

Abstract
The diversity in substrate recognition spectra exhibited by various β-lactamases can result from one or a few mutations in the active-site area. Using Escherichia coli TEM-1 β-lactamase as a template that efficiently hydrolyses penicillins, we performed site-saturation mutagenesis simultaneously on two opposite faces of the active-site cavity. Residues 104 and 105 as well as 238, 240, and 244 were targeted to verify their combinatorial effects on substrate specificity and enzyme activity and to probe for cooperativity between these residues. Selection for hydrolysis of an extended-spectrum cephalosporin, cefotaxime (CTX), led to the identification of a variety of novel mutational combinations. In vivo survival assays and in vitro characterization demonstrated a general tendency toward increased CTX and decreased penicillin resistance. Although selection was undertaken with CTX, productive binding (KM) was improved for all substrates tested, including benzylpenicillin for which catalytic turnover (kcat) was reduced. This indicates broadened substrate specificity, resulting in more generalized (or less specialized) variants. In most variants, the G238S mutation largely accounted for the observed properties, with additional mutations acting in an additive fashion to enhance these properties. However, the most efficient variant did not harbor the mutation G238S but combined two neighboring mutations that acted synergistically, also providing a catalytic generalization. Our exploration of concurrent mutations illustrates the high tolerance of the TEM-1 active site to multiple simultaneous mutations and reveals two distinct mutational paths to substrate spectrum diversification.
Keywords: TEM-1 β-lactamase, combinatorial mutagenesis, protein engineering, antibiotic resistance, extended-spectrum β-lactamase, plasticity
Introduction
The rapid evolution of the hydrolase specificity in β-lactamases is an important contributor to antibiotic resistance worldwide. β-Lactamases have developed ever-increasing sequence diversity, with decades of penicillin and cephalosporin antibiotic use providing the required selective pressure. The appearance and combination of point mutations have resulted in varied recognition spectra for β-lactam antibiotics of earlier generations. More recently developed cephalosporins were designed to elude this process but, increasingly, resistant mutants are reported. Such extended-spectrum β-lactamases (ESBLs) efficiently hydrolyze β-lactam antibiotics not normally recognized by native β-lactamases. The amino acids responsible for ESBL activity are generally near the substrate-binding site and do not directly contribute to catalysis.1 Residues at positions 104, 238, and 240 (numbering according to Ambler et al.2) are frequently associated with ESBL resistance and have been previously characterized by mutagenesis in several class A β-lactamases.3–5 Mutation G238S6 appears to expand the active-site cavity to accommodate substrates such as the extended-spectrum cephalosporin, cefotaxime (CTX), which harbors a large, branched R1 substituent (see Fig. 1).7–10 Mutation E104K also results in ESBL activity, through the possible formation of an electrostatic bond between the side chain of Lys-104 and the bulky R1 substituent of an extended-spectrum cephalosporin.3,4,11 Similarly, the E240K substitution has been proposed to improve binding of the extended-spectrum cephalosporins ceftazidime (CAZ) and CTX.7
Figure 1.
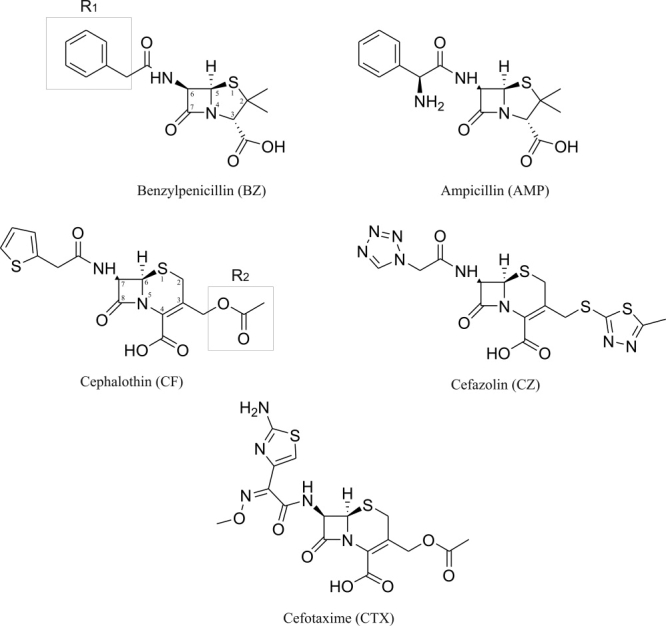
β-lactam antibiotics used in this study. BZ and AMP are classical penicillins, CF and CZ are first-generation cephalosporins, whereas CTX is an extended-spectrum cephalosporin. The location of R1 and R2 substituents is identified to highlight differences between substrates. Note the bulkiness of R1 from CTX relative to CF and CZ.
TEM-1 β-lactamase belongs to the serine hydrolase class A family and is the most prevalent member found among gram-negative bacteria.12 Many drug-resistant mutants of TEM-1, both clinically identified and laboratory-generated, have been characterized.1,6,13,14 Importantly, the acquisition of resistance toward extended-spectrum cephalosporins has generally resulted in a concomitant decrease of resistance toward classical substrates such as penicillins and first-generation cephalosporins.3,15,16 For example, mutant G238S increased TEM-1 resistance toward CTX 8-fold with a concomitant 4-fold decrease in ampicillin (AMP) resistance.5
Here, we investigate the potential for increased recognition of an extended-spectrum cephalosporin by targeted sequence variation in TEM-1. The combination of mutations in β-lactamases can yield multiplicative effects as opposed to simple additive effects on enzyme activity. Such synergy has been observed for the double mutants G238S:E240K8 and E104M:G238S16 toward CAZ and CTX hydrolysis, respectively. The E104M:G238S variant has the characteristic that the two mutations belong to opposite faces of the active-site cavity (see Fig. 2). These residues may form contacts with distinct atoms of the substrate such that the effect of their concurrent mutation cannot be predicted. To verify if further, similarly successful solutions to CTX-resistance frequently result from multiple, simultaneous active-site mutations, we explored concurrent mutations of a restricted subset of residues in close proximity to the substrate and which include known solutions to CTX resistance. We also investigated whether switching the substrate profile was general or whether it was mutation-specific. To this end, we mutated Glu-104 as well as its neighbor Tyr-105, which is moderately conserved in class A β-lactamases.17 On the opposite face of the active-site cavity, we mutated Gly-238 and Glu-240 as well as Arg-244. The latter is partially conserved in class A β-lactamases2 and its mutation has led to inhibitor resistance,6,13 although no changes in substrate spectrum recognition have been reported upon its mutation. Because of its close three-dimensional proximity to residues Gly-238 and Glu-240, we included Arg-244 in the combinatorial mutation scheme, to verify whether the effect of mutating it may be different in the context of simultaneously mutated neighboring residues. In addition, by mutating opposite faces of the active site simultaneously, we probed the extent to which mutations that contact different functional groups of the substrate work in concert to modify substrate recognition.
Figure 2.
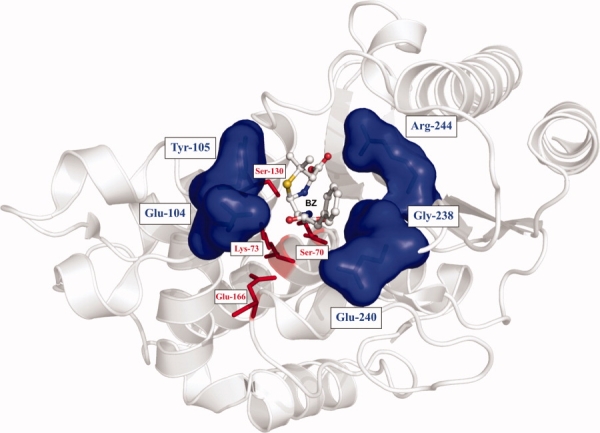
Schematic representation of the active site of TEM-1 β-lactamase. Residues targeted for saturation mutagenesis (Glu-104, Tyr-105, Gly-238, Glu-240, and Arg-244) are represented in blue according to their Connolly surface, defined by a 1.6 Å solvent radius. These amino acids belong to two distinct faces of the active-site cavity. Residues known to be involved in the catalytic mechanism (Ser-70, Lys-73, Ser-130, and Glu-166) are colored in red. The cocrystallized BZ is shown in ball-and-sticks representation and is displayed to illustrate its proximity to the targeted active-site positions. PDB coordinates 1FQG. [Color figure can be viewed in the online issue, which is available at www.interscience.wiley.com.]
Selection of the mutated TEM-1 variants yielded a variety of combinatorial mutants with increased extended-spectrum cephalosporin resistance and decreased penicillin resistance. Although mutants displayed increased affinity toward all substrates, turnover was enhanced only for CTX and considerably reduced for penicillins. Overall, our results show that the active site of TEM-1 β-lactamase tolerates a variety of multiple simultaneous amino acid substitutions, indicating active-site plasticity that allows for substrate spectrum diversification.
Results
Selection of resistant mutants from the combinatorial library
To gain a better understanding of the role of specific residues giving rise to ESBLs, TEM-1 residues 104, 105, 238, 240, and 244 (see Fig. 2) were combinatorially mutated using a semi-random saturation mutagenesis approach.18 Prior to selection, DNA sequencing of 36 randomly picked clones from the library revealed no unexpected mutations. The randomized nucleotides diverged quantitatively from the expected distribution although all expected nucleotides were encoded at a frequency ensuring adequate randomization of the five target residues.
Selection against CTX yielded 34 resistant clones, several of which were redundant (Table I). The double mutant EYSKR* was the most prevalent variant selected, representing over one-third of the sequenced population. The 4 double-, 3 triple-, and 4 quadruple-mutant combinations identified include nine novel mutational combinations, as well as two previously characterized combinations (EYSKR and EYSRR).7,19
Table I.
TEM-1 β-Lactamase Mutants Selected on Media Containing 250 ng/mL CTX
| Mutated positionsa | |||||
|---|---|---|---|---|---|
| 104 | 105 | 238 | 240 | 244 | Frequency of occurrence |
| E | Y | G | E | R | 0 (WT TEM-1) |
| E | Y | S | K | R | 14 |
| E | Y | S | R | R | 4 |
| Q | W | S | E | R | 4 |
| E | Y | S | T | R | 3 |
| V | N | S | T | R | 2 |
| R | S | S | T | R | 2 |
| E | Y | N | H | R | 1 |
| P | H | S | E | R | 1 |
| R | W | S | E | R | 1 |
| S | W | S | S | R | 1 |
| V | N | S | L | R | 1 |
Bold-type identifies mutations relative to WT TEM-1.
The opposite active-site walls 104-105 and 238-240-244 exhibited different mutational patterns in the selected variants (Table I). Roughly two-thirds of all variants displayed native amino acids at positions 104 and 105 while position 238 was mutated in all cases. In fact, with the sole exception of variant EYNHR, all selected mutants encoded the G238S mutation, which is sufficient to confer extended-spectrum antibiotic resistance on its own.6 Similarly, position 240 was mutated in all but three cases. Interestingly, all selected mutants carried the native arginine at position 244, suggesting that this residue is intolerant to mutation in the context of selection against CTX.
We selected a subset of the mutants for further characterization, with the goal of obtaining a broad amino acid divergence at the five mutated positions. The novel variants VNSLR, SWSSR, PHSER, and EYNHR were thus chosen for characterization by in vivo antibiotic resistance and enzyme kinetics. Mutant VNSTR was also characterized for comparison to VNSLR, as they differ only at codon 240 (Leu or Thr).
Bacterial antibiotic susceptibility
Minimal inhibitory concentrations (MICs) were determined for E. coli XL1-Blue expressing the TEM-1 mutants VNSTR, VNSLR, SWSSR, PHSER, and EYNHR to assess the effects of the multiple mutations on bacterial antibiotic resistance (Table II). E. coli expressing or lacking wild-type (WT) TEM-1 served as controls. The point-mutant E240H was also created and its MICs determined to assess the impact of this mutation, which is contained in the variant EYNHR. MICs were obtained for two classical penicillins (benzylpenicillin (BZ) and AMP), two first-generation cephalosporins (cephalothin (CF) and cefazolin (CZ)), and the extended-spectrum cephalosporin CTX that was used during selection.
Table II.
Minimum Inhibitory Concentrations of E. coli XL1-Blue Expressing Wild-Type TEM-1 or Mutant β-Lactamases
| Substrates (μg/mL) | |||||
|---|---|---|---|---|---|
| β-Lactamase variants | CTX | AMP | BZ | CF | CZ |
| Nonea | 0.125 | 64 | 250 | 32 | 8 |
| WT TEM-1 (EYGER)b | 0.125 | 10,000 | 10,000 | 125 | 64 |
| VNSLR | 0.5 | 250 | 500 | 64 | 64 |
| PHSER | 0.5 | 250 | 500 | 125 | 32 |
| EYNHR | 0.5 | 500 | 500 | 64 | 16 |
| SWSSR | 0.75 | 1,000 | 1,000 | 64 | 32 |
| VNSTR | 2 | 250 | 1,000 | 64 | 32 |
| E240Hc (EYGHR) | 0.125 | 1,000 | 1,000 | 32 | 32 |
E. coli XL1-Blue
The five-letter nomenclature identifies mutants according to the one-letter code of the amino acids at positions 104, 105, 238, 240, and 244, respectively. Bold-type identifies mutations relative to WT TEM-1. Variants are listed according to increasing CTX MIC.
Control point-mutant, not obtained by selection against CTX.
The concentrations inhibiting bacterial growth were higher for penicillins (64–10,000 μg/mL) than for cephalosporins (0.125–125 μg/mL). This result was not unexpected as cephalosporins were developed to counteract the resistance observed with standard penicillins.20,21 This difference was yet more pronounced for CTX, which belongs to a subsequent generation of cephalosporins that are not efficiently hydrolyzed by WT TEM-1.22 As a consequence, the WT MIC for CTX was no higher than for the negative control (Table II).
MICs for CTX were significantly increased for the five characterized combinatorial mutants, up to one order of magnitude, consistent with the fact that the variants had been selected against CTX (Table II). The sensitivity of all combinatorial mutants toward first-generation cephalosporins (CF and CZ) remained in the same range as WT TEM-1: a maximum 2-fold decrease for CF was observed. Importantly, these CTX-resistant variants all exhibited an important reduction in resistance toward penicillin substrates. For both AMP and BZ, MIC values dropped by at least one order of magnitude relative to WT TEM-1.
Despite their different mutations, the MICs observed for the five characterized combinatorial variants were similar, with only 2- to 4-fold differences amongst each other. This suggests that TEM-1 can withstand various combinations of multiple mutations in the vicinity of the active site while maintaining a certain level of resistance, albeit lower than WT TEM-1 in the case of penicillins. The E240H point mutant, which had not been identified upon selection for CTX resistance, was created as a control to compare the impact of that mutation alone to its contribution within mutant EYNHR. In contrast to the combinatorial variants, point mutant E240H showed no increase in CTX hydrolysis and significantly decreased MICs for all other substrates tested.
Enzyme kinetics
Steady-state kinetic parameters (kcat and KM) were determined for the five selected mutants as well as for WT TEM-1 and point-mutant E240H, toward BZ, CF, CZ, and CTX (Table III). The low molar extinction coefficient of AMP (900 M−1 cm−1; Ref.8) resulting in a weak signal-to-noise ratio, as well as deviations from linear kinetics (lag phase) precluded spectrophotometric characterization of its hydrolysis. In agreement with previous reports,5,24 WT TEM-1 exhibited low CTX turnover (kcat) and poor productive CTX binding (KM), resulting in weak catalytic efficiency (kcat/KM) toward CTX hydrolysis (Table III). Comparison of catalytic efficiencies of the selected variants relative to the WT (Table III) yielded high values for CTX and low values for BZ. Importantly, the greatest differences lie in comparing all selected variants to the WT; differences among the selected variants were much less important for any given parameter (see Fig. 3), consistent with the results of in vivo resistance experiments (Table II). Interestingly, while all selected variants displayed similar improvements in KM relative to WT toward CTX (extended-spectrum cephalosporin) and CF (first- generation cephalosporin), the relative turnover rate increased only for CTX hydrolysis, resulting in greater improvements in relative catalytic efficiency toward CTX (Table III).
Table III.
Kinetic Parameters for β-Lactam Hydrolysis by Wild-Type and Mutant TEM-1 β-Lactamases Containing Mutation G238S or Related to Variant EYNHR
| Substrates | TEM-1 variants | kcat (s−1) | kcat relative to WT | KM (μM) | KM relative to WT | kcat/KM relative to WT | kcat/KM relative to G238S |
|---|---|---|---|---|---|---|---|
| CTX | TEM-1 WT (EYGERa) | 0.74 ± 0.1b | 1 | 840 ± 160 | 1 | 1 | 0.02 |
| VNSTR | 23 ± 7 | 31 | 72 ± 13 | 0.09 | 360 | 6.9 | |
| VNSLR | 20 ± 2 | 27 | 28 ± 7 | 0.03 | 810 | 16 | |
| SWSSR | 20 ± 2 | 27 | 40 ± 7 | 0.05 | 570 | 11 | |
| PHSER | 61 ± 6 | 82 | 110 ± 9 | 0.13 | 610 | 12 | |
| G238S (EYSER)c | 3.8 | 0.08 | 52 | 1 | |||
| EYNHR | 55 ± 10 | 74 | 61 ± 9 | 0.07 | 1020 | — | |
| G238N (EYNER)c | 1.4 | 0.16 | 10 | — | |||
| E240H (EYGHR) | 2.7 ± 0.8 | 3.6 | 620 ± 190 | 0.74 | 5 | — | |
| CF | TEM-1 WT (EYGERb) | 77 ± 4 | 1 | 180 ± 28 | 1 | 1 | 0.84 |
| VNSTR | 10 ± 0.3 | 0.13 | 16 ± 0.6 | 0.09 | 1.5 | 1.3 | |
| VNSLR | 16 ± 0.5 | 0.21 | 5 ± 1 | 0.03 | 7.4 | 6.2 | |
| SWSSR | 24 ± 3 | 0.31 | 10 ± 5 | 0.06 | 5.6 | 4.7 | |
| PHSER | 31 ± 6 | 0.40 | 7 ± 2 | 0.04 | 10 | 8.5 | |
| G238S (EYSER)c | 0.03 | 0.02 | 1.2 | 1 | |||
| EYNHR | 31 ± 5 | 0.40 | 11 ± 1 | 0.06 | 6.5 | — | |
| G238N (EYNER)c | 0.13 | 0.29 | 0.44 | — | |||
| E240H (EYGHR) | 63 ± 7 | 0.82 | 23 ± 10 | 0.13 | 6.3 | — | |
| CZ | TEM-1 WT (EYGERb) | 69 ± 13 | 1 | 130 ± 9 | 1 | 1 | 0.80 |
| VNSTR | 38 ± 7 | 0.55 | 83 ± 7 | 0.67 | 0.87 | 0.69 | |
| VNSLR | 41 ± 1 | 0.59 | 28 ± 2 | 0.22 | 2.8 | 2.2 | |
| SWSSR | 50 ± 9 | 0.72 | 55 ± 14 | 0.44 | 1.7 | 1.4 | |
| PHSER | 88 ± 4 | 1.3 | 61 ± 9 | 0.49 | 2.6 | 2.1 | |
| G238S (EYSER)d | 0.85 | 0.68 | 1.3 | 1 | |||
| EYNHR | 82 ± 9 | 1.2 | 120 ± 23 | 0.98 | 1.3 | — | |
| G238N (EYNER) | NDe | ND | ND | — | |||
| E240H (EYGHR) | 118 ± 6 | 1.7 | 95 ± 24 | 0.73 | 2.3 | — | |
| BZ | TEM-1 WT (EYGERb) | 1660 ± 230 | 1 | 62 ± 21 | 1 | 1 | 50 |
| VNSTR | 19 ± 1 | 0.01 | 17 ± 7 | 0.27 | 0.04 | 2.0 | |
| VNSLR | 29 ± 2 | 0.02 | 15 ± 2 | 0.24 | 0.07 | 3.5 | |
| SWSSR | ND | ND | ND | ND | ND | ND | |
| PHSER | 20 ± 4 | 0.01 | 31 ± 6 | 0.50 | 0.02 | 1.2 | |
| G238S (EYSER)c | 0.003 | 0.16 | 0.02 | 1 | |||
| EYNHR | 82 ± 9 | 0.02 | 106 ± 18 | 1.7 | 0.01 | — | |
| G238N (EYNER)c | 0.02 | 0.12 | 0.19 | — | |||
| E240H (EYGHR) | 2000 ± 420 | 1.2 | 420 ± 110 | 6.8 | 0.17 | — |
The five-letter nomenclature identifies mutants according to the one-letter code of the amino acids at positions 104, 105, 238, 240, and 244, respectively. Bold-type identifies mutations relative to WT TEM-1.
Mean value ± standard deviation.
Data from Ref.5; values relative to WT were calculated using the WT values reported in the same study.
Data from Ref.23; values relative to WT were calculated using the WT values reported in the same study.
ND, not determined.
Figure 3.
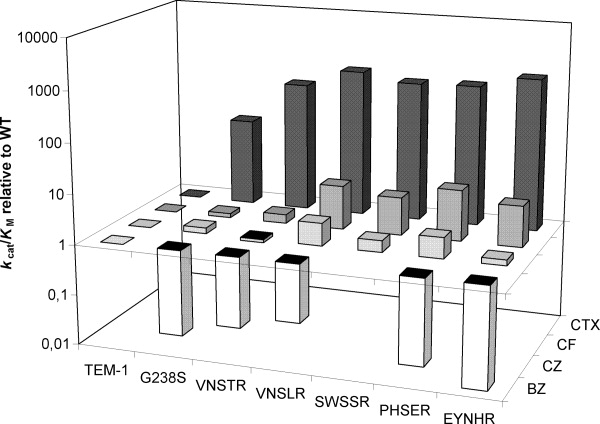
Variation in the relative catalytic efficiencies of the mutant β-lactamases relative to WT TEM-1, plotted on a logarithmic scale. The greatest increase in activity was observed toward the extended-spectrum cephalosporin, CTX, used in the selection. Activity toward two first-generation cephalosporins, CF and CZ, increased only slightly relative to TEM-1. However, the important loss of activity toward the native substrate, BZ, was a negative trade-off to the increased CTX activity.
Impact of additional mutations in the context of G238S
Point mutant G238S is known to enhance extended-spectrum hydrolysis.5,6 To discern the effects of the additional mutations relative to the G238S point mutation, kinetic parameters of the selected variants were compared with those determined for G238S under similar conditions (Table III). For the combinatorial variants that include mutation G238S, the relative catalytic efficiencies (kcat/KM) for the four substrates tested were generally improved relative to the G238S mutant as a result of the additional mutations at positions 104, 105, and 240. The most important increases were seen for CTX hydrolysis, consistent with this extended-spectrum cephalosporin having been used in the selection, and for hydrolysis of the first-generation cephalosporin, CF.
The relative catalytic efficiencies for CTX hydrolysis were improved ∼10-fold relative to the G238S mutant. In the variants including G238S, 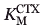 was similar to that of G238S, whereas
was similar to that of G238S, whereas 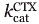 was increased by roughly one order of magnitude relative to G238S. The increase in catalytic efficiency (kcat/KM) thus resulted from increased turnover. This differs from the effect of mutation G238S relative to WT TEM-1, where increased CTX hydrolysis resulted primarily from improved . Thus, the additional mutations had a modest additive effect toward CTX hydrolysis in the context of mutation G238S, and the nature of their effect on hydrolysis differed from that of mutation G238S.
was increased by roughly one order of magnitude relative to G238S. The increase in catalytic efficiency (kcat/KM) thus resulted from increased turnover. This differs from the effect of mutation G238S relative to WT TEM-1, where increased CTX hydrolysis resulted primarily from improved . Thus, the additional mutations had a modest additive effect toward CTX hydrolysis in the context of mutation G238S, and the nature of their effect on hydrolysis differed from that of mutation G238S.
Variant PHSER showed the greatest improvement in (∼80-fold enhancement). This variant also exhibited the greatest reduction of BZ hydrolysis, where 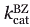 = 1% of WT, as for variant VNSTR. Variants VNSLR and VNSTR differ only by the substitution of the native Glu240 by Leu or Thr, respectively. Overall, mutant VNSLR exhibited among the best productive affinities (KM) and catalytic efficiencies (kcat/KM) for most substrates tested. VNSTR and VNSLR exhibited 10- to 30-fold improvements in their (increased) and (decreased) relative to WT. KM decreases of the same order were observed for the first-generation cephalosporin CF but smaller KM decreases (1.5- to 5-fold) were seen for CZ as well as for BZ. However, the kcat for hydrolysis of both first-generation cephalosporins decreased similarly by a maximum of 8-fold and dropped to 1–2% of WT activity for BZ. Importantly, the additional mutations did not reduce the overall catalytic efficiency (kcat/KM) of BZ or CZ hydrolysis relative to mutant G238S. Thus, the impact of the additional mutations is greatest toward CTX hydrolysis and important (except for mutant VNSTR) for CF hydrolysis.
= 1% of WT, as for variant VNSTR. Variants VNSLR and VNSTR differ only by the substitution of the native Glu240 by Leu or Thr, respectively. Overall, mutant VNSLR exhibited among the best productive affinities (KM) and catalytic efficiencies (kcat/KM) for most substrates tested. VNSTR and VNSLR exhibited 10- to 30-fold improvements in their (increased) and (decreased) relative to WT. KM decreases of the same order were observed for the first-generation cephalosporin CF but smaller KM decreases (1.5- to 5-fold) were seen for CZ as well as for BZ. However, the kcat for hydrolysis of both first-generation cephalosporins decreased similarly by a maximum of 8-fold and dropped to 1–2% of WT activity for BZ. Importantly, the additional mutations did not reduce the overall catalytic efficiency (kcat/KM) of BZ or CZ hydrolysis relative to mutant G238S. Thus, the impact of the additional mutations is greatest toward CTX hydrolysis and important (except for mutant VNSTR) for CF hydrolysis.
Resistance of variants excluding G238S
The double mutant EYNHR harbors the G238N mutation rather than G238S. This combinatorial variant displayed the highest relative catalytic efficiency (kcat/KM) for CTX hydrolysis (Table III), confirming that mutations of Gly238 to residues other than serine can provide high resistance.5 The G238N mutation has previously been shown to be approximately one order of magnitude less efficient in extended-spectrum cephalosporin hydrolysis than G238S although it provides a catalytic efficiency 10- and 5-fold greater than WT toward hydrolysis of CTX and CAZ, respectively.5 This indicates that the additional mutation, E240H, contributed to the resistance phenotype of variant EYNHR.
The previously unreported point mutant E240H was not identified during selection against CTX, suggesting that it does not, by itself, offer a high level of resistance. We created it and determined that it displayed a slight improvement in catalytic efficiency toward the three cephalosporins tested and a small decrease of efficiency toward BZ, but the differences were much smaller than for the double mutant EYNHR (Table III). Both mutations contributed to the decreased . In fact, the ΔΔG for kcat/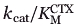 of the double mutant EYNHR (−4.1 kcal/mol) relative to either point mutant G238N (−1.4 kcal/mol) or E240H (−0.9 kcal/mol) indicates that the mutations behave in a synergistic manner toward CTX hydrolysis (rather than an additive manner25), which is also reflected by the difference in the kcat values.
of the double mutant EYNHR (−4.1 kcal/mol) relative to either point mutant G238N (−1.4 kcal/mol) or E240H (−0.9 kcal/mol) indicates that the mutations behave in a synergistic manner toward CTX hydrolysis (rather than an additive manner25), which is also reflected by the difference in the kcat values.
Discussion
In this work, we systematically investigated the potential cooperative effects of amino acid substitutions at five positions belonging to two opposite active-site faces of TEM-1 β-lactamase. Selection against CTX allowed identification of several active TEM-1 mutants displaying a variety of substitutions at four of the five targeted positions. Contrary to our expectations, we saw no evidence of cooperativity between the two faces of the active site: mutations identified in addition to the highly effective G238S mutation procured only small, additive increases to CTX hydrolysis efficiency, with no particular pattern related to either of the two faces.
Gly-238
Earlier studies focusing exclusively on the TEM-1 237-240 region identified ESBL mutants harboring either a serine or an asparagine at position 238, consistent with our results.19 The fact that no further sequence variations were identified in this study immediately indicates that the concurrent mutations on the opposite face (104-105) did not importantly modify the outcome at position 238. Thus, the two faces can be mutated relatively independently of each other, illustrating the important adaptive capacity of the active site. Approximately 20% of natural TEM ESBL isolates include the G238S mutation; TEM-19 is the clinically relevant G238S point mutant. The G238N and the G238D substitutions are found in combination with E104K and one other mutation (see http://lahey.org/studies/temtable.asp), but do not occur alone in isolates. Although the G238N mutation provides only for modest ESBL activity and is approximately one order of magnitude less efficient in extended-spectrum cephalosporin hydrolysis than G238S, it provides a catalytic efficiency one order of magnitude greater than WT toward hydrolysis of CTX and CAZ (Table III; Ref.5). The observation of the G238S and G238N mutations in all our CTX-resistant variants suggests that their functional effect on CTX hydrolysis is unmatched by any other mutation at this position.
Substitution of the native Gly238 has been suggested to displace the β3 strand,7,9 expanding the active-site cavity to accommodate bulkier substrates (i.e., extended-spectrum β-lactam antibiotics). Although elements of kinetic evidence support an alternative shift of the Ω-loop,5,24 analysis of crystal structures of ESBL mutants has substantiated the β3 strand displacement hypothesis.9,10 Considering the modest increase of CTX hydrolysis resulting from additional mutations at positions 104, 105, and 240 relative to mutant G238S, it appears likely that the position 238-dependent displacement of the β3 strand is an independent and compulsory structural requirement of CTX hydrolysis in TEM-1 that is expected to be present in most, if not all of the selected mutants.
Glu-240
Position 240 displayed diverse substitutions in the selected CTX-resistant variants (Glu, Arg, Lys, Thr, Ser, His, and Leu), consistent with other TEM ESBLs (http://lahey.org/studies/temtable.asp). The most frequent mutation observed in natural isolates, E240K, was identified only in our most frequently selected variant: EYSKR, which is the doubly mutated, clinically relevant TEM-7126 (Table I). The E240K mutation may aid binding of extended-spectrum cephalosporins through electrostatic or hydrogen bonding interactions.3,7 The diversity observed confirms that mutations other than E240K are compatible with CTX hydrolysis, when combined with mutations at positions 104, 105, and/or 238. Among the mutations, all but Leu have hydrogen-bonding capacity, suggesting that this feature is important. However, the previous selection of mutant E240G,27 which also displays an increased catalytic efficiency toward CTX,28 indicated that a H-bonding side-chain is not essential. Indeed, for the three cephalosporins tested, variant VNSLR (containing mutation E240L) exhibited slightly better KM values (∼2.5-fold reduction) relative to VNSTR (containing mutation E240T; Table III), in the context of identical mutations at positions 104, 105, and 238. This comparison demonstrates that the ESBL phenotype can be observed either in the presence of hydrophobic bulk (Leu) or of a hydrophilic side-chain (Thr) at position 240.
Arg-244
Among the five randomized positions, only Arg-244 remained invariable. This conservation was not entirely unexpected because Arg-244 mutations in TEM-1 are not associated with ESBL activity, but rather with inhibitor resistance.6,13 We nonetheless randomized this residue to evaluate whether changes in its environment resulting from neighboring, concomitant mutations would allow its contribution to the ESBL phenotype. Its strict conservation is consistent with its proposed role in substrate stabilization29 and suggests that the role of Arg-244 in CTX recognition cannot be compensated by a mutation (or combinations of mutations) at positions 104, 105, 238, or 240.
Tyr-105
With few exceptions, aromatic and small amino acids are conserved at position 105 in class A β-lactamases, a structural and functional requirement that has been rationalized by the necessity to form a stable ligand binding surface within the TEM-1 active-site.17 The mutations selected at position 105 in this study (Tyr, Trp, His, Asn, and Ser) are consistent with the necessity for this residue to be small or planar to prevent steric clashes with the substrate. Mutations at position 104 and/or on the opposite face at positions 238 and 240 did not broaden the variety at position 105, demonstrating that its role in binding and catalysis cannot be readily compensated by neighboring mutations. Interestingly, the native Tyr-105 was selected only in combination with the native Glu-104 (Table I), suggesting that they constitute a favored pair and act in an interdependent fashion. Mutation of either allowed mutation of the second, consistent with results of a combinatorial study of TEM-1 residues 103-105.14
Glu-104
Among class A β-lactamases, position 104 displays a modest variety of side chains, either hydrophobic (Val or Pro), polar (Ser) or charged (Glu).2,14,30 In TEM-1, Glu-104 is involved in positioning the catalytically significant SDN loop (residues 130-132).4 In natural ESBL TEM isolates, only the E104K mutation is observed, and is present in >25% of cases (http://lahey.org/studies/temtable.asp). The E104K mutation has been suggested to hydrogen-bond with the oxime substituent of CTX and to form an electrostatic contact with the oxime carboxylate of CAZ.3 Although the E104K mutation increases hydrolysis of extended-spectrum cephalosporins, it is not sufficient to confer in vivo resistance unless combined with G238S, yielding the clinically significant double mutant E104K: G238S (TEM-15).31,32 Although we sequenced the E104K mutation among the non-selected clones, we did not observe it in any CTX-selected mutant, suggesting that other E104 replacements offer similar or greater catalytic advantages in this combinatorial context. In fact, the selected mutants displayed a variety of amino acids at position 104 (Gln, Val, Arg, Pro, Ser), always concurrently with mutation G238S as well as a mutation at position 105, and did not produce large changes in kinetic parameters. This suggests that, once the threshold for CTX resistance has been met via the G238S mutation, the concurrent presence of a mutation at position 105 broadens the mutational permissivity at position 104, giving rise to a chemically diverse set of CTX-resistant environments that is not restricted to the E104K mutation.
The fair chemical diversity observed at positions 104 and 240 did not result in large differences in kinetic parameters. This may result from both positions being solvent-exposed and relatively unconstrained. Because residues 104 and 238 are involved in loop positioning and because residue 240 is the immediate neighbor of 238 (TEM-1 has no residue 239), their mutation may also contribute to active-site expansion, favoring binding of bulkier substrates as observed in BlaC β-lactamase from Mycobacterium tuberculosis.33 We propose that replacements at positions 104 and 240 mainly fulfill steric requirements that support productive binding of CTX, rather than to provide specific contacts with the substrate. Our results are consistent with active-site expansion both through displacement of the β3-strand (mutations G238S or G238N) and with flexibility of residues 104 and 240, which could open the active site from opposite faces of the cavity and allow expansion.
Catalytic properties of the combinatorial mutants
Upon simultaneous mutation, residues may behave either in an independent or in an interdependent fashion toward protein function.34 In TEM-1, synergy has been observed toward CAZ hydrolysis for mutant G238S:E240K,8 harboring neighboring mutations, and toward CTX hydrolysis for mutant E104M: G238S,16 where the mutations are on opposite faces of the active site. Additivity has been reported for mutant E104M:G238S (mutations on opposite faces) toward cefuroxime hydrolysis (second-generation cephalosporin).16
Synergy in neighboring mutations
The double mutant EYNHR (G238N:E240H) that does not harbor the G238S mutation conferred an important 1000-fold improvement in catalytic efficiency relative to WT TEM-1. As the G238N and E240H mutations accounted only for a 10-fold and a 5-fold increase, respectively, in CTX hydrolysis efficiency (Table III), the G238N:E240H combination behaved in a synergistic manner. The synergy upon KM was marked: G238N provided only a 6-fold decrease5 and E240H had a negligible effect on , which was reduced 14-fold in EYNHR relative to WT (Table III). In addition, kcat was increased only 3.6-fold in E240H and hardly modified in G238N,5 whereas the G238N:E240H combination exhibited a 74-fold kcat increase. Thus, both productive binding and turnover were synergistically enhanced by the combination of the neighboring mutations, in a manner that was not predictable from the contributing point mutants.
Variant EYNHR also displayed decreased penicillin resistance. This was attributed to the 50-fold decrease in for G238N,5 equivalent to the effect on in EYNHR and consistent with the observation that E240H had little effect on . G238N provided mild improvements in KM toward penicillins5 while the E240H mutation resulted in a 6.8-fold increase in relative to WT. It thus appears that G238N attenuated the effect of E240H in EYNHR, which displayed only a 1.7-fold increase in . The G238N5 and E240H variants both provided mild improvements in 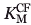 , resulting in a 16-fold improvement in EYNHR. The combined mutations of EYNHR resulted in the most pronounced switch in the substrate profile among the selected variants, to strongly disfavor penicillin hydrolysis in favor of cephalosporins (see Fig. 3). The neighboring G238N:E240H mutations were sufficient to extend the substrate spectrum with no mutation at the opposite face of the active site (residues 104/105).
, resulting in a 16-fold improvement in EYNHR. The combined mutations of EYNHR resulted in the most pronounced switch in the substrate profile among the selected variants, to strongly disfavor penicillin hydrolysis in favor of cephalosporins (see Fig. 3). The neighboring G238N:E240H mutations were sufficient to extend the substrate spectrum with no mutation at the opposite face of the active site (residues 104/105).
Additivity of mutations within the active-site cavity of TEM-1
In most of the variants selected in this study, the major contributor to increased CTX hydrolysis efficiency was mutation G238S. The quadruple mutant SWSSR (E104S:Y105W:G238S:E240S) encodes the previously characterized E104S and Y105W mutations. The former procures a modest increase in kcat/ relative to WT4 while the latter, taken alone, does not.17 The added thermodynamic contribution of E104S with G238S, on the opposite face of the active site, can account for the effect of the quadruple mutant, suggesting an additive effect of the mutations across the active-site cavity. Interestingly, the 6-fold increase in MICCTX for SWSSR relative to WT was more modest than the 130-fold increase observed for the SHV-1 β-lactamase double mutant D104S:G238S.11 Mutations Y105W and/or E240S in SWSSR may have a damping effect on CTX resistance; alternatively, the different context of the mutations in TEM-1 relative to SHV-1 may modulate the effect of those specific mutations. Nonetheless, the trend seen for the TEM-1 variant SWSSR and the SHV-1 variant D104S:G238S indicates that the TEM-1 active site tolerates multiple simultaneous mutations.
The triple mutant PHSER (E104P:Y105H:G238S) combines the previously characterized E104P and Y105H mutations, on one face of the active site, with G238S on the opposite face. Mutant E104P is advantageous toward the affinity and hydrolysis of various substrates (including CTX),4 whereas mutant Y105H displays WT-like levels of resistance toward penicillins and first-generation cephalosporins.17 As with SWSSR, the combined mutations in PHSER resulted in a modest increase in cephalosporin hydrolysis relative to G238S, again supporting an additive effect of mutations on the two faces of the active site. In SHV-1, MICs of mutant D104P:G238S revealed a cephalosporin resistance one to two orders of magnitude above WT, very similar to the triple mutant PHSER. However, the SHV-1 variant was practically unchanged in its MICAMP11 while PHSER showed an important decrease in AMP resistance (Table II). The antagonistic effect toward AMP hydrolysis may result from mutation Y105H or from the specific context in which these mutations arise.
The quadruple mutants VNSTR and VNSLR (E104V:Y105N:G238S:E240T/L) share three mutations. On one face of the active site, E104V, which has been observed in a CTX-hydrolyzing β-lactamase from Kebsiella oxytoca,35 was coupled with the highly active Y105N mutation.17 On the opposite face, G238S was combined with either the novel E240L replacement (VNSLR), or the E240T substitution (VNSTR). The latter mutation has been observed in other class A β-lactamases2,36 but not characterized. Comparison with G238S again supports additivity between mutations on the two faces of the active site, as hydrolytic efficiency was improved for most substrates.
Thus, a variety of multiple active-site mutants, each including the G238S mutation and chosen from a larger panel of similar selected variants (Table I), display improvements in CTX hydrolysis efficiency that are consistent with additive effects of mutations on the same face or on the opposite face of the active site. Some mutations have little effect on the kinetics, indicating tolerance to a variety of chemical groups and sterics within the immediate vicinity of the bound substrate. Synergistic effects on catalysis were also seen, but only in variant EYNHR, between neighboring residues (238 and 240). Within our sample, improved CTX hydrolysis was readily achieved via a variety of solutions.
Promiscuity, robustness, and plasticity in substrate binding
Our results illustrate that TEM-1 tolerates several combinations of mutations in the active site with conservation or improvement of hydrolysis toward a variety of substrates. It has been noted that mutants at position 238, alone and in combination with substitutions at position 104, lose much activity toward the penicillins AMP and BZ while increasing their efficiency toward extended-spectrum cephalosporins.19,23,31 Similarly, in this study, all variants selected against CTX showed a two- to three-order of magnitude improvement of catalytic efficiency toward CTX hydrolysis, a conservation or slight improvement in the hydrolysis of first-generation cephalosporins and a one- to two-order of magnitude decrease in BZ hydrolysis (Table III). Similar trends have been observed in other TEM ESBL variants but are not limited to this family of enzymes.3,23,37–39 Thus, while selection was directed exclusively toward CTX hydrolysis, the evolution of this promiscuous binding function (CTX binding)40 remained compatible with recognition of first-generation cephalosporins. Being relatively innocuous toward enzyme function, the additional mutations at positions 104, 105 and 240 indicate a degree of robustness in TEM-1,41 which tolerated multiple active-site mutations and still allowed recognition of related compounds. Such emergence of a new phenotype (CTX resistance) by the introduction of a small number of mutations has been defined as enzyme plasticity.42 This trait can allow enzymes to recognize a broad range of substrates. It has been observed in many protein families43 and confirms that enzyme active sites have evolved to work as cooperative entities that do not simply rely on a restricted set of amino acids for catalysis.44 In different enzymatic systems displaying plasticity, one to four mutations at the active site (as in the present case) can increase a promiscuous activity up to 1000-fold while reducing the native activity of the enzyme less than 5-fold,42 although heavier trade-offs are also observed (see Ref.45 and references therein). The mutants characterized herein displayed a promiscuous activity (CTX hydrolysis) increased up to 1000-fold while the native function (BZ hydrolysis) was reduced up to 100-fold.
In most directed evolution experiments, trade-offs that compromise the original activity are generally weak and yield “generalized” enzymes that conserve significant native activity while gaining the second. Strong trade-offs are often seen in dual selections as a greater loss of overall activity occurs.42 Our selection for CTX hydrolysis was done at the cost of weak to moderate trade-offs, where the native, BZ-hydrolyzing activity was reduced but remained significant (see Fig. 4). This is consistent with the fact that mutations introduced by rational or semi-random methods (as in the present case) generally show greater negative trade-offs relative to random mutagenesis studies.42 Rational and semi-random methods generally target active-site residues, whereas random methods also yield beneficial mutations located far from the active site; the latter may be less likely to induce negative trade-offs on activity. In this study, the selected mutants apparently evolved until the generalized stage, allowing binding and catalysis of many substrates rather than becoming specialized CTX-hydrolyzing β-lactamases (see Fig. 4). We note that they remain less specialized than other engineered β-lactamase mutants.27,46
Figure 4.
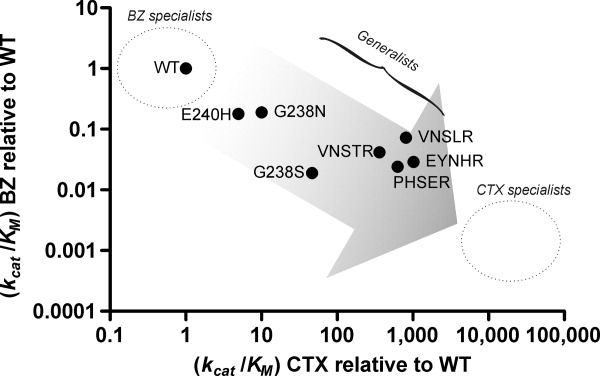
Generalization of the mutant β-lactamase activities. The logarithm of catalytic efficiencies of WT and mutant β-lactamases toward the native penicillin substrate, and the extended-spectrum cephalosporin used in selection, CTX, were related. The gray-scale gradient represents a hypothetical route leading from a specialized enzyme (BZ specialist; light gray) toward a new specialization for another substrate (CTX specialist; dark gray); enzymes displaying intermediate efficiencies toward both substrates map between the two extremes: these show “generalized” activity. Here, WT TEM-1 was the most BZ-specialized, displaying high BZ hydrolysis and low CTX hydrolysis. The point mutants, E240H, G238N, and G238S, and the combinatorial mutants, VNSTR, VNSLR, PHSER, EYNHR, were successively more generalized, where the combinatorial mutants were more efficient than the point mutants toward CTX hydrolysis. Figure based on Ref.42; the data was plotted using GraphPad Prism 5 and the graphics overlayed with CorelDRAW X3.
The capacity of an active site to accommodate mutations is a function of the relation between the targeted residues: by maintaining a degree of independence between key residues, mutations will not necessarily be deleterious for enzymatic activity. Such weakly linked residues can lead to robust systems, which may serve as starting points for evolution of new enzymatic functions. According to Wagner,41 an enzyme that can perform its native function following multiple neutral mutations should possess the necessary characteristics for emergence of a promiscuous activity. The significant drop in penicillin-hydrolysis of the selected TEM-1 variants may indicate that the native and promiscuous activities are mutually exclusive, because of structural requirements for the specific recognition and hydrolysis of each substrate. The additive catalytic efficiency enhancements relative to mutant G238S were modest, suggesting that the mutated residues behave independently. However, mutant EYNHR displayed mutational synergy, illustrating that complex effects can prevail depending on the identity of the residues. Globally, we observed no evidence of mutational linkage between the two active-site faces we modified, despite some previous evidence to this effect (mutant E104M:G238S, Ref.16), suggesting that these two areas of the active site are not highly interdependent. This may be an evolutionary strategy to limit loss of activity that would otherwise occur upon mutation of highly linked residues. Nonetheless, evidence of some degree of linkage between positions 104 and 105 sets a restriction to the robustness of the active site.
Methods
Reagents
All enzymes were purchased from MBI Fermentas (Burlington, ON) or New England Biolabs (Mississauga, ON). Nitrocefin was purchased from Calbiochem (Mississauga, ON), chloramphenicol (Chl) from A&C American Chemicals (Montréal, QC), AMP from Bioshop Canada (Burlington, ON), while BZ, CF, CZ, CTX, and Fast-Flow DEAE-Sepharose were obtained from Sigma-Aldrich (Oakville, ON).
Bacterial strains and plasmids
Escherichia coli strain SS320 (F− lacI22 lacZ pro-48 met-90 trpA trpR his-85 rpsL azi-9 gyrA λ− P1S) was used for site-directed mutagenesis and E. coli strain XL1-Blue (supE44, hsdR17, recA1, endA1, gyrA46, thi, relA1 lac− F′ [proAB+ lacIq lacZΔM15 Tn10(tetr)]) was used for the propagation and expression of plasmid DNA. Plasmid pQE32Chl17 was used for protein expression. The chloramphenicol acetyltransferase gene replaces the blaTEM-1 gene as the selectable marker, allowing plasmid maintenance using 12.5 μg/mL Chl. The blaTEM-1 gene was then subcloned under control of the IPTG-inducible promoter, as previously described,17 and mutated. The resulting constructs harbor a 6-histidine tag that is N-terminal to the native TEM-1 signal peptide. Upon processing of the signal peptide during expression and maturation in E. coli, the histidine tag is also removed.
Oligonucleotides and library creation
Oligonucleotide primers used for mutagenesis were synthesized by Alpha DNA (Montréal, QC) or Integrated DNA Technologies (Coralville, IA). PCR reactions were performed with a PTC-200 DNA Engine Thermal Cycler (Bio-Rad, Mississauga, ON).
The five positions selected for mutagenesis were initially split into two libraries: library 104-105 and library 238-240-244. Library 104-105 was constructed using the Site-Overlap Extension (SOE) method,47 whereas library 238-240-244 was constructed using the Megaprimer technique.48 In all cases, the positions to be mutated were encoded by the 5′-NNS-3′ codon (coding strand), allowing 32 possible codons and all 20 amino acids. The 861-bp blaTEM-1 gene was PCR-amplified from pBR322 (lacking the V84I and A184V mutations)49 using the previously described terminal primers BamHITEMF and TEMHindIIIR, containing the BamHI and HindIII restriction sites respectively.17 Library 104-105 used degenerate primer Lib104-105NNSR (5′-GATGCTTTTCTGTGACTGGTGASNNSNNAACCAAGTCATTCTGAGAATAGT-3′) and primer Lib104-105F (5′-TCACCAGTCACAGAAAAGCATC-3′), whereas library 238-240-244 used degenerate primer Lib238,240,244NNSF (5′-GATAAATCTGGAGCCNNSN NSCGTGGGTCTNNSGGTATCATTGCAGCA-3′). The point mutant E240H was synthesized using primers E240H-F (5′-CTGATAAATCTGGAGCCGGTCACCGTGG GTCTCGCGGTATC-3′) and E240H-R (5′-GATACCGCGAGACCCACGGTGACCGGCTCCAGATTTATCAG-3′). It should be noted that there is no residue 239 in TEM-1 according to the Ambler numbering scheme.2 Digestion of the full-length amplicons with BamHI/HindIII was followed by subcloning into BamHI/HindIII-digested and shrimp alkaline phosphatase-treated pQE32Chl. The ligated DNA was electroporated into E. coli SS320. Plasmid DNA was harvested and libraries 104-105 and 238-240-244 were combined via the TEM-1 internal PstI restriction site for transformation into E. coli XL1-Blue, generating the combinatorial library 104-105-238-240-244. Prior to selection, library quality was assessed by DNA sequencing. Plasmid DNA was prepared as previously described.50 Sequencing of 34 clones was performed by the dideoxy chain termination method using the Thermo Sequenase Cycle Sequencing Kit (USB Corporation, Cleveland, OH) with a dye-labeled primer and a Long ReadIR 4200 Li-Cor automated DNA sequencer (Li-Cor Biotechnology, Lincoln, NB).
Selection of cefotaxime-resistant variants
The 104-105-238-240-244 combinatorial library was plated on Luria-Bertani (LB) medium containing 250 ng/mL CTX, thus inhibiting WT growth. The theoretical library is comprised of ∼3.4 × 107 DNA variants (32 possible codons per mutated position = 325) encoding 3.2 × 106 unique protein variants (20 possible amino acids per mutated position = 205). Overall, less than 1% of the whole library was screened. CTX-resistant colonies were individually picked for identification of mutations by DNA sequencing.
Antibiotic susceptibility
Minimum inhibitory concentrations (MICs) were determined by broth microdilutions according to Cantu et al.19 Briefly, 1 × 104 E. coli XL1-Blue CFUs were inoculated into 96-well microtiter plates containing 100 μL of LB media and appropriate dilutions of the antibiotic being tested. The ranges tested were as follows: 250–10,000 μg/mL BZ, 64–10,000 μg/mL AMP, 32–125 μg/mL CF, 8–64 μg/mL CZ, and 0.125–2 μg/mL CTX. Each MIC determination was performed in triplicate. The plates were examined visually and the lowest antibiotic concentration at which bacterial growth was inhibited was recorded as the MIC.
β-Lactamase expression and purification
A 10-mL overnight culture of each selected mutant of interest was used to inoculate 1 L of LB without antibiotics. Cells were propagated at 37°C with agitation until A600 nm = 0.6–0.8. Protein expression was induced with 1 mM isopropyl 1-thio-β-d-galactopyranoside (IPTG) at 37°C for an additional 3 h until the culture reached late log phase. Cells were pelleted by centrifugation (20 min, 6000g, 4°C), resuspended in 10–15 mL of 10 mM Tris-Cl buffer pH 7.0 and separated into 1 mL aliquots. A gentle lysis of the outer membrane of E. coli was performed by four 2-min freeze-thaw cycles using a dry ice/ethanol bath and a 37°C water bath. After centrifugation (15 min, 20,000g, 4°C), the supernatant was pooled (∼15 mL).
Purification of mutants was performed as previously described,17 with the following modifications. Purifications were undertaken at room temperature on a Pharmacia LKB high performance liquid chromatography system (Pharmacia LKB Technology, Uppsala, Sweden). Following sample application, the DEAE-Sepharose column (1.6 cm × 30 cm) was washed with 4.5 column volumes (CV) of 10 mM Tris-Cl buffer pH 7.0, the gradient to 150 mM Tris-Cl pH 7.0 was over 6.5 CV, and the column wash was performed over 3 CV. The column was regenerated by applying ≥6 CV of 1 M Tris-Cl pH 7.0. A mock purification was run by injecting 10 mM Tris-Cl buffer as a sample, followed by a nitrocefin assay and SDS-PAGE, confirming that no β-lactamase activity was carried over between runs. Fractions containing β-lactamase were identified by a nitrocefin hydrolysis assay and SDS-polyacrylamide gel electrophoresis with Coomassie Brilliant Blue staining. Analysis of gel patterns with Scion Image (National Institute of Health, http://rsb.info.nih.gov/nih-image) allowed determination of the yield and purity. Enzyme concentration was determined by Bradford assay using the Bio-Rad protein assay solution and normalized according to its purity.
Enzyme kinetics
KM and kcat for BZ, CF, CZ, and CTX were determined at room temperature in 50 mM sodium phosphate buffer pH 7.0. The following extinction coefficients and concentration ranges were used: Δɛ232 nm = 1100 M−1 cm−1 for BZ (50–300 μM),51 Δɛ262 nm = 7960 M−1 cm−1 for CF (20–250 μM),51, Δɛ260 nm = 7900 M−1 cm−1 for CZ (20–250 μM),52 and Δɛ264 nm = 7250 M−1 cm−1 for CTX (25–250 μM).5 Substrate hydrolysis was monitored according to initial steady-state velocities for six substrate concentrations generally flanking the KM values (where allowed by the extinction coefficients and the KM values) using a Cary 100 Bio UV–visible spectrophotometer (Varian Canada, Montréal, QC). The kinetic parameters were determined from the rates of hydrolysis calculated from the initial linear portion of the curve and fitted to a Lineweaver-Burk (1/V vs. 1/[S]) plot. In most cases, initial rates were also analyzed with the software GraphPad Prism (GraphPad Software, San Diego, CA) by a non-linear regression curve corresponding to the Michaelis-Menten equation to verify results from linear analyses.
Changes in transition-state stabilization energy for kcat/KM were calculated as ΔΔG = −(RT ln kcat/KM)mutant/ (RT ln kcat/KM)WT (R is the gas constant and T is the absolute temperature).25 According to double-mutant cycles, ΔΔG for a multiple mutant is roughly equivalent to the sum of ΔG for each point-mutant when the effects of mutations are additive, indicative of functional independence of those residues. When the absolute value of ΔΔG is significantly smaller than the sum for the single mutants, the mutations are considered to be antagonistic. When ΔΔG is significantly larger than the sum for the single mutants, the mutations are considered to be synergistic.
Conclusions
All the novel TEM-1 variants described herein displayed improved affinities toward all antibiotics investigated relative to the WT TEM-1, consistent with functional diversification and adaptability. This demonstrates that the active site of TEM-1 can sustain multiple simultaneous substitutions to broaden its substrate spectrum and thus allow a better fit of the aminothiazole moiety as hypothesized by Bush and coworkers.3 Our results support the hypothesis that active-site cavities can tolerate a relatively large number of mutations that confer “evolvability” toward related substrates and highlight different strategies to achieve substrate generalization.
Acknowledgments
The authors thank Jeffrey Keillor and Serguei Chteinberg for helpful comments, and Christopher Clouthier for critical reading of the manuscript.
Glossary
Abbreviations:
- AMP
ampicillin
- BZ
benzylpenicillin
- CAZ
ceftazidime
- CF
cephalothin
- Chl
chloramphenicol
- CTX
cefotaxime
- CV
column volume
- CZ
cefazolin
- ESBL
extended-spectrum β-lactamase
- MIC
minimum inhibitory concentration.
Footnotes
The five-letter nomenclature identifies mutants according to the one-letter code of the amino acids at positions 104, 105, 238, 240 and 244, respectively.
References
- 1.Matagne A, Lamotte-Brasseur J, Frère JM. Catalytic properties of class A β-lactamases: efficiency and diversity. Biochem J. 1998;330:581–598. doi: 10.1042/bj3300581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ambler RP, Coulson AF, Frère JM, Ghuysen JM, Joris B, Forsman M, Levesque RC, Tiraby G, Waley SG. A standard numbering scheme for the class A β-lactamases. Biochem J. 1991;276:269–270. doi: 10.1042/bj2760269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sowek JA, Singer SB, Ohringer S, Malley MF, Dougherty TJ, Gougoutas JZ, Bush K. Substitution of lysine at position 104 or 240 of TEM-1pTZ18R β-lactamase enhances the effect of serine-164 substitution on hydrolysis or affinity for cephalosporins and the monobactam aztreonam. Biochemistry. 1991;30:3179–3188. doi: 10.1021/bi00227a004. [DOI] [PubMed] [Google Scholar]
- 4.Petit A, Maveyraud L, Lenfant F, Samama JP, Labia R, Masson JM. Multiple substitutions at position 104 of β-lactamase TEM-1: assessing the role of this residue in substrate specificity. Biochem J. 1995;305(Part 1):33–40. doi: 10.1042/bj3050033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cantu C, III, Palzkill T. The role of residue 238 of TEM-1 β-lactamase in the hydrolysis of extended-spectrum antibiotics. J Biol Chem. 1998;273:26603–26609. doi: 10.1074/jbc.273.41.26603. [DOI] [PubMed] [Google Scholar]
- 6.Fisher JF, Meroueh SO, Mobashery S. Bacterial resistance to β-lactam antibiotics: compelling opportunism, compelling opportunity. Chem Rev. 2005;105:395–424. doi: 10.1021/cr030102i. [DOI] [PubMed] [Google Scholar]
- 7.Huletsky A, Knox JR, Levesque RC. Role of Ser-238 and Lys-240 in the hydrolysis of third-generation cephalosporins by SHV-type β-lactamases probed by site-directed mutagenesis and three-dimensional modeling. J Biol Chem. 1993;268:3690–3697. [PubMed] [Google Scholar]
- 8.Venkatachalam KV, Huang W, LaRocco M, Palzkill T. Characterization of TEM-1 β-lactamase mutants from positions 238 to 241 with increased catalytic efficiency for ceftazidime. J Biol Chem. 1994;269:23444–23450. [PubMed] [Google Scholar]
- 9.Orencia MC, Yoon JS, Ness JE, Stemmer WP, Stevens RC. Predicting the emergence of antibiotic resistance by directed evolution and structural analysis. Nat Struct Biol. 2001;8:238–242. doi: 10.1038/84981. [DOI] [PubMed] [Google Scholar]
- 10.Nukaga M, Mayama K, Hujer AM, Bonomo RA, Knox JR. Ultrahigh resolution structure of a class A β-lactamase: on the mechanism and specificity of the extended-spectrum SHV-2 enzyme. J Mol Biol. 2003;328:289–301. doi: 10.1016/s0022-2836(03)00210-9. [DOI] [PubMed] [Google Scholar]
- 11.Bethel CR, Hujer AM, Hujer KM, Thomson JM, Ruszczycky MW, Anderson VE, Pusztai-Carey M, Taracila M, Helfand MS, Bonomo RA. Defining the role of Asp104 in the SHV β-Lactamase. Antimicrob Agents Chemother. 2006;50:4124–4131. doi: 10.1128/AAC.00848-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ambler R. The structure of β-lactamases. Philos Trans R Soc Lond B Biol Sci. 1980;289:321–331. doi: 10.1098/rstb.1980.0049. [DOI] [PubMed] [Google Scholar]
- 13.Knox JR. Extended-spectrum and inhibitor-resistant TEM-type β-lactamases. Mutations, specificity, and three-dimensional structure. Antimicrob Agents Chemother. 1995;39:2593–2601. doi: 10.1128/aac.39.12.2593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Huang WZ, Petrosino J, Hirsch M, Shenkin PS, Palzkill T. Amino acid sequence determinants of β-lactamase structure and activity. J Mol Biol. 1996;258:688–703. doi: 10.1006/jmbi.1996.0279. [DOI] [PubMed] [Google Scholar]
- 15.Palzkill T, Botstein D. Identification of amino acid substitutions that alter the substrate specificity of TEM-1 β-lactamase. J Bacteriol. 1992;174:5237–5243. doi: 10.1128/jb.174.16.5237-5243.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Viadiu H, Osuna J, Fink AL, Soberón X. A new TEM β-lactamase double mutant with broadened specificity reveals substrate-dependent functional interactions. J Biol Chem. 1995;270:781–787. [PubMed] [Google Scholar]
- 17.Doucet N, De Wals P-Y, Pelletier JN. Site-saturation mutagenesis of Tyr-105 reveals its importance in substrate stabilization and discrimination in TEM-1 β-lactamase. J Biol Chem. 2004;279:46295–46303. doi: 10.1074/jbc.M407606200. [DOI] [PubMed] [Google Scholar]
- 18.Chica RA, Doucet N, Pelletier JN. Semi-rational approaches to engineering enzyme activity: combining the benefits of directed evolution and rational design. Curr Opin Biotechnol. 2005;16:378–384. doi: 10.1016/j.copbio.2005.06.004. [DOI] [PubMed] [Google Scholar]
- 19.Cantu C, III, Huang W, Palzkill T. Selection and characterization of amino acid substitutions at residues 237-240 of TEM-1 β-lactamase with altered substrate specificity for aztreonam and ceftazidime. J Biol Chem. 1996;271:22538–22545. doi: 10.1074/jbc.271.37.22538. [DOI] [PubMed] [Google Scholar]
- 20.Page MI. The mechanisms of reactions of β-lactam antibiotics. In: Bethel D, editor. Advances in physical organic chemistry. London: Academic Press; 1987. pp. 165–270. [Google Scholar]
- 21.Bush K, Mobashery S. How β-lactamases have driven pharmaceutical drug discovery. From mechanistic knowledge to clinical circumvention. Adv Exp Med Biol. 1998;456:71–98. [PubMed] [Google Scholar]
- 22.Medeiros AA. Evolution and dissemination of β-lactamases accelerated by generations of β-lactam antibiotics. Clin Infect Dis. 1997;24:S19–S45. doi: 10.1093/clinids/24.supplement_1.s19. [DOI] [PubMed] [Google Scholar]
- 23.Raquet X, Lamotte-Brasseur J, Fonze E, Goussard S, Courvalin P, Frère JM. TEM β-lactamase mutants hydrolysing third-generation cephalosporins. A kinetic and molecular modelling analysis. J Mol Biol. 1994;244:625–639. doi: 10.1006/jmbi.1994.1756. [DOI] [PubMed] [Google Scholar]
- 24.Saves I, Burlet-Schiltz O, Maveyraud L, Samama JP, Prome JC, Masson JM. Mass spectral kinetic study of acylation and deacylation during the hydrolysis of penicillins and cefotaxime by β-lactamase TEM-1 and the G238S mutant. Biochemistry. 1995;34:11660–11667. doi: 10.1021/bi00037a003. [DOI] [PubMed] [Google Scholar]
- 25.Wells JA. Additivity of mutational effects in proteins. Biochemistry. 1990;29:8509–8517. doi: 10.1021/bi00489a001. [DOI] [PubMed] [Google Scholar]
- 26.Rasheed JK, Anderson GJ, Queenan AM, Biddle JW, Oliver A, Jacoby GA, Bush K, Tenover FC. TEM-71, a novel plasmid-encoded, extended-spectrum β-lactamase produced by a clinical isolate of Klebsiella pneumoniae. Antimicrob Agents Chemother. 2002;46:2000–2003. doi: 10.1128/AAC.46.6.2000-2003.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zaccolo M, Gherardi E. The effect of high-frequency random mutagenesis on in vitro protein evolution: a study on TEM-1 β-lactamase. J Mol Biol. 1999;285:775–783. doi: 10.1006/jmbi.1998.2262. [DOI] [PubMed] [Google Scholar]
- 28.Lenfant F, Petit A, Labia R, Maveyraud L, Samama JP, Masson JM. Site-directed mutagenesis of β-lactamase TEM-1. Investigating the potential role of specific residues on the activity of Pseudomonas-specific enzymes. Eur J Biochem. 1993;217:939–946. doi: 10.1111/j.1432-1033.1993.tb18324.x. [DOI] [PubMed] [Google Scholar]
- 29.Zafaralla G, Manavathu EK, Lerner SA, Mobashery S. Elucidation of the role of arginine-244 in the turnover processes of class A β-lactamases. Biochemistry. 1992;31:3847–3852. doi: 10.1021/bi00130a016. [DOI] [PubMed] [Google Scholar]
- 30.Tranier S, Bouthors AT, Maveyraud L, Guillet V, Sougakoff W, Samama JP. The high resolution crystal structure for class A β-lactamase PER-1 reveals the bases for its increase in breadth of activity. J Biol Chem. 2000;275:28075–28082. doi: 10.1074/jbc.M003802200. [DOI] [PubMed] [Google Scholar]
- 31.Lenfant F, Labia R, Masson JM. Probing the active site of β-lactamase R-TEM1 by informational suppression. Biochimie. 1990;72:495–503. doi: 10.1016/0300-9084(90)90073-p. [DOI] [PubMed] [Google Scholar]
- 32.Mabilat C, Courvalin P. Development of “oligotyping” for characterization and molecular epidemiology of TEM β-lactamases in members of the family Enterobacteriaceae. Antimicrob Agents Chemother. 1990;34:2210–2216. doi: 10.1128/aac.34.11.2210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang F, Cassidy C, Sacchettini JC. Crystal structure and activity studies of the Mycobacterium tuberculosis β-lactamase reveal its critical role in resistance to β-lactam antibiotics. Antimicrob Agents Chemother. 2006;50:2762–2771. doi: 10.1128/AAC.00320-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mildvan AS. Inverse thinking about double mutants of enzymes. Biochemistry. 2004;43:14517–14520. doi: 10.1021/bi048052e. [DOI] [PubMed] [Google Scholar]
- 35.Reynaud A, Peduzzi J, Barthelemy M, Labia R. Cefotaxime-hydrolysing activity of the β-lactamase of Klebsiella oxytoca D488 could be related to a threonine residue at position 140. FEMS Microbiol Lett. 1991;65:185–192. doi: 10.1016/0378-1097(91)90301-p. [DOI] [PubMed] [Google Scholar]
- 36.Chan PT. Nucleotide sequence of the Staphylococcus aureus PC1 β-lactamase gene. Nucleic Acids Res. 1986;14:5940. doi: 10.1093/nar/14.14.5940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bush K, Singer SB. Biochemical characteristics of extended broad spectrum β-lactamases. Infection. 1989;17:429–433. doi: 10.1007/BF01645566. [DOI] [PubMed] [Google Scholar]
- 38.Bouthors AT, Delettre J, Mugnier P, Jarlier V, Sougakoff W. Site-directed mutagenesis of residues 164, 170, 171, 179, 220, 237 and 242 in PER-1 β-lactamase hydrolysing expanded-spectrum cephalosporins. Protein Eng. 1999;12:313–318. doi: 10.1093/protein/12.4.313. [DOI] [PubMed] [Google Scholar]
- 39.Hujer AM, Hujer KM, Helfand MS, Anderson VE, Bonomo RA. Amino acid substitutions at Ambler position Gly238 in the SHV-1 β-lactamase: exploring sequence requirements for resistance to penicillins and cephalosporins. Antimicrob Agents Chemother. 2002;46:3971–3977. doi: 10.1128/AAC.46.12.3971-3977.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Aharoni A, Gaidukov L, Khersonsky O, Mc QGS, Roodveldt C, Tawfik DS. The ‘evolvability’ of promiscuous protein functions. Nat Genet. 2005;37:73–76. doi: 10.1038/ng1482. [DOI] [PubMed] [Google Scholar]
- 41.Wagner A. Robustness, evolvability, and neutrality. FEBS Lett. 2005;579:1772–1778. doi: 10.1016/j.febslet.2005.01.063. [DOI] [PubMed] [Google Scholar]
- 42.Khersonsky O, Roodveldt C, Tawfik DS. Enzyme promiscuity: evolutionary and mechanistic aspects. Curr Opin Chem Biol. 2006;10:498–508. doi: 10.1016/j.cbpa.2006.08.011. [DOI] [PubMed] [Google Scholar]
- 43.Todd AE, Orengo CA, Thornton JM. Plasticity of enzyme active sites. Trends Biochem Sci. 2002;27:419–426. doi: 10.1016/s0968-0004(02)02158-8. [DOI] [PubMed] [Google Scholar]
- 44.Peracchi A. Enzyme catalysis: removing chemically ‘essential’ residues by site-directed mutagenesis. Trends Biochem Sci. 2001;26:497–503. doi: 10.1016/s0968-0004(01)01911-9. [DOI] [PubMed] [Google Scholar]
- 45.Kelly RM, Leemhuis H, Gatjen L, Dijkhuizen L. Evolution toward small molecule inhibitor resistance affects native enzyme function and stability, generating acarbose-insensitive cyclodextrin glucanotransferase variants. J Biol Chem. 2008;283:10727–10734. doi: 10.1074/jbc.M709287200. [DOI] [PubMed] [Google Scholar]
- 46.Stemmer WP. Rapid evolution of a protein in vitro by DNA shuffling. Nature. 1994;370:389–391. doi: 10.1038/370389a0. [DOI] [PubMed] [Google Scholar]
- 47.Ho SN, Hunt HD, Horton RM, Pullen JK, Pease LR. Site-directed mutagenesis by overlap extension using the polymerase chain reaction. Gene. 1989;77:51–59. doi: 10.1016/0378-1119(89)90358-2. [DOI] [PubMed] [Google Scholar]
- 48.Sarkar G, Sommer SS. The “megaprimer” method of site-directed mutagenesis. Biotechniques. 1990;8:404–407. [PubMed] [Google Scholar]
- 49.Bolivar F, Rodriguez RL, Greene PJ, Betlach MC, Heyneker HL, Boyer HW. Construction and characterization of new cloning vehicles. II. A multipurpose cloning system. Gene. 1977;2:95–113. [PubMed] [Google Scholar]
- 50.Sambrook J, Fritsch EF, Maniatis T. Molecular cloning: a laboratory manual. 2nd ed. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory; 1989. [Google Scholar]
- 51.Bouthors AT, Dagoneau-Blanchard N, Naas T, Nordmann P, Jarlier V, Sougakoff W. Role of residues 104, 164, 166, 238 and 240 in the substrate profile of PER-1 β-lactamase hydrolysing third-generation cephalosporins. Biochem J. 1998;330(Part 3):1443–1449. doi: 10.1042/bj3301443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tribuddharat C, Moore RA, Baker P, Woods DE. Burkholderia pseudomallei class A β-lactamase mutations that confer selective resistance against ceftazidime or clavulanic acid inhibition. Antimicrob Agents Chemother. 2003;47:2082–2087. doi: 10.1128/AAC.47.7.2082-2087.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]