 40 V for 17 h at 15°C.
Acknowledgments
The authors thank Dr. Carl Frieden (Washington University in St. Louis) for use of the fluorine cryoprobe and comments on the manuscript, and Dr. Gregory DeKoster for his assistance with the SMRC experiments. They also thank Dr. Jose Rizo Rey (UT-Southwestern) and Dr. Jacob Schaefer (Washington University) for helpful comments on the manuscript. J.F.E. was supported by a Research Sites for Educators postdoctoral fellowship. K.L.K., J.H., and D.E.A were funded by the intramural research program of NIDDK.
References
- 1.Collier RJ, Young JA. Anthrax toxin. Annu Rev Cell Dev Biol. 2003;19:45–70. doi: 10.1146/annurev.cellbio.19.111301.140655. [DOI] [PubMed] [Google Scholar]
- 2.Duesbery NS, et al. Proteolytic inactivation of MAP-kinase-kinase by anthrax lethal factor. Science. 1998;280:734–737. doi: 10.1126/science.280.5364.734. [DOI] [PubMed] [Google Scholar]
- 3.Leppla SH. Anthrax toxin edema factor: a bacterial adenylate cyclase that increases cyclic AMP concentrations of eukaryotic cells. Proc Natl Acad Sci USA. 1982;79:3162–3166. doi: 10.1073/pnas.79.10.3162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bradley KA, Mogridge J, Mourez M, Collier RJ, Young JA. Identification of the cellular receptor for anthrax toxin. Nature. 2001;414:225–229. doi: 10.1038/n35101999. [DOI] [PubMed] [Google Scholar]
- 5.Scobie HM, Rainey GJ, Bradley KA, Young JA. Human capillary morphogenesis protein 2 functions as an anthrax toxin receptor. Proc Natl Acad Sci USA. 2003;100:5170–5174. doi: 10.1073/pnas.0431098100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lacy DB, Wigelsworth DJ, Melnyk RA, Harrison SC, Collier RJ. Structure of heptameric protective antigen bound to an anthrax toxin receptor: a role for receptor in pH-dependent pore formation. Proc Natl Acad Sci USA. 2004;101:13147–13151. doi: 10.1073/pnas.0405405101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lacy DB, Lin HC, Melnyk RA, Schueler-Furman O, Reither L, Cunningham K, Baker D, Collier RJ. A model of anthrax toxin lethal factor bound to protective antigen. Proc Natl Acad Sci USA. 2005;102:16409–16414. doi: 10.1073/pnas.0508259102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Melnyk RA, Hewitt KM, Lacy DB, Lin HC, Gessner CR, Li S, Woods VL, Jr, Collier RJ. Structural determinants for the binding of anthrax lethal factor to oligomeric protective antigen. J Biol Chem. 2006;281:1630–1635. doi: 10.1074/jbc.M511164200. [DOI] [PubMed] [Google Scholar]
- 9.Abrami L, Liu S, Cosson P, Leppla SH, van der Goot FG. Anthrax toxin triggers endocytosis of its receptor via a lipid raft-mediated clathrin-dependent process. J Cell Biol. 2003;160:321–328. doi: 10.1083/jcb.200211018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Benson EL, Huynh PD, Finkelstein A, Collier RJ. Identification of residues lining the anthrax protective antigen channel. Biochemistry. 1998;37:3941–3948. doi: 10.1021/bi972657b. [DOI] [PubMed] [Google Scholar]
- 11.Nassi S, Collier RJ, Finkelstein A. PA63 channel of anthrax toxan extended beta-barrel. Biochemistry. 2002;41:1445–1450. doi: 10.1021/bi0119518. [DOI] [PubMed] [Google Scholar]
- 12.Katayama H, Janowiak BE, Brzozowski M, Juryck J, Falke S, Gogol EP, Collier RJ, Fisher MT. GroEL as a molecular scaffold for structural analysis of the anthrax toxin pore. Nat Struct Mol Biol. 2008;15:754–760. doi: 10.1038/nsmb.1442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Krantz BA, Trivedi AD, Cunningham K, Christensen KA, Collier RJ. Acid-induced unfolding of the amino-terminal domains of the lethal and edema factors of anthrax toxin. J Mol Biol. 2004;344:739–756. doi: 10.1016/j.jmb.2004.09.067. [DOI] [PubMed] [Google Scholar]
- 14.Krantz BA, Melnyk RA, Zhang S, Juris SJ, Lacy DB, Wu Z, Finkelstein A, Collier RJ. A phenylalanine clamp catalyzes protein translocation through the anthrax toxin pore. Science. 2005;309:777–781. doi: 10.1126/science.1113380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Krantz BA, Finkelstein A, Collier RJ. Protein translocation through the anthrax toxin transmembrane pore is driven by a proton gradient. J Mol Biol. 2006;355:968–979. doi: 10.1016/j.jmb.2005.11.030. [DOI] [PubMed] [Google Scholar]
- 16.Blaustein RO, Koehler TM, Collier RJ, Finkelstein A. Anthrax toxchannel-forming activity of protective antigen in planar phospholipid bilayers. Proc Natl Acad Sci USA. 1989;86:2209–2213. doi: 10.1073/pnas.86.7.2209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Miller CJ, Elliott JL, Collier RJ. Anthrax protective antigen: prepore-to-pore conversion. Biochemistry. 1999;38:10432–10441. doi: 10.1021/bi990792d. [DOI] [PubMed] [Google Scholar]
- 18.Rainey GJ, Wigelsworth DJ, Ryan PL, Scobie HM, Collier RJ, Young JA. Receptor-specific requirements for anthrax toxin delivery into cells. Proc Natl Acad Sci USA. 2005;102:13278–13283. doi: 10.1073/pnas.0505865102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wigelsworth DJ, Krantz BA, Christensen KA, Lacy DB, Juris SJ, Collier RJ. Binding stoichiometry and kinetics of the interaction of a human anthrax toxin receptor, CMG2, with protective antigen. J Biol Chem. 2004;279:23349–23356. doi: 10.1074/jbc.M401292200. [DOI] [PubMed] [Google Scholar]
- 20.Scobie HM, Marlett JM, Rainey GJ, Lacy DB, Collier RJ, Young JA. Anthrax toxin receptor 2 determinants that dictate the pH threshold of toxin pore formation. PLoS ONE. 2007;2:e329. doi: 10.1371/journal.pone.0000329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Santelli E, Bankston LA, Leppla SH, Liddington RC. Crystal structure of a complex between anthrax toxin and its host cell receptor. Nature. 2004;430:905–908. doi: 10.1038/nature02763. [DOI] [PubMed] [Google Scholar]
- 22.Shindyalov IN, Bourne PE. Protein structure alignment by incremental combinatorial extension (CE) of the optimal path. Protein Eng. 1998;11:739–747. doi: 10.1093/protein/11.9.739. [DOI] [PubMed] [Google Scholar]
- 23.DeLano WL. Pymol v. 099. 2006. Available at http://pymol.sourceforge.net.
- 24.Arac D, Murphy T, Rizo J. Facile detection of protein-protein interactions by one-dimensional NMR spectroscopy. Biochemistry. 2003;42:2774–2780. doi: 10.1021/bi0272050. [DOI] [PubMed] [Google Scholar]
- 25.Dulubova I, Khvotchev M, Liu S, Huryeva I, Sudhof TC, Rizo J. Munc18-1 binds directly to the neuronal SNARE complex. Proc Natl Acad Sci USA. 2007;104:2697–2702. doi: 10.1073/pnas.0611318104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Guan R, Dai H, Rizo J. Binding of the Munc13-1 MUN domain to membrane-anchored SNARE complexes. Biochemistry. 2008;47:1474–1481. doi: 10.1021/bi702345m. [DOI] [PubMed] [Google Scholar]
- 27.Liu S, Leung HJ, Leppla SH. Characterization of the interaction between anthrax toxin and its cellular receptors. Cell Microbiol. 2007;9:977–987. doi: 10.1111/j.1462-5822.2006.00845.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Eichler JF, Cramer JC, Kirk KL, Bann JG. Biosynthetic incorporation of fluorohistidine into proteins in E. coli: a new probe of macromolecular structure. ChemBioChem. 2005;6:2170–2173. doi: 10.1002/cbic.200500249. [DOI] [PubMed] [Google Scholar]
- 29.Wimalasena DS, Cramer JC, Janowiak BE, Juris SJ, Melnyk RA, Anderson DE, Kirk KL, Collier RJ, Bann JG. Effect of 2-fluorohistidine labeling of the anthrax protective antigen on stability, pore formation, and translocation. Biochemistry. 2007;46:14928–14936. doi: 10.1021/bi701763z. [DOI] [PubMed] [Google Scholar]
- 30.Bann JG, Frieden C. Folding and domain-domain interactions of the chaperone PapD measured by 19 F NMR. Biochemistry. 2004;43:13775–13786. doi: 10.1021/bi048614u. [DOI] [PubMed] [Google Scholar]
- 31.Frieden C, Hoeltzli SD, Bann JG. The preparation of 19F-labeled proteins for NMR studies. Methods Enzymol. 2004;380:400–415. doi: 10.1016/S0076-6879(04)80018-1. [DOI] [PubMed] [Google Scholar]