

Abstract
Conformational changes play important roles in the regulation of many enzymatic reactions. Specific motions of side chains, secondary structures, or entire protein domains facilitate the precise control of substrate selection, binding, and catalysis. Likewise, the engineering of allostery into proteins is envisioned to enable unprecedented control of chemical reactions and molecular assembly processes. We here study the structural effects of engineered ionizable residues in the core of the glutathione-S-transferase to convert this protein into a pH-dependent allosteric protein. The underlying rational of these substitutions is that in the neutral state, an uncharged residue is compatible with the hydrophobic environment. In the charged state, however, the residue will invoke unfavorable interactions, which are likely to induce conformational changes that will affect the function of the enzyme. To test this hypothesis, we have engineered a single aspartate, cysteine, or histidine residue at a distance from the active site into the protein. All of the mutations exhibit a dramatic effect on the protein's affinity to bind glutathione. Whereas the aspartate or histidine mutations result in permanently nonbinding or binding versions of the protein, respectively, mutant GST50C exhibits distinct pH-dependent GSH-binding affinity. The crystal structures of the mutant protein GST50C under ionizing and nonionizing conditions reveal the recruitment of water molecules into the hydrophobic core to produce conformational changes that influence the protein's active site. The methodology described here to create and characterize engineered allosteric proteins through affinity chromatography may lead to a general approach to engineer effector-specific allostery into a protein structure.
Keywords: conformational switches, pH sensor, allostery, dynamics, protein design
Introduction
One of the most exceptional characteristics of life is the high degree of control exercised by most of its biochemical processes. Conformational changes and protein dynamics are of crucial importance to many biochemical functions and reactions (see, for example, Refs. 1–4). The intimate relationship between structure and dynamics enables communication and control among these different processes. Such changes, however, usually involve complex shifts and motions of residues that have challenged our ability to predict or design allostery into proteins.5
Allosteric functions of many natural proteins are facilitated by large-scale as well as small-scale conformational changes. Hemagglutinin of the influenza virus, for example, contains a spring-loaded conformational switch mechanism that can be triggered specifically upon acidification to facilitate the fusion of viral membrane and host cell membranes. Helices within the core of the protein reassemble into an entirely new structure.6,7 This irreversible large-scale refolding process of the protein involves reorientations of existing helices as well as coil-to-helix transitions of specific sequences. Small conformational changes, on the other hand, only involve minor reorientations of side chains, loop structures, or displacements of secondary structures upon binding of an effector molecule. Such changes can, however, lead to substantial hinge-bending motions between domains of a protein as in Hsp70 and adenylate kinase.8,9
Many of the responsive functions of natural proteins display a high degree of complexity and specificity that are also of prime interest for the engineering of nanoscale sensors, assemblies, and catalytic devices (see, for example, Refs. 10–12). Even though the first successful attempts have been made in engineering large-scale conformational switch peptides that transition from coil to helix or vice versa,13,14 these peptides may exhibit likelihood to oligomerize or have limited solubility. Small-scale conformational changes, on the other hand, may be more likely to sustain solubility while facilitating precisely controlled allosteric functions because of the essentially unchanged surface properties of the molecule.
In this study we tested the possibility of engineering pH-responsive, small-scale allosteric properties into a protein. Specifically, we placed chargeable amino acids into the core of a protein structure with the goal of controlling its function via pH. The ionizable residues are designed to operate as pH-sensors that, when charged, will invoke unfavorable interactions in the protein thereby compromising its function. The motivation for the design is, in part, based on a previous observation of a pH-induced structural change of the ATP synthase. Subunit c of this integral membrane protein carries a single aspartate residue in the center of the hydrophobic region.15 A pH-induced charging of this residue facilitates a re-orientation of helices within the subunit to transport protons from the cytoplasmic site across the membrane. Inspired by these observations, we hypothesized that such a hydrophobically buried charge may also induce conformational changes when engineered into other proteins. As the target of our study, we selected the protein glutathione-S-transferase (GST). GST proteins catalyze the conjugation of reduced glutathione (GSH) to a broad spectrum of compounds including many exogenous toxic substances. GSTs from different organisms can be divided into at least 13 different classes that share a similar fold.16 Whereas the native enzymes have not been shown to perform cooperative catalysis, reports of engineered mutant proteins suggest that both active sites of the homodimers may communicate cooperatively.17–19 The GST of Schistosoma japonicum consists of 214 amino acids and displays a strong binding affinity to its natural substrate glutathione as well as its derivatives. Glutathione-affinity chromatography thus provides an advantageous experimental system for the engineering and characterization of newly engineered allosteric response mechanisms.
Design strategy
We have focused our study on the three amino acids aspartate, cysteine, and histidine. All of these residues are short with respect to other similarly charged residues and have therefore relatively limited degrees of conformational freedom. Aspartate is isosteric to leucine and can therefore easily be substituted into its hydrophobic core. Histidine, on the other hand, bears a relatively bulky side chain that could possibly replace phenylalanine, tyrosine, or tryptophan residues. Both of these charged residues exhibit very unique protonation kinetics of their side chains. Whereas aspartic acids become negatively charged at a pH greater than 4.0, histidine will become positively charged at a pH below 6.04 (see Supp. Info.). Additionally, we also tried cysteine with regard to its protonation kinetics. The thiol of this residue will become charged at a pH of 8.3 or greater. As a charged group usually induces destabilizing effects on a protein structure, 20–22 it may also compromise its function. It is therefore anticipated that the charged state of either of these residues can also be used to control the function of the protein selectively via the pH of the surrounding solution.
Within the scope of this study, we have selected position 50 of GST in order to test the pH effects of these residues in a hydrophobic environment (Fig. 1). The native enzyme carries a leucine residue in this position, which is in close proximity to the other hydrophobic residues Trp41, Leu48, Pro53, Leu55, Tyr57, Ile59 and the methyl group of Thr66. As this position is located in the smaller domain of the enzyme and at a distance from the active site, it serves as an advantageous target site for the study of charge-induced conformational changes. Because of the protein's strong affinity to GSH, we employ affinity chromatography to characterize charge-induced allosteric effects on GSH binding. In the uncharged state of the mutant residue, the protein is expected to bind glutathione sepharose. Conversely, in the charged state, the protein is expected to elute from the column.
Figure 1.
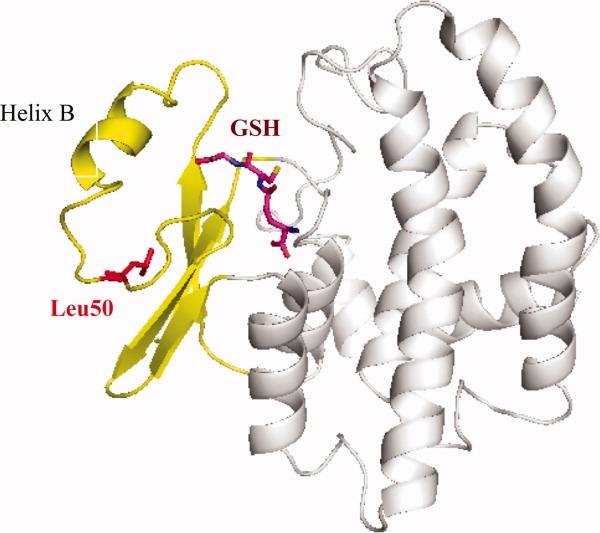
Ribbon representation of the overall structure of wild-type GST in the presence of glutathione (GSH, shown as stick model in purple). The smaller amino terminal domain of the protein is colored in yellow with Leu50 highlighted in red. This residue is pointing toward the hydrophobic core of this smaller domain of the protein. It is located at ∼10 Å distance from the substrate binding site. Any pH-sensitive effects of the Asp50, Cys50, or the His50 mutants, therefore, must be transmitted through conformational changes to the binding site. This figure as well as Figures 4 and 5 were prepared with the program PyMol (DeLano Scientific LLC, http://pymol.sourceforge.net).
Results
Solution behavior and thermal stabilities of the mutant proteins
The cloning and purification of GST50C, GST50H, and GST50D has resulted in soluble protein as determined by SDS-PAGE of the Ni2+ affinity-purified protein. Size-exclusion chromatography confirmed the monodisperse and dimeric state of all mutant proteins at all pH values relevant to this study (data not shown). Systematic circular dichroism thermal melting studies revealed that the mutant proteins are destabilized with respect to the wild-type protein. The native protein exhibits a melting temperature of approximately Tm = 57°C, whereas all mutant proteins are significantly destabilized (Fig. 2 and Supp. Info. Table 2). Mutant GST50H is moderately destabilized at 6°C, whereas GST50D is destabilized the most at ∼15°C. Surprisingly, thermal denaturations of GST50C revealed a melting temperature of Tm = 46°C under ionizing (pH 9.5) as well as nonionizing conditions (pH 6.4) (Fig. 2). The significantly elongated shape of the denaturation curve at high pH, however, suggests significantly altered cooperative unfolding of the mutant.
Figure 2.
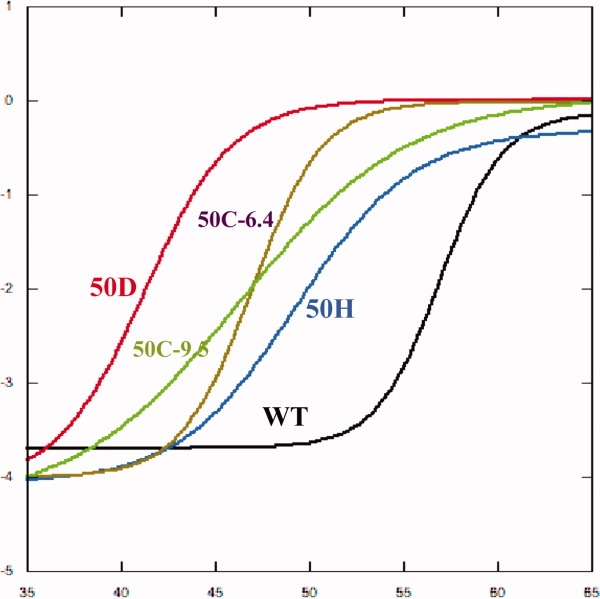
Thermal melts of the mutant proteins by circular dichroism spectroscopy. The concentrations of all proteins were set to equal concentration prior to the melts to a common optical density (OD280 nm = 0.24). All thermal melts were carried out between 25 and 80°C. (The unique thermal transitions between 25 and 65°C are shown here.) As can be seen in the transition temperatures, all mutant proteins are significantly destabilized compared with the wild type by at least 6°C or more. GST50D is the most destabilized protein by ∼15°C.
GSH-affinity chromatography
To evaluate the pH-dependent binding behavior of the mutant proteins to glutathione, the mutants were subjected to GSH-affinity chromatography. For this purpose, the proteins were allowed to bind to the GSH matrix under high- or low-pH starting conditions prior to the application of the pH elution gradient. The mutant proteins were then systematically subjected to a low-to-high pH gradient (pH 5.5–9.5) as well as to a high-to-low elution gradient (or pH 9.5–5.5).
As expected, the wild-type protein bound with high affinity to the GSH matrix at all pH values below 8.5. At a pH higher than 9, however, the protein eluted as a broad peak with a pH maximum at pH 9.2 [Fig. 3(A)]. The wild-type protein, therefore, loses its affinity to GSH only at high pH. At all other pH values between 5.5 and 8.5, the protein remains bound to the GSH-resin.
Figure 3.

Elution profile of wild-type (A) and mutant GST50C (B) in response to pH change. The elution of the proteins was monitored by absorption at λ = 280 nm, and the pH was monitored at the immediate exit of the column. (A) pH-dependent elution profile of the wild-type protein from the GSH column. The dashed lines describe the actual pH as a result of the linearly applied buffer gradient from pH 6.0 to 9.5 (red) and from pH 9.5 to 6.0 (black). The wild type can only be loaded onto the column at low pH (red solid line). The immobilized protein elutes from the matrix at pH 9.3 (black solid line). (B) In contrast to the wild type, GST50C elutes at pH 8.3. Whereas the high-to-low pH-change did not result in any binding or elution of the protein, the low-to-high elution resulted in a sharp elution peak. The allosteric pH-responsive binding behavior of this mutant was completely reversible. Upon lowering of the pH of the eluted protein solution, the protein could directly be reapplied to the column for another round of pH-triggered elution. The elution of the proteins was monitored by absorption at λ = 280 nm. Analysis of the matrix after elution suggested that GST50C elution was as complete. Comparison of the binding and elution behaviors of GST50C is therefore quantitative and similar to those of the wild type.
The substitution of aspartate into the hydrophobic core had the most dramatic effect on the protein's affinity to bind GSH. At all pH values tested, GST50D has lost its affinity entirely. The predicted proton pKα for the Asp side chain is 3.9, suggesting that this amino acid remains charged under all conditions tested. The circular dichroism spectra indicate that the secondary structure content of this protein has remained the same compared with that of the wild type (Fig. S1 in Supp. Info.), suggesting that the mutation has not caused significant denaturations.
In contrast to the GST50D mutant, GST50H exhibits the opposite extreme binding behavior. This mutant appears to bind strongly to GSH at all pH values tested. The side chain pKα of an isolated histidine imidazole is 6.04, which would imply that this mutant should elute within the tested pH-range. Systematic applications of a pH gradient on the GSH-immobilized protein demonstrated though that this mutant failed to elute from the matrix within the applied pH range. Even attempts to elute the protein with the addition of 20 mM soluble GSH have failed as well, further demonstrating its altered binding affinity. Only high concentrations of soluble GSH of above 100 mM (see in Supp. Info. Fig. S2) triggered the elution of the protein from the GSH-matrix, confirming its high substrate specificity. As the protein remains completely soluble as a homodimer at all pH values tested, unspecific binding or precipitation to the column matrix due to denaturation can be ruled out. Therefore, mutants GST50H and GST50D exhibit two extreme binding affinities that resulted from the introductions of the residues. Even though these mutations are introduced at a distance from the active site, they produce conformational changes that significantly alter the binding affinity of the protein.
With respect to the other mutants, GST50C had a different pH-dependent binding behavior. At a pH below 8.0, this mutant bound stably to the GSH resin. At a pH of 8.3, however, the protein eluted sharply and completely off the column [Fig. 3(B)]. As the affinity of the eluted protein could be restored with the acidification of the eluted protein solution pH, its responsive binding behavior proved to be fully reversible. Additionally, the bound protein can also be eluted with the addition of soluble GSH, demonstrating its sustained and specific binding affinity to GSH.
Crystallography
To ascertain the structural consequences of the mutations with respect to pH, we have determined the crystal structures of the mutant proteins GST50H and GST50C. Data collection statistics are given in Table I. Both mutants were crystallized by microseeding with wild-type crystals of space group P43212. This crystal form is particularly interesting as it exposes the entire active site domain of the protein to nearby solvent channels. It was hypothesized that possible conformational changes of the active site in response to the mutations may be observed in this symmetry in particular.
Table I.
Crystallographic Data
| GST50H | GST50C75 | GST50C95 | |
|---|---|---|---|
| pH | 7.5 | 7.5 | 9.5 |
| Space group | P43212 | P43212 | P43212 |
| X-ray source | SSRL 11-1 | Cu | Cu |
| Wavelength | 0.979 | 1.5418 | 1.5418 |
| Cell dimensions (Å) | 93.500 | 91.180 | 92.680 |
| 93.500 | 91.180 | 92.680 | |
| 58.70 | 57.530 | 57.820 | |
| Molecules per asymmetric unit | 1 | 1 | 1 |
| Resolution (Å) | 19.8–2.5 | 19.5–1.8 | 20.0–2.3 |
| High-resolution bin | 2.5–2.7 | 1.8–1.95 | 2.3–3.0 |
| Rsym (high-resolution bin) | 6.1 (18.8) | 9.4 (39.1) | 6.1 (15.9) |
| Completeness (%) | 98.5 (96.1) | 99.84 (71.3) | 84 (76.5) |
| Number of reflection (test set) | 16,403 (1278) | 34,548 (2712) | 17,967 (868) |
| Rwork/Rtest | 23.9/31.1 | 22.7/25.7 | 22.1/29.1 |
| Δbond | 0.0078 | 0.00724 | 0.0073 |
| Δangle | 1.40 | 1.408 | 1.3297 |
| PDBid | 3D0Z | 3CRT | 3CRU |
Rsym gives the agreement between equivalent intensities. Values in parentheses indicate the R values for the high-resolution bin. Rwork and Rtest are the crystallographic residuals following refinement with CNS. Δbond and Δangle give the average discrepancy of bond lengths and bond angles from ideal values.
The most dramatic structural changes in response to the mutation were found in the histidine mutant. As can be seen in Figures 4 and 7, the overall B values of the structure have increased dramatically. The collection of several datasets from different crystals confirmed that the increased flexibility is an intrinsic property of this mutant protein. In particular, Helix B and the loop residues between residues 41 and 49 had greatly increased B values that are almost twofold higher than the remainder of the structure or the wild type (Figs. 4 and 7). In addition to the increased B values, the electron density in the vicinity of the mutation site becomes very weak and vanishes entirely between residues 47 and 49, suggesting that statistical or thermal disorder of this part of the structure had increased substantially. Comparison of the B values of the 50H mutant to that of wild type suggests that despite the overall increase of B values, the size and location of the individual mobile parts of the structure are similar (see Fig. 7). The core residues 16, 71, 98, and 156, for example, show similarly low B values compared with the wild-type structure, whereas the flexible loops and secondary structures of the wild type (PDBid: 1UA5) also remain more flexible in the mutant structure. Superpositioning of this structure onto GST50H between residues 80 and 200 suggests that Helix B moves as a rigid body by about 1 Å (Fig. 5). The tightly bound substrate molecule reveals relatively low B values that are similar to the protein's core values suggesting strong interactions with the protein. Even though we have attempted to obtain diffraction data from crystals soaked at different pH values, no significant resulting structural changes were observed (data not shown). In summary, the most dramatic structural changes of this mutant are the increased B values of Helix B and the strongly enhanced flexibility of the loop structure bearing the mutation.
Figure 4.
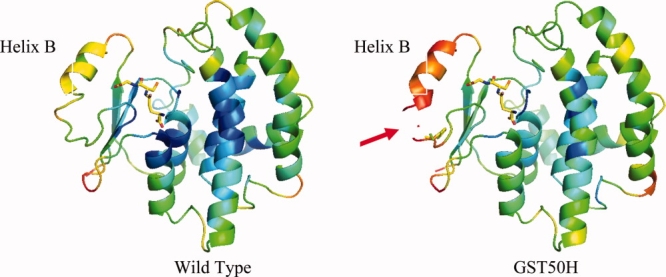
Ribbon diagram of the crystal structures of the wild-type protein and the GST50H mutant. The rainbow color-coding represents the B value distribution of the backbone atoms of the structures (blue = low B values, red = high B values). In the crystal structures, the highest conformational flexibility is observed in Helix B. The average B value for the main-chain atoms of this structural segment averages to about B = 60 Å2 (see also Fig. 7). With the introduction of a histidine at position 50, the mobility of this helix is dramatically increased by almost a factor of 2 (see also Fig. 7). Despite this structural flexibility, the affinity of this mutant to bind GSH has increased dramatically as well. The corresponding density of the substrate was clearly visible in the electron density maps and could be refined to full occupancy.
Figure 7.
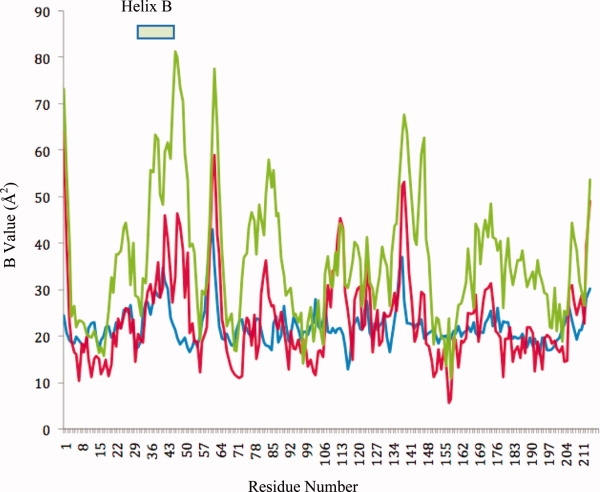
B value distribution of the wild-type (blue) (PDBid: 1UA5, isomorphous structure) and the mutant structures GST50C high (red) and GST50H (green). The B values of the Cα atoms with respect to the residues numbers are shown. In general, the B values for most parts of the structure vary between 10 and 50 Å2 in the different mutants. The flexibility between different loop structures correlates strongly. The most dramatic difference was observed in the Helix B region between the wild type, GST50C, and GST50H. In the structure of the latter, the backbone B values have increased to 60 Å2 or higher, with the highest values observed at above 80 Å2 for the C-terminal end of that helix. Consequently, no reliable electron density was observed between residues 46 and 49.
Figure 5.
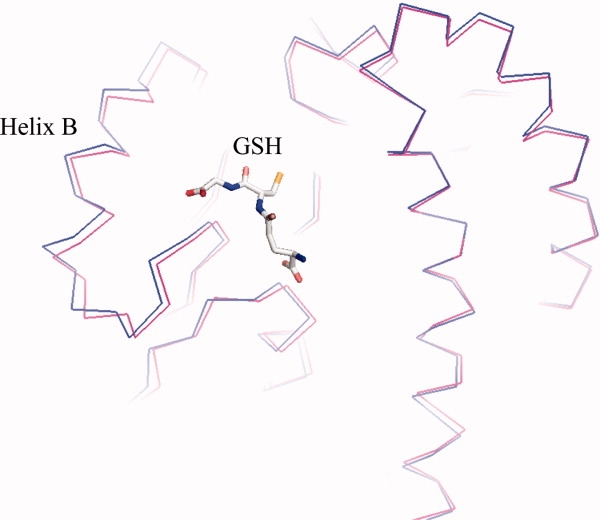
Structural superposition of the GST wild-type (red) and the GST50H (blue) mutant. Both structures are shown as a Cα—Cα backbone representation. The bound glutathione molecule is shown as a stick model in the active site of the proteins. The superpositioning was performed on residues 80–214. The structures show a significant discrepancy between Helices B. In comparison to the wild type, this helix of the mutant shifts by about 1 Å and rolls by about 1.5°.
The overall structure of GST50C at the pH of crystallization (pH 7.5) remains indistinguishable from that of the wild type, which is demonstrated by the low root-mean-square-deviation of 0.4 Å between the backbone atoms between both structures. The mutation site, however, can clearly be identified by the smaller side chain as well as by a distinct anomalous density caused by the scattering of the sulfur atom of this residue (data not shown). Because of the void-space created by this large-to-small substitution, a new water molecule was observed in proximity to the thiol (water molecule WAT 3 in the structure 3CRT).
To visualize the effects of high pH on the structure of this mutant, the crystals were soaked into mother liquor at pH 9 or higher. As described earlier, at this pH the protein has lost its affinity to bind to its substrate. The corresponding electron densities are shown in Figure 6(A). In the crystal structure, new and strong positive electron density was observed in the immediate vicinity of the cysteine, suggesting the recruitment of additional water molecules around this residue. In contrast to other solvent molecule densities, this density appears large and elongated and could possibly account for two or three water molecules [Fig. 6(A)]. In addition to this new electron density, the position of the new water molecule identified in the pH 7.5-structure moved toward the thiol group as well. Superpositions of the structures at pH 7.5 and at pH 9.0 highlight a distinct motion of the loop structure carrying the mutation [Fig. 6(B)]. Residue Leu48 was shifted outwards by more than 1 Å in the pH 9.0-structure, suggesting that the additional density observed in the vicinity of the thiol group has pushed this residue aside.
Figure 6.
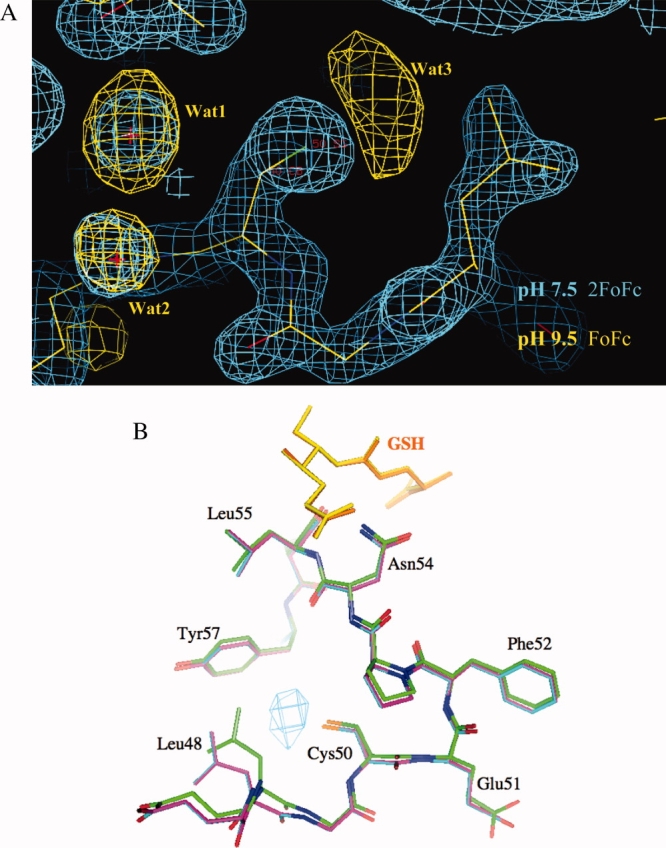
(A) Deletion electron density of GST50C of the vicinity of the mutation site. The data for these densities were collected at pH 7.5 (substrate binding mode, 2FoFc map) and at pH 9.5 (substrate releasing mode, FoFc map), shown in blue and yellow, respectively. No water molecules were included in the map calculation. Water molecules Wat1 and Wat2 are present at both pH values as can be seen in the corresponding blue and yellow densities. The strong density for Wat3, however, can only be identified in the high-pH data. This density stands out by 4.5 sigma above the mean, indicating a strongly bound molecule. The extended triangular shape of this density suggests that possibly two or three water molecules are coordinating to the thiol group of Cys50. (B) Superposition of the high- and low-pH structures of GST50C. With the recruitment of water to the thiol group, Leu48 is forced to move by ∼1.5 Å away from the thiol group. This shift also includes backbone atoms. The substrate molecules are shown in yellow and orange. The superposition was calculated for residues 80–200. The entire GSH-binding domain of the protein was therefore not included in the calculation. The electron density for the corresponding water molecule is shown as a FoFo deletion map contoured at 6 sigma above the mean. For this calculation, the difference structure factors of the pH 7.5 data and the pH 9.5 data were used. These structural consequences are likely to contribute to the changed affinity of this mutant to bind substrate at high pH. Upon reversing the pH from pH 9.5 back to 7.5, the Wat3 density is no longer visible in the structure, confirming the reversible binding affinity of this mutant. This figure was prepared with the Program O (41).
The active sites of the structures at both pH 7.5 and 9.0 contained a bound GSH molecule at full occupancy. Additional data sets of other crystals of this mutant collected at different pH values confirmed this finding. As these crystals could only be grown in the presence of GSH at pH 7.5 (i.e., GSH binding conditions), the crystal packing may have prevented a release of the substrate. Consequently, the observed shifts of Leu48 and the neighboring structures, also, do not appear to be sufficient to cause dissociation of the substrate from the protein in the crystal. This may suggest that further conformational changes may be necessary to release the substrate from the protein. In agreement with our solution studies, we could also demonstrate that the structural shifts were fully reversible. When a crystal that was previously soaked at high pH was transferred back into a low-pH solution, the broad electron density of the newly recruited water molecules disappeared.
In summary, the pH-dependent chromatographic binding experiments as well as the structural investigations strongly suggest that at pH 9.0 the new thiol of GST50C is in a charged state. The binding behavior and structural responses to pH change also suggest that the pKα of this mutant residue is consistent with the experimental pKα of a free cysteine thiol. In mutant GST50H, the pKα of the new histidine may have shifted to a new value as no pH-dependent response to GSH binding within the tested pH-range could be observed.
Discussion
The engineering of allostery into GST was based on the hypothesis that destabilizing mutations can be utilized to control the function of a protein. It was anticipated that the binding of specific effector molecules, in this case protons, can be used directly to control stability and thus the function of the protein. The charged residues therefore serve two distinct purposes: The charged group of a residue is used as a sensor (1) to detect a specific environmental pH and (2) to trigger conformational changes that impact the protein's function.
Previous studies have shown how destabilizing mutations can influence a protein's functional site from a distance. Matthews and coworkers, for example, have shown that specifically engineered polar and charged residues in the core of T4 lysozyme dramatically deteriorate the protein's stability and function.20,23 Even though the overall fold of the enzyme was maintained, the protein underwent a substantial loss of function as a result. Similarly, the engineering of small-to-large mutations into the core of a protein also resulted in severely destabilized protein structures. With the introduction of a tryptophan residue into the hydrophobic core of T4 lysozyme, for example, the surrounding residues rearrange to adopt new conformations to accommodate the bulky residue.24 The resulting conformational rearrangements dissipate throughout the entire C-terminal domain of the protein. These studies therefore demonstrate that a destabilizing mutation may not necessarily exhibit structural changes only at the mutation site but also at a distance.
The burial of polar or charged amino acids in the hydrophobic core is usually detrimental to the stability of a protein structure. Studies on lysozyme and staphylococcus nuclease22 have shown that the introduction of an aspartate or a lysine residue generally decreased the protein's stability significantly. Conversely, the removal of naturally occurring charged residues from the core was also shown to potentially increase a protein's stability.25 The stability profile of the staphylococcal V66K mutant thereby strongly reflected the proton dynamics of the ɛ-amino group. The structural environments of the newly introduced lysine residues of staphylococcal nuclease as well as lysozyme conferred a large shift of their respective pKα values by ∼4 units. Consequently, the resulting pH-dependent stability profiles of these proteins were also shifted accordingly. Both of these studies highlight the importance of the specific structural environment on the protonation dynamics of a charged residue. Solvent accessibility, salt-bridges as well as the dielectric constant of the protein interior are likely to contribute to the shifted pKα values. As such the environmental effects are difficult to rationalize, and thus the prediction of allosteric behavior of newly introduced charges in a core of a protein has also remained challenging.
Structural changes induced by pH
Our observations on mutant GST50H therefore further demonstrate the importance of the structural environment to the effective protonation dynamics as similar environmental effects may have influenced the pKα of the new histidine. Based on the observations of an isolated histidine residue, one would predict that any pH-induced change in substrate affinity would occur at pH 6.0 or lower. At all pH values tested, however, this mutant maintains a very strong affinity to its substrate. GST50H may therefore also exemplify a mutant with a significantly shifted pKα as result of the new environment. QM/QD-based predictions of the side chain pKα values26 in the mutant proteins confirm these observations. For both mutant proteins, GST50D and GST50C, the program predicted pKα values of the mutant side chains that are very similar to the reported literature values. Only the pKα of the new histidine is predicted to be much lower, by more than 3 pH units. Most other histidine residues maintained values closer to literature (Supp. Info. Table 3). These findings are consistent with a previous study that also reported a buried histidine to have a shifted pKα (rat GST isoenzyme 3-327). The large decrease of the pKα of that residue was also attributed to the hydrophobic environment of this residue. In GST50H, the only other histidines that exhibited shifted pKα values interact directly with other hydroxyl or charged groups (Supp. Info. Table 3). Around the mutation site, the only nearby polar residue to His50 is Thr66. In the wild-type structure, this residue points its side chain methyl group toward Leu50 to maintain the hydrophobic core. A rotation of its hydroxyl toward the imidazole seems possible and would further contribute to a loss of hydrophobic groups around residue 50. In the crystal structure, a reorientation of the side chain was not observed, but because of the increased flexibility and the weak electron density it cannot be ruled out.
A significantly lowered pKα of the histidine would indicate that this residue is likely to exist in an uncharged state at all pH values tested here. As the uncharged state is mostly compatible with hydrophobic surroundings, it would explain the protein's sustained affinity for glutathione.
Inspection of the crystal structure suggests that the histidine side chain only barely fits into the hydrophobic core of this mutant. Despite the extremely weak electron density for residues 46–49, the density for the mutated histidine is clearly resolved. A superposition of the mutation site onto the wild type shows that the coordinates have remained very similar, suggesting that sufficient space is available to accommodate the new residue. However, as histidine is ∼1.3 Å longer than the native leucine side chain, its larger size may contact Tyr57 directly and thus contribute to a decreased thermal stability rather than a shifted protonation equilibrium. Thus the tight steric fit of His50 within the hydrophobic core as well as its polar nature is likely to contribute synergistically to a destabilized structure. The dramatically increased B-values of the structures surrounding the mutation site are also consistent with this observation (Fig. 4). Even though no additional water molecules are observed, the increased flexibility is likely to permit solvent molecules to temporarily penetrate this part of the structure.
The cysteine substitution into the same site, on the other hand, produced a protein with GSH-binding behavior that is entirely consistent with its side chain pKα. As a smaller residue replaces a larger one, additional space has become available to accommodate a solvent molecule. With the proximity of a water molecule, the thiol group may be able to maintain its intrinsic pKα. Since the structural changes of this mutant are most pronounced around the site of mutation, we suspect that the protonation behavior of the newly introduced cysteine is responsible for the pH-dependent binding behavior. Contribution of other residues to the altered binding behavior, however, cannot be excluded.
Possible applications
Affinity chromatography is one of the most effective methods to isolate target proteins from complex protein mixtures such as cell lysates. It is also frequently used to immobilize bait proteins in order to copurify and identify other specifically interacting proteins for further biophysical characterizations.28 Even though interacting proteins can be isolated efficiently and on a large scale, unspecific interactions of some proteins with the column matrix may lead to unwanted contaminations during the purification process. To minimize such unspecific binding events, tandem affinity chromatography is frequently employed by selecting protein complexes via two successive rounds of affinity chromatography. As the elution of proteins with any of the commercially available tags often requires dialysis or tag-digestion in order to prepare the eluted protein for the next round affinity chromatography, this procedure is rather time-consuming.29
Mutant GST50C may present a new type of affinity purification tool as its elution can be controlled reversibly with the pH of the buffer. Consequently, a single-tag GST50C-fusion protein can be submitted to multiple rounds of GSH affinity chromatography directly. Upon a first elution at high pH, the protein can be directly applied onto a second column by simply lowering the pH of the protein solution to the permissible (GSH-binding) value. With the addition of the corresponding acidic buffer rather than dialysis, the reversible switch mechanism permits a swift and efficient way to isolate protein complexes.
With the introduction of a buried cysteine into the enzyme it may also be possible to utilize other thiol-binding compounds to control the binding activity of the enzyme. For this purpose, it is necessary to identify compounds that are capable of entering the domain's core and specifically bind to the newly introduced thiol. With the recent discovery of cysteine-containing allosteric sites in caspases, natural allosteric mechanisms were identified which are responsive to such thiol-binding agents. Even though the thiols are positioned ∼15 Å away from the active site of the enzyme, the binding of these compounds compromised the activity of the enzyme allosterically.30,31 Whereas systematic incubations of GST50C with reducing agents such a β-mercaptoethanol and GSH do not appear to affect the binding affinity of the enzyme, preliminary results suggest that mercuriations or nitrosations can compromise binding (to be published).
Even though the GST protein was successfully converted into a pH-responsive protein via the newly introduced residues, it needs to be emphasized that the altered binding affinities or the newly acquired allosteric functions may not only be the direct result of these mutations. Rather, the dramatically altered GSH-binding behavior could also have been caused indirectly by structural or dynamic changes within the protein causing other native residues to suddenly become pH-responsive. GST50H, for example, exhibits dramatically increased B-values, which could possibly have shifted pKα values of other residues to new values. Even though this is not observed with theoretical calculations of protonation behaviors of other residues using QM/QD simulations,26 structural changes may have been assumed by the protein, which are not visible in the current X-ray structures. Predicting the effects of the histidine accurately may therefore also be challenged by the difficulties to include the increased dynamic behavior of the protein in the calculations (see, for example, Ref.32). Whether or not the changed GSH-binding affinities are caused directly or indirectly by the mutations, the use of affinity chromatography clearly demonstrates its potential to identify and engineer new allosteric functions into a protein. Soluble, as well as specifically responsive, proteins can be identified and characterized efficiently with this tool.
Protein design studies have shown most dramatically how challenging the engineering of catalytic functions into proteins is (see, for example, Refs. 33–35). These studies demonstrate that the precise constellation of catalytic residues and dynamic motions of a protein contribute greatly to the catalytic efficiency of an enzyme. Consequently, even minor conformational changes, such as the structural changes induced by the charging of an engineered residue, can therefore effectively be utilized to control a protein's function artificially. Even though this study clearly reveals the feasibility of this rational, it also demonstrates its challenges. Mutant GST50H, for example, fails to exhibit responsive dissociation of the substrate at a histidine's specific pKα. At this point it is also not clear as to how this mutant binds GSH with higher affinity despite its dramatically increased flexibility. We suggest that it is the combination of changed dynamical and structural effects that contribute to a shift of the pKα value and its increased affinity.
On the other hand, however, the structural influences of additional mutations can, in principle, be explored in order to refine the protonation equilibrium of an engineered pH-switch protein. Specifically, we hypothesize that additional mutations elsewhere in a structure maybe utilized to refine allosteric effects of the protein. For example, there is no natural amino acid available with a side chain pKα of 7.0 or 5.5. With the introduction of additional random mutations, however, it may be possible to specifically select proteins that exhibit the desired pH-dependent responses (i.e. at pH 7.0 or 5.5) via selective affinity chromatography. The unpredictable effects of rationally engineered mutations may thus be overcome by selecting functional proteins from a switch-protein library that carry additional random mutations. This hypothesis was recently tested by the use of combinatorial experiments with the goal to customize pH-sensitive allosteric functions (Chapleau RR, Sagermann M. Manuscript in preparation).
There are many metabolic processes inside a cell that are controlled and regulated by specifically interacting effector molecules and the deciphering of the underlying molecular mechanisms that enable allosteric communications are thus of pivotal importance. Understanding responsive functions may not only be beneficial to the understanding of diseases caused by malfunctioning allosteric proteins (see, for example, Ref.36) but also to the development of novel pharmaceutical allosteric modulators.37
Materials and Methods
The vector, pGEX-4T3, encoding GST from Schistosoma japonicum was purchased from GE Healthcare. The substitutions of aspartate, cysteine, and histidine residues were performed by two-stage PCR on the vector template using the following primers:
Aspartate: CAAAAAGTTTGAATTGGGTGATGAGTTTCCCAATCTTCC,
Aspartate_anti: GGAAGATTGGGAAACTCATCACCCAATTCAAACTTTTTG,
Cysteine: CAAAAAGTTTGAATTGGGTTGCGAGTTTCCCAATCTTCC,
Cysteine_anti: GGAAGATTGGGAAACTCGCAACCCAATTCAAACTTTTTG,
Histidine: CAAAAAGTTTGAATTGGGTCATGAGTTTCCCAATCTTCC,
Histidine_anti: GGAAGATTGGGAAACTCATGACCCAATTCAAACTTTTTG.
Cloning and overexpression
The mutated gene sequences were cloned into a TOPO pET151 (Invitrogen) vector to enable Ni2+ affinity chromatography using blunt-end cloning according to the manufacturer's protocol. Successfully cloned vectors were transformed into BL21-AI cells for overexpression. Overnight cultures of the mutant bacteria were prepared in LB broth at 37°C, and 60 mL of the culture was used to inoculate 3 L of LB broth in the presence 1 mM ampicillin. Cells were grown to mid-log phase before induction with 100 μM IPTG and 200 μM (+) arabinose. Induction was carried out for 3 h at 28°C. Cells were harvested by centrifugation at 6000 rpm in a Beckman JA10 rotor. Cells were resuspended in 50 mM Tris-HCl pH 8.0, 100 mM NaCl (also referred to as Standard Buffer), for lysis with a French-Press at 4°C. Subsequently, lysed cell homogenates were then centrifuged at 17,000 rpm using a Beckman JA20 rotor for 20 min. The supernatant was then subjected to purification by Ni2+ affinity chromatography following standard protocols (Novagen, IMAC). The purified protein was dialyzed against 50 mM Tris-HCl at pH 8.0, 100 mM NaCl, and 1 mM EDTA overnight at 4°C. To remove the His6-tag, proteolytic digestion with TEV was carried out overnight at room temperature. After the completion of the digest, the protease, noncleaved proteins, and liberated His6-tags were removed by subsequent application to Ni2+ affinity chromatography.
Spectroscopy
The secondary structure content was determined via circular dichroism (CD) spectroscopy on an Olis-CD spectrometer. The concentrations of all mutants were set equal to the same optical absorption of OD(280 nm) = 0.24. CD spectra between 200 and 250 nm were obtained at 0.5 nm steps and averaged. The thermal stability of the proteins was determined by following the CD signal at 221 nm with respect to temperature. The temperature was changed in 1°C increments and allowed to equilibrate for 1 min before each data point was measured for 30 s, three times, and averaged.
Binding activity to GSH
The use of Ni2+ affinity chromatography allowed for the isolation of mutant proteins without exposure to GSH. Prior to the affinity selection experiments, all mutant proteins and the GSH affinity matrix were dialyzed and equilibrated into the corresponding loading buffers. Elutions of the protein with respect to systematically changed pH conditions were analyzed with GSTrap FF columns. The elution of proteins was monitored by absorption at λ = 222 nm and λ = 280 nm on an Äkta Purifier (GE Healthcare). New affinity columns were used for each mutant protein and the measurement was repeated at least three times.
Crystallography
TEV-cleaved GST mutants were dialyzed into Standard Buffer and concentrated to at least 10 mg/mL. Crystallizations were carried out with commercially available screens (Hampton Research) as well as custom-made screens. Even though the wild-type protein crystallized in four different crystal forms, only very few and small crystals of the mutant proteins were obtained that were too small for diffraction studies. Seeding experiments, however, produced crystals of sufficient quality. For the studies described here, we have selected wild-type crystal form P43212 for the seeding trials of the mutant proteins. It was anticipated that induced conformational changes may be visualized in this crystal form in particular (see the “Results” section). The two mutant proteins, GST50C and GST50H, were crystallized in this symmetry with 50 mM Tris (pH 7.5), 20% PEG 8000, 100 mM ammonium sulfate, 10 mM GSH, and β-mercaptoethanol. Even though the circular dichroism spectra of GST50D exhibits highly similar secondary structure content to that of the wild type (Fig. S1 in Supp. Info.), it did not crystallize. GST50C crystals were grown at pH 7.5, whereas the GST50H were grown at pH 6.4. To perform data collections at the respective pH values of interest, crystals were soaked in the corresponding buffer solutions for 24 h prior to the measurements.
Data collections of rapidly frozen crystals were carried out on a Rigaku FR-E X-ray generator, equipped with Osmic HR focusing mirrors and a Raxis 4+ imaging plate detector, on a Bruker Microstar H generator equipped with a PT135 CCD detector, and at SSRL beamline 11-1 equipped with a MAR Mosaic CCD325 detector. Data was processed with either Proteum/Saint (Bruker AXS) or XDS38,39 software (Table I). The structures were determined by isomorphous replacement using the PDB structure 1GNE (residues 1–214) as starting model.40 Model building and refinement were carried out using COOT,41 O,42 and CNS,43 respectively. In all cases, datasets from several different crystals of the mutants were obtained and refined with similar results. Data with the best integration and scaling statistics are presented in the “Results” section.
Conclusion
We report here the successful engineering of a pH-responsive enzyme switch. With the introduction of charges into the hydrophobic core of GST, regulation of substrate binding can be controlled precisely with the pH of the surrounding solution. Specifically, we show that a charged residue can significantly change the structure as well as the dynamics within the protein structure. In the case of mutant GST50C, the specific recruitment of water molecules by the charged group is accompanied by induced structural changes that influence the function of the active site allosterically. As this study is entirely based on the protein's potential to bind to its substrate, studies on the allosteric effect of pH on this mutants's enzymatic activity confirm these results. These results will be reported elsewhere.
The use of proteins for the creation of responsive biomaterials, catalysts, and devices may enable the engineering of unprecedented allosteric functions into macromolecules. The ability to construct such objects with molecular precision is envisioned to revolutionize manufacturing, permitting materials properties and device performance to be greatly improved. Smart materials are of particular interest, as they may enable precise control of function via external stimuli such as pH, temperature, and stress. The reprogramming of GST into a pH responsive protein is a first step in the design of such new materials. Even though the altered binding properties of mutant GST50H demonstrate the difficulties to design pKα-dependent switches into a protein, the use of affinity chromatography exemplifies the possibility to custom-select functional pH-switches among different mutant proteins. The use of destabilizing mutations and affinity chromatography as described here may thus lead to an effective and generally applicable strategy to identify and characterize responsive variants of an otherwise nonresponsive protein.
Acknowledgments
The authors are most grateful for the expert help on the data collection at SSRL and Bruker (Matt Benning, Madison). Furthermore, we they also thank Joshua Fields for help with the circular dichroism measurements and Dr. Akashi Ohtaki for helpful comments on the manuscript.
References
- 1.Horovitz A, Willison KR. Allosteric regulation of chaperonins. Curr Opin Struct Biol. 2005;15:646–651. doi: 10.1016/j.sbi.2005.10.001. [DOI] [PubMed] [Google Scholar]
- 2.Vetter IA, Wittinghofer A. The guanidine nucleotide binding switch in three dimensionss. Science. 2001;294:1299–1304. doi: 10.1126/science.1062023. [DOI] [PubMed] [Google Scholar]
- 3.Hatley ME, Lockless SW, Gibson SK, Gilman GA, Ranganathan R. Allosteric determinants in guanine nucleotide-binding proteins. Proc Natl Acad Sci USA. 2003;100:14445–14450. doi: 10.1073/pnas.1835919100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kuriyan J. Allostery and coupled sequence variation in nuclear hormone receptors. Cell. 2004;116:354–356. doi: 10.1016/s0092-8674(04)00125-4. [DOI] [PubMed] [Google Scholar]
- 5.Swain JF, Gierasch LM. The changing landscape of protein allostery. Curr Opin Struct Biol. 2006;16:102–108. doi: 10.1016/j.sbi.2006.01.003. [DOI] [PubMed] [Google Scholar]
- 6.Bullough PA, Hughson FM, Skehel JJ, Wiley DC. Structure of influenza haemagglutinin at the pH of membrane fusion. Nature. 1994;371:37–43. doi: 10.1038/371037a0. [DOI] [PubMed] [Google Scholar]
- 7.Carr C, Kim PS. A spring-loaded mechanism for the conformational change of influenza hemagglutinin. Cell. 1993;73:823–832. doi: 10.1016/0092-8674(93)90260-w. [DOI] [PubMed] [Google Scholar]
- 8.Zhang Y, Zuiderweg ER. The 70-kDa heat shock protein chaperone nucleotide-binding domain in solution unveiled as a molecular machine that can reorient its functional subdomains. Proc Natl Acad Sci USA. 2004;101:10272–10277. doi: 10.1073/pnas.0401313101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schlauderer GJ, Proba K, Schulz GE. Structure of a mutant adenylate kinase ligated with an ATP-analogue showing domain closure over ATP. J Mol Biol. 1996;256:223–227. doi: 10.1006/jmbi.1996.0080. [DOI] [PubMed] [Google Scholar]
- 10.Feynman RP. There is plenty of room at the bottom. Eng Sci (Caltech) 1960;23:22–36. [Google Scholar]
- 11.Fairman R, Akerfedt KS. Peptides as novel smart materials. Curr Opin Struct Biol. 2005;15:453–463. doi: 10.1016/j.sbi.2005.07.005. [DOI] [PubMed] [Google Scholar]
- 12.Zhang S. Fabrication of novel biomaterials through molecular self-assembly. Nat Biotechnol. 2003;21:1171–1178. doi: 10.1038/nbt874. [DOI] [PubMed] [Google Scholar]
- 13.Pandya MJ, Cerasoli E, Joseph A, Stoneman RG, Waite E, Woolfson DN. Sequence and structural duality: designing peptides to adopt two stable conformations. J Am Chem Soc. 2004;126:17016–17024. doi: 10.1021/ja045568c. [DOI] [PubMed] [Google Scholar]
- 14.Ambroggio XL, Kuhlman B. Computational design of a single amino acid sequence that can switch between two distinct protein folds. J Am Chem Soc. 2005;128:1154–1161. doi: 10.1021/ja054718w. [DOI] [PubMed] [Google Scholar]
- 15.Rastogi VK, Girvin ME. Structural changes linked to proton translocation by subunit c of the ATPsynthase. Nature. 2004;402:263–268. doi: 10.1038/46224. [DOI] [PubMed] [Google Scholar]
- 16.Sheehan D, Meade G, Foley VM, Dowd CA. Structure, function and evolution of glutathione transferases: implications for classification of non-mammalian members of an ancient enzyme superfamily. Biochem J. 2001;360:1–16. doi: 10.1042/0264-6021:3600001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Andújar-Sánchez M, Smith AW, Clemente-Jimenez JM, Rodriguez-Vico F, Las Heras-Vazquez FJ, Jara-Pérez V, Cámara-Artigas A. Crystallographic and thermodynamic analysis of the binding of S-octylglutathione to the Tyr 7 to Phe mutant of glutathione S-transferase from Schistosoma japonicum. Biochemistry. 2005;44:1174–1183. doi: 10.1021/bi0483110. [DOI] [PubMed] [Google Scholar]
- 18.Ricci G, Lo Bello M, Caccuri AM, Pastore A, Nucetelli M, Parker MW, Grederici F. Site-directed mutagenesis of human glutathione transferase P1-1. J Biol Chem. 1995;270:1243–1248. doi: 10.1074/jbc.270.3.1243. [DOI] [PubMed] [Google Scholar]
- 19.Wongsantichon J, Ketterman AJ. An intersubunit lock-and-key ‘clasp’ motif in the dimer interface of Delta class glutathione transferase. Biochem J. 2006;394:135–144. doi: 10.1042/BJ20050915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dao-pin S, Anderson DE, Baase WA, Dahlquist FW, Matthews BW. Structural and thermodynamic consequences of burying a charged residue within the hydrophobic core of T4 lysozyme. Biochemistry. 1991;30:11521–11529. doi: 10.1021/bi00113a006. [DOI] [PubMed] [Google Scholar]
- 21.Ladbury J, Wynn ER, Thomson JA, Sturtevant JM. Substitution of charged residues into the hydrophobic core of Escherichia coli thioredoxin results in a change in heat capacity of the native protein. Biochemistry. 1995;34:2148–2152. doi: 10.1021/bi00007a007. [DOI] [PubMed] [Google Scholar]
- 22.Stites WE, Gittis AG, Lattman EE, Shortle D. In a staphylococcal nuclease mutant the side-chain of a lysine replacing valine 66 is fully buried in the hydrophobic core. J Mol Biol. 1991;221:7–14. doi: 10.1016/0022-2836(91)80195-z. [DOI] [PubMed] [Google Scholar]
- 23.Blaber M, Lindstrom JD, Gassner N, Xu J, Heinz DW, Matthews BW. Energetic cost and structural consequences of burying a hydroxyl group within the core of a protein determined from Ala→Ser and Val→Thr substitutions in T4 lysozyme. Biochemistry. 1993;32:11363–11373. doi: 10.1021/bi00093a013. [DOI] [PubMed] [Google Scholar]
- 24.Baldwin EP, Hajiseyedjavadi O, Baase WA, Matthews BW. The role of backbone flexibility in the accommodation of variants that repack the core of T4 lysozyme. Science. 1993;262:1715–1718. doi: 10.1126/science.8259514. [DOI] [PubMed] [Google Scholar]
- 25.Kajander T, Kahn PC, Passila SH, Cohen DC, Lehtiö L, Adolfsen W, Warwicker J, Schell U, Goldman A. Buried charged surface in proteins. Structure. 2000;8:1203–1214. doi: 10.1016/s0969-2126(00)00520-7. [DOI] [PubMed] [Google Scholar]
- 26.Li H, Robertson AD, Jensen JH. Very fast empirical prediction and interpretation of protein pKa values. Proteins. 2005;61:704–721. doi: 10.1002/prot.20660. [DOI] [PubMed] [Google Scholar]
- 27.Zhang P, Graminskyi GF, Armstrong RN. Are the histidine residues of glutathione-S-transferase important in catalysis? J Biol Chem. 1991;266:19475–19479. [PubMed] [Google Scholar]
- 28.Chen GI, Gingras AC. Affinity-purification mass spectrometry (AP-MS) of serine/threonine phosphatases. Methods. 2007;42:298–305. doi: 10.1016/j.ymeth.2007.02.018. [DOI] [PubMed] [Google Scholar]
- 29.Rigaut G, Shevchenko A, Rutz B, Wilm M, Mann M, Séraphin B. A generic protein purification method for protein complex characterization and proteome exploration. Nat Biotechnol. 1999;17:1030–1032. doi: 10.1038/13732. [DOI] [PubMed] [Google Scholar]
- 30.Hardy JA, Lam J, Nguyen JT, O'Brien T, Wells JA. Discovery of an allosteric site in the caspases. Proc Natl Acad Sci USA. 2004;101:12461–12466. doi: 10.1073/pnas.0404781101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Scheer JM, Romanowski MJ, Wells JA. A common allosteric site and mechanism in caspases. Proc Natl Acad Sci USA. 2006;103:7595–7600. doi: 10.1073/pnas.0602571103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Baran KL, Chimenti MS, Schlessman JL, Fitch CA, Herbst KJ, Garcia-Moreno BE. Electrostatic effects in a network of polar and ionizable groups in staphylococcal nuclease. J Mol Biol. 2008;379:1045–1062. doi: 10.1016/j.jmb.2008.04.021. [DOI] [PubMed] [Google Scholar]
- 33.Bolon DN, Voigt CA, Mayo SL. De novo design of biocatalysts. Curr Opin Chem Biol. 2002;6:125–129. doi: 10.1016/s1367-5931(02)00303-4. [DOI] [PubMed] [Google Scholar]
- 34.Lippow SM, Tidor B. Progress in computational protein design. Curr Opin Biotechnol. 2007;18:305–311. doi: 10.1016/j.copbio.2007.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jiang L, Althoff EA, Clemente FR, Doyle L, Röthlisberge D, Zanghellini A, Gallaher AL, Betker JL, Tanaka F, Barbas CF, III, Hilvert D, Houk KN, Stoddard BL, Baker D. De novo computational design of retro-aldol enzymes. Science. 2008;319:1387–1391. doi: 10.1126/science.1152692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liu J, Nussinov R. Allosteric effects in the marginally stable von Hippel-Lindau tumor suppressor protein and allostery-based rescue mutant design. Proc Natl Acad Sci USA. 2008;105:901–906. doi: 10.1073/pnas.0707401105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hardy JA, Wells JA. Searching for new allosteric sites in enzymes. Curr Opin Struct Biol. 2004;14:706–715. doi: 10.1016/j.sbi.2004.10.009. [DOI] [PubMed] [Google Scholar]
- 38.Kabsch W. Evaluation of single-crystal X-ray diffraction data from a position-sensitive detector. J Appl Crystallogr. 1988;21:916–924. [Google Scholar]
- 39.Kabsch W. Automatic processing of rotation diffraction data from crystals of initially unknown symmetry and cell constants. J Appl Crystallogr. 1993;26:795–800. [Google Scholar]
- 40.Lim K, Ho JX, Keeling K, Gilliland GL, Ji X, Ruker F, Carter DC. Three-dimensional structure of Schistosoma japonicum glutathione S-transferase fused with a six-amino acid conserved neutralizing epitope of gp41 from HIV. Protein Sci. 1994;3:2233–2244. doi: 10.1002/pro.5560031209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Collaborative Computational Project Number 4. The CCP4 suite: programs for protein crystallography. Acta Crystallogr D Biol Cryst. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 42.Jones A. Acta Cryst A. 1991;47:110–119. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- 43.Brünger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang J-S, Kuszewski J, Nilges M, Pannu NS. Crystallography & NMR system: a new software suite for macromolecular structure determination. Acta Crystallogr D. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]