

Abstract
The polyproteins of coronaviruses are cleaved by viral proteases into at least 15 nonstructural proteins (Nsps). Consisting of five domains, Nsp3 is the largest of these (180–210 kDa). Among these domains, the so-called X-domain is believed to act as ADP-ribose-1″-phosphate phosphatase or to bind poly(ADP-ribose). However, here we show that the X-domain of Infectious Bronchitis Virus (strain Beaudette), a Group-3 coronavirus, fails to bind ADP-ribose. This is explained on the basis of the crystal structure of the protein, determined at two different pH values. For comparison, we also describe the crystal structure of the homologous X-domain from Human Coronavirus 229E, a Group-1 coronavirus, which does bind ADP-ribose.
Keywords: X-domain, macrodomain, nonstructural protein 3, coronavirus, SARS, ADP-ribose, X-ray structure, ADP-ribose-1″-phosphate phosphatase
Introduction
Coronaviruses are positive-stranded RNA viruses with a genome size of 27–32 kb. Their enveloped, spherical virions have a diameter of 100–160 nm and carry prominent spike proteins that give the surface of the particle a crown-like appearance in the electron microscope, hence the name coronaviruses. The genera Torovirus and Coronavirus are members of the family Coronaviridae. This family belongs to the order Nidovirales, together with the Arterivirdae and Roniviridae families. Nidoviruses have similar genome organization and replication strategies but they differ in morphology, genome size, and several other biological properties.1
Coronaviruses have been subdivided into three main groups based on serological and genetic properties. Group 1 includes the human coronaviruses 229E (HCoV 229E) and NL63 (HCoV NL63), the porcine Transmissible Gastroenteritis Virus (TGEV), and feline coronavirus (FCoV). Although a Group-3 coronavirus was recently isolated from a Beluga whale,2 members of this subgroup generally infect birds, for example, the Infectious Bronchitis Virus (IBV) which can cause serious economic loss in the chicken industry.3 The human coronaviruses OC43 (HCoV OC43) and HKU1 (HCoV HKU1), Mouse Hepatitis Virus (MHV), and bovine coronavirus (BCoV) belong to Group 2. The 2003 outbreak of severe acute respiratory syndrome (SARS), caused by a coronavirus that was classified as an outlier in Group 2,4 renewed the interest in the coronavirus family.
HCoV 229E and HCoV OC43 are responsible for 10–30% of all common colds. HCoV NL63 causes respiratory tract illness in children below the age of 1 year and in immunocompromised adults.5
The coronavirus genome contains several open reading frames (ORFs). The two overlapping ORFs 1a and 1b at the 5′ end comprise two thirds of the genome. ORF1a encodes the polyprotein 1a (pp1a) and ORF1a and 1b together code for pp1ab. Synthesis of the latter depends on a (−1) ribosomal frameshift during translation, which occurs just upstream of the ORF1a stop codon. In most coronaviruses, the polyproteins are processed by two papain-like proteases (PL1pro and PL2pro) and the main proteinase (Mpro), all of which are encoded within ORF1a. The resulting 15 (IBV) or 16 nonstructural proteins (Nsps) are associated with genomic and/or subgenomic RNA synthesis (replication and transcription, respectively). Some of them may also be involved in interactions with host-cell components.
Several of the Nsps of SARS-CoV have had their three-dimensional structures determined within structural proteomics programs (see Refs.6 and7 for reviews). However, structural information on other coronavirus Nsps is limited: Of HCoV 229E, the only structures known are those of the main proteinase (Nsp5; Ref.8) and of Nsp9.9 Among the IBV Nsps, only the structure of the main proteinase10 is known so far. As we will show here for the X-domain of Nsp3, complementary structural information on other coronaviruses than SARS-CoV can make a major contribution to understand the function of the Nsps of the entire family.
With a molecular mass of 180–210 kDa, the multidomain Nsp3 is the largest cleavage product of pp1a and pp1ab. Its domain organization is more or less conserved among the coronaviruses. Several highly hydrophobic stretches in the C-terminal Y domain anchor Nsp3 to intracellular membranes.11–13 Immuno-EM studies revealed that most of Nsp3 localizes to convoluted membranes, which are part of a reticulovesicular network of modified endoplasmic reticulum that is thought to support the viral replication/transcription complexes.14
The N-terminal domain of Nsp3 comprises 150–240 amino-acid residues and is called acidic domain in view of the high number of Asp and Glu residues in its carboxy-terminal third. The first papain-like protease (PL1pro) is located downstream of the acidic domain. Along with the PL2pro, which is further downstream, the PL1pro is responsible for the processing of three (IBV: two) cleavage sites containing LXGG↓X consensus sequences in the N-terminal third of the coronaviral polyproteins. Interestingly, the PL1pro is missing in SARS-CoV,4 and in IBV, it is catalytically inactive because of amino-acid replacements in the catalytic center.13
C-terminal to the PL1pro of most coronaviruses, there is the so-called X-domain, which was first described by Gorbalenya et al.15 and was later proposed to be an ADP-ribose-1″-phosphate phosphatase (Appr-1″-pase).4 Appr-1″-pases are involved in tRNA splicing by removing the phosphate group of adenosine diphosphate ribose 1″-phosphate, thus producing adenosine diphosphate ribose (ADP-ribose).16 Appr-1″-pase activity has been demonstrated in vitro for the isolated X-domains of HCoV 229E17 and SARS-CoV.18,19 The structure of the SARS-CoV Nsp3 X-domain (SARS-X) has been determined by Saikatendu et al.,18 and, in complex with ADP-ribose, by Egloff et al.19
The fold of the SARS-X was found to be similar to that of the so-called macrodomain. This name goes back to the noncanonical domain in the histone macroH2A.20 The macrodomain fold has also been found in X-ray structures of proteins from the thermophilic organism Archaeoglobus fulgidus (AF1521 protein; Refs.21 and22), yeast (Ymx7 protein; Ref.23), and E. coli (Er58 protein).
Here, we describe the crystal structures of the X-domains of the coronavirus Group-1 member HCoV 229E (229E-X) and the Group-3 coronavirus IBV (strain Beaudette; IBV-X), and compare them with the structure of the homologous SARS-X. We also investigate the binding properties for ADP-ribose and show that 229E-X binds this product of Appr-1″-pase activity, whereas IBV-X (strain Beaudette) fails to do so.
Results
ADP-ribose binding studies
Using both isothermal titration calorimetry (ITC; Fig. 1) and zone-interference gel electrophoresis (ZIGE; results not shown), recombinant 229E-X was found to bind ADP-ribose with a KD of 28.9 ± 2.6 μM. This compares well to the value of 24 μM determined for SARS-X.19 In contrast, IBV-X (strain Beaudette) failed to show any binding of ADP-ribose using either technique (data not shown).
Figure 1.
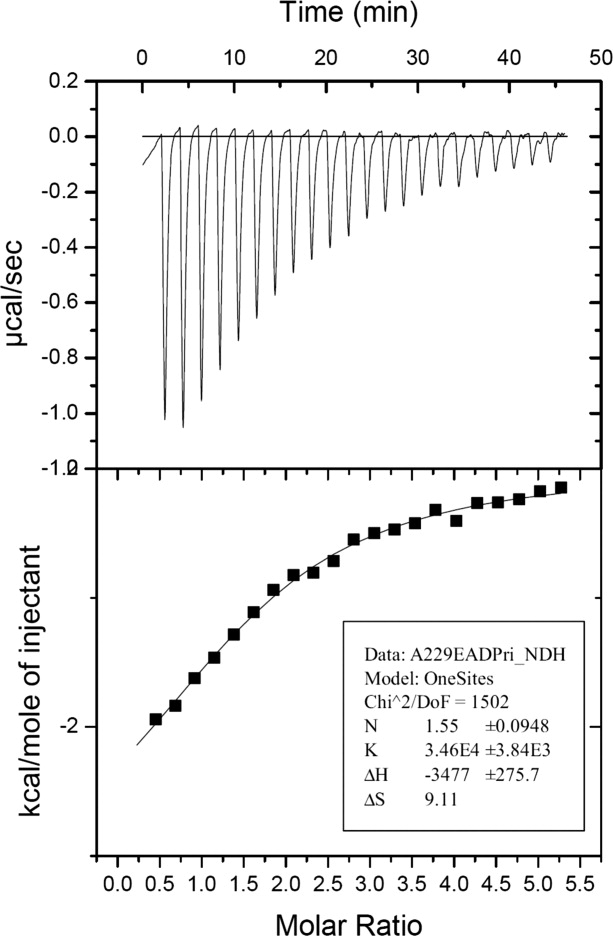
Isothermal titration calorimetry profile for the binding of ADP-ribose to HCoV-229E X-domain.
Crystallization and structure determination
We determined the crystal structure of IBV-X (strain Beaudette) at two pH values, 5.6 and 8.5. The crystals were grown under different conditions, using ammonium sulfate and polyethylene glycol (PEG) 4000, respectively, as precipitants (see Table I). The low-pH crystals displayed space group P322, with a = 78.2 Å and c = 81.7 Å, whereas the high-pH crystals belonged to space group C2221, with a = 42.2 Å, b = 79.8 Å, and c = 99.7 Å. The crystals diffracted X-rays to Bragg spacings of 2.1 Å and 1.6 Å, respectively. Both crystal forms are different from the IBV-X crystals reported very recently by Wei et al.24 In what follows, our low-pH and high-pH forms of the IBV-X will be abbreviated by IBV-X5.6 and IBV-X8.5, respectively.
Table I.
Crystallization, Data Collection, and Refinement
| HCoV-229E X-domain | IBV X-domain | IBV X-domain | |
|---|---|---|---|
| Crystallization conditions | 20% PEG 8000 | 30% PEG 4000 | 1.8M (NH4)2SO4 |
| 0.1M Tris | 0.1M Tris | 0.1M Na-citrate pH 5.6 | |
| pH 8.5 | pH 8.5 | 0.2M K-,Na-tartrate | |
| 5% MPD | 0.2M MgCl2 | ||
| Data collection | |||
| Wavelength (Å) | 1.04 | 0.808 | 0.808 |
| Resolution (Å) | 24.22–1.78 (1.88–1.78) | 40.00–1.60 (1.64–1.60) | 31.28–2.10 (2.21–2.10) |
| Space group | P21 | C2221 | P322 |
| Unit-cell parameters | |||
| a (Å) | 33.56 | 42.21 | 78.20 |
| b (Å) | 65.89 | 79.81 | 78.20 |
| c (Å) | 38.02 | 99.74 | 81.70 |
| α (°) | 90 | 90 | 90 |
| β (°) | 110.1 | 90 | 90 |
| γ (°) | 90 | 90 | 120 |
| Solvent content (%, v/v) | 33.73 | 43.12 | 66.88 |
| Overall reflections | 101,730 | 158,270 | 503,497 |
| Unique reflections | 14,479 (1946) | 22,201 (1448) | 17,123 (2231) |
| Multiplicity | 4.2 (3.8) | 7.1 (6.2) | 11.5 (10.4) |
| Completeness (%) | 96.9 (89.9) | 97.9 (97.1) | 98.4 (89.4) |
| Rmergea (%) | 5.2 (15.5) | 10.3 (24.4) | 7.9 (40.1) |
| I/σ(I) | 19.6 (7.8) | 21.7 (6.8) | 25.6 (6.5) |
| Refinement | |||
| Resolution (Å) | 24.22–1.78 | 40.00–1.60 | 31.28–2.10 |
| Rcrystb | 0.165 | 0.169 | 0.199 |
| Rfreeb | 0.205 | 0.203 | 0.231 |
| r.m.s.d. from ideal geometry | |||
| Bonds (Å) | 0.013 | 0.011 | 0.022 |
| Angles (°) | 1.284 | 1.349 | 1.896 |
| Protein atoms | 1283 | 1325 | 1335 |
| Solvent atoms | 146 | 212 | 75 |
| Heteroatoms | 20 | ||
| Ramachandran plot | |||
| Most favored (%) | 91.7 | 92.5 | 92.1 |
| Additionally allowed (%) | 6.9 | 6.8 | 6.6 |
| Generously allowed (%) | 0.7 | 0.0 | 0.7 |
| Disallowed regions (%) | 0.7 | 0.7 | 0.7 |
Values in parentheses are for the highest resolution shell.
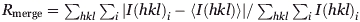 , where I(hkl) is the intensity of reflection hkl and 〈I(hkl)〉 is the average intensity over all equivalent reflections.
, where I(hkl) is the intensity of reflection hkl and 〈I(hkl)〉 is the average intensity over all equivalent reflections.
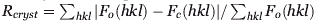 Rfree was calculated for a test set of reflections (6%, 5%, and 6%, respectively) omitted from the refinement.
Rfree was calculated for a test set of reflections (6%, 5%, and 6%, respectively) omitted from the refinement.
In addition, we crystallized 229E-X, in order to compare the structures of X-domains from a Group-1 (HCoV 229E) and a Group-3 (IBV, strain Beaudette) coronavirus, along with the known structure18,19 of the corresponding domain of SARS coronavirus, which belongs to Group 2b.25 The 229E-X crystals were grown from 20% PEG 8000, in the presence of 5% 2-methyl-2,4-pentanediol (MPD) at pH 8.5; they displayed space group P21 with a = 33.6 Å, b = 65.9 Å, c = 38.0 Å, and β = 110.1°. The diffraction limit of these crystals was at 1.8 Å resolution.
All structures were determined by molecular replacement. In case of 229E-X, our structure of the X-domain of HCoV NL63 (NL63-X; Piotrowski et al., in preparation) was used as a search model, whereas solving the structures of both forms of IBV-X required the construction of a combined search model that made use of fragments of SARS-X18 as well as of eukaryotic macrodomains (see Materials & Methods). In view of the relatively low sequence identity between IBV-X and the other coronaviral X-domains (19% to SARS-X, 22% to 229E-X, 25% to NL63-X), and the structural deviations that we will describe later, it is no surprise, in retrospect, that straightforward molecular replacement did not work in this case.
Quality of the structural models
All three X-domain structures described here are well defined, with crystallographic R factors ranging from 0.16 to 0.20 (Rfree values from 0.20 to 0.23) (see Table I). The resolutions of the structures reach 1.6 Å for IBV-X8.5, 1.8 Å for 229E-X, and 2.1 Å for IBV-X5.6.
The HCoV-229E X-domain consists of 165 amino-acid residues and its gene construct coding carried an additional 28 residues (including a His6 sequence and a Tobacco Etch Virus (TEV) protease cleavage site) at the N-terminus. Because cleavage of the purification tag led to precipitation of 229E-X, uncleaved protein was used for crystallization. None of the extra residues have visible electron density, whereas all 165 residues of the authentic X-domain could be observed in the maps. Alternative conformations were detected for Asp 10, Lys 56, Asp 100, and Cys 157.
In case of IBV-X, removal of the N-terminal purification tag yielded soluble protein. As a result, four extra amino-acid residues (GAMA) were still present at the N-terminus of the X-domain. The X-domain of IBV (strain Beaudette) consists of 172 amino-acid residues. When crystallization was performed at pH 8.5 (IBV-X8.5), all amino-acid residues could be modeled except -3GAMAPATCE5 and Lys 172, for which electron density was lacking. Alternative conformations in this structure could be detected for Lys 51, His 72, Met 138, Glu 148, Thr 151, and Ser 160. Lys 31 has unusual ϕ/ψ angles. Located in a two-residue turn between helix α1 and strand β3 (loop L4), this residue has well-defined electron density; its side-chain interacts with Asp 28.
In the IBV X-domain structure derived from crystals grown at pH 5.6 (IBV-X5.6), all amino-acid residues except -3GAMAP1 could be modeled, although the density was well defined only from residue 6 onward. For Asp 135, an alternative conformation was refined. For crystallization at pH 5.6, 200 mM K-,Na-tartrate was used. During refinement of the structure, difference density for two tartrate (TLA) molecules emerged. One molecule is located at loop L10 and is hydrogen-bonded to the backbone NH of Asp 117 and the side-chain of Arg 77. This tartrate is also involved in a crystal contact with Ser 47 and Gly 48 of a neighboring molecule. The second tartrate molecule is located at L5 and the N-terminus of α2, where it interacts with the backbone amides of His 45, Gly 46, and Ser 47. In spite of the involvement of tartrate in crystal contacts, isomorphous crystals could also be grown in the absence of this additive, but diffracted only to a Bragg spacing of 2.4 Å.
In the final Fourier maps for IBV-X5.6, three regions of density could not be interpreted. The first of these is located on the twofold axis of symmetry, near the carboxy-terminal Lys 172 and the Phe 7-Tyr 12 segment, as well as Arg 144 and Phe 167. It could represent two sets of twofold disordered glycerol molecules, as glycerol was used as a cryoprotectant, but this model was not stable during refinement. The second unexplained density is surrounded by Leu 159-Glu 162, as well as Pro 81-Gln 88 of a symmetry-related molecule. The third noninterpreted density is located in the interior of the domain, close to His 45. It could represent a second conformation of this histidine, but this model could not be refined either.
Overall structures
The overall structures of 229E-X and IBV-X are similar to one another, even though the sequence identity between the two is only 22% (see Fig. 2). They are also similar to the known structure of SARS-X (34% and 19%, respectively). The structures show the so-called macrodomain fold, consisting of a central, mostly parallel β-sheet flanked by three α-helices on one side and three on the other (see Fig. 3). Along the polypeptide chain, the order of regular secondary structure elements is β1-β2-α1-β3-α2-α3-β4-β5-α4-β6-α5-β7-α6. The β-sheet topology is β1-β2-β7-β6-β3-β5-β4, with the external strands β1 and β4 being antiparallel to their neighbors. Interestingly, β1 is missing in IBV-X. As a result, the positions of the N-terminal residues are very different (15.0 Å between Leu 3 of 229E-X and the corresponding Ala 2 of IBV-X5.6) in the two structures.
Figure 2.
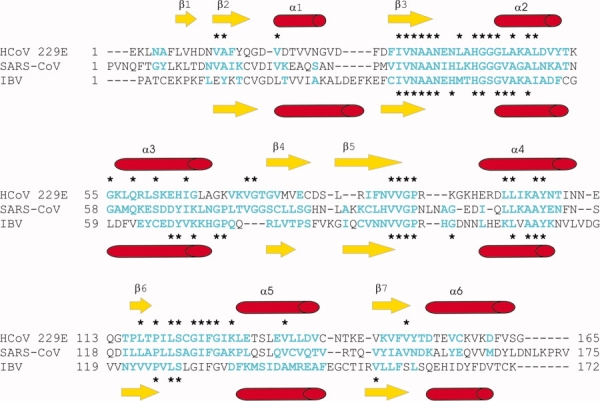
Structure-based sequence alignment of the X-domains of HCoV 229E and IBV (strain Beaudette) with the homologue in SARS-CoV. Secondary structure elements of the HCoV-229E X-domain and the IBV X-domain are represented above and below the alignment, respectively. Amino acid residues labeled in cyan have been included in the calculation of the r.m.s.d. value. Asterisks above and below the alignment indicate aligned and conserved amino acid residues.
Figure 3.
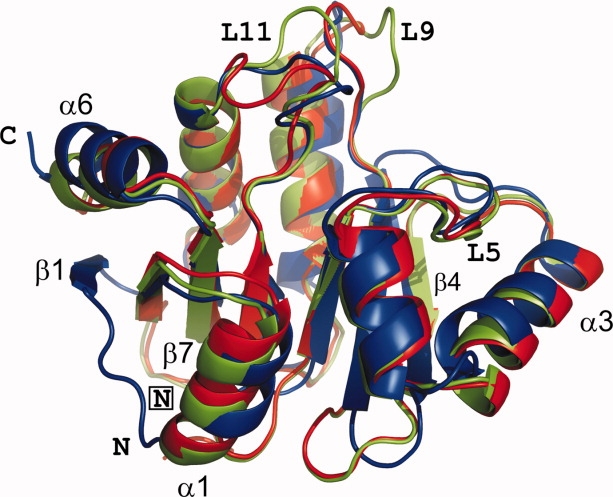
Ribbon-representation of the superimposed X-domains of HCoV 229E (blue) and IBV (strain Beaudette) (green, pH 8.5; red, pH 5.6). N- and C-termini are indicated; the symbol “N” is framed for IBV-X.
The overall r.m.s. deviation between the X-domains of HCoV 229E and IBV (strain Beaudette) is 1.81 Å and 1.72 Å for IBV-X5.6 and IBV-X8.5, respectively, for 146 Cα pairs out of 165 compared. The low-pH and high-pH forms of the IBV X-domain display a low overall r.m.s.d. of 0.46 Å, the only significant difference being caused by crystal contacts in loop region L11 connecting β6 and α5, with deviations of up to 11 Å at Phe 132 and Gly 133. This region is involved in an extensive crystal contact in the low-pH but not the high-pH form. In 229E-X, this loop makes only weak, if any, crystal contacts and adopts a conformation similar to that seen in SARS-X19 (see Fig. 3).
The putative ligand-binding site
Coronaviral X-domains have been proposed to bind ADP-ribose, either as product of an ADP-ribose-1″-phosphate phosphatase (Appr-1″-pase) activity,17 or as part of poly(ADP-ribose) binding.19 As described earlier, we found by ZIGE and ITC that 229E-X binds ADP-ribose, but IBV-X (strain Beaudette) fails to do so. To rationalize this observation on the basis of our crystal structures, we will inspect the putative binding sites for ADP-ribose in the two proteins in detail and compare them with the crystal structure of SARS-X in complex with ADP-ribose.19
In the latter structure, the adenine of ADP-ribose is recognized by a 2.9 Å hydrogen bond from the exocyclic NH2 group of the base to the carboxylate of Asp 23 [Fig. 4(A)]. This aspartate residue is highly conserved in coronavirus X-domains, including HCoV 229E and IBV. In the structures of IBV-X and 229E-X, both of which lack a bound ADP-ribose, this aspartate is oriented away from the binding site. In SARS-X, the adenine ring of ADP-ribose is sandwiched between the side-chains of Asn 157 [L13 (β7-α6) loop], Val 50 [L5 (β3-α2) loop], and Ile 24 [see Fig. 4(A)]. In IBV-X (strain Beaudette), Asn 157 is replaced by Leu 159, whereas Val 50 is conserved (Val 49), and Ile 24 becomes Leu 19. In 229E-X, Asn 157 is replaced by Tyr 151, the side-chain of which is oriented along the putative binding site for ADP-ribose but could easily reorient to interact with the adenine ring when present. Val 50 is replaced by Leu 45, and Ile 24 by Val 19.
Figure 4.
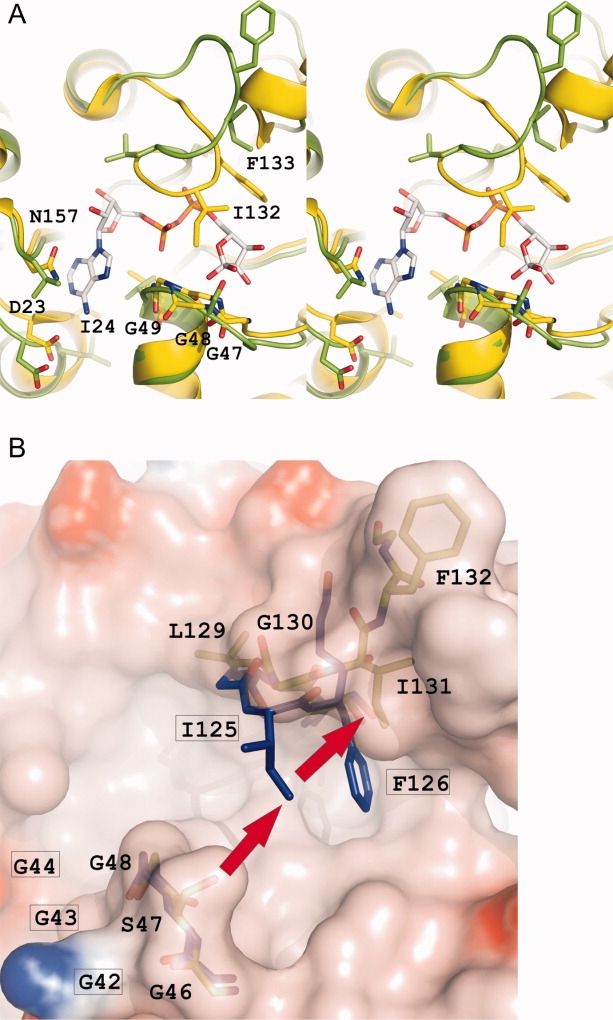
A: Stereo representation showing the binding of ADP-ribose to the X-domain of SARS-CoV,19 compared with the equivalent site in IBV-X8.5. The ADP-ribose molecule and the amino acid residues involved in the binding are represented as sticks. Carbon atoms of the ADP-ribose are colored white, carbon atoms of the protein are colored yellow and green, for SARS-X and IBV-X, respectively. Nitrogens and oxygens are colored blue and red, respectively. B: Residues 42GGG44 and 122SCGIFG127 (L11) of 229E-X (sticks, blue carbon atoms), likely involved in binding ADP-ribose, superimposed onto the corresponding site (46GSG48 and 128SLGIFG133) in IBV-X8.5 (transparent surface colored according to electrostatic potential, green sticks). Residue labels for 229E-X are framed. Note that Ile 131, and with it, the entire loop L11, in IBV-X is “pushed away” (red arrows) due to steric hindrance from Ser 47, thereby destroying the binding site for ADP-ribose.
In 229E-X, Glu 155 could interact with the hydroxy groups of the adenosine ribose when present (as it does in HCoV-NL63 X-domain; Piotrowski et al., in preparation), but in SARS-X, this glutamate becomes Leu 161 and in IBV-X (strain Beaudette), His 163, both of which are not capable of interacting with the proximal ribose.
Further along the binding site for ADP-ribose in SARS-X, the cleft is lined by three consecutive glycines [nos. 47, 48, and 49; at the C-terminus of the β3-α2 (L5) loop] on one side and the 129SAGIFG134 segment on the other [Fig. 4(A)]. The amide of Gly 48 makes a 3.0 Å hydrogen bond to the O1″ of the distal ribose and Phe 133 packs against this sugar on its opposite face. The backbone amide groups of Ser 129, Gly 131, Ile 132, and Phe 133 donate hydrogen bonds to the phosphate oxygens. Most importantly, the side-chain of Ile 132 is in close contact (3.5 Å) with Gly 48, thereby forming a “bridge” over the binding cleft.19 Even in the absence of ADP-ribose, this close interaction exists in SARS-X (4.1 Å; Ref.18). The “triple glycine” sequence is conserved in most coronavirus X-domains, with a few exceptions, including IBV (strain Beaudette, but not all IBV strains) and MHV, which have the second glycine replaced by Ser and Ala, respectively, and TGEV and FCoV, which have Met and Val, respectively, instead of the first glycine. The replacement of the “central glycine” by serine is correlated with dramatic structural differences between the X-domains of IBV (strain Beaudette) and SARS-CoV: as the close contact to the conserved isoleucine (no. 131 in IBV-X) is no longer possible, the bridge across the binding cleft collapses, and the entire β6-α5 (L11) loop carrying the 128SLGIFG133 segment is pushed away (see Fig. 4). As a consequence, the Cα atom of Ile 131 in the high-pH form of IBV-X is located 5.9 Å away from the position of the corresponding Ile 132 in SARS-X [Fig. 4(A)]. In the low-pH form, this distance is 4.4 Å, but as described earlier, the β6-α5 (L11) loop is involved in a crystal contact in this structure.
As a consequence of the conformational changes of the L11 loop in IBV-X8.5 when compared with SARS-X (and 229E-X), at least five hydrogen bonds to the phosphates and the distal ribose, as modeled into the structure, are lost in IBV-X (strain Beaudette). Furthermore, along with Ile 131, Phe 132 is displaced from the binding cleft in IBV-X [by ∼6.5 Å for its Cα, compared with SARS-X and 229E-X, see Fig. 4(B)]. Through a mutational study, this phenylalanine was identified as one of only two amino-acid residues essential for the Appr-1″-pase activity of SARS-X.19 (The other was Asn 41, which is present in both 229E-X and IBV-X, in the same orientation as in SARS-X). In view of the conformational changes described, it is no surprise that IBV-X (strain Beaudette) does not bind ADP-ribose to a significant extent.
In 229E-X, Ile 125 and Gly 43 are positioned opposite to one another, similarly to the situation in SARS-X, but their distance is >6 Å, so that no close contact exists [see Fig. 4(B)]. Yet, the hydrogen bonds between the main-chain amides of the 122SCGIFG127 and the phosphates as well as the distal ribose of ADP-ribose can all be made, as demonstrated in the structure of the highly similar X-domain of human coronavirus NL63 with ADP-ribose (Piotrowski et al., in preparation). In agreement with this, we were able to measure significant binding of ADP-ribose to 229E-X (see Fig. 1).
Discussion
The crystal structures of the X-domains of HCoV 229E and IBV (strain Beaudette), a Group-1 and a Group-3 coronavirus, reveal high similarity to the corresponding domain of SARS-CoV, which belongs to Group 2b.25 They are also similar to the macrodomains of Archaeoglobus fulgidus (AF1521; Ref.21), the noncanonical histone macroH2A,26,27 and the yeast protein Ymx7.23 In fact, the closest structural homologue to IBV-X is the macrodomain protein Er58 of E. coli (r.m.s.d. 1.54 Å; PDB ID 1SPV). Like IBV-X, this protein also lacks the first β-strand (β1); however, it should be able to bind ADP-ribose, as its triple-Gly sequence is conserved (whereas in IBV-X strain Beaudette, Gly 47 is replaced by serine). IBV-X shares 20% sequence identity with AF1521, 15% with the N-terminal domain of Ymx7, and 15% with Er58.
Many Nsps, including domains of Nsp3, have been shown to nonspecifically bind to RNA (see Ref.7, for a review). This has also been discussed for the X-domain of SARS-CoV. However, while the latter is basic (calculated pI = 8.1), this is not true for the X-domains of HCoV 229E (pI = 6.1) and IBV (pI = 5.7), making a general nucleic acid-binding function less likely.
Both 229E-X and SARS-X, as well as the X-domain of TGEV, another Group-1 coronavirus, have been shown to display Appr-1″-pase activity.17–19,28 However, by mutating Asn 36, which is essential for the Appr-1″-pase function, it was demonstrated that this enzymatic activity is not important for the HCoV-229E viability in cell culture.17 In eukaryotes, the Appr-1″-pase activity is involved in tRNA splicing.29,30 Likewise, the X-domain through its Appr-1″-pase activity was implicated in the regulation of viral RNA synthesis.4 In the tRNA metabolic pathway, the Appr-1″-pase works in concert with ADP-ribose 1″,2″-cyclic phosphodiesterase (CPD). A similar cooperation was envisioned for a fraction of coronaviruses which encode a CPD homologue.4,31,32 However, the turnover constant, kcat, measured for the Appr-1″-pase activity of isolated coronaviral X-domains, is unusually low (5–20 min−1; Ref.28) for an enzyme involved in metabolic regulation. Although the kcat for the full-length Nsp3 remains unknown, this observation was used to question a direct role of the coronavirus X-domain in viral RNA synthesis.19
If the X-domain does not participate in coronavirus RNA replication, its involvement in the regulation of virus-host interaction has to be considered. Eriksson et al.33 have shown that mice infected with mutant MHV, which carried the aforementioned Asn 41→Ala mutation (SARS-X numbering) in the X-domain, exhibited reduced cytokine production, decreased liver pathogenesis, and increased interferon-α production. We also note that other domains of Nsp3 are involved in interaction with host-cell components. The PLpro of SARS-CoV has been shown to block type-I interferon production in the host.34 The SARS-unique domain (SUD) of SARS-CoV Nsp3 has been proposed to counteract host-cell apoptosis at the translational level by binding to oligo(G) stretches in the 3′UTR of the mRNA of the apoptosis-related protein Bbc3, or to interrupt type-I interferon production by binding to similar regions of mRNA coding for signaling proteins in the MAP-kinase cascade.35 For SARS-X, Egloff et al.19 have demonstrated poly(ADP-ribose)-binding properties, suggesting that the domain may play a role in signaling pathways involving this polymer.
Poly(ADP-ribosylation) is an important posttranslational modification catalyzed by the enzyme family of the poly(ADP-ribose) polymerases (PARPs). The prototype of the family, PARP-1, is involved in the recognition of DNA damage. Binding to DNA single-strand and double-strand breaks stimulates PARP-1 to catalyze the synthesis of linear and branched polymers of ADP-ribose from the substrate nicotinamide-adenine-dinucleotide (NAD+), with concomitant release of nicotinamide.36 PARP-1 is also a major regulator of apoptosis; this is, at least in part, because of its function as a coactivator of NF-κB-dependent gene expression.37,38 PARPs are primarily localized in the nucleus, but certain members of the family appear to act in the cytoplasm, at least partly.39
Several viruses have evolved mechanisms to counteract the apoptotic activity of PARPs. The viral protein R (Vpr) of HIV-1 has been reported to form a ternary complex with the glucocorticoid receptor and PARP-1, thereby preventing translocation of the latter to the nucleus.40 Poliovirus, SARS coronavirus, and Infectious Bronchitis Virus have been shown to induce proteolytic cleavage of PARP-1.41–43 We speculate that the viral X-domains could also have an adverse affect on the activity of cytoplasmic PARPs, either by binding to the poly(ADP-ribose) chains that PARPs not only attach to target proteins but also to themselves, or by direct interaction. Such a direct interaction between the parent macrodomain, that of histone macroH2A, and nuclear PARP-1 has recently been shown to decrease the catalytic activity of the enzyme.44,45
Our data show that the IBV-X (strain Beaudette) fails to bind ADP-ribose. This observation is explained by the three-dimensional structure of the protein. The replacement by serine of the central glycine (Gly 47) of the triple-Gly sequence that forms part of the ADP-ribose binding site, pushes away the opposite Ile 131 and leads to a major conformational change of the loop (L11) carrying this residue (see Fig. 4), resulting in the loss of as many as five hydrogen bonds and one hydrophobic interaction with ADP-ribose. Therefore, we conclude that not all coronavirus X-domains bind ADP-ribose or poly(ADP-ribose) and suggest that either the X-domain in IBV (strain Beaudette) is a relict of evolution, or it has another function remaining to be characterized.
Materials and Methods
Domain prediction, cloning, expression, and purification
The X-domains of HCoV 229E and IBV (corresponding to pp1a/pp1ab residues 1270–1434 and 1005–1176, respectively) were predicted in Nsp3 using pan-coronavirus multiple sequence alignment generated with support from the Viralis software platform (Gorbalenya et al., unpublished) and previously characterized X-domains.17–19 Domains were PCR-amplified from viral RNA or cDNA and cloned into the pET-M11 vector and expressed with an N-terminal extension (MKHHHHHHPMSDYDIPTTENLYFQGAMA), including a His6 tag for purification and a TEV protease cleavage site (ENLYFQ↓G) for removal of the tag. The two proteins were produced and purified in similar ways. The respective plasmid was transformed into the competent E. coli BL21 (DE3) strain. Cultivation in 1× TY medium was performed at 37°C. Gene expression was induced by addition of 0.5 mM IPTG when cells had reached an optical density of 0.5 at 660 nm. Expression was carried out at 20°C for 5 h. Cells were harvested by centrifugation for 15 min at 7200g and 4°C. The cell pellets were resuspended in 50 mM Tris, 300 mM NaCl, 20 mM imidazole, 10% glycerol, pH 8.0, for 229E-X and 50 mM Tris, 300 mM NaCl, 20 mM imidazole, pH 8.0, for IBV-X, each including 10 mg lysozyme and one Complete EDTA-free protease inhibitor cocktail tablet (Roche Diagnostics, Mannheim, Germany). Cells were broken by French press. After ultracentrifugation (150,000g, 1 h, 4°C) of the sample, the supernatant was applied to a His-Trap FF 5-mL column (GE Healthcare, Freiburg, Germany) with a flow rate of 1 mL/min. After washing with the buffer mentioned earlier, the protein was eluted with a linear gradient of 20–500 mM imidazole. For IBV-X, the buffer was changed to cleavage buffer (50 mM Tris, 0.5 mM EDTA, 1 mM dithiothreitol (DTT), pH 8.0) by size exclusion on a Superdex 75 column (GE Healthcare, Freiburg, Germany). TEV protease and X-domain carrying an N-terminal His6 tag were combined to a molar ratio of 1 : 40 (protease: tagged IBV-X) and the sample was incubated for 16 h at 20°C. To separate tag-free IBV-X from the His6 tag and the TEV protease, the protein solution was applied to a His-Trap FF 5-mL column (GE Healthcare, Freiburg, Germany) with a flow rate of 1 mL/min. In case of 229E-X, the His6-tag-free protein tended to precipitate after incubation with the TEV protease at 20°C and even at 4°C. Therefore, purification of this X-domain was carried out without removal of the His6 tag and the protein was applied to the Superdex 75 column (GE Healthcare, Freiburg, Germany) for size exclusion and buffer exchange (10 mM Tris, 100 mM NaCl, 1 mM DTT, pH 7.5) directly after Ni2+-NTA affinity chromatography. IBV-X was treated in the same way after the cleavage. For further details, see Piotrowski et al.46
Crystallization, structure determination, and refinement
Crystallization screening was performed using a Phoenix robot (Dunn Labortechnik, Thelenberg, Germany) employing the sitting-drop vapor-diffusion technique. Drops containing 0.26 μL protein solution and 0.28 μL reservoir solution were equilibrated against 85 μL reservoir solution. Initial hits were optimized the same way.
IBV X-domain
Protein crystals appeared within 1 day at 12°C and a protein concentration of 11–13 mg/mL. Two different crystal forms could be observed when PEG 4000 or 8000 (at pH 7.5 or 8.5) and ammonium sulfate (at pH 5.6) were used as precipitants, respectively. When PEG was used as precipitant, crystal growth required the presence of 0.2M Mg2+, a PEG concentration of 20–30%, and pH 7.5 or 8.5. When ammonium sulfate was used instead, crystal growth was restricted to a narrow pH range around 5.6. The quality of the latter crystals improved upon addition of 200 mM K-,Na-tartrate to the crystallization experiment (see Table I for details). For diffraction data collection of crystals grown from PEG 4000, no additional cryoprotectant was needed, whereas 30% glycerol had to be added as cryoprotectant when (NH4)2SO4 was used as precipitant.
HCoV-229E X-domain
For crystallization, a protein concentration of 10–13 mg/mL was used. Protein crystals appeared within 2 days at 12°C when PEG 400, 4000, 6000, 8000, or 10,000 in a concentration of 10–30% was used as precipitant at pH 7.5 to 8.5 (see Table I for details). The cryoprotectant was 30% PEG 400.
Collection and processing of diffraction data
X-ray diffraction data from crystals of IBV-X were collected using synchrotron radiation of λ = 0.808 Å at beamline X13 (University of Hamburg - University of Lübeck - EMBL) at DESY (Deutsches Elektronen-Synchrotron), Hamburg, Germany. Data collection from crystals of 229E-X was carried out using synchrotron radiation (λ = 1.04 Å) at beamline I911, MAX-lab, Lund, Sweden. Diffraction data for IBV-X8.5 were processed, reduced, and scaled with DENZO/SCALEPACK (HKL Suite; Ref.47). Data for 229E-X and IBV-X5.6 were processed with MOSFLM48 and scaled with the programm SCALA from the CCP4 suite.49
Structure determination and refinement
The structures were determined by molecular replacement using the CCP4 program Phaser.50–52 For HCoV 229E, the X-domain of HCoV NL63 (Piotrowski et al., in preparation; PDB ID 2VRI) was used as a search model. In case of IBV-X5.6, molecular replacement was not straightforward and required the usage of eight superimposed coordinate sets. This ensemble included structures of similar fold as predicted by FFAS03 (Ref.53; PDB codes: 1YD9, 1ZR3, 2DX6, 1SPV, 1ZR5, 2ACF) and previously solved structures of the X-domain from HCoV NL63 (Piotrowski et al., in preparation) and HCoV 229E (this communication). Truncation of flexible regions in the search model was crucial to obtain a clear molecular replacement solution. To improve the relatively poor overall electron density of the initial model, protein backbone conformations were remodeled, ambiguous side-chains were changed to alanine, and two helices were adjusted manually. The model was completed using ARP/wARP.54,55 The structure of IBV-X8.5 was determined by molecular replacement using the structure of IBV-X5.6 as a search model. Models were built into electron density using COOT,56 and refined by REFMAC5.57 Structure superimposition and calculation of r.m.s. deviations were carried out using ALIGN.58
Atomic coordinates have been deposited with the Protein Data Bank (PDB ID of 229E-X: 3EJG, IBV-X8.5: 3EJF, and IBV-X5.6: 3EKE).
Zone-interference gel electrophoresis
The ZIGE experiment was based on Abrahams et al.60 and optimized for the conditions used here. A 1% agarose gel in 1× TBE buffer (1.08 g Tris, 0.55 g boric acid, 0.093 g Na-EDTA in 100 mL Aqua dest., pH 8.3 adjusted using acetic acid) was prepared with large slots for the ligand solution and small slots for the protein/ligand solution.61 After polymerization, the large slots were filled with 100 μL of ADP-ribose solution (100 μM or 1 mM) including 10% DMSO/bromophenol blue (BPB). The small slots were filled with 10 μL of the protein/ADP-ribose solution with a protein concentration of 10 μM, an ADP-ribose concentration of 100 μM or 1 mM, and an additional 10% DMSO/BPB. The electrophoresis was performed in 0.5× TBE buffer at 100 mA and 4°C for 1 h. Afterward, the gel was incubated in denaturing solution (3.5% α-sulfosalycylic acid, 10% trichloroacetic acid) for 10 min, followed by incubation in washing buffer (8% acetic acid, 15% ethanol) for an additional 10 min. The gel was stained by Coomassie Brilliant Blue (0.25% Coomassie Brilliant Blue, 15% ethanol, 8% acetic acid, 10% methanol) for 20 min and destained. The figures were created using Pymol.59
Isothermal titration calorimetry
Isothermal titration calorimetry was performed at 25°C using a VP-ITC instrument (Microcal, Milton Keynes, United Kingdom). Reactions were carried out in 20 mM Tris, 100 mM NaCl, 10 mM MgCl2, 1 mM DTT, pH 7.5, for 229E-X and in TBE buffer, pH 8.3, for IBV-X, and successive injections of 1.5 mM ADP-ribose for 229E-X and 700 μM for IBV-X. The protein concentration was 33 μM for 229E-X and 50 μM for IBV-X.
Note added in proof
After acceptance and typesetting of this publication, we became aware of a paper by Xu et al., e-published on Nov 05, 2008, by J. Virol. online ahead of print, that describes a crystal structure of the X-domain of IBV strain M41 in complex with ADP-ribose. In contrast to the X-domain of IBV strain Beaudette, which is the subject of the present paper, residue 47 is a glycine rather than a serine in the protein of IBV strain M41. Thus, the canonical triple-glycine sequence seen in other coronavirus X-domains is conserved and ADP-ribose binding is not prevented, in contrast to the X-domain of IBV strain Beaudette. The observations described in the Xu et al. publication thus support our conclusions.
Acknowledgments
The authors thank Ulrike Langerwisch and Silke Schmidtke for expert technical support, Jeroen R. Mesters for discussions, Arie Geerlof and Paul Tucker for an expression construct of TEV protease, John Ziebuhr and Paul Britton for providing HCoV-229E and IBV reagents, respectively, as well as Dmitry Samborskiy and Alexander Kravchenko for Viralis support. RH thanks the Fonds der Chemischen Industrie for continuous support.
References
- 1.Gorbalenya AE, Enjuanes L, Ziebuhr J, Snijder EJ. Nidovirales: evolving the largest RNA virus genome. Virus Res. 2006;117:17–37. doi: 10.1016/j.virusres.2006.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mihindukulasuriya KA, Wu G, St Leger J, Nordhausen RW, Wang D. Identification of a novel coronavirus from a beluga whale by using a panviral microarray. J Virol. 2008;82:5084–5088. doi: 10.1128/JVI.02722-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cavanagh D. Coronavirus avian infectious bronchitis virus. Vet Res. 2007;38:281–297. doi: 10.1051/vetres:2006055. [DOI] [PubMed] [Google Scholar]
- 4.Snijder EJ, Bredenbeek PJ, Dobbe JC, Thiel V, Ziebuhr J, Poon LLM, Guan Y, Rozanov M, Spaan WJM, Gorbalenya AE. Unique and conserved features of genome and proteome of SARS-coronavirus, an early split-off from the coronavirus group 2 lineage. J Mol Biol. 2003;331:991–1004. doi: 10.1016/S0022-2836(03)00865-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.van der Hoek L, Pyrc K, Jebbink MF, Vermeulen-Oost W, Berkhout RJM, Wolthers KC, Wertheim-van Dillen PME, Kaandorp J, Spaargaren J, Berkhout B. Identification of a new human coronavirus. Nat Med. 2004;10:368–373. doi: 10.1038/nm1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bartlam M, Yang H, Rao Z. Structural insights into SARS coronavirus proteins. Curr Opin Struct Biol. 2005;15:664–672. doi: 10.1016/j.sbi.2005.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hilgenfeld R, Tan J, Chen S, Shen X, Jiang H. Structural proteomics of emerging viruses: the examples of SARS-CoV and other coronaviruses. In: Sussman JL, Silman I, editors. Structural proteomics and its impact on the life sciences. Singapore: World Scientific; 2008. pp. 361–433. [Google Scholar]
- 8.Anand K, Ziebuhr J, Wadhwani P, Mesters JR, Hilgenfeld R. Coronavirus main proteinase (3CLpro) structure: basis for design of anti-SARS drugs. Science. 2003;300:1763–1767. doi: 10.1126/science.1085658. [DOI] [PubMed] [Google Scholar]
- 9.Ponnusamy R, Moll R, Weimar T, Mesters JR, Hilgenfeld R. Variable oligomerization modes in coronavirus non-structural protein 9. J Mol Biol. 383:1081–1096. doi: 10.1016/j.jmb.2008.07.071. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Xue X, Yu H, Yang H, Xue F, Wu Z, Shen W, Li J, Zhou Z, Ding Y, Zhao Q, Zhang XC, Liao M, Bartlam M, Rao Z. Structures of two coronavirus main proteases: implications for substrate binding and antiviral drug design. J Virol. 2008;82:2515–2527. doi: 10.1128/JVI.02114-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Oostra M, Hagemeijer MC, van Gent M, Bekker CPJ, te Lintelo EG, Rottier PJM, de Haan CAM. Topology and membrane anchoring of the coronavirus replication complex: not all of the hydrophobic domains of nsp3 and nsp6 are membrane-spanning. J Virol. doi: 10.1128/JVI.01219-08. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kanjanahaluethai A, Chen Z, Jukneliene D, Baker SC. Membrane topology of murine coronavirus replicase nonstructural protein 3. Virology. 2007;36:391–401. doi: 10.1016/j.virol.2006.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ziebuhr J, Thiel V, Gorbalenya AE. The autocatalytic release of a putative virus transcription factor from its polyprotein precursor involves two paralogous papain-like proteases that cleave the same peptide bond. J Biol Chem. 2001;276:33220–33232. doi: 10.1074/jbc.M104097200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Knoops K, Kikkert M, van den Worm SHE, Zevenhoven-Dobbe JC, van der Meer Y, Koster AJ, Mommaas AM, Snijder EJ. SARS-Coronavirus replication is supported by a reticulovesicular network of modified endoplasmic reticulum. PLoS Biol. 2008;6:e226. doi: 10.1371/journal.pbio.0060226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gorbalenya AE, Koonin EV, Lai MM-C. Putative papain-related thiol proteases of positive-strand RNA viruses. Identification of rubi- and aphthovirus proteases and delineation of a novel conserved domain associated with proteases of rubi-, alpha- and coronaviruses. FEBS Lett. 1991;288:201–205. doi: 10.1016/0014-5793(91)81034-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Martzen MR, McCraith SM, Spinelli SL, Torres FM, Fields S, Grayhack EJ, Phizicky EM. A biochemical approach for identifying genes by the activity of their products. Science. 1999;286:1153–1155. doi: 10.1126/science.286.5442.1153. [DOI] [PubMed] [Google Scholar]
- 17.Putics Á, Filipowicz W, Hall J, Gorbalenya AE, Ziebuhr J. ADP-ribose-1″-monophosphatases: a conserved coronavirus enzyme that is dispensable for viral replication in tissue culture. J Virol. 2005;79:12721–12731. doi: 10.1128/JVI.79.20.12721-12731.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Saikatendu KS, Joseph JS, Subramanian V, Clayton T, Griffith M, Moy K, Velasquez J, Neuman BW, Buchmeier MJ, Stevens RC, Kuhn P. Structural basis of severe acute respiratory syndrome coronavirus ADP-ribose-1″-phosphate dephosphorylation by a conserved domain of nsP3. Structure. 2005;13:1665–1675. doi: 10.1016/j.str.2005.07.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Egloff M-P, Malet H, Putics Á, Heinonen M, Dutartre H, Frangeul A, Gruez A, Campanacci V, Cambillau C, Ziebuhr J, Ahola T, Canard B. Structural and functional basis for ADP-ribose and poly(ADP-ribose) binding by viral macro domains. J Virol. 2006;80:8493–8502. doi: 10.1128/JVI.00713-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pehrson JR, Fried VA. MacroH2A, a core histone containing a large nonhistone region. Science. 1992;257:1398–1400. doi: 10.1126/science.1529340. [DOI] [PubMed] [Google Scholar]
- 21.Allen MD, Buckle AM, Cordell SC, Löwe J, Bycroft M. The crystal structure of AF1521 a protein from Archaeoglobus fulgidus with homology to the non-histone domain of macroH2A. J Mol Biol. 2003;330:503–511. doi: 10.1016/s0022-2836(03)00473-x. [DOI] [PubMed] [Google Scholar]
- 22.Karras GI, Kustatscher G, Buhecha HR, Allen MD, Pugieux C, Sait F, Bycroft M, Ladurner AG. The macro domain is an ADP-ribose binding module. EMBO J. 2005;24:1911–1920. doi: 10.1038/sj.emboj.7600664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kumaran D, Eswaramoorthy F, Studier FW, Swaminathan S. Structure and mechanism of ADP-ribose-1″-monophosphatase (Appr-1″-pase), a ubiquitous cellular processing enzyme. Protein Sci. 2005;14:719–726. doi: 10.1110/ps.041132005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wei L, Chen C, Zhao Q, Li C, Cong L, Xu X, Ma Y, Liao M, Xu Y, Rao Z. Purification, crystallization and preliminary crystallographic analysis of avian infectious bronchitis virus nsp3 ADRP domain. Acta Crystallogr Sect F Struct Biol Cryst Commun. 2008;64:802–804. doi: 10.1107/S1744309108024391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gorbalenya AE, Snijder EJ, Spaan WJ. Severe acute respiratory syndrome coronavirus phylogeny: toward consensus. J Virol. 2004;78:7863–7866. doi: 10.1128/JVI.78.15.7863-7866.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chakravarthy S, Gundimella SK, Caron C, Perche PY, Pehrson JR, Khochbin S, Luger K. Structural characterization of the histone variant macroH2A. Mol Cell Biol. 2005;25:7616–7624. doi: 10.1128/MCB.25.17.7616-7624.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kustatscher G, Hothorn M, Pugieux C, Scheffzek K, Ladurner AG. Splicing regulates NAD metabolite binding to histone macroH2A. Nat Struct Mol Biol. 2005;12:624–625. doi: 10.1038/nsmb956. [DOI] [PubMed] [Google Scholar]
- 28.Putics Á, Gorbalenya AE, Ziebuhr J. Identification of protease and ADP-ribose 1″-monophosphatase activities associated with transmissible gastroenteritis virus non-structural protein 3. J Gen Virol. 2006;87:651–656. doi: 10.1099/vir.0.81596-0. [DOI] [PubMed] [Google Scholar]
- 29.Culver GM, Consaul SA, Tycowski KT, Filipowicz W, Phizicky EM. tRNA splicing in yeast and wheat germ. J Biol Chem. 1994;40:24928–24934. [PubMed] [Google Scholar]
- 30.Shull NP, Spinelli SL, Phizicky EM. A highly specific phosphatase that acts on ADP-ribose 1″-phosphate, a metabolite of tRNA splicing in Saccharomyces cerevisiae. Nucleic Acids Res. 2005;33:650–660. doi: 10.1093/nar/gki211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schwarz B, Routledge E, Siddell SG. Murine coronavirus nonstructural protein ns2 is not essential for virus replication in transformed cells. J Virol. 1990;64:4784–4791. doi: 10.1128/jvi.64.10.4784-4791.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sperry SM, Kazi L, Graham RL, Baric RS, Weiss SR, Denison MR. Single-amino-acid substitutions in open reading frame (ORF) 1b-nsp14 and ORF 2a proteins of the coronavirus mouse hepatitis virus are attenuating in mice. J Virol. 2005;79:3391–3400. doi: 10.1128/JVI.79.6.3391-3400.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Eriksson KK, Cervantes-Barragan L, Ludewig B, Thiel V. A coronavirus replicase function impacts on mouse hepatitis virus-induced liver pathology. 2007. In the Third European Congress of Virology, Nürnberg, September 1–5; Abstract HBV032.
- 34.Devaraj SG, Wang N, Chen Z, Chen Z, Tseng M, Barretto N, Lin R, Peters CJ, Tseng C-TK, Baker SC, Li K. Regulation of IRF-3-dependent innate immunity by the papain-like protease domain of the severe acute respiratory syndrome coronavirus. J Biol Chem. 2007;282:32208–32221. doi: 10.1074/jbc.M704870200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tan J, Kusov Y, Mutschall D, Tech S, Nagarajan K, Hilgenfeld R, Schmidt CL. The “SARS-unique domain” (SUD) of SARS coronavirus is an oligo(G)-binding protein. Biochem Biophys Res Commun. 2007;364:877–882. doi: 10.1016/j.bbrc.2007.10.081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Benjamin RC, Gill DM. Poly(ADP-ribose) synthesis in vitro programmed by damaged DNA. J Biol Chem. 1981;255:10502–10508. [PubMed] [Google Scholar]
- 37.Hassa PO, Hottiger MO. The functional role of poly(ADP-ribose)polymerase 1 as novel coactivator of NF-κB in inflammatory disorders. Cell Mol Life Sci. 2002;59:1534–1553. doi: 10.1007/s00018-002-8527-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chang WJ, Alvarez-Gonzalez R. The sequence-specific DNA binding of NF-κB is reversibly regulated by the automodification reaction of poly(ADP-ribose) polymerase 1. J Biol Chem. 2001;276:47664–47670. doi: 10.1074/jbc.M104666200. [DOI] [PubMed] [Google Scholar]
- 39.Nguewa PA, Fuertes MA, Valladares B, Alonso C, Pérez JM. Poly(ADP-ribose) polymerases: homology, structural domains and functions. Novel therapeutical applications. Prog Biophys Mol Biol. 2005;88:143–172. doi: 10.1016/j.pbiomolbio.2004.01.001. [DOI] [PubMed] [Google Scholar]
- 40.Muthumani K, Choo AY, Zong W-X, Madesh M, Hwang DS, Premkumar A, Thieu KP, Emmanuel J, Kumar S, Thompson CB, Weiner DB. The HIV-1 Vpr and glucocorticoid receptor complex is a gain-of-function interaction that prevents the nuclear localization of PARP-1. Nature Cell Biol. 2006;8:170–179. doi: 10.1038/ncb1352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Calandria C, Irurzun A, Barco A, Carrasco L. Individual expression of poliovirus 2Apro and 3Cpro induces activation of caspase-3 and PARP cleavage in HeLa cells. Virus Res. 2004;104:39–49. doi: 10.1016/j.virusres.2004.02.042. [DOI] [PubMed] [Google Scholar]
- 42.Zhang L, Wei L, Jiang D, Wang J, Cong X, Fei R. SARS-CoV nucleocapsid protein induced apoptosis of COS-1 mediated by the mitochondrial pathway. Artif Cells Blood Substit Immobil Biotechnol. 2007;35:237–253. doi: 10.1080/10731190601188422. [DOI] [PubMed] [Google Scholar]
- 43.Zhong Y, Tam JP, Liu DX. Functional characterization of Mcl-1 and Bak in coronavirus-induced apoptosis. In 9th International Conference on Nidoviruses; June 22–27; Oxford. 2008. Abstract P45. [Google Scholar]
- 44.Ouararhni K, Hadj-Slimane R, Ait-Si-Ali S, Robin P, Mietton F, Harel-Bellan A, Dimitrov S, Hamiche A. The histone variant mH2A1.1 interferes with transcription by down-regulating PARP-1 enzymatic activity. Genes Dev. 2006;20:3324–3336. doi: 10.1101/gad.396106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nusinow DA, Hernández-Muñoz I, Fazzio TG, Shah GM, Kraus WL, Panning B. Poly(ADP-ribose) polymerase 1 is inhibited by a histone H2A variant, macroH2A, and contributes to silencing of the inactive X chromosome. J Biol Chem. 2007;282:12851–12859. doi: 10.1074/jbc.M610502200. [DOI] [PubMed] [Google Scholar]
- 46.Piotrowski Y, Ponnusamy R, Glaser S, Daabach A, Moll R, Hilgenfeld R. Production of coronavirus nonstructural proteins in soluble form for crystallization. In: Cavanagh D, editor. SARS- and other coronaviruses. Vol. 454. Totowa, NJ: Humana Press; 2008. pp. 139–159. Methods in molecular biology. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 48.Leslie AGW. Joint CCP4 + ESF-EAMCB newsletter on protein crystallography, No. 26: Recent changes to the MOSFLM package for processing film and image plate data. 1992. pp. 27–33. Davesbury Laboratory, Warrington, UK.
- 49.Collaborative Computational Project, Number 4. The CCP4 suite: programms for protein crystallography. Acta Crystallogr Sect D Biol Crystallogr. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 50.Read RJ. Pushing the boundaries of molecular replacement with maximum likelihood. Acta Crystallogr Sect D Biol Crystallogr. 2001;57:1373–1382. doi: 10.1107/s0907444901012471. [DOI] [PubMed] [Google Scholar]
- 51.Storoni LC, McCoy AJ, Read RJ. Likelihood-enhanced fast rotation functions. Acta Crystallogr Sect D Biol Crystallogr. 2004;60:432–438. doi: 10.1107/S0907444903028956. [DOI] [PubMed] [Google Scholar]
- 52.McCoy AJ, Grosse-Kunstleve RW, Storoni LC, Read RJ. Likelihood-enhanced fast translation functions. Acta Crystallogr Sect D Biol Crystallogr. 2005;61:458–464. doi: 10.1107/S0907444905001617. [DOI] [PubMed] [Google Scholar]
- 53.Jaroszewski L, Rychlewski L, Li Z, Li W, Godzik A. FFAS03: a server for profile-profile sequence alignments. Nucleic Acids Res. 2005;33:W284–W288. doi: 10.1093/nar/gki418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lamzin VS, Wilson KS. Automated refinement of protein models. Acta Crystallogr Sect D Biol Crystallogr. 1993;49:129–149. doi: 10.1107/S0907444992008886. [DOI] [PubMed] [Google Scholar]
- 55.Perrakis A, Sixma TK, Wilson KS, Lamzin VS. wARP: improvement and extension of crystallographic phases by weighted averaging of multiple refined dummy atomic models. Acta Crystallogr Sect D Biol Crystallogr. 1997;53:448–455. doi: 10.1107/S0907444997005696. [DOI] [PubMed] [Google Scholar]
- 56.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr Sect D Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 57.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr Sect D Biol Crystallogr. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 58.Cohen GE. ALIGN: a programm to superimpose protein coordinates, accounting for insertions and deletions. J Appl Crystallogr. 1997;30:1160–1161. [Google Scholar]
- 59.DeLano WL. The PyMOL Molecular Graphics System. San Carlos, CA, USA: DeLano Scientific; 2002. [Google Scholar]
- 60.Abrahams JP, Kraal B, Bosch L. Zone-interference gel electrophoresis: a new method for studying weak protein-nucleic acid complexes under native equilibrium conditions. Nucleic Acids Res. 1988;16:10099–10108. doi: 10.1093/nar/16.21.10099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Matthes N, Mesters JR, Coutard B, Canard B, Snijder EJ, Moll R, Hilgenfeld R. The non-structural protein Nsp10 of mouse hepatitis virus binds zinc ions and nucleic acids. FEBS Lett. 2006;580:4143–4149. doi: 10.1016/j.febslet.2006.06.061. [DOI] [PMC free article] [PubMed] [Google Scholar]