

Abstract
To identify protein split sites quickly, a selection procedure by using chloramphenicol acetyl transferase (CAT) as reporter was introduced to search for folded protein fragments from libraries generated by random digestion and reassembly of the target gene, which yielded an abundant amount of DNA fragments with controllable lengths. Experimental results of tryptophan synthase alpha subunit (TSα) and TEM-1 β-lactamase agreed well with what the literature has reported. The solubility of these fragments correlated roughly with the minimum inhibitory concentrations of the CAT fusions. The application of this dissection protocol to protein fragment complementation assay (PCA) was evaluated using aminoglycoside-3′-phosphotransferase I (APH(3′)-I) as a model protein. Three nearly bisectional sites and a number of possible split points were identified, and guided by this result, four novel pairs of fragments were tested for complementation. Three out of four pairs partially restored the APH activity with the help of leucine zippers, and a truncated but active APH(3′)-I (Δ1–25) was also found. Finally, the weakly active APH(3′)-I-(1–253)NZ/CZ (254–271) containing a short 18 residue tag was further improved by error-prone PCR, and a best mutant was obtained showing a fourfold improvement after just one round of evolution. These results demonstrate that protein random dissection based on the CAT selection can provide an efficient search for protein breakage points and guide the design of fragments for protein complementation assay. Furthermore, more active fragment pairs can be achieved with the classical directed evolution approach.
Keywords: protein random dissection, chloramphenicol acetyl transferase, protein fragment complementation, directed evolution
Abbreviations
- APH(3′)-I
aminoglycoside-3′-phosphotransferase I
- APH(3′)-IIa
aminoglycoside-3′-phosphotransferase IIa
- CAT
chloramphenicol acetyl transferase
- MIC
minimum inhibitory concentration
- PCA
protein fragment complementation assay
- SDS-PAGE
sodium dodecyl sulfate polyacrylamide gel electrophoresis
- TSα
tryptophan synthase alpha subunit
Introduction
Proteins are made of single or multiple functional domains that often can fold independently. More recent evidences show that they can be fragmented into smaller folded units,1,2 which is relevant to protein evolution,3,4 re-design,5–7 fragment complementation,8–11 and protein structure-function relationship.2,12 However, current approaches for identifying protein breakage points have various limitations. Experimental methods employing proteolysis or chemical cleavage of purified proteins access only limited sites.13,14 A different strategy employed by several laboratories is to create random fragment libraries by random primer polymerase chain reaction (PCR) method,15 deoxyuridine incorporation method,12,16 or DNase I fragmentation,17,18 then search for properly folded polypeptides using a reporter protein such as green fluorescent protein (GFP) or an antibody.16 Folded units can be probed independently of structure and function by these approaches.12,15,16 However, the quality of fragment libraries constructed needs to be further improved because of preferential amplification, biased digestion, low efficiency, and the difficulty in controlling the lengths of fragments.
We previously developed a method combining gene fragmentation by DNase I with an additional reassembly step to generate a random fragment library, in which a pool of DNA fragments ranging from 50 bp up to the full gene size can be produced from a small amount of template DNA.19,20 The advantage of this protocol is that an abundant amount of short gene fragments of a desirable size can be easily obtained with a reassembly PCR step using an appropriate number of cycles. In this current study, the previously used reporter protein, GFP, was replaced by chloramphenicol acetyl transferase (CAT) to allow for quicker selection of soluble protein fragments by simply evaluating chloramphenicol resistance of fragment-CAT fusion proteins.21,22 Also, improvements were introduced that led to generation of gene fragments concentrating in the range of 250–500 bp, which covered the most likely folded units in the range of about 80–170 aa.17,23 Experimental results for tryptophan synthase alpha subunit (TSα) and TEM-1 β-lactamase agreed well with what the literature has reported.9,10,24 Further dissection of aminoglycoside-3′-phosphotransferase I (APH(3′)-I) yielded three nearly bisectional sites and a number of interesting cleavable points.
Furthermore, the application of this modified protocol to protein fragment complementation assay (PCA) was evaluated. PCA has been widely used in simple and rapid detection of in vivo protein–protein interaction for biochemical pathway mapping as well as drug discovery.25 A traditional method for PCA is to rationally cleave the target protein into two or more fragments based on an analysis of its structure,8,9 which, however, is often unattainable for many proteins. Protein bisection using an incremental truncation assay10,11,26 represents an improvement but requires a functionality-based selection assay, which limits its utility. Because PCA is based on the complementary action of normally two independently expressed protein fragments,25 it is reasonable that more active heterodimers will be obtained when complementation occurs at the boundaries of independently folded units. To this end, four breakage sites from APH(3′)-I dissection with levels of different solubility were tested for complementation with the help of leucine zippers.11,27 Three out of the four pairs were found to restore partial activity, including the pair APH(3′)-I-(1–253)NZ/CZ(254–271). A truncated but active APH(3′)-I (Δ1–25) was also found, with activity as high as 50% of the wild type in terms of the minimum inhibitory concentration (MIC). These illustrate that possible sites for protein complementation can be probed directly by simply evaluating the solubility of corresponding fragments. Finally, the short tag containing 18 amino acid residues in the pair APH(3′)-I-(1–253)NZ/CZ(254–271) was not reported previously, and as it has the potential to be a useful PCA reporter, random mutagenesis was further performed on the pair to improve its activity.
Results
Construction of selection vectors
pCAT-1 and pCAT-2 (pCAT-2d) were constructed to allow for insertion of gene fragments upstream of the CAT gene cat (see Fig. 1). The corresponding soluble protein fragments were isolated based on the levels of chloramphenicol resistance conferred by the downstream CAT protein.21 As shown in Figure 1, two different linkers, WPGSPA and AGSSAAGSGS,19,22 were used in pCAT-1 and pCAT-2 (pCAT-2d), respectively. On the other hand, plasmid pCY-T7 with two T7 promoters and linkers was constructed to evaluate the complementation of protein fragments.
Figure 1.
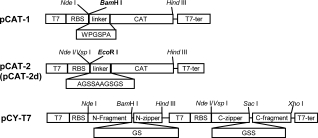
Vectors constructed in this study. Vectors pCAT-1, pCAT-2, and pCAT-2d used for selection of soluble fragments are derivatives of pET30a(+), linker sequences (boxed) are placed upstream of the CAT gene, and an internal BamH I site for pCAT-1 and EcoR I site for pCAT-2 are used for insertion of gene fragments, respectively. The pCAT-2d vector was created by deleting the ATG in the cat gene of pCAT-2. The pCY-T7, a derivative of pTWin 1, contains two T7 promoters which control the expression of the N-terminal and C-terminal fragments, respectively. Linker sequences between zippers and inserted fragments are shown (boxed). T7, T7 promoter; RBS, ribosome binding site; T7-ter, T7-terminator.
Fragment library preparation
Amplified TSα gene trpA, TEM-1 β-lactamase gene bla, and APH(3′)-I gene aph were digested by DNase I, respectively, to generate a pool of short DNA segments (∼50 bp). Residual templates were removed to ensure correct reassembly of short segments to produce DNA fragments with random ends but controllable lengths. Here a mixture of DeepVentR® polymerase plus rTaq polymerase was used in reassembly to minimize mutation and to create blunt fragment ends. Fragment sizes were adjusted by the number of PCR cycles and starting short DNA segment concentrations. A size distribution ranging from 50 bp up to the full gene size but enriched around 250–500 bp was achieved after 10–30 cycles for reassembly of 4.5–9.0 ng/μL of segments, depending on the nature of the template (see Fig. 2). These fragments were phosphorylated and ligated with digested and dephosphorylated selection vectors, and then transformed into E.coli BL21 (DE3) cells for selection of folded fragments based on the levels of chloramphenicol resistance, which were then confirmed by colony PCR of the inserted gene fragments and subsequently sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) analysis of the expressed protein fragment-CAT fusions. The library size was normally about 1.0 × 105.
Figure 2.
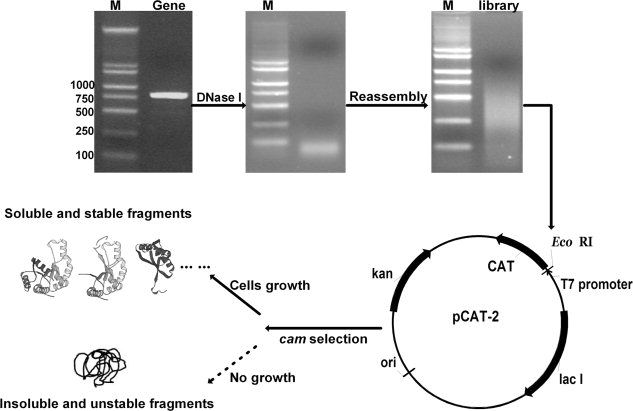
Experimental procedure for protein random dissection and selection to search for soluble fragments of a target protein. A target gene was PCR amplified and digested with DNase I, and the gene segment libraries were generated by reassembly of the smaller digestion products. Folded protein fragments were selected by using CAT as a reporter. DNA was analyzed by 1.2% agarose gel. Lane M, DNA ladder.
Linker effect on the isolation of folded fragments
The two linkers, WPGSPA and AGSSAAGSGS, led to a dramatic difference for TSα fragment library selection. As shown in Table I, the linker WPGSPA used in pCAT-1 clearly led to slower cell growth on selective plates and smaller insertions. Also, about 130 out of 3000 randomly picked colonies could grow out using the pCAT-2 plasmid, compared to 63 for the pCAT-1 vector. But most strikingly, no in-frame fragment was found from 12 randomly selected resistant colonies when the linker WPGSPA was used, consistent with results from an SDS-PAGE analysis of the same colonies (data not shown). Therefore, the linker AGSSAAGSGS was chosen in this study.
Table I.
Comparisons of Two Selection Vectors With Different Linkers
| Resistant Variantsc | |||||
|---|---|---|---|---|---|
| Vectors | Colonies pickeda | Conditionsb | <250 bp | >250 bp | In-frame ratio |
| pCAT-1 | ∼3000 | 30°C for 36 h | 54 | 9 | 0/12 |
| pCAT-2 | ∼3000 | 30°C for 24 h | 85 | 45 | 3/12 |
Colonies were randomly picked for characterization of linker effects.
Incubation for 24 h was sufficient for colonies to appear in the case of pCAT-2.
The insertion fragment sizes were determined by colony PCR using two flanking primers. The in-frame ratio was determined by randomly picked 12 chloramphenicol resistant cells for DNA sequencing.
Selection of folded TSα fragments
Using pCAT-2, seven folded TSα fragments and 11 split sites were obtained [Fig. 3(A)]. Interestingly, all these polypeptides contained a region from residues 73 to 137. TSα fragment-CAT fusions were expressed, analyzed by 12% SDS-PAGE, and compared to the corresponding MIC values (Table II, also see Methods and Materials). The solubility of the fragments in the CAT fusion form was found to be consistent with their MICs determined in this study. Furthermore, the solubility of fragments expressed alone without the CAT protein (but with a C-terminal His6 tag) also roughly correlated with the MICs determined for the corresponding fragment-CAT fusions (Fig. S1 in the supplementary data), suggesting the solubility of isolated fragments could be characterized by the MICs of the CAT fusion proteins.
Figure 3.
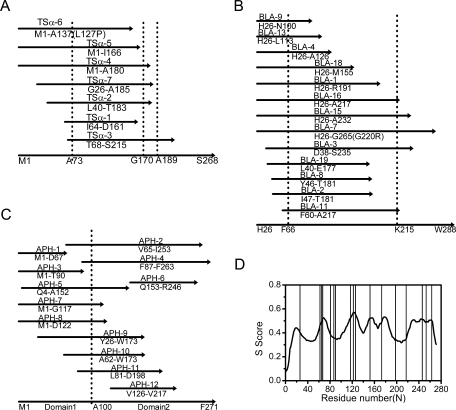
Coverage plots of non-redundant sets of fragments identified by the protein random dissection and selection method, arrayed against the parental sequences (lower arrow lines). (A) Fragments of TSα deduced from the sequences of inserts contained in fragment-CAT fusion clones. The three dot lines represent previously reported breakage points at residues A73, G170, and A189. A substitution L127P was found in fragment TSα-6. (B) Fragments obtained from TEM-1 β-lactamase dissection. Domain boundaries of TEM-1 β-lactamase at residues F66 and K215 are shown in dot lines. A substitution G220R was found in fragment BLA-7. (C) Identified fragments from APH(3′)-I random dissection. The presumed domain boundary is marked by 9 dot line. (D) The score curve of APH(3′)-I generated by the STAR predictor. Solid lines, sites obtained in this work.
Table II.
MICa Determined with Chloramphenicol for Fragment-CAT Fusions at 30°C
| Fragments obtained | Fragment sequence | Solubilityb | MIC of fragment-CAT fusion (μg/mL) |
|---|---|---|---|
| TSα-1 | I64-D161 | ++ | 400 |
| TSα-2 | L40-T183 | +++ | 800 |
| TSα-3 | T68-S215 | + | 200 |
| TSα-4 | M1-A180 | + | 200 |
| TSα-5 | M1-I166 | +++ | 800 |
| TSα-6 | M1-A137(L127P)c | ++ | 400 |
| TSα-7 | G26-A185 | + | 200 |
| BLA-1 | H26-R191 | ++ | 400 |
| BLA-2 | I47-T181 | +++ | 800 |
| BLA-3 | D38-S235 | +++ | 800 |
| BLA-4 | H26-A126 | + | 200 |
| BLA-7 | H26-G265(G220R)d | +++ | 800 |
| BLA-8 | Y46-T181 | ++ | 400 |
| BLA-9 | H26-N100 | ++ | 400 |
| BLA-11 | F60-A217 | ++ | 400 |
| BLA-13 | H26-L113 | ++ | 400 |
| BLA-15 | H26-A232 | ++ | 400 |
| BLA-16 | H26-A217 | ++++ | >800 |
| BLA-18 | H26-M155 | ++ | 400 |
| BLA-19 | L40-E177 | ++ | 400 |
| APH-1 | M1-D67 | +++ | 800 |
| APH-2 | V65-I253 | ++ | 400 |
| APH-3 | M1-T90 | +++ | 800 |
| APH-4 | F87-F263 | ++++ | 800 |
| APH-5 | Q4-A152 | + | 200 |
| APH-6 | Q153-R246 | ++ | 400 |
| APH-7 | M1-G117 | ++++ | 800 |
| APH-8 | M1-D122 | +++ | 400 |
| APH-9 | Y26-W173 | ++ | 400 |
| APH-10 | A62-W173 | ++ | 400 |
| APH-11 | L81-D198 | +++ | 800 |
| APH-12 | V126-V217 | ++ | 400 |
Cells were patched on LB agar plates with 0.5mM IPTG and different concentrations of chloramphenicol to determine MIC values. The MIC values for BL21(DE3)/pET30a and CAT were 40 μg/mL and > 800 μg/mL, respectively.
The solubility was classified based on the soluble expression levels of fragment-CAT fusions (protein expression was induced with 0.2 mM IPTG for 6 h at 23°C), judging by SDS-PAGE analyses: +, no detectable expression; ++, low expression; +++, medium expression; ++++, high expression; +++++, excellent, CAT alone.
A mutation L127P was found in this fragment.
A mutation G220R was found in this fragment.
Among the 11 breakage sites obtained, seven (I64, T68, D161, I166, A180, T183, A185) were close (within 10 residues, similarly hereinafter) to the well-known breakage points previously identified (residues A73, G170, and A189)10,13 (Table IIIa). Moreover, sites L40, A137, and D161 were close to the possible split sites E42, E135, R145, and P155 reported by Yamagishi and co-workers using the incremental truncation assay.10 Only two sites (N108, K120) reported in the aforementioned work were missed, but then two new sites (G26, S215) were obtained in this study, suggesting that the breakage points were not as strictly unique as previously reported. Taken together, the protein random dissection based on CAT selection can be used in searching for protein split sites of TSα, yielding comparable results with other methods but the procedure outlined here was much simpler.
Table III.
Split Sites Obtained in This Work and Previously Reported
| Group 1a | Group 2 | |
|---|---|---|
| (a) TSα | ||
| Sites obtained in this work | L40, I64, T68, A137, D161, I166, A180, T183, A185 | G26, S215, |
| Previously reported resultsb | E42, A73, E135, R145, P155, G170, R188, P192, | N108, K120, |
| (b) TEM-1 beta-lactamase | ||
| Sites obtained in this work | Y46, I47, F60, N100, E177, T181, R191, A217,A232, S235 | D38, L40, L113, A126, M155, G265 |
| Previously reported sitesc | F66, G196, E197, L198, K215 | |
| Sites used in SCHEMA recombinationd | G41, R65, M69, Y105, S130, N132, T149, R161, D163, D176, D179, T189, L190, V216, G218, P226, S266 | S70, K73, W165 |
| (c) APH(3′)-I | ||
| Sites obtained in this work | A62, V65, D67, L81, | Y26, F87, T90, G117, D122, V126, A152, W173, D198, V217, R246, I253, F263, |
| Previously reported sitese | D64 (E59 in APH(3′)-IIa), T76-F78 (A71-G74 in APH(3′)-IIa) | G103(G99 in APH(3′)-IIa) |
Group 1: Sites within 10 residues of the previously reported sites; Group 2: sites identified differently in this work and other reports.
Sites reported by Yamagishi and co-workers.10
Selection of folded TEM-1 β-lactamase fragments
When pCAT-2 was used for TSα fragment selection, 75% of chloramphenicol resistant cells were false-positives as these fragments were not in-frame (Table I). The start codon ATG in the cat gene of pCAT-2 was subsequently deleted to reduce the false positive cells (see pCAT-2d in 1).22 Sequencing of random samples revealed that the in-frame ratios were improved to about 60% for both TEM-1 β-lactamase and APH(3′)-I fragment selection using pCAT-2d. Using this modified vector, 13 folded fragments were generated from TEM-1 β-lactamase dissection, aligned to the template sequence as shown in Fig.3(B). The dissection results of TEM-1 β-lactamase showed four hot spots: around L40, E177-T181, A217, and A232-S235. Closer inspection showed that, among the 16 breakage sites obtained (Table III b), F60 and A217 were near the sub-domain boundaries around F66 and K215.28 Site R191 was close to the breakage points G196, E197, and L198 previously identified by complementation.9,24. Thirteen new sites (D38, L40, Y46, I47, N100, L113, A126, M155, E177, T181, A232, S235, and G265) were obtained in this study. Very interestingly, several of these breakage points (D38, L40, A126, and G265) were very much similar to the recombination sites of TEM-1 β-lactamase (at residues G41, N132, and S266) predicted by SCHEMA, a structure-based computational algorithm based on multiple templates4,29 (http://www.che.caltech.edu/groups/fha/code) and then confirmed experimentally. Furthermore, sites L113 and M155 obtained in our CAT selection were close to the peak regions around G116 and T149 judged by SCHEMA to be harmful for recombination but later proved experimentally to be useful recombination sites.30,32 As recombination of homologous proteins likely requires the exchanged fragments to be more independently folded or less disrupted by exchange, these useful recombination sites were considered to be likely breakage points in this study. Overall, 16 out of 19 reported recombinant sites for TEM-1 β-lactamase were found in our CAT selection. A rough correlation was again observed for the solubility and the MIC value for TEM-1 β-lactamase fragments in the CAT fusion form (Table II).
Application to APH(3′)-I dissection
Encouraged by the dissection results of these two proteins with very different structures (TSα is an α/β barrel monomeric protein whereas TEM-1 β-lactamase is an open-face-β-sandwich structure consisting of two domains), we further applied the dissection method to APH(3′)-I. The start and end points of 12 folded fragments obtained were illustrated in Figure 3(C). Interestingly, three nearly bisectional points were obtained and around at residues V65-D67, F87-T90, and A152-Q153. The latter two were not reported before. Furthermore, sites A62, V65-D67, and L81 were similar to the reported sites E59 and A71-G74 in aminoglycoside-3′-phosphotransferase IIa (APH(3′)-IIa) (corresponding to D64 and T76-F78 in APH(3′)-I), which shares a 31.4% homology with APH(3′)-I at the amino acid level.11 Only one previously reported site (G99 in APH(3′)-IIa, corresponding to G103 in APH(3′)-I) was missed in this work,31 the closest sites in our work being T90 and G117. However, additional breakage points were again found at Y26, G117, D122, V126, W173, D198, V217, R246, I253, and F263.
Fragment complementation for APH(3′)-I
To test the complementation of APH(3′)-I fragments for possible use in PCA, a dual T7 promoter plasmid, pCY-T7, was used in which leucine zippers were introduced to facilitate complementation11,27 (see Fig. 1). Leucine zippers have been shown to assist the reassembly of fragments of GFP, TEM-1 β-lactamase, and APH(3′)-IIa.9,11,27 It was also reported that a short linker between fragment and the leucine zipper could lead to higher activity for APH(3′)-IIa,11 thus in this study leucine zippers and short linkers GS and GSS were used for APH(3′)-I fragment complementation (see Fig. 1). Sites for complementation were chosen based on the solubility of the fragment-CAT fusions (Table II). Sites T90 (in APH-3) and G117 (in APH-7) were chosen as APH-3, APH-7, and the closely related APH-4 (with site F87) and APH-11 (with site L81) were more soluble than others. Sites A62, V65, and D67 were identical to the reported ones,11 and thus were not further tested. A152 was not chosen because of the poor solubility of APH-5. Furthermore, D198 was in the ATP binding pocket, while W173 and V217 were close to the presumed kanamycin binding sites D168, E169, and R219 in APH(3′)-I (corresponding to D160, E161, and R211 of APH(3′)-IIa),33 and F263 was only 8 residues away from the C-terminus. Lastly, D122, V126, and R246 were close to G117 and I253, thus these sites were not chosen. The remaining two sites, Y26 and I253, were also chosen for testing. During the actual sub-cloning, the corresponding gene segments for the intended N-terminal and C-terminal fragments were pooled respectively, and then inserted into the dual promoter vector pCY-T7 in sequel.
At first, the activity of individual APH(3′)-I fragments with leucine zippers was characterized. All fragments could not confer resistance to kanamycin above the background level (12.5 μg/mL) except for APH(3′)-I-CZ(26–271), which showed a MIC of 100 μg/mL at 37°C in the absence of IPTG and 800 μg/mL at 23°C in the presence of IPTG, which was about 50% of the wild type. The deletion of the leucine zipper slightly lowered the MIC level (Table IV). On the other hand, the MIC of APH(3′)-I-(1–25)NZ/CZ(26–271) was higher at all conditions, suggesting residues 1–25 of APH(3′)-I was not critical but helpful for the APH activity. At both 23 and 37°C, heterodimers also partially restored the APH activity at T90, I253 but not at G117 in the presence of 0.2 mM IPTG (Table IV). The weak APH(3′)-I-(1–253)NZ/CZ(254–271) activity suggests that the C-terminal helix with 18 amino acids of APH(3′)-I can serve as an essential part for complementation.
Table IV.
MIC of Kanamycin for APH(3′)-I Fragment Pairs
| MIC of kanamycin (μg/mL)b | ||||
|---|---|---|---|---|
| Heterodimersa | 23°C | 37°C | ||
| IPTG | 0 mM | 0.2 mM | 0 mM | 0.2 mM |
| APH(3′)-I | >1600 | ndc | >1600 | nd |
| BL21(DE3)/pCY-T7 (Control) | 12.5 | 12.5 | 12.5 | 12.5 |
| APH(3′)-I-(26–271) | 100 | 800 | 25 | 100 |
| APH(3′)-I-CZ(26–271) | 400 | 800 | 100 | 25 |
| APH(3′)-I-(1–25)NZ/CZ(26–271) | 800 | 800 | 200 | 200 |
| APH(3′)-I-(1–90)NZ/CZ(91–271) | 25 | 100 | 25 | 100 |
| APH(3′)-I-(1–117)NZ/CZ(118–271) | 12.5 | 12.5 | 12.5 | 12.5 |
| APH(3′)-I-(1–253)NZ/CZ(254–271) | 25 | 100 | 25 | 50 |
| APH(3′)-I-EP-21d | ||||
| (N18S, C137G, R145H, L313Q, Q331R) | 100 | 400 | 200 | 100 |
| APH(3′)-I-(1–117)NZ/CZ(26–271) | 800 | 800 | 800 | 800 |
| APH(3′)-I-(1–117)NZ/CZ(91–271) | 200 | 50 | 400 | 400 |
| APH(3′)-I-(1–253)NZ/CZ(26–271) | 400 | 400 | 400 | 50 |
Heterodimers were named by their N-terminal and C-terminal fragments, for example, APH(3′)-I-(1-117)NZ/CZ(26–271) consisted of the APH(3′)-I-(1-117) fragment with the leucine zipper attached to its C terminus, and the APH(3′)-I-(26–271) fragment with the leucine zipper attached to its N-terminus.
Minimum inhibitory concentrations of kanamycin were determined in 96 well plates; concentrations of kanamycin tested were 12.5, 25, 50, 100, 200, 400, 800, 1600 μg/mL.
nd: not detected.
The best mutant obtained in the evolution of APH(3′)-I-(1–253)NZ/CZ(254–271) with five mutations listed.
Surprisingly, several heterodimers containing overlapping sequences showed much higher activity (Table IV), which were obtained unintentionally as the gene segments were not inserted into pCY-T7 in a pair-wise fashion but were pooled together before insertion. For example, APH(3′)-I-(1–117)NZ/CZ(26–271) showed a MIC of 800 μg/mL, which was eightfold of APH(3′)-I-(26–271) and fourfold of APH(3′)-I-(1–25)NZ/CZ(26–271) at 37°C. Heterodimers conferred such high MIC values were not found in APH(3′)-IIa fragment complementation previously reported, where active pairs containing overlapping sequences were also obtained but with lower MIC values (the best one showing a MIC of 100 μg/mL).11 It was notable that APH(3′)-I-(1–117)NZ/CZ(118–271) conferred no resistance above background but APH(3′)-I-(1–117)NZ/CZ(91–271) partially restored resistance. What is also interesting is that APH(3′)-I-(1–117)NZ/CZ(91–271) showed a fourfold activity of that for APH(3′)-I-(1–90)NZ/CZ(91–271). Finally, low temperatures seemed to favor complementation as heterodimers showed higher activity when the expression was induced with 0.2 mM IPTG and at 23°C, except for APH(3′)-I-(1–117)NZ/CZ(91–271) which was more active when expressed at 37°C (Table IV).
Directed evolution for APH(3′)-I-(1–253)NZ/CZ(254–271)
The novel APH(3′)-I-(1–253)NZ/CZ(254–271) was selected to undergo random mutagenesis to improve its activity. Error-prone PCR was performed to create a library with 1.8 × 105 transformants. Cells were selected on LB plates supplemented with 400 μg/mL of kanamycin and 0.2 mM IPTG. Of the six colonies that grew at 37°C, five were found to be false positives bearing the full-length aph gene. Likewise, 6/12 clones that grew at 23°C contained the reconstituted APH(3′)-I. The source of the full length gene was unclear but this was observed similarly before.11 The best mutant, namely APH(3′)-I-EP-21, showed a fourfold improvement compared with the starting pair after just one round of evolution at 23°C. Sequencing revealed three amino acids substitutions in the N-terminal fragment (N18S, C137G, and R145H), one mutation in the C-terminal fragment (Q331R) and one within the CZ sequence (L313Q) (IV). Interestingly, the N18S, R145H, and Q331R substitutions could be found in the natural APH family.33
Discussion
In this study, a modified protein random dissection method combined with selection using CAT to search for folded fragments for identification of protein split sites was established. Experimental results of TSα and TEM-1 β-lactamase agreed well with what was reported in the literature, and yielded a wealth of information on the protein breakage points for APH(3′)-I, demonstrating that this method had the potential to be a general strategy to search protein split sites quickly and likely exhaustively, without requiring the structural knowledge of the target protein. Furthermore, three APH(3′)-I fragment pairs partially restored the activity with the help of leucine zippers, which may be useful for use in PCA. Therefore, active heterodimers can be obtained when complementation occurs at split sites identified from protein random dissection, and this provides a significant guide for PCA design. Nonetheless, it should be noted that for any such pairs to be useful in a PCA assay, further tests for spontaneous complementation and reversibility, and possibly further mutagenesis are required.34
For APH(3′)-I, it is noteworthy that a number of more active heterodimers contain short or long overlapping sequences (Table IV), an observation consistent with other reports.35–38 It has been proposed that the redundant polypeptide chains are pushed away,36 but the reason for promoting complementation by these extra chains remains to be investigated. Notably, these heterodimers, such as APH(3′)-I-(1–117)NZ/CZ(91–271), restore much higher activity (more than 25% of the parental protein) than those APH(3′)-IIa pairs previously reported (less than 10% of the parental protein).11
Exploitation of possible protein split sites by our method has some advantages over the others. First, the procedure is facile, and which is similar to DNA shuffling protocol.39 And using a reassembly step helps to control the fragment size distribution by simply adjusting the number of PCR cycles. As judged from the agarose gel analysis of reassembled DNA samples (see Fig. 2), gene fragment libraries constructed in this study were enriched at 250–500 bp, a range of sizes where domain or smaller folded units mostly likely reside. Colony PCR results of chloramphenicol resistant cells containing fragment-CAT fusions also revealed that the gene fragments in these cells centered at 250–500 bp (see Fig. 4). In addition, sequencing of resistant clones revealed ∼93.6% (74/79) folded fragments in libraries were unique, indicating no fragment was amplified preferentially. The use of CAT (with deletion of the initiation codon ATG) also significantly facilitates screening, independent of protein structure or function.
Figure 4.
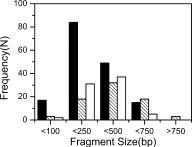
Histograms for the size distribution frequency (N) of fragments determined by colony PCR with flanking primers from chloramphenicol resistant cells harboring fragment-CAT fusions. Black bars: TSα fragments; hatched bars: TEM-1 β-lactamase fragments; white bars: APH(3′)-I fragments.
In addition to various experimental assays,11,15,16,29 in recent years two computational algorithms, SCHEMA and the successor STAR,4,40 were also developed to predict possible recombination sites. As these sites are the boundaries at which the fragments can be exchanged, they are thus considered to be likely breakage points in this work. We have chosen STAR to systematically search for such possible sites for comparison. As shown in Figure 3(D), the STAR score curve for APH(3′)-I generated by the predictor has five clear valleys which are presumably to contain possible recombination and thus likely spilt sites. Among the dissection sites of APH(3′)-I obtained in this work [solid lines in Figure 3(D)], only five sites (L81, F87, T90, D198, and V217) are within the STAR-predicted valleys, while 12/17 sites are away from the valleys, and in particularly 9/17 are close to the peaks. A similar pattern of discrepancy has been observed for TSα and TEM-1 β-lactamase between experimentally determined split sites and predicted recombination sites (Fig. S2 in supplementary data). Nonetheless, these two computational methods provide complementary guides in search for possible split sites for a target protein.
Folded fragments for the three proteins dissected in this work were mapped onto the primary amino acids sequence of the parental proteins (see Figure 3). A structural analysis shows that split points from TSα and TEM-1 β-lactamase are predominant near the surface of the proteins and tend to cluster within loops (∼90% of the sites are on the surface and half of which within loops).38,41 Split sites within secondary structures might introduce more severe disruption to a protein structure, therefore these sites should be carefully analyzed for further application in protein complementation. The fact that APH(3′)-I-(1-117)NZ/CZ(118–271) showed no activity is presumably due to the position of G117 in one of the helices.
Our study shows that the active heterodimers can be obtained when proteins are split at the boundaries of folded units. In fact, the MIC values (or the solubility) of fragment-CAT fusions roughly correlate with the activity levels of complementary APH(3′)-I fragment pairs. For example, APH(3′)-I-(1–90)NZ/CZ(91–271) shows higher activity than APH(3′)-I-(1–253)NZ/CZ(254–271), this is consistent with the higher MIC values of APH-3(1–90)-CAT and APH-4(87–263)-CAT (Table II). Thus possible sites for complementation can be probed directly by evaluating the solubility of folded fragments obtained, and in this case, the MIC values of the fragment-CAT fusions.
Materials and Methods
Materials
Restriction enzymes and DNA polymerases were purchased from New England Biolabs (Beverly, MA) or Takara (Dalian, China). Oligonucleotides were synthesized by Sangon (Shanghai, China) or Takara. The kits for DNA purification, gel recovery, and plasmid mini-prep were all from Tiangen (Beijing, China) or QIAgen (Valencia, CA). DNA Sequencing was performed by Takara or by Sanboyuanzhi (Beijing, China). Isopropylthio-β-d-galactoside (IPTG) was obtained from Takara. E.coli BL21 (DE3) and plasmid pET30a(+) were obtained from Novagen (Wisconsin, USA). Plasmids pTWin 1 and pACYC184 were from New England Biolabs.
Construction of selection vectors
The CAT gene cat was amplified from pACYC184 vector using DeepVentR® polymerase (New England Biolabs) with forward primer 5′-TCTCGTACATATGGGATCCTGGCCTGGCAGCCCCGCTATGGAGAAAAAAATCACTGGATATAC-3′ and reverse primer 5′-AGACCAAGCTTTTACGCCCCGCCCTGCCACTC-3′ (Nde I, BamH I and Hind III sites were in bold and italic, and the linker sequence was underlined). Purified PCR products were then restricted and inserted between the Nde I and Hind III sites of pET30a(+) plasmid to yield the pCAT-1 with the WPGSPA linker.22 As there is an EcoR I site in the cat gene, which was used for subsequent fragment library insertion, site-directed mutagenesis was performed to mutate GAATTC to GAATTT by an overlap extension PCR procedure. Purified PCR products were again restricted and inserted between the BamH I and Hind III site of pET30a-linker-GFP vector which contained an AGSSAAGSGS linker,19 yielding pCAT-2. pCAT-2 vector was further modified by deleting the start codon ATG of cat gene to generate pCAT-2d plasmid.
Vector pCY-T7 was designed to allow for co-expression of two protein fragments tagged by a leucine zipper sequence, both under the separate control of a T7 promoter. The leucine zippers were designed by DNAworks (http://www.DNAworks.net), and chemically synthesized. The sequences of the N-terminal leucine zipper (NZ) and C-terminal leucine zipper (CZ) are SAQLKKELQANKKELAQLKWELQALKKELAQ and MASAQLEKKLQALEKKLAQLEWKNQALEKKLAQ, respectively.11,27 The full-length NZ and CZ were inserted into pET30a(+) to yield pET30a-NZ (between BamH I & Hind III sites) and pET30a-CZ (between Nde I (VspI) and Sac I sites), respectively. To construct the two-promoter plasmid pCY-T7, the sequence between the T7 promoter and Xho I of pET30a-CZ was amplified and digested with Hind III & Xho I, and the sequence between the Nde I and Hind III sites in the pET30a-NZ vector was restricted and gel purified, then these two fragments were directly ligated into the Nde I and Xho I sites of the pTwin 1 vector, yielding plasmid pCY-T7.
Fragment library preparation
The trpA gene coding for TSα was amplified from the genome of E.coli XL-1 blue cells (Stratagene, LaJolla, CA). Fragmentation and reassembly of trpA gene were performed as described,19 except that residual templates were removed by passing the reaction mixture through a Microcon YM-100 (Millipore, Billerica, MA) and recovered segments were resuspended at a concentration of 4.5–9.0 ng/μL, then rTaq polymerase (Takara) and DeepVentR® polymerase were added at 2.5 units each per 100 μL reaction mixture for reassembly. The backbone vector was digested with BamH I for pCAT-1 and EcoR I for pCAT-2, respectively, then blunt-ended with T4 DNA polymerase (New England Biolabs) in the presence of 0.1 mM each dNTP, and purified with gel purification kit. Dephosphorylation was carried out twice for the blunt-end vectors with shrimp alkaline phosphatase (Promega, Madison, WI). The gene fragments and the backbone vector were ligated at 12°C for 16 h in the presence of 5% PEG 8000, and then electroporated into E. coli BL21 (DE3) competent cells. Fragment libraries for TEM-1 β-lactamase (the bla gene was amplified from the pUC18 vector) and APH(3′)-I (the aph gene was amplified from pET30a(+) vector) were prepared following the same protocol except using pCAT-2d as the selection vector.
Isolation of folded fragments
Transformed E. coli BL21 (DE3) cells were plated onto LB agar medium supplemented with 50 μg/mL kanamycin and grown overnight at 37°C. Cells were scraped and plated to LB plates containing 40 μg/mL chloramphenicol and 0.5 mM IPTG. After incubation at 30°C for about 36 h, pre-selected cells were re-patched onto the LB plates with various concentrations of chloramphenicol (40, 100, 200, 400, and 800 μg/mL) and 0.5 mM IPTG. MIC of chloramphenicol was determined by visually inspection for bacterial growth after 24 h at 30°C. Then cells were picked and tested with colony PCR using primers flanking the fragment inserts. Fragments larger than 200 bp were randomly picked for sequencing.
Protein fragment complementation
For each chosen site of APH(3′)-I, N-terminal fragments were PCR amplified and subcloned into the Nde I and BamH I sites of pCY-T7, then the C-terminal fragments were subsequently inserted into the Sac I and Xho I sites of the same plasmid, yielding pCY-T7-APHNC. Active heterodimers were selected on 25 μg/mL kanamycin and 0.2 mM IPTG at 23°C. Kanamycin resistant clones were mini-preped and re-transformed into BL21 (DE3) cells to confirm their activity which was further characterized by MIC. To determine MICs, single colonies were inoculated into LB medium supplemented with ampicillin (50 μg/mL) and grown overnight at 37°C. Then it was diluted 50-fold with LB medium supplemented with ampicillin and grown at 37°C for 1.5 h (OD600 nm = 0.5–0.6). Two μl of these suspensions were added to 198 μL of LB medium in sterile microtiter plate wells containing serial dilutions of kanamycin (12.5–1600 μg/mL) in the presence or absence of 0.2 mM IPTG. The innocula were allowed to grow at 37°C or 23°C for 24 h before being visually inspected for bacterial growth.
Directed evolution
Error-prone PCR was performed as follows in a 100 μL reaction: 20 fmol templates, 0.3 μM forward and reverse primers, 7 mM MgCl2, 0.2 mM MnCl2, 200 mM dATP and dGTP, 300 mM dTTP and dCTP, 1×PCR buffer with MgCl2 and 5 U of Taq polymerase (Takara). Reactions were heated to 94°C for 2 min and 30 cycles of 1 min at 94°C, 1 min at 57°C and 80 s at 72°C. PCR products were digested with Nde I and EcoR I, cloned back into the pTwin 1 vector and transformed into E.coli BL21 (DE3). Pre-selected mutants were re-transformed to confirm their activity. Mutants with higher MICs were sequenced to identify mutations.
Acknowledgments
The authors would like to thank Zhen Cai and Pan Li for their technical help.
Supplemental material
References
- 1.Ladurner A, Itzhaki L, Gay G, Fersht A. Complementation of peptide fragments of the single domain protein chymotrypsin inhibitor 2. J Mol Biol. 1997;273:317–329. doi: 10.1006/jmbi.1997.1303. [DOI] [PubMed] [Google Scholar]
- 2.Zitzewitz J, Gualfetti P, Perkons I, Wasta S, Matthews C. Identifying the structural boundaries of independent folding domains in the α subunit of tryptophan synthase, a β/α barrel protein. Protein Sci. 1999;8:1200–1209. doi: 10.1110/ps.8.6.1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hayes F, Hallet B, Cao YH. Insertion mutagenesis as a tool in the modification of protein function. J Biol Chem. 1997;272:28833–28836. doi: 10.1074/jbc.272.46.28833. [DOI] [PubMed] [Google Scholar]
- 4.Voigt CA, Martinez C, Wang ZG, Mayo SL, Arnold FH. Protein building blocks preserved by recombination. Nat Struct Biol. 2002;9:553–558. doi: 10.1038/nsb805. [DOI] [PubMed] [Google Scholar]
- 5.Hopfner KP, Kopetzki E, Kresse GB, Bode W, Huber R, Engh RA. New enzyme lineages by subdomain shuffling. Proc Natl Acad Sci USA. 1998;95:9813–9818. doi: 10.1073/pnas.95.17.9813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tsuji T, Onimaru M, Yanagawa H. Towards the creation of novel proteins by block shuffling. Comb Chem High Throughput Screening. 2006;9:259–269. doi: 10.2174/138620706776843237. [DOI] [PubMed] [Google Scholar]
- 7.Seitz T, Bocola M, Claren J, Sterner R. Stabillsation of a (βα)8-barrel protein designed from ldentical half barrels. J Mol Biol. 2007;372:114–129. doi: 10.1016/j.jmb.2007.06.036. [DOI] [PubMed] [Google Scholar]
- 8.Pelletier JN, Campbell-Valois FX, Michnick SW. Oligomerization domain-directed reassembly of active dihydrofolate reductase from rationally designed fragments. Proc Natl Acad Sci USA. 1998;95:12141–12146. doi: 10.1073/pnas.95.21.12141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Galarneau A, Primeau M, Trudeau LE, Michnick SW. β-Lactamase protein fragment complementation assays as in vivo and in vitro sensors of protein-protein interactions. Nat Biotechnol. 2002;20:619–622. doi: 10.1038/nbt0602-619. [DOI] [PubMed] [Google Scholar]
- 10.Hiraga K, Yamagishi A, Oshima T. Mapping of unit boundaries of a proteexhaustive search for permissive sites for duplication by complementation analysis of random fragment libraries of tryptophan synthase α subunit. J Mol Biol. 2004;335:1093–1104. doi: 10.1016/j.jmb.2003.11.029. [DOI] [PubMed] [Google Scholar]
- 11.Paschon DE, Patel ZS, Ostermeier M. Enhanced catalytic efficiency of aminoglycoside phosphotransferase (3′)-IIa achieved through protein fragmentation and reassembly. J Mol Biol. 2005;353:26–37. doi: 10.1016/j.jmb.2005.08.026. [DOI] [PubMed] [Google Scholar]
- 12.Dyson MR, Perera RL, Shadbolt SP, Biderman L, Bromek K, Murzina NV, McCafferty J. Identification of soluble protein fragments by gene fragmentation and genetic selection. Nucleic Acids Res. 2008;36:e51. doi: 10.1093/nar/gkn151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Higgins W, Fairwell T, Miles EW. An active proteolytic derivative of the α subunit of tryptophan synthase. Identification of the site of cleavage and characterization of the fragments. Biochemistry. 1979;18:4827–4835. doi: 10.1021/bi00589a010. [DOI] [PubMed] [Google Scholar]
- 14.Li AQ, Sowder RC, Henderson LE, Moore SP, Garfinkel DJ, Fisher RJ. Chemical cleavage at aspartyl residues for protein identification. Anal Chem. 2001;73:5395–5402. doi: 10.1021/ac010619z. [DOI] [PubMed] [Google Scholar]
- 15.Kawasaki M, Inagaki F. Random PCR-based screening for soluble domains using green fluorescent protein. Biochem Biophys Res Commun. 2001;280:842–844. doi: 10.1006/bbrc.2000.4229. [DOI] [PubMed] [Google Scholar]
- 16.Reich S, Puckey LH, Cheetham CL, Harris R, Ali AAE, Bhattacharyya U, Maclagan K, Powell KA, Prodromou C, Pearl LH, Driscoll PC, Sawa R. Combinatorial domain hunting: an effective approach for the identification of soluble protein domains adaptable to high-throughput applications. Protein Sci. 2006;15:2356–2365. doi: 10.1110/ps.062082606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cochrane D, Webster C, Masih G, McCafferty J. Identification of natural ligands for SH2 domains from a phage display cDNA library. J Mol Biol. 2000;297:89–97. doi: 10.1006/jmbi.2000.3561. [DOI] [PubMed] [Google Scholar]
- 18.Prodromou C, Sawa R, Driscoll PC. DNA fragmentation-based combinatorial approaches to soluble protein expression. I. Generating DNA fragment libraries. Drug Discov Today. 2007;12:931–938. doi: 10.1016/j.drudis.2007.08.012. [DOI] [PubMed] [Google Scholar]
- 19.Li S, Cai Z, Chen Y, Lin ZL. Dissection of SARS coronavirus spike protein into discrete folded fragments. Tsinghua Sci Technol. 2006;11:49–53. doi: 10.1016/S1007-0214(06)70222-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lin ZL, Li S. Identification of viral peptide fragments for vaccine development. In: Hicks BW, Chen Y, editors. Methods in molecular biology series: viral applications of the green fluorescent protein. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Maxwell KL, Mittermaier AK, Forman-Kay JD, Davidson AR. A simple in vivo assay for increased protein solubility. Protein Sci. 1999;8:1908–1911. doi: 10.1110/ps.8.9.1908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sieber V, Martinez C, Arnold F. Libraries of hybrid proteins from distantly related sequences. Nat Biotechnol. 2001;19:456–460. doi: 10.1038/88129. [DOI] [PubMed] [Google Scholar]
- 23.Marsden RL, McGuffin LJ, Jones DT. Rapid protein domain assignment from amino acid sequence using predicted secondary structure. Protein Sci. 2002;11:2814–2824. doi: 10.1110/ps.0209902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wehrman T, Kleaveland B, Her JH, Balint RF, Blau HM. Protein–protein interactions monitored in mammalian cells via complementation of β-lactamase enzyme fragments. Proc Natl Acad Sci USA. 2002;99:3469–3474. doi: 10.1073/pnas.062043699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Michnick SW, Ear PH, Manderson EN, Remy I, Stefan E. Universal strategies in research and drug discovery based on protein-fragment complementation assays. Nat Rev Drug Discov. 2007;6:569–582. doi: 10.1038/nrd2311. [DOI] [PubMed] [Google Scholar]
- 26.Tafelmeyer P, Johnsson N, Johnsson K. Transforming a (β/α)8-barrel enzyme into a split-protein sensor through directed evolution. Chem Biol. 2004;11:681–689. doi: 10.1016/j.chembiol.2004.02.026. [DOI] [PubMed] [Google Scholar]
- 27.Ghosh I, Hamilton AD, Regan L. Antiparallel leucine zipper-directed protein reassembly: Application to the green fluorescent protein. J Am Chem Soc. 2000;122:5658–5659. [Google Scholar]
- 28.Jelsch C, Mourey L, Masson JM, Samama JP. Crystal structure of Escherichia coli TEM1 β-lactamase at 1.8 Å resolution. Proteins: Struct Funct Bioinf. 1993;16:364–383. doi: 10.1002/prot.340160406. [DOI] [PubMed] [Google Scholar]
- 29.Hiraga K, Arnold FH. General method for sequence-independent site-directed chimeragenesis. J Mol Biol. 2003;330:287–296. doi: 10.1016/s0022-2836(03)00590-4. [DOI] [PubMed] [Google Scholar]
- 30.Meyer MM, Hochrein L, Arnold FH. Structure-guided SCHEMA recombination of distantly related β-lactamases. Protein Eng Des Sel. 2006;19:563–570. doi: 10.1093/protein/gzl045. [DOI] [PubMed] [Google Scholar]
- 31.Michnick SW, Remy I, Campbell-Valois FX, Vallee-Belisle A, Pelletier JN. Detection of protein-protein interactions by protein fragment complementation strategies. Methods Enzymol. 2000;328:208–230. doi: 10.1016/s0076-6879(00)28399-7. [DOI] [PubMed] [Google Scholar]
- 32.Meyer MM, Silberg JJ, Voigt CA, Endelman JB, Mayo SL, Wang ZG, Arnold FH. Library analysis of SCHEMA-guided protein recombination. Protein Sci. 2003;12:1686–1693. doi: 10.1110/ps.0306603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nurizzo D, Shewry SC, Perlin MH, Brown SA, Dholakia JN, Fuchs RL, Deva T, Baker EN, Smith CA. The crystal structure of aminoglycoside-3′-phospho-transferase-IIa, an enzyme responsible for antibiotic resistance. J Mol Biol. 2003;327:491–506. doi: 10.1016/s0022-2836(03)00121-9. [DOI] [PubMed] [Google Scholar]
- 34.Stagljar I, Korostensky C, Johnsson N, te Heesen S. A genetic system based on split-ubiquitin for the analysis of interactions between membrane proteins in vivo. Proc Natl Acad Sci USA. 1998;95:5187–5192. doi: 10.1073/pnas.95.9.5187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Taniuchi H, Anfinsen CB. Simultaneous formation of two alternative enzymology active structures by complementation of two overlapping fragments of staphylococcal nuclease. J Biol Chem. 1971;246:2291–2301. [PubMed] [Google Scholar]
- 36.RR, Taniuchi H. Formation of a biologically active, ordered complex from two overlapping fragments of cytochrome c. J Biol Chem. 1977;252:1367–1374. [PubMed] [Google Scholar]
- 37.Matsuyama S, Kimura E, Mizushima S. Complementation of two overlapping fragments of SecA, a protein translocation ATPase of Escherichia coli, allows ATP binding to its amino-terminal region. J Biol Chem. 1990;265:8760–8765. [PubMed] [Google Scholar]
- 38.Ostermeier M, Nixon AE, Shim JH, Benkovic SJ. Combinatorial protein engineering by incremental truncation. Proc Natl Acad Sci USA. 1999;96:3562–3567. doi: 10.1073/pnas.96.7.3562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lorimer I, Pastan I. Random recombination of antibody single-china FV sequences after fragmentation with DNase I in the presence of Mn2+ Nucleic Acids Res. 1995;23:3067–3068. doi: 10.1093/nar/23.15.3067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bauer DC, Boden M, Thier R, Gillam EM. STAR: predicting recombination sites from amino acid sequence. BMC Bioinf. 2006;7:437. doi: 10.1186/1471-2105-7-437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Graf R, Schachman HK. Random circular permutation of genes and expressed polypeptide chains: application of the method to the catalytic chains of aspartate transcarbamoylase. Proc Natl Acad Sci USA. 1996;93:11591–11596. doi: 10.1073/pnas.93.21.11591. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.