

Abstract
Checkpoint kinase 2 (Chk2), a ser/thr kinase involved in the ATM-Chk2 checkpoint pathway, is activated by genomic instability and DNA damage and results in either arrest of the cell cycle to allow DNA repair to occur or apoptosis if the DNA damage is severe. Drugs that specifically target Chk2 could be beneficial when administered in combination with current DNA-damaging agents used in cancer therapy. Recently, a novel inhibitor of Chk2, NSC 109555, was identified that exhibited high potency (IC50 = 240 nM) and selectivity. This compound represents a new chemotype and lead for the development of novel Chk2 inhibitors that could be used as therapeutic agents for the treatment of cancer. To facilitate the discovery of new analogs of NSC 109555 with even greater potency and selectivity, we have solved the crystal structure of this inhibitor in complex with the catalytic domain of Chk2. The structure confirms that the compound is an ATP-competitive inhibitor, as the electron density clearly reveals that it occupies the ATP-binding pocket. However, the mode of inhibition differs from that of the previously studied structure of Chk2 in complex with debromohymenialdisine, a compound that inhibits both Chk1 and Chk2. A unique hydrophobic pocket in Chk2, located very close to the bound inhibitor, presents an opportunity for the rational design of compounds with higher binding affinity and greater selectivity.
Keywords: checkpoint kinase inhibitor, structure-assisted drug design, cocrystal structure
Introduction
Checkpoint kinase 2 (Chk2) is a tumor suppressor protein that participates in the regulation of the cell cycle in response to DNA damage by phosphorylating a number of downstream targets involved in cell cycle arrest, DNA damage repair, and apoptosis.1–3 Chk2 is activated primarily by either ATM or DNA-PK (also ATR and hMPS1) via phosphorylation of Thr68 in the SQ/TQ cluster domain of Chk2,4 resulting in homodimerization followed by trans-activating autophosphorylation of Thr383 and Thr3875,6 and cis-phosphorylation of Ser516.7 Crystal structures of the Chk2 kinase domain in complex with ADP and debromohymenialdisine (DBQ) revealed a novel dimeric arrangement of kinase molecules in which the activation loops are exchanged between adjacent kinase domains to facilitate the trans-autophosphorylation mechanism.6,8 On activation, Chk2 phosphorylates a number of downstream targets involved in cell cycle arrest such as Cdc25A, Cdc25C, BRCA1, and/or proteins that play a functional role in apoptosis such as p53, PML, and E2F1 [for review see Refs. 9 and10].
There are two main rationales for the development of Chk2 inhibitors.11 First, studies have shown that Chk2 is activated in precancerous lesions with genomic instability12,13 and cancer cells grown in culture.14,15 Moreover, Chk2 has been shown to play an important role in the release of survivin, a protein implicated in tumor survival, from the mitochondria following DNA damage,16 and in tumor cell adaptation to changes that result from the cycling nature of hypoxia and reoxygenation found in solid tumors.17 Thus, a selective inhibitor of Chk2 is of considerable interest as it may result in the death of cells with endogenous activation of Chk2 in response to Chk2 inhibition. Second, selective inhibition of Chk2 in p53-deficient tumor cells may increase their sensitivity to chemotherapeutics and radiation by targeting the G2 checkpoint.9,18–20 Previously, it has been shown that down-regulation of Chk2 in p53-defective tumor cells results in enhanced apoptotic activity in response to ionizing radiation.21 Thus, targeted inhibition of Chk2 could potentially result in an increase in the therapeutic effects of DNA-targeting agents in p53-defective tumors.9,20,22 Additionally, because of the role of p53 in the initiation of apoptosis in response to DNA damage, inhibition of Chk2 in normal cells may also protect normal tissues,1,23 which may reduce side effects due to chemotherapy or radiation therapy.24
Because only a few specific inhibitors of Chk2 have been identified to date,24–30 the discovery of additional inhibitors remains of considerable interest. A novel inhibitor of Chk2, NSC 109555 [Fig. 1(A)], was recently identified by our laboratories using a high-throughput screening assay.29 NSC 109555 is an ATP-competitive inhibitor that represents a new chemotype for a Chk2 inhibitor with high potency (IC50 = 240 nM) and selectivity. Seeking to understand the mode of inhibition of Chk2 by NSC 109555, we cocrystallized the catalytic domain of Chk2 with the inhibitor and refined the structure to a resolution of 2.05 Å. These results provide detailed structural insight into the protein–inhibitor interactions and establish an opportunity for structure-assisted optimization of NSC 109555 and the development of new inhibitors of Chk2.
Figure 1.
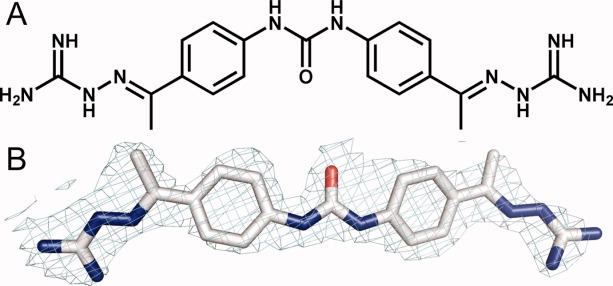
A: Chemical structure of NSC 109555. B: The coordinates of NSC 109555 superimposed on the final 2Fo − Fc electron density maps at 2.05 Å resolution contoured at the 1σ level.
Results and Discussion
Crystallization and overall structure
Initial cocrystallization trials searching for diffraction quality crystals of the Chk2 catalytic domain in complex with NSC 109555 failed to yield single crystals or did not result in any crystals at all. Thus, streak seeding was used to obtain single crystals of the Chk2-NSC 109555 complex using previously grown Chk2-ADP crystals as a seed source. The crystals diffracted X-rays to 2.05 Å resolution and the structure was solved by molecular replacement using the coordinates of the Chk2-ADP complex as a search model (PDB code: 2CN5). The resulting 2Fo − Fc and Fo − Fc electron density maps enabled unambiguous placement of NSC 109555 into the ATP-binding pocket of Chk2 [Fig. 1(B)]. The refined structure exhibited a working R-factor of 0.22 and R-free of 0.25. The Ramachandran plot obtained from an analysis of the coordinates resulted in more than 90% of the residues in the most-favored region and no residues in the disallowed region. X-ray data collection and refinement statistics are summarized in Table I.
I.
Data Collection and Model Refinement Statistics
| Parameter | Chk2-NSC 109555 |
|---|---|
| Data collection | |
| X-ray source | 22-ID, SER-CAT |
| Space group | P3221 |
| Cell dimensions | |
| a = b (Å) | 90.9 |
| c (Å) | 93.6 |
| Wavelength (Å) | 1.0 |
| Resolution (Å)a | 50–2.05 (2.12–2.05) |
| Total reflections | 180,774 |
| Unique reflections | 28,429 |
| Completeness (%) | 99.9 (99.9) |
| Redundancy | 7.4 (7.3) |
| Rsym | 0.062 (0.541) |
| I/σ (I) | 25.9 (4.1) |
| Model refinement statistics | |
| Resolution (Å) | 50–2.05 |
| Unique reflections used | 26,962 |
| Rwork/Rfreeb | 0.22/0.25 |
| Molecules/A.U. | |
| Protein | 1 |
| Water | 133 |
| NSC 109555 | 1 |
| Nitrate | 1 |
| Mean B factor (Å2) | |
| Protein | 42.9 |
| Water | 49.3 |
| NSC 109555 | 57.6 |
| Nitrate | 43.2 |
| RMSDs | |
| Bond length (Å) | 0.016 |
| Bond angle (°) | 1.6 |
| Ramachandran plots | |
| Most favored (%) | 91.1 |
| Additionally allowed (%) | 8.9 |
| Generously allowed (%) | 0 |
| Disallowed (%) | 0 |
| PDB code | 2W0J |
A.U., asymmetric unit.
Values for the highest-resolution shell of data are given in parentheses.
Calculated with 5% of the data.
Binding mode of NSC 109555
The biochemical characterization of NSC 109555 indicated that it is a potent competitive inhibitor of ATP binding, exhibiting an IC50 of 240 nM.29 The cocrystal structure of Chk2 with NSC 109555 confirms that the inhibitor interacts with the ATP-binding pocket in an elongated manner (see Fig. 2) but more importantly, the mode of binding differs from what had been previously predicted by molecular modeling.29 In the binding mode predicted by computational methods, the inhibitor adopted a “horse-shoe” conformation with hydrogen bonds to Leu226, Asp311, Asp328, and Glu351. The electron density maps, however, clearly reveal that NSC 109555 is anchored to the ATP-binding pocket in an extended conformation via hydrogen bonding of the guanidinium terminus to Glu273, which is located on the C-α helix (see Fig. 3). Additional interactions with the ATP-binding pocket occur via water-mediated hydrogen bonds between the carbonyl group of the urea functional group of the inhibitor with the backbone carbonyl of Glu302 and the amide of Met304. The aryl moieties of the phenyl guanidinohydrazone also contribute to binding affinity via van der Waals interactions with Val 234, Lys249, Ile299, Leu301, and Leu354 resulting in highly favorable surface complementarity between the inhibitor and the ATP-binding pocket. The cocrystal structure reveals the presence of a hydrophobic pocket in Chk2 which is directly accessible by the methyl group adjacent to the guanidylhydrazone moiety that is bound to Glu273 [Fig. 4(A)]. The size and location of this cavity is such that it could accommodate additional substituents on the methyl group.
Figure 2.

Crystal structure of the Chk2 catalytic domain in complex with NSC 109555 (PDB code: 2W0J). A: The protein is illustrated in green ribbons and the binding mode of the inhibitor is shown in space-filling spheres with carbon (gray), nitrogen (blue), and oxygen (red). B: A 90° rotation with respect to the vertical axis illustrating the orientation of the inhibitor in the hinge region of Chk2.
Figure 3.
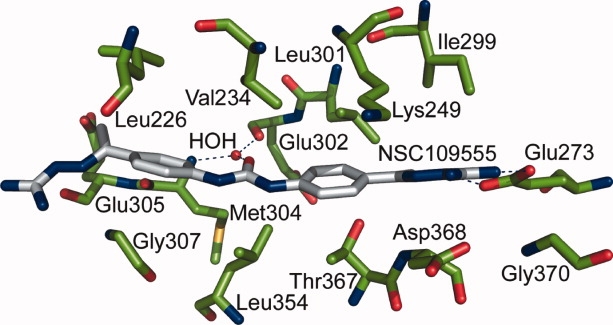
The binding mode of NSC 109555 in the Chk2 ATP-binding pocket (PDB code: 2W0J). The enzyme residues are depicted in green stick form whereas the inhibitor is depicted in gray stick form. Nitrogen atoms are depicted in blue and oxygen in red. The blue dashes represent hydrogen bonds.
Figure 4.

A: Illustration of the surface area of the Chk2 ATP-binding pocket (gray) with bound NSC 109555 (yellow stick model) highlighting the location of the hydrophobic pocket (PDB code: 2W0J). B: The surface area of the Chk1 ATP-binding pocket (gray) with bound inhibitor ABO (green stick) superimposed with the coordinates of the Chk2-NSC 109555 (yellow stick) coordinates (PDB code: 2C3K). Nitrogen atoms are depicted in blue and oxygen in red.
Insights into structure–activity relationships for NSC 109555
Prior structure–activity relationship (SAR) studies of NSC 109555 using structural analogs (see Table II in Ref. 29) suggested that the guanidylhydrazone and urea groups are important for the activity of NSC 109555. With the availability of the cocrystal structure, it is now possible to correlate these studies with important aspects of the protein–inhibitor interactions. For example, compounds NSC 177944 (meta-substituted analog) and NSC 177941 (meta-substituted analog with a urea to thiourea substitution) were both inactive against Chk2. The meta-substituted analog would result in different orientations of the urea and guanidylhydrazone, thereby disrupting the water-mediated hydrogen bonds between the urea carbonyl and the backbone of Glu302 and Met304, and would also disrupt the binding of the guanidinium terminus to Glu273, which appears to be a critical residue for anchoring the inhibitor in the active site. Furthermore, the van der Waals interactions between the protein and both aryl groups in NSC 109555 appear to be structurally important, as replacement of these groups by a different linker, such as in NSC 67931 (a single phenyl group) or NSC 69432 (an aliphatic group), renders the molecules inactive against Chk2. The urea functional group also appears to be an important structural feature because replacing it with a thiourea group, as in the case of NSC 177940, results in an inactive compound. This can be rationalized in terms of the less electronegative character of the C=S bond which could potentially disrupt the water-mediated hydrogen bonds to Glu302 and Met304. Although this molecule is missing one of the terminal guanidylhydrazone groups, the second guanidylhydrazone group does not appear to make any strong contacts with the ATP-binding pocket in the crystal structure.
Comparison with the Chk2-DBQ and Chk2-ADP structures
To gain additional insight into the high specificity of NSC 109555 for Chk2, we compared the cocrystal structure with the previously determined structure of Chk2 in complex with DBQ at 2.70 Å resolution.6 DBQ is an ATP-competitive inhibitor with an IC50 below 183 nM for Chk2 but it is not specific for Chk2.10,25,29 Overall, the global structures of the protein molecules in the two cocrystal structures are almost identical. The binding mode of DBQ (see Fig. 5) differs from that of NSC 109555 in that it exhibits direct hydrogen bonding interactions with the backbone of the hinge residues Glu302 and Met304 and an additional extensive hydrogen bonding network involving residues Glu308 and Asn352. In contrast, there are no direct hydrogen bonds between NSC 109555 and the hinge region. Rather, there are water-mediated hydrogen bonds between the urea carbonyl and the backbone of the hinge residues Glu302 and Met304. Furthermore, there are no polar or water-mediated interactions between DBQ and the ATP-binding pocket, unlike NSC 109555, which exhibits both water-mediated hydrogen bonds and a polar interaction between the guanidinium terminus and Glu273.
Figure 5.
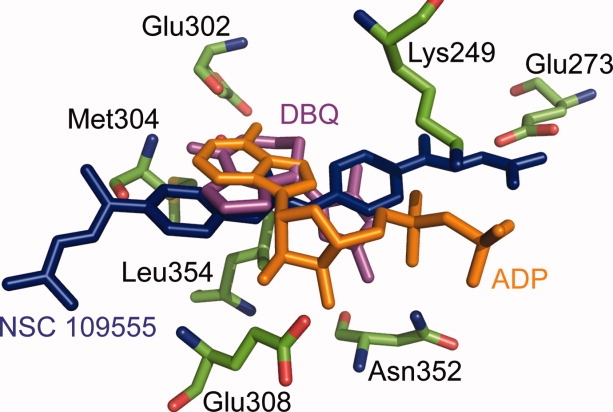
An overlay of the coordinates of the Chk2-NSC 109555 (PDB code: 2W0J), Chk2-ADP (PDB code: 2CN5), and Chk2-DBQ (PDB code: 2CN8) crystal structures comparing the orientations of NSC 109555 (blue stick), ADP (orange stick), and DBQ (purple stick) in the ATP-binding pocket of Chk2 (green stick). The coordinates for the amino acid side chains are for the refined model of Chk2 in complex with NSC 109555.
Another significant difference between the structures is the orientation of Lys249. In the structures of Chk2 in complex with ADP and DBQ, the Lys249 residue is positioned closer to Glu273. However, on binding of NSC 109555 the side chain of Lys249 shifts approximately 3.9 Å away from Glu273 such that the aliphatic portion of the former residue packs against the aryl ring of NSC 109555, thereby eliminating its interaction with the latter residue. These two residues are strictly conserved among kinases and the lysine plays an important structural role by coordinating the α- and β-phosphates of ATP. A common feature among kinases is a salt-bridge between these two residues that couples the conformation of the C-α helix with nucleotide binding.31 Another difference between the structures is that the glycine-rich phosphate binding loop (P-loop) is disordered in the Chk2-NSC 109555 complex (residues 229–231 lack electron density) whereas the same loop is well defined in the Chk2-ADP complex. Interestingly, the P-loop is also disordered in the Chk2-DBQ complex. This suggests that this loop undergoes some dynamic changes in Chk2 on inhibitor binding.
Structural insights into Chk2 selectivity
The IC50 of NSC 109555 for Chk2 is 240 nM whereas its IC50 for Chk1 is greater than 10 μM.29 NSC 109555 also exhibits high selectivity for Chk2 against a panel of 20 kinases.29 The availability of the cocrystal structure of Chk2 in complex with NSC 109555 enabled us to identify structural features that may contribute to the high selectivity of NSC 109555 for Chk2 over Chk1 and other kinases. In general, the ATP-binding pockets of Chk2 and Chk1 are highly conserved, but there are some notable differences. The most significant of these are in the hinge loop where Chk2 residues Leu303, Met304, and Glu305 are replaced by Tyr, Cys, and Ser in Chk1, respectively. Additionally, residue Thr367 of Chk2 (located proximal to the aryl ring of NSC 109555) is a serine in Chk1. Portions of the glycine-rich P-loop in Chk2, which is located directly above the inhibitor, are disordered (residues 229–231), whereas this loop is well-defined in the structure of Chk1. There are also sequence differences between Chk2 and Chk1 in this glycine-rich loop (P-loop). The P-loop contains a sequence motif that is highly conserved among kinases, GXGXΦG, where Φ is usually a Tyr or Phe residue.31 In Chk2, the Φ position is occupied by Cys231 whereas in Chk1 this residue is a bulkier tyrosine. The P-loop is known to be conformationally flexible, which allows it to accommodate inhibitors that induce large structural distortions in this loop by interacting with the conserved aromatic Φ residue.31 Indeed, a structural alignment of Chk2 and Chk1 [apo form (PDB code:1IA8) and in complex with an inhibitor (PDB code: 2C3K)] reveals differing conformations of this loop which results in different shapes of the ATP-binding pockets in the two proteins. The structural features of the Chk1 ATP-binding pocket also reveal that steric hindrance of Glu55 (Glu 273 in Chk2) by the neighboring Tyr20 and Lys38 residues may prevent the guanidinium terminus of NSC 109555 from forming hydrogen bonds with Glu55, which would likely reduce the binding affinity [Fig. 4(B)].
Seeking further insight into the high selectivity that the inhibitor exhibits for Chk2, we also examined crystal structures of other kinases that were screened for inhibition by NSC 109555.29 The conformation of NSC 109555 that is observed in the cocrystal structure with Chk2 did not fit into the ATP-binding pockets of any of these kinases without encountering some type of steric impediment. For example, the guanidinium terminus was sterically hindered from optimally interacting with the homologous glutamate residues (Glu273 in Chk2) in all of the structures examined, although in some instances small changes in the positioning of side chains near the glutamate would make it more accessible. Also, the orientation of the C-α helix on which Glu273 is located differs slightly in some structures when compared with Chk2, thereby altering the orientation of the glutamate side chain. In certain examples such as C-Met (PDB code: 1RLW), AKT (PDB code: 2GU8), and p38 (PDB code: 1KV1) kinases, against which NSC 109555 was found to be poorly active (IC50 > 10 μM), the inhibitor had a very poor fit to the ATP-binding pocket due to multiple steric constraints.32–34 This analysis is, of course, qualitative in nature as there are more influences on inhibitor binding than just steric interactions. For instance, overall protein dynamics may influence the selectivity of kinases for inhibitor binding, as has been observed in some instances.31 The conformation of the P-loop also differs significantly in these enzymes, which may influence access of the inhibitor to the ATP-binding pocket. Interestingly, all of these kinases have either a Phe or Tyr residue in the Φ position of the GXGXΦG motif in the P loop, in contrast to Chk2, which has a smaller cysteine residue with different chemical properties. Structural alignment of Chk2 with the ser/thr kinase PIM-1 (PDB code: 2OBJ) illustrates the highly conserved nature of their ATP-binding pockets, with the notable exception of Pro123 in the hinge region of PIM-1, which corresponds to Met304 in Chk2. This Pro residue is unable to maintain a hydrogen bond network to the urea group of NSC 109555 because it lacks the amide hydrogen, but the inhibitor could potentially maintain the guanidinium–glutamate interaction.35 Nevertheless, NSC 109555 was found to be poorly active against PIM-1 kinase (IC50 >10 μM).
Implications for drug design
The determination of the cocrystal structure of the catalytic domain of Chk2 in complex with NSC 109555 presents a new opportunity for the structure-assisted design of novel Chk2 inhibitors based on a highly potent and selective lead compound. The combination of high-throughput screening and structural biology has proven to be a powerful tool for guiding the optimization of the pharmacological properties of lead compounds in the design of specific protein kinase inhibitors.36–38 Among the important features discovered from the cocrystal structure reported herein are the water-mediated hydrogen bonds between the urea group of NSC 109555 and the hinge region of Chk2, the van der Waals interactions between the aryl moiety of the phenyl guanidinohydrazone and the ATP-binding pocket, the polar interactions between the guanidinium terminus and Glu273, and the hydrophobic pocket adjacent to Glu273.
It will be of particular interest to ascertain whether the binding affinity of the inhibitor can be increased by optimizing its interactions with the hydrophobic pocket. This can be explored by examining the effects of new substituents on the methyl group of NSC 109555, such as branched and cyclic alkanes that may fill this pocket and interact via hydrophobic packing. This may present an opportunity to modulate the specificity of inhibitors as well, because the shape and degree of hydrophobicity of this pocket varies quite a bit among kinases.36,37 For instance, in Chk2 the pocket is surrounded entirely by hydrophobic residues. Although the corresponding pocket in Chk1 is also primarily hydrophobic, one polar residue, Asn59 (Leu 277 in Chk2), is also present.39 In other kinases, access to this pocket is entirely blocked. For example, when the structure of Chk2 is compared with those of Zap (PDB code: 1U59) and EGFR (PDB code: 1XKK), it is self evident that full access to this pocket is impeded by two bulkier methionine residues in the latter two enzymes whereas the analogous residues are Leu277 and Leu301 in Chk2.40,41 Leu301 in Chk2 corresponds to the “gatekeeper” residue in many kinases,42 which has been found to form contacts with bound inhibitors and is poorly conserved. Accordingly, differences in this position appear to account for the selectivity of many kinase inhibitors by modulating access to the hydrophobic pocket.43 Indeed, inhibitors that were designed to interact with the homologous pocket in Chk1 showed increased binding affinity and selectivity for Chk1.39 Thus, this pocket is of considerable interest for the design of new inhibitors with greater potency and specificity for Chk2.
Another issue that may now be addressed is the exploration of alternate functional replacements for the guanidine moiety of NSC 109555. The highly basic nature of the guanidine group (pKa ∼ 12) may reduce cellular permeability and so a functional replacement that maintains the hydrogen bonding with Glu273 but has a lower pKa would be highly desirable.
Conclusions
The crystal structure of the catalytic domain of Chk2 in complex with a highly potent and selective inhibitor, NSC 109555, has been solved and provides a new template and chemotype for structure-assisted design of novel Chk2 inhibitors. The structure reveals binding interactions between NSC 109555 and the ATP-binding pocket that differ significantly from those observed in the previously reported structure of Chk2 in complex with DBQ, an inhibitor that does not exhibit selectivity for Chk2. A hydrophobic pocket near Glu273 of the ATP-binding pocket could potentially be exploited to improve the selectivity and binding affinity of the inhibitor by incorporating new functional groups aimed at optimizing the interactions between the inhibitor and the pocket. The design of specific inhibitors of protein kinases continues to remain a significant challenge. However, these crystallographic results provide new information that will facilitate the rational design of novel Chk2 inhibitors that may not only be useful therapeutic agents for the treatment of cancer, but also as molecular tools for biological studies of Chk2. To this end, we are continuing to develop and test additional analogs of NSC 109555.
Methods
Cloning, protein expression, and purification
The catalytic domain of human Chk2 (Ser210-Glu531) was amplified from the full-length cDNA clone by the polymerase chain reaction (PCR) using the following oligonucleotide primers: 5′-GAG AAC CTG TAC TTC CAG GGT GGT GGT GGT TCT CAT ATG TCA GTT TAT CCT AAG GCA TTA AGA G-3′ and 5′-GGG GAC CAC TTT GTA CAA GAA AGC TGG GTT ATT ACT CGG CAC CCT CGG CTT CCC CTT CAC-3′ (primer R). The PCR amplicon was subsequently used as a template for a second PCR with the following primers: 5′-GGG GAC AAG TTT GTA CAA AAA AGC AGG CTC GGA GAA CCT GTA CTT CCA G-3′ and primer R. The amplicon from the second PCR was inserted by recombinational cloning into the entry vector pDONR201 (Invitrogen, Carlsbad, CA) and the nucleotide sequence confirmed experimentally. The open reading frame encoding the Chk2 catalytic domain (Ser210-Glu531), now preceded by an in-frame recognition site for tobacco etch virus (TEV) protease and a seven amino acid spacer (ENLYFQGGGGSHM), was moved by recombinational cloning into the destination vector pDEST-HisMBP44 to create pDZ1927. This plasmid produces the human Chk2 catalytic domain as a fusion to the C-terminus of E. coli maltose binding protein (MBP) with an intervening TEV protease recognition site. The MBP contains an N-terminal hexahistidine tag for affinity purification by immobilized metal affinity chromatography. The fusion protein was expressed in the E. coli strain BL21-CodonPlus (DE3)-RIL (Stratagene, La Jolla, CA). Cells were grown to mid-log phase (OD600 ∼ 0.5) at 37°C in Luria broth containing 100 μg/mL ampicillin, 30 μg/mL chloramphenicol, and 0.2% glucose. Overproduction of the fusion protein was induced with isopropyl-β-d-thiogalactopyranoside at a final concentration of 1 mM for 4 h at 30°C. The cells were pelleted by centrifugation and stored at −80°C.
All purification procedures were performed at 4–8°C. Ten grams of E. coli cell paste were suspended in 150 mL ice-cold 50 mM HEPES (pH 7.5), 200 mM NaCl, 25 mM imidazole buffer (buffer A) containing 1 mM benzamidine HCl (Sigma Chemical Company, St. Louis, MO) and complete EDTA-free protease inhibitor cocktail tablets (Roche Molecular Biochemicals, Indianapolis, IN). The cells were lysed with an APV-1000 homogenizer (Invensys, Roholmsvej, Denmark) at 10,000 psi and centrifuged at 30,000g for 30 min. The supernatant was filtered through a 0.22 μm polyethersulfone membrane and applied to a 12 mL Ni-NTA superflow column (Qiagen, Valencia, CA) equilibrated in buffer A. The column was washed to baseline with buffer A and then eluted with a linear gradient of imidazole to 250 mM. Fractions containing recombinant His6-MBP-Chk2 catalytic domain were pooled, concentrated using an Amicon YM30 membrane (Millipore Corporation, Bedford, MA), diluted with 50 mM HEPES (pH 7.5) 200 mM NaCl buffer to reduce the imidazole concentration to about 25 mM and digested overnight at 4°C with His6-tagged TEV protease.45 The digest was applied to a 30 mL Ni-NTA superflow column equilibrated in buffer A and the catalytic domain emerged in the column effluent. The column effluent was incubated overnight with 10 mM dithiothreitol, concentrated using an Amicon YM10 membrane, and applied to a HiPrep 26/60 Sephacryl S-100 HR column equilibrated with 25 mM Tris (pH 7.2), 150 mM NaCl, 2 mM tris(2-carboxyethyl)phosphine buffer. The peak fractions containing recombinant catalytic domain were pooled and concentrated to 35–45 mg/mL (estimated at 280 nm using a molar extinction coefficient of 32,890 M−1 cm−1). Aliquots were flash-frozen in liquid nitrogen and stored at −80°C. The final product was judged to be >90% pure by sodium dodecyl sulfate-polyacrylamide gel electrophoresis. The molecular weight was confirmed by electrospray ionization mass spectrometry.
Crystallization and data collection
All crystallization reagents were obtained from Hampton Research (Aliso Viejo, CA). NSC 109555 was supplied by the Drug Synthesis and Chemistry Branch, Developmental Therapeutics Program, National Cancer Institute, Rockville, MD, dissolved in neat DMSO and stored at −80°C. To obtain diffraction quality crystals of Chk2 in complex with NSC 109555, streak seeding was necessary. First, crystals of Chk2 in complex with ADP were grown as previously described and were used as seed sources.6 A solution of 10 mg/mL Chk2 in 25 mM Tris pH 7.2, 150 mM NaCl, 2 mM TCEP - was incubated with 1 mM NSC 109555 (dissolved in neat DMSO) such that the final DMSO concentration in the protein–inhibitor mixture was 10% v/v. The mixture was incubated at room temperature for 30 min followed by an additional incubation for 1.5 h at 4°C. Insoluble compound was removed by centrifugation before crystallization setup. Crystals were grown using the hanging-drop vapor diffusion technique. A 1:1 ratio of protein–inhibitor solution and well solution (0.1M HEPES pH 7.8, 0.1M magnesium nitrate, 14% w/v polyethylene glycol 3350, and 16% v/v ethylene glycol) were mixed and the drops were sealed over 1 mL of well solution. The tray was incubated at 4°C overnight and the drops were streak-seeded the following day by transferring microseeds from the Chk2-ADP crystal to the drops with a whisker. Crystals grew to final dimensions of 0.3 mm × 0.1 mm × 0.1 mm within 3–7 days. The crystals were removed from the drop with a litho-loop and were immediately flash frozen in liquid nitrogen. X-ray diffraction data were collected from a single crystal held at approximately 100 K at beamline 22-ID (SER-CAT) of the Advanced Photon Source. A data set at 1.0 Å wavelength consisting of 120 images was collected using a 1.0° oscillation angle and a 3 s exposure time. The data were integrated and scaled using HKL3000.46 The crystals for the Chk2-NSC 109555 complex belong to space group P3221 with unit cell dimensions a = b = 90.9, c = 93.6 Å and contain 1 molecule in the asymmetric unit (Matthew's coefficient 3.1, 59.7% solvent content).47
The Chk2-NSC 109555 complex structure was solved by molecular replacement using the MOLREP program from the CCP4 suite.48 The coordinates of the Chk2-ADP structure (pdb code: 2CN5) were used as a search model after removing all solvent and ligand molecules.6 Cross-rotation and translational searches for 1 molecule in the asymmetric unit were conducted using a resolution range of 15–2.5 Å followed by rigid body refinement with REFMAC5. The model was rebuilt and corrected using cross-validated σa-weighted 2Fo − Fc and Fo − Fc maps49 with COOT50 and refined with REFMAC5. The progress of the refinement was monitored by setting aside 5% of the reflections for use in calculation of the R-free value.51 The coordinates for the NSC 109555 molecule and appropriate chemical restraints and topology files were prepared using the Dundee PRODRG52 server and the location of the inhibitor was determined using the Fo − Fc maps contoured at 3σ level. Water molecules were added with COOT after the working R-factor dropped below 0.3 and were refined with REFMAC5. The model was refined to a working R-factor of 0.22 and an R-free of 0.25. Model validation was performed using MolProbity.53 The coordinates and structure factors for the Chk2 catalytic domain in complex with NSC 109555 were deposited in the Protein Data Bank with accession code 2W0J. All molecular superpositions and figures were made using Pymol (Delano Scientific LLC, Castro City, CA).
Acknowledgments
Electrospray mass spectrometry experiments were conducted on the LC/ESMS instrument maintained by the Biophysics Resource in the Structural Biophysics Laboratory, Center for Cancer Research, National Cancer Institute at Frederick. X-ray diffraction data were collected at the Southeast Regional Collaborative Access Team (SER-CAT) beamline 22-ID at the Advanced Photon Source, Argonne National Laboratory. Supporting institutions may be found at http://www.ser-cat.org/members.html. Use of the Advanced Photon Source was supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under contract no. W-31-109-Eng-38.
Glossary
Abbreviations
- ADP
adenosine diphosphate
- ATM
ataxia telangectasia mutated protein
- ATP
adenosine triphosphate
- ATR
ataxia telangectasia-related protein; Chk1, checkpoint kinase 1
- Chk2
checkpoint kinase 2
- DBQ
debromohymenialdisine
- DNA-PK
DNA-dependent protein kinase
- NSC
National Screening Collection
- PML
promyelocytic leukemia protein
- p53
tumor protein 53.
References
- 1.Hirao A, Kong YY, Matsuoka S, Wakeham A, Ruland J, Yoshida H, Liu D, Elledge SJ, Mak TW. DNA damage-induced activation of p53 by the checkpoint kinase Chk2. Science. 2000;287:1824–1827. doi: 10.1126/science.287.5459.1824. [DOI] [PubMed] [Google Scholar]
- 2.Matsuoka S, Rotman G, Ogawa A, Shiloh Y, Tamai K, Elledge SJ. Ataxia telangiectasia-mutated phosphorylates Chk2 in vivo and in vitro. Proc Natl Acad Sci USA. 2000;97:10389–10394. doi: 10.1073/pnas.190030497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hirao A, Cheung A, Duncan G, Girard PM, Elia AJ, Wakeham A, Okada H, Sarkissian T, Wong JA, Sakai T, De Stanchina E, Bristow RG, Suda T, Lowe SW, Jeggo PA, Elledge SJ, Mak TW. Chk2 is a tumor suppressor that regulates apoptosis in both an ataxia telangiectasia mutated (ATM)-dependent and an ATM-independent manner. Mol Cell Biol. 2002;22:6521–6532. doi: 10.1128/MCB.22.18.6521-6532.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ahn JY, Schwarz JK, Piwnica-Worms H, Canman CE. Threonine 68 phosphorylation by ataxia telangiectasia mutated is required for efficient activation of Chk2 in response to ionizing radiation. Cancer Res. 2000;60:5934–5936. [PubMed] [Google Scholar]
- 5.Ahn JY, Li X, Davis HL, Canman CE. Phosphorylation of threonine 68 promotes oligomerization and autophosphorylation of the Chk2 protein kinase via the forkhead-associated domain. J Biol Chem. 2002;277:19389–19395. doi: 10.1074/jbc.M200822200. [DOI] [PubMed] [Google Scholar]
- 6.Oliver AW, Paul A, Boxall KJ, Barrie SE, Aherne GW, Garrett MD, Mittnacht S, Pearl LH. Trans-activation of the DNA-damage signalling protein kinase Chk2 by T-loop exchange. EMBO J. 2006;25:3179–3190. doi: 10.1038/sj.emboj.7601209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wu X, Chen J. Autophosphorylation of checkpoint kinase 2 at serine 516 is required for radiation-induced apoptosis. J Biol Chem. 2003;278:36163–36168. doi: 10.1074/jbc.M303795200. [DOI] [PubMed] [Google Scholar]
- 8.Oliver AW, Knapp S, Pearl LH. Activation segment exchange: a common mechanism of kinase autophosphorylation? Trends Biochem Sci. 2007;32:351–356. doi: 10.1016/j.tibs.2007.06.004. [DOI] [PubMed] [Google Scholar]
- 9.Pommier Y, Weinstein JN, Aladjem MI, Kohn KW. Chk2 molecular interaction map and rationale for Chk2 inhibitors. Clin Cancer Res. 2006;12:2657–2661. doi: 10.1158/1078-0432.CCR-06-0743. [DOI] [PubMed] [Google Scholar]
- 10.Antoni L, Sodha N, Collins I, Garrett MD. CHK2 kinase: cancer susceptibility and cancer therapy—two sides of the same coin? Nat Rev Cancer. 2007;7:925–936. doi: 10.1038/nrc2251. [DOI] [PubMed] [Google Scholar]
- 11.Pommier Y, Sordet O, Rao VA, Zhang H, Kohn KW. Targeting chk2 kinase: molecular interaction maps and therapeutic rationale. Curr Pharm Des. 2005;11:2855–2872. doi: 10.2174/1381612054546716. [DOI] [PubMed] [Google Scholar]
- 12.Bartkova J, Horejsi Z, Koed K, Kramer A, Tort F, Zieger K, Guldberg P, Sehested M, Nesland JM, Lukas C, Orntoft T, Lukas J, Bartek J. DNA damage response as a candidate anti-cancer barrier in early human tumorigenesis. Nature. 2005;434:864–870. doi: 10.1038/nature03482. [DOI] [PubMed] [Google Scholar]
- 13.Gorgoulis VG, Vassiliou LV, Karakaidos P, Zacharatos P, Kotsinas A, Liloglou T, Venere M, Ditullio RA, Jr, Kastrinakis NG, Levy B, Kletsas D, Yoneta A, Herlyn M, Kittas C, Halazonetis TD. Activation of the DNA damage checkpoint and genomic instability in human precancerous lesions. Nature. 2005;434:907–913. doi: 10.1038/nature03485. [DOI] [PubMed] [Google Scholar]
- 14.DiTullio RA, Jr., Mochan TA, Venere M, Bartkova J, Sehested M, Bartek J, Halazonetis TD. 53BP1 functions in an ATM-dependent checkpoint pathway that is constitutively activated in human cancer. Nat Cell Biol. 2002;4:998–1002. doi: 10.1038/ncb892. [DOI] [PubMed] [Google Scholar]
- 15.Bartek J, Lukas J. Chk1 and Chk2 kinases in checkpoint control and cancer. Cancer Cell. 2003;3:421–429. doi: 10.1016/s1535-6108(03)00110-7. [DOI] [PubMed] [Google Scholar]
- 16.Ghosh JC, Dohi T, Raskett CM, Kowalik TF, Altieri DC. Activated checkpoint kinase 2 provides a survival signal for tumor cells. Cancer Res. 2006;66:11576–11579. doi: 10.1158/0008-5472.CAN-06-3095. [DOI] [PubMed] [Google Scholar]
- 17.Freiberg RA, Hammond EM, Dorie MJ, Welford SM, Giaccia AJ. DNA damage during reoxygenation elicits a Chk2-dependent checkpoint response. Mol Cell Biol. 2006;26:1598–1609. doi: 10.1128/MCB.26.5.1598-1609.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhou BB, Anderson HJ, Roberge M. Targeting DNA checkpoint kinases in cancer therapy. Cancer Biol Ther. 2003;2:S16–S22. [PubMed] [Google Scholar]
- 19.Zhou BB, Sausville EA. Drug discovery targeting Chk1 and Chk2 kinases. Prog Cell Cycle Res. 2003;5:413–421. [PubMed] [Google Scholar]
- 20.Levesque AA, Eastman A. p53-based cancer therapies: is defective p53 the Achilles heel of the tumor? Carcinogenesis. 2007;28:13–20. doi: 10.1093/carcin/bgl214. [DOI] [PubMed] [Google Scholar]
- 21.Yu Q, Rose JH, Zhang H, Pommier Y. Antisense inhibition of Chk2/hCds1 expression attenuates DNA damage-induced S and G2 checkpoints and enhances apoptotic activity in HEK-293 cells. FEBS Lett. 2001;505:7–12. doi: 10.1016/s0014-5793(01)02756-9. [DOI] [PubMed] [Google Scholar]
- 22.Sharma V, Hupp CD, Tepe JJ. Enhancement of chemotherapeutic efficacy by small molecule inhibition of NF-κB and checkpoint kinases. Curr Med Chem. 2007;14:1061–1074. doi: 10.2174/092986707780362844. [DOI] [PubMed] [Google Scholar]
- 23.Takai H, Naka K, Okada Y, Watanabe M, Harada N, Saito S, Anderson CW, Appella E, Nakanishi M, Suzuki H, Nagashima K, Sawa H, Ikeda K, Motoyama N. Chk2-deficient mice exhibit radioresistance and defective p53-mediated transcription. EMBO J. 2002;21:5195–5205. doi: 10.1093/emboj/cdf506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Arienti KL, Brunmark A, Axe FU, McClure K, Lee A, Blevitt J, Neff DK, Huang L, Crawford S, Pandit CR, Karlsson L, Breitenbucher JG. Checkpoint kinase inhibitors: SAR and radioprotective properties of a series of 2-arylbenzimidazoles. J Med Chem. 2005;48:1873–1885. doi: 10.1021/jm0495935. [DOI] [PubMed] [Google Scholar]
- 25.Curman D, Cinel B, Williams DE, Rundle N, Block WD, Goodarzi AA, Hutchins JR, Clarke PR, Zhou BB, Lees-Miller SP, Anderson RJ, Roberge M. Inhibition of the G2 DNA damage checkpoint and of protein kinases Chk1 and Chk2 by the marine sponge alkaloid debromohymenialdisine. J Biol Chem. 2001;276:17914–17919. doi: 10.1074/jbc.M100728200. [DOI] [PubMed] [Google Scholar]
- 26.Yu Q, La Rose J, Zhang H, Takemura H, Kohn KW, Pommier Y. UCN-01 inhibits p53 up-regulation and abrogates γ-radiation-induced G(2)-M checkpoint independently of p53 by targeting both of the checkpoint kinases, Chk2 and Chk1. Cancer Res. 2002;62:5743–5748. [PubMed] [Google Scholar]
- 27.Sharma V, Tepe JJ. Potent inhibition of checkpoint kinase activity by a hymenialdisine-derived indoloazepine. Bioorg Med Chem Lett. 2004;14:4319–4321. doi: 10.1016/j.bmcl.2004.05.079. [DOI] [PubMed] [Google Scholar]
- 28.Carlessi L, Buscemi G, Larson G, Hong Z, Wu JZ, Delia D. Biochemical and cellular characterization of VRX0466617, a novel and selective inhibitor for the checkpoint kinase Chk2. Mol Cancer Ther. 2007;6:935–944. doi: 10.1158/1535-7163.MCT-06-0567. [DOI] [PubMed] [Google Scholar]
- 29.Jobson AG, Cardellina JH, 2nd, Scudiero D, Kondapaka S, Zhang H, Kim H, Shoemaker R, Pommier Y. Identification of a bis-guanylhydrazone [4,4′-diacetyldiphenylurea-bis(guanylhydrazone); NSC 109555] as a novel chemotype for inhibition of Chk2 kinase. Mol Pharmacol. 2007;72:876–884. doi: 10.1124/mol.107.035832. [DOI] [PubMed] [Google Scholar]
- 30.Neff DK, Lee-Dutra A, Blevitt JM, Axe FU, Hack MD, Buma JC, Rynberg R, Brunmark A, Karlsson L, Breitenbucher JG. 2-Aryl benzimidazoles featuring alkyl-linked pendant alcohols and amines as inhibitors of checkpoint kinase Chk2. Bioorg Med Chem Lett. 2007;17:6467–6471. doi: 10.1016/j.bmcl.2007.09.098. [DOI] [PubMed] [Google Scholar]
- 31.Huse M, Kuriyan J. The conformational plasticity of protein kinases. Cell. 2002;109:275–282. doi: 10.1016/s0092-8674(02)00741-9. [DOI] [PubMed] [Google Scholar]
- 32.Pargellis C, Tong L, Churchill L, Cirillo PF, Gilmore T, Graham AG, Grob PM, Hickey ER, Moss N, Pav S, Regan J. Inhibition of p38 MAP kinase by utilizing a novel allosteric binding site. Nat Struct Biol. 2002;9:268–272. doi: 10.1038/nsb770. [DOI] [PubMed] [Google Scholar]
- 33.Schiering N, Knapp S, Marconi M, Flocco MM, Cui J, Perego R, Rusconi L, Cristiani C. Crystal structure of the tyrosine kinase domain of the hepatocyte growth factor receptor c-Met and its complex with the microbial alkaloid K-252a. Proc Natl Acad Sci USA. 2003;100:12654–12659. doi: 10.1073/pnas.1734128100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lin X, Murray JM, Rico AC, Wang MX, Chu DT, Zhou Y, Del Rosario M, Kaufman S, Ma S, Fang E, Crawford K, Jefferson AB. Discovery of 2-pyrimidyl-5-amidothiophenes as potent inhibitors for AKT: synthesis and SAR studies. Bioorg Med Chem Lett. 2006;16:4163–4168. doi: 10.1016/j.bmcl.2006.05.092. [DOI] [PubMed] [Google Scholar]
- 35.Kumar A, Mandiyan V, Suzuki Y, Zhang C, Rice J, Tsai J, Artis DR, Ibrahim P, Bremer R. Crystal structures of proto-oncogene kinase Pim1: a target of aberrant somatic hypermutations in diffuse large cell lymphoma. J Mol Biol. 2005;348:183–193. doi: 10.1016/j.jmb.2005.02.039. [DOI] [PubMed] [Google Scholar]
- 36.Toledo LM, Lydon NB, Elbaum D. The structure-based design of ATP-site directed protein kinase inhibitors. Curr Med Chem. 1999;6:775–805. [PubMed] [Google Scholar]
- 37.Scapin G. Structural biology in drug design: selective protein kinase inhibitors. Drug Discov Today. 2002;7:601–611. doi: 10.1016/s1359-6446(02)02290-0. [DOI] [PubMed] [Google Scholar]
- 38.Fischer PM. The design of drug candidate molecules as selective inhibitors of therapeutically relevant protein kinases. Curr Med Chem. 2004;11:1563–1583. doi: 10.2174/0929867043365062. [DOI] [PubMed] [Google Scholar]
- 39.Foloppe N, Fisher LM, Francis G, Howes R, Kierstan P, Potter A. Identification of a buried pocket for potent and selective inhibition of Chk1: prediction and verification. Bioorg Med Chem. 2006;14:1792–1804. doi: 10.1016/j.bmc.2005.10.022. [DOI] [PubMed] [Google Scholar]
- 40.Jin L, Pluskey S, Petrella EC, Cantin SM, Gorga JC, Rynkiewicz MJ, Pandey P, Strickler JE, Babine RE, Weaver DT, Seidl KJ. The three-dimensional structure of the ZAP-70 kinase domain in complex with staurosporine: implications for the design of selective inhibitors. J Biol Chem. 2004;279:42818–42825. doi: 10.1074/jbc.M407096200. [DOI] [PubMed] [Google Scholar]
- 41.Wood ER, Truesdale AT, McDonald OB, Yuan D, Hassell A, Dickerson SH, Ellis B, Pennisi C, Horne E, Lackey K, Alligood KJ, Rusnak DW, Glimer TM, Shewchuk L. A unique structure for epidermal growth factor receptor bound to GW572016 (Lapatinib): relationships among protein conformation, inhibitor off-rate, and receptor activity in tumor cells. Cancer Res. 2004;64:6652–6659. doi: 10.1158/0008-5472.CAN-04-1168. [DOI] [PubMed] [Google Scholar]
- 42.Buchanan SG. Protein structure: discovering selective protein kinase inhibitors. Targets. 2003;2:101–108. [Google Scholar]
- 43.Noble ME, Endicott JA, Johnson LN. Protein kinase inhibitors: insights into drug design from structure. Science. 2004;303:1800–1805. doi: 10.1126/science.1095920. [DOI] [PubMed] [Google Scholar]
- 44.Tropea JE, Cherry S, Nallamsetty S, Bignon C, Waugh DS. A generic method for the production of recombinant proteins in Escherichia coli using a dual hexahistidine-maltose-binding protein affinity tag. Methods Mol Biol. 2007;363:1–19. doi: 10.1007/978-1-59745-209-0_1. [DOI] [PubMed] [Google Scholar]
- 45.Kapust RB, Tozser J, Fox JD, Anderson DE, Cherry S, Copeland TD, Waugh DS. Tobacco etch virus protease: mechanism of autolysis and rational design of stable mutants with wild-type catalytic proficiency. Protein Eng. 2001;14:993–1000. doi: 10.1093/protein/14.12.993. [DOI] [PubMed] [Google Scholar]
- 46.Minor W, Cymborowski M, Otwinowski Z, Chruszcz M. HKL-3000: the integration of data reduction and structure solution–from diffraction images to an initial model in minutes. Acta Crystallogr D Biol Crystallogr. 2006;62:859–866. doi: 10.1107/S0907444906019949. [DOI] [PubMed] [Google Scholar]
- 47.Matthews BW. Solvent content of protein crystals. J Mol Biol. 1968;33:491–497. doi: 10.1016/0022-2836(68)90205-2. [DOI] [PubMed] [Google Scholar]
- 48.Potterton E, Briggs P, Turkenburg M, Dodson E. A graphical user interface to the CCP4 program suite. Acta Crystallogr D Biol Crystallogr. 2003;59:1131–1137. doi: 10.1107/s0907444903008126. [DOI] [PubMed] [Google Scholar]
- 49.Read RJ. Model phases: probabilities and bias. Methods Enzymol. 1997;277:110–128. doi: 10.1016/s0076-6879(97)77009-5. [DOI] [PubMed] [Google Scholar]
- 50.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 51.Brunger AT. Free R value: a novel statistical quantity for assessing the accuracy of crystal structures. Nature. 1992;355:472–475. doi: 10.1038/355472a0. [DOI] [PubMed] [Google Scholar]
- 52.Schuttelkopf AW, van Aalten DM. PRODRG: a tool for high-throughput crystallography of protein-ligand complexes. Acta Crystallogr D Biol Crystallogr. 2004;60:1355–1363. doi: 10.1107/S0907444904011679. [DOI] [PubMed] [Google Scholar]
- 53.Davis IW, Leaver-Fay A, Chen VB, Block JN, Kapral GJ, Wang X, Murray LW, Arendall WB, III, Snoeyink J, Richardson JS, Richardson DC. MolProbity: all-atom contacts and structure validation for proteins and nucleic acids. Nucleic Acids Res. 2007;35:W375–W383. doi: 10.1093/nar/gkm216. [DOI] [PMC free article] [PubMed] [Google Scholar]