

Abstract
Hinge motions are important for molecular recognition, and knowledge of their location can guide the sampling of protein conformations for docking. Predicting domains and intervening hinges is also important for identifying structurally self-determinate units and anticipating the influence of mutations on protein flexibility and stability. Here we present StoneHinge, a novel approach for predicting hinges between domains using input from two complementary analyses of noncovalent bond networks: StoneHingeP, which identifies domain-hinge-domain signatures in ProFlex constraint counting results, and StoneHingeD, which does the same for DomDecomp Gaussian network analyses. Predictions for the two methods are compared to hinges defined in the literature and by visual inspection of interpolated motions between conformations in a series of proteins. For StoneHingeP, all the predicted hinges agree with hinge sites reported in the literature or observed visually, although some predictions include extra residues. Furthermore, no hinges are predicted in six hinge-free proteins. On the other hand, StoneHingeD tends to overpredict the number of hinges, while accurately pinpointing hinge locations. By determining the consensus of their results, StoneHinge improves the specificity, predicting 11 of 13 hinges found both visually and in the literature for nine different open protein structures, and making no false-positive predictions. By comparison, a popular hinge detection method that requires knowledge of both the open and closed conformations finds 10 of the 13 known hinges, while predicting four additional, false hinges.
Keywords: hinge bending, conformational change, flexibility, rigidity theory, ProFlex, FIRST, DomDecomp, domain identification
Introduction
Flexibility is critical to both the structure and function of proteins and impacts areas from protein folding1 to prion propagation2 to structure-based drug design.3,4 Hinge motions can be particularly important for interactions between proteins and small molecules by exposing the interaction surface to the ligand.5 Therefore, predicting hinges has the potential to enhance structure-based ligand discovery.
Large-scale conformational changes such as hinge motion can be divided into three classes on the basis of size: fragment, domain, and subunit.6,7 Fragment motion refers to movements of small regions such as surface loops, whereas domain motion generally involves large conformational changes between covalently linked domains. Subunit motion refers to movement between polypeptide chains, often associated with allostery.6,7 Here, we focus on identifying hinges between domains, which can be considerably harder to analyze and predict than fragment motions. Even within this class of motion, hinges have been defined in different ways. Here, we define a hinge as a region of localized, internal motion of the main chain between two domains of a protein, rather than between a domain and a flexible region, such as a loop.
The StoneHinge method, presented here, computes the consensus of two network-based hinge predictors, StoneHingeP and StoneHingeD. StoneHingeP identifies overconstrained and underconstrained regions in the bond network, corresponding to mutually rigid or flexible regions. All residues intervening between a pair of rigid domains are defined as the hinge. These hinges may have rigid inclusions such as β turns or short helices, as the movement of these substructures can also contribute to hinge motion. This definition allows for disseminated motions, where domain movement is caused by small changes spread over a number of residues. Such hinges are known for a number of proteins, including Bence-Jones protein,8 lysine/arginine/ornithine (LAO) binding protein,8 and T4 lysozyme.9,10 For instance, a twelve-residue, strap-like hinge connects domains in the LAO binding protein.8 Other hinge-prediction methods such as FlexProt11 and StoneHingeD (presented here) define hinges as pivot points between two consecutive residues. StoneHingeD defines each hinge as a fixed point between connected regions undergoing large-scale opening and closing modes in DomDecomp.12 Such pivot-like hinges are known for proteins including adenylate kinase,13 inorganic pyrophosphatase,14 and ribose binding protein.15
To account for backbone flexibility during ligand docking, programs such as FlexDock have been developed to sample hinge rotations.3 FlexDock partitions a protein into rigid regions with intervening hinges and docks the other molecule against each rigid region. The success of FlexDock for protein-protein docking at CAPRI (http://www.ebi.ac.uk/msd-srv/capri/) demonstrates the utility of incorporating hinge motion.3 However, this approach requires that the hinges first be defined by another method.
Identifying hinges
Hinge detectors, such as FlexProt,11 require two conformations of the protein and analyze which residues stay mutually rigid between the conformers or change in conformation; the latter are identified as hinges. On the other hand, hinge predictors such as the StoneHinge algorithms presented here identify the locations of hinges given a single structure as input. Such methods are more widely applicable and focus more on the intrinsic flexibility of the protein rather than on ligand–induced changes.
A number of other domain and hinge prediction methods are available, using a variety of criteria to identify the domains bordering a hinge, such as compactness,16,17 structural redundancy,17 abundance of interdomain contacts,18–20 presence of hydrophobic cores,21,22 distribution of electron density,23 and interdomain versus intradomain potential energy.24 Alternatively, hinge prediction can be based on sequence statistics25 or assessed by normal mode analysis.26,12
StoneHinge
StoneHinge predicts hinges based on the consensus between StoneHingeP and StoneHingeD results. StoneHingeP is unique in using constraint counting from rigidity theory, as implemented in ProFlex, as the basis for predictions. ProFlex (successor to the FIRST method27) analyzes flexibility using a three-dimensional constraint counting algorithm that decomposes a protein structure into rotatable and nonrotatable bonds. This analysis is based on bond rotational constraints placed by covalent and noncovalent bonds in the network. The noncovalent constraints are reminiscent of the elastic interactions in normal mode analysis or the contacts analyzed in geometric approaches, both of which also represent the tertiary interactions stabilizing the protein. StoneHingeP analyzes the ProFlex results to identify the energy at which the protein structure first decomposes into two rigid regions (domains) of significant size, connected by a flexible hinge. If two domains containing at least 20 residues are not found, then hinge motion is not predicted.
StoneHingeD uses the DomDecomp Gaussian Network Model (GNM) normal mode analysis to identify domains.12 A reduced protein representation is used, modeling favorable contacts as an elastic network of springs connecting pairs of alpha carbons.28 StoneHingeD defines hinges by identifying residues that are fixed points, or undergo the least motion, along the direction of maximal lowest-frequency (largest-scale) motion in the DomDecomp analysis.
The consensus predictor, StoneHinge, assigns the residues of a StoneHingeD prediction as a consensus hinge if they fall within five residues of a StoneHingeP prediction. StoneHingeD is used to assign hinge residues because it was observed during training (see Methods section) to be more sensitive to the precise location of hinges, whereas requiring consensus with StoneHingeP greatly reduces false-positive hinge predictions. StoneHinge consensus results are presented for a series of protein structures solved both in open and closed conformations, allowing assessment of the effects of input conformation on prediction. StoneHinge predictions are then compared with hinges reported in the literature, as well as hinges selected by visual inspection of protein motion between the open and closed states29 using the Molecular Motions Database morph viewer.30,31 Open conformations (normally a ligand-free form of the protein) may allow more accurate predictions, as the domains are usually separated in space. This makes distinguishing the domains easier, both visually and in the bond network analysis. However, StoneHinge predictions from the open and closed states are typically comparable, and the method yields more specific predictions of interdomain hinges than either of its component methods. StoneHinge predictions are then compared with those of the popular hinge detector, FlexProt (not to be confused with the ProFlex constraint-counting method), which performs domain superposition between open and closed states to locate any hinges.11,32
Results
Following initial tests on training data (see Methods section), StoneHinge was run on nine protein structures in both open and closed conformations (Table I). The resulting predictions are summarized alongside the literature-reported and visually-assigned29 hinge residues, along with hinges predicted using each of the component methods, StoneHingeP and StoneHingeD, and those detected by the FlexProt domain superposition method (Table II and Fig. 1). Sensitivity of the hinge predictions was assessed against the intersection of the literature and visually-assigned hinges, referred to as the gold standard hinges (see Methods section).
Table I.
Hinged and Nonhinged (Control) Proteins Analyzed by StoneHinge
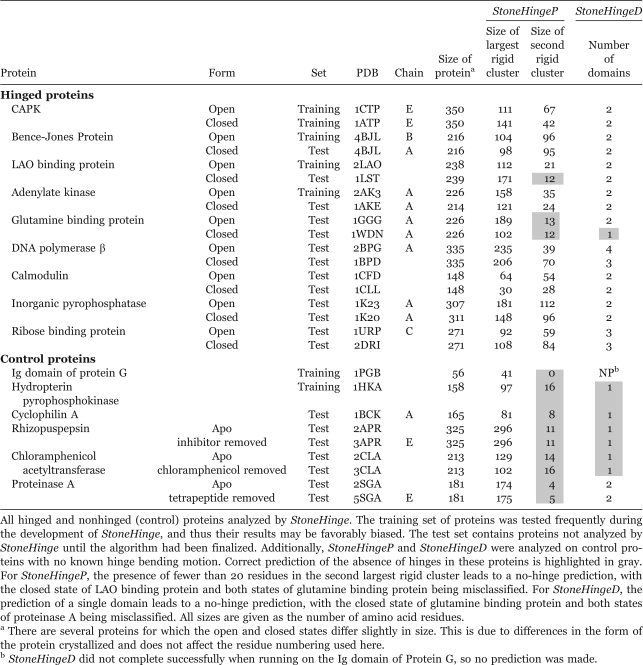
Table II.
Hinges Predicted by StoneHinge, StoneHingeP, StoneHingeD, and FlexProt
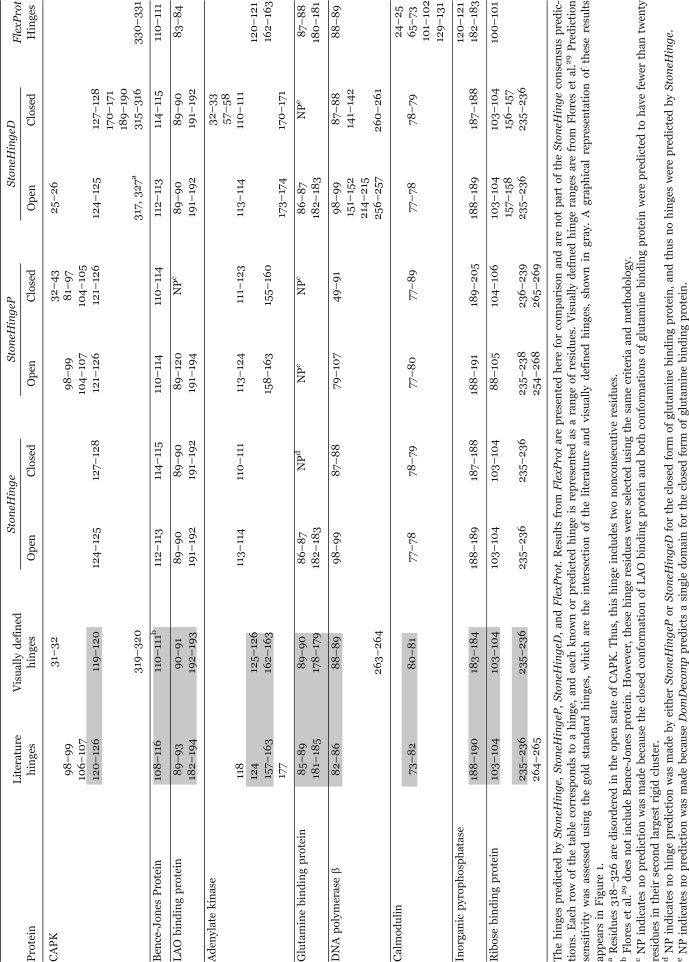
Figure 1.
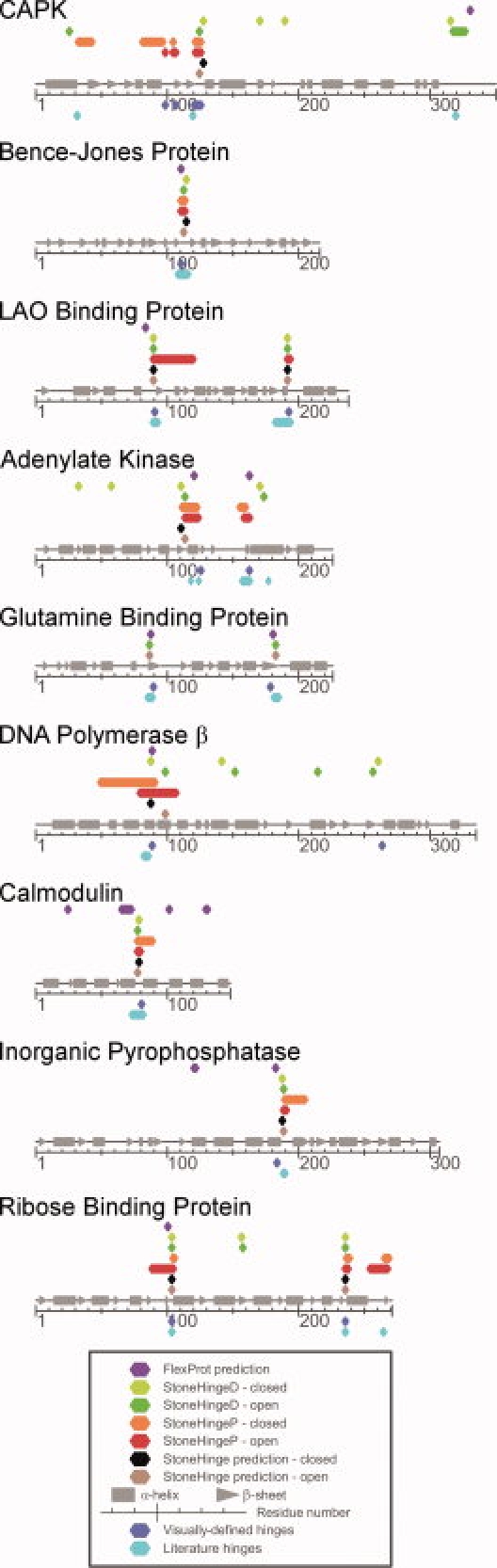
A visual representation of the hinges predicted by StoneHinge compared to the visually defined and literature hinges as well as those detected by FlexProt. Each line represents the protein sequence from N-terminal (left) to C-terminal (right), with residue numbers appearing below and secondary structure denoted above. Colored diamonds represent hinge residues reported by each method.
Cyclic AMP dependent protein kinase
The hinge motion of cyclic AMP dependent protein kinase (CAPK) involves multiple stretches of the backbone and was defined in the literature by molecular dynamics analysis.33 StoneHinge predictions in both the open and closed states match the gold standard hinge. FlexProt predicts residues 330–331, which are 10 residues from a visually-defined hinge that is spatially adjacent to the literature hinges.
Bence-Jones protein
The two domains involved in hinge motion are easily distinguished by eye. There is a single stretch of the backbone passing between them, and the domains remain slightly separated in the closed conformation. The predictions of all programs on the open and closed states match the literature hinge, which was assigned using a variant of Siddiqui and Barton's method.8
Lysine/arginine/ornithine binding protein
The literature hinge comprises two backbone segments.8 StoneHinge predictions agree with the literature and visually-assigned hinges for both the open and closed states. For the closed state, in which the StoneHingeP component made no prediction, StoneHingeD predictions were used (following the protocol described in Fig. 3). FlexProt reports a hinge close in sequence to the first literature hinge, but based on visual inspection, it is located in a rigid region distal from the hinge.
Figure 3.
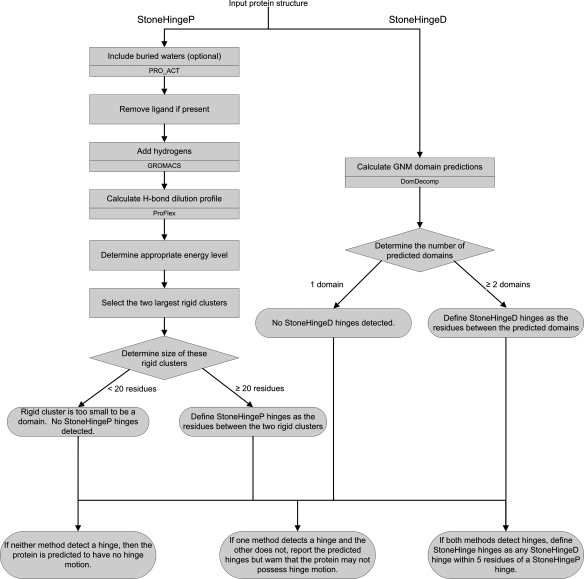
A flow chart of the StoneHinge algorithm. All steps except the optional inclusion of buried water molecules are performed automatically by StoneHinge. For all steps that make use of an external program, the program name is noted in the lower half of the box. Details are given in the Methods section.
Adenylate kinase
Here, the hinge region again incorporates two segments of the backbone, with two additional hinges identified in the literature but not by visual inspection.13 StoneHinge predictions from both the open and the closed states miss the gold standard hinges, while detecting one of the literature-only hinges. FlexProt identifies both of the gold standard hinges. Adenylate kinase presents a challenging case in which the literature and visual hinge definitions disagree on two hinges, and in which the StoneHinge component predictors also do not show clear consensus.
Glutamine binding protein
During this hinge motion, which involves two segments of the backbone, the second domain swings approximately 90°. In both the open and closed states, a face of this domain lies against the first domain. By observing this motion, the two domains can be distinguished clearly. However, because the two domains fold together, StoneHingeP predicts the majority of the protein as belonging to a single large domain, and therefore predicts no hinges. This also occurs for StoneHingeD predictions on the closed state, but it correctly predicts hinges in the open structure. Because StoneHingeP makes no predictions on the open structure, StoneHingeD predictions are used alone for StoneHinge (see Fig. 3). Both the consensus StoneHinge and FlexProt hinge identifications agree well with the visually and crystallographically defined hinges for the open state,34 but StoneHinge misses both hinges in the closed state.
DNA polymerase β
A protease sensitive hinge region between residues 82 and 86 comprises the single literature hinge.35 However, visual inspection identified three domains that move relative to each other, with less motion in the second hinge at residues 263–264.29 Because StoneHingeP identifies hinges between two rigid domains, it misses the secondary hinge. The length of the primary hinge is significantly overpredicted in the open and closed forms, due to the inclusion of α helices bordering the hinge. StoneHingeD correctly predicts both hinges but also predicts two hinges in the middle of the second domain. This case particularly indicates the strengths of using StoneHingeP and StoneHingeD together to pinpoint the location of hinges that are in agreement, and cancel extraneous predictions. FlexProt does not detect the secondary hinge, but it accurately detects the first.
Calmodulin
This is an atypical hinge that consists of an α helix which partially unwinds in the center, as determined using NMR36 and computational analyses37,38 as well as visual examination of the conformational transition.29 StoneHinge predictions on both forms of the protein match this hinge, whereas FlexProt identifies four hinges, one of which is adjacent to the correct hinge.
Inorganic pyrophosphatase
This hinge includes one segment of backbone, and the two domains are easily distinguished in the open conformation. The literature hinge is based on a comparison of crystal structures.14 StoneHinge predictions from the open and closed structures agree with the literature and visually-defined hinge. FlexProt detects this hinge but also identifies an extra one.
Ribose binding protein
Here, the hinge involves multiple stretches of backbone. The literature hinges are based on the comparison of the closed structure with several open structures.15 StoneHinge predictions agree with the two gold standard hinges. FlexProt detects the first hinge region, but misses the second. The predictions for ribose binding protein are mapped onto the three-dimensional structure of the protein in Figure 2, with the literature and visually-defined hinges appearing in panel (G) for comparison.
Figure 2.
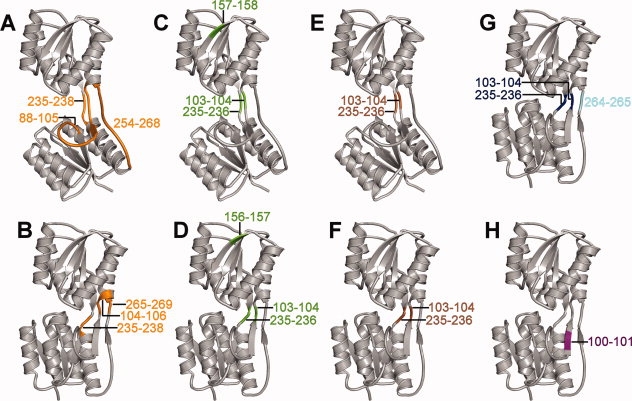
Ribose binding protein hinge predictions highlighted in color on the backbone, represented as a ribbon diagram. Shown are the StoneHingeP predictions on the (A) open and (B) closed conformations, StoneHingeD predictions on the (C) open and (D) closed conformations, and StoneHinge consensus predictions on the (E) open and (F) closed conformations. (G) The gold standard hinges (consensus of literature and visually assigned hinges) are highlighted in blue, with an additional literature-defined hinge shown in cyan. (H) Hinges detected by FlexProt superposition between open and closed conformations are displayed. This figure was generated using PyMol.39
Summary of StoneHinge consensus predictions
All gold standard (consensus of literature and visually-assigned) hinges were predicted by StoneHinge in the open states of the nine proteins, except for two in adenylate kinase. The same hinges were missed in the closed state of adenylate kinase, and two were missed in the closed glutamine binding protein; however, in the open state of this protein, both were predicted. No extra hinges were predicted in any protein.
Negative controls
To assess whether StoneHinge would falsely predict intradomain loops as hinges, the algorithm was tested on six proteins with low main-chain RMSD values between ligand-bound and ligand-free structures, indicating no significant backbone motion.40,41 Both the apo and ligand-bound structures were tested for three of the proteins. As with all predictions, ligands were removed first. These negative controls are listed in Table II, along with their StoneHingeP rigid cluster (predicted domain) sizes and StoneHingeD domain count. For all six proteins, StoneHingeP predicted the second rigid domain to have fewer than 20 residues, thus correctly predicting no hinge motion (see Methods section). Similarly, StoneHingeD predicted no hinges in all cases except for Protein A, due to identifying at most one domain in these proteins. Therefore, the consensus StoneHinge prediction for all proteins except Protein A indicated no hinges.
Discussion
The results of applying StoneHingeP, StoneHingeD, and StoneHinge to 27 structures (seven of which were used in training, with the other 20 reserved for testing), indicate they are effective predictors. StoneHingeP was shown to be highly sensitive: nearly all hinges in the gold standard set were identified for the structures in which StoneHingeP made a prediction. As shown in Figure 1, any additional predictions corresponded to a hinge either reported in the literature or determined by visual inspection. However, in both DNA polymerase β structures and the open form of LAO binding protein, StoneHingeP significantly overpredicted the lengths of hinges. These overpredictions were caused by predicting several smaller rigid regions in place of the second rigid domain. Similarly, in glutamine binding protein and closed LAO binding protein, StoneHingeP missed the hinges due to not identifying the second rigid domain. Predicting more flexibility than is apparent from experimental results is typically due to irregular secondary structure or suboptimal hydrogen-bond stereochemistry, which weakens the network of hydrogen bonds analyzed by ProFlex constraint counting. StoneHingeD, the other component algorithm, was shown to be a precise predictor of known hinges. Each predicted hinge was specified as a pair of residues, although additional hinges were predicted for DNA polymerase β, ribose binding protein, and the closed structures of CAPK and adenylate kinase.
StoneHingeP is also notable as the only algorithm using rigidity theory to locate hinges. Thus, it is likely to provide useful and independent information when used in consensus predictions, as shown by StoneHinge. Its consensus of StoneHingeD and StoneHingeP results was both sensitive and specific. All predictions corresponded to a gold standard hinge, and all gold standard hinges were correctly predicted, excepting adenylate kinase and the closed form of glutamine binding protein. Additionally, StoneHinge runs quickly due to the integer constraint counting algorithm of StoneHingeP and the reduced protein representation of StoneHingeD. It takes roughly 2 min to analyze a 300-residue protein on a Pentium IV, including determining the optimal energy level. The algorithm is also completely automated and does not require human intervention to interpret the results. As such, it is suitable for high-throughput use.
Using StoneHinge to guide flexible docking
As with other hinge predictors, StoneHinge results can guide the sampling of protein main-chain flexibility during ligand docking. One approach is to use StoneHinge together with FlexDock, which can dock ligands into proteins with prespecified hinges.3 As only the location of the hinge must be specified, not the direction of motion, predictions from StoneHinge are suitable for use with FlexDock. Additionally, the network of rotatable bonds derived by StoneHinge can also be used with ROCK,42 which generates a panel of protein conformations by sampling the hinge angles within stereochemically favorable ranges. Ligands can then be independently docked into each protein conformation using SLIDE,43 as was demonstrated for fully flexible docking of the cyclic peptide cyclosporin with its receptor, cyclophilin A.44 StoneHingeP already provides the input files needed by ROCK, making the process easy to automate.
As an alternative sampling method, the ProFlex component of StoneHingeP can be combined with a rotations and translations of blocks normal mode analysis to sample conformation changes in proteins45 as input to docking. The output of StoneHinge can also be used as a starting point for MBO(N)D molecular dynamics simulations.46 By dividing the protein into rigid bodies and flexible regions (e.g., using the StoneHinge predictions), this algorithm reduces the computational time required for molecular dynamics simulations. We also foresee other advances and applications of the StoneHinge algorithm, including recognizing and sampling active-site loop motion.
Methods
StoneHinge determines the consensus of the StoneHingeP and StoneHingeD automated hinge predictions, which are described in greater detail in this section.
StoneHingeP overview
StoneHingeP uses ProFlex27 to predict a protein's rigid clusters, which are groups of atoms that are constrained by the bond network and do not move relative to each other. However, one rigid cluster may move relative to another, like two stones linked by a tether. These clusters may range in size from a few atoms to nearly the size of the entire protein. StoneHingeP prepares the input, runs the rigidity analysis module of ProFlex, and determines the appropriate energy level cutoff for hydrogen bonds and salt bridges to include in the network. The residue ranges within the largest two rigid clusters are then defined and filtered by StoneHingeP, based on whether they meet the domain size criterion, and the hinge residues between the domains are identified, possibly including rigid motifs such as turns. Figure 3 shows a flowchart of the algorithm, with each step explained below. StoneHinge performs all necessary preparation and analysis steps without intervention. This makes StoneHinge easy to use and enables automatic analysis of a database of structures.
Protein preparation
Including buried waters
ProFlex works best when internal (entirely buried) water molecules are present in the structure but surface-bound water molecules are removed. The hydrogen bonds to internal waters can be important for the protein structure, whereas including surface waters tends to lead to overestimation of the rigidity of the protein.27 All predictions presented here are made with internal water molecules only, as determined by PRO_ACT.47 Predictions were also run using no water molecules (data not shown), as is done on the automated StoneHinge server. Although the overall flexibility analysis changed slightly, the hinge predictions were largely the same. Negligible differences have also been noted in a previous ProFlex-based analysis following removal of structural water from the Ras-Raf complex.48
Removing ligands
All inhibitors and/or cofactors are removed from the structure before the prediction, as they tend to cross-link and rigidify the domains relative to each other. Ligand removal also enables identification of the intrinsic, rather than ligand-induced, flexibility of the protein. StoneHinge automatically removes all nonpolypeptide inhibitors by stripping heteroatom records from the PDB file. However, the protein will remain in a ligand-bound conformation, at least in terms of side-chain orientations. As this can influence the network of interactions analyzed by StoneHinge, we recommend analyzing ligand-free, open structures when possible. StoneHinge also removes metal ions by default, as bonds between a metal ion and the protein are difficult to deduce automatically from the structure. However, for biologically relevant bound metals, neglecting them may result in increased flexibility, affecting the hinge predictions. It is possible to avoid this by preparing the bond network using the standalone version of ProFlex (available by contacting ProFlex@sol.bch.msu.edu), but this procedure has not yet been automated as part of the StoneHinge predictions.
Adding hydrogen atoms
ProFlex requires polar hydrogen atoms to be present in the structure, as they are used to calculate hydrogen-bond energies. StoneHingeP calls GROMACS49 to add polar hydrogen atoms to the protein structure and optimize their positions by steepest descent energy minimization for 100 2-femtosecond steps. GROMACS is freely available under the GNU Public License from http://www.gromacs.org.
Calculating the hydrogen-bond dilution profile
ProFlex calculates a hydrogen-bond dilution profile,50 which StoneHingeP analyzes to identify an energy level optimal for determining domains and intervening hinges (Supp. Info. Fig. 1). To calculate the profile, the protein's hydrogen bonds are broken one by one, from weakest to strongest, and the constraint counting algorithm is run after each bond is broken. This simulates incremental thermal denaturation of the structure, as the calculated temperature rises and hydrogen bonds weaker than the current energy level are broken. The protein commonly appears as a single large rigid region when the simulated temperature is low and very weak hydrogen bonds and salt bridges are included. The structure then gradually breaks into two or more rigid regions (often corresponding to the known native state), before going through a cooperative phase transition to a completely flexible chain as the simulated temperature rises.51 Hydrophobic interactions are maintained throughout the process, as the strength of these interactions increases somewhat with modest increases in temperature.52
Domain and hinge prediction using StoneHingeP
Selecting an energy level and domain size
There is no single energy level that corresponds to the native state of all proteins, in terms of rigid and flexible regions. This may be a result of the different conditions and forcefields under which protein structures are determined. Because our goal is to predict hinges, we wish to identify an energy level in which the protein contains at least two rigid domains of substantial size (20 or more residues). Rigid clusters of this size typically correspond to supra-secondary structures (e.g., two packed helices, rather than one long helix), whereas smaller rigid clusters do not. The appropriateness of this size threshold was assessed using the training set (Table I) and fixed before running StoneHingeP on the test set proteins. StoneHingeP identifies the energy level at which the second-largest rigid cluster is maximal in size, measured by the number of residues with mutually rigid backbones. This typically occurs when the protein has just relaxed from a single large rigid cluster into two clusters, each potentially representing a domain. If more than one energy level has a second-largest rigid cluster of the same size, the more rigid (lower-temperature) state of the protein is selected. The two largest rigid clusters are then checked against the 20-residue size criterion. If either cluster is smaller than this, then no hinge is predicted.
Identifying hinges in StoneHingeP
Hinges are predicted as the residues intervening between two rigid clusters meeting the aforementioned criteria. Predicted hinges can be one or multiple stretches of residues, as rigid clusters do not necessarily consist of contiguous amino acids. For example, in the open form of adenylate kinase (Supp. Info. Fig. 1), the largest rigid cluster (called cluster 1 or the first rigid domain) is predicted as residues 5 through 12, 17 through 113, 163 through 195, and 198 through 217. These residues are proximal in space, but not contiguous in sequence. The second-largest rigid cluster (cluster 2 or the second rigid domain) is predicted as residues 124 through 158. Thus, in primary structure, cluster 2 occurs between two regions of cluster 1, and the hinges are predicted as the two stretches of residues between these clusters (residues 113 through 124 and 158 through 163). This corresponds to one flexible region of the backbone passing from rigid cluster 1 to rigid cluster 2, then a second flexible region later in sequence passing from rigid cluster 2 back to cluster 1. If cluster 2 also contained discontinuous sequence, as in the case of cAMP dependent protein kinase, then more than two hinges could be predicted.
Training and test sets
During development, StoneHingeP was frequently tested against closed and open CAPK; the open forms of Bence-Jones protein, lysine-arginine-ornithine binding protein, and adenylate kinase; and two proteins without hinges, the immunoglobulin domain of protein G and hydropterin pyrophosphokinase. The training results led to adjustments in the domain size threshold and the determination of appropriate energy levels for analysis. Thus, the StoneHingeP results of these training proteins may be favorably biased. StoneHingeP was also run on additional proteins once the algorithm was finalized. The results on this test set may be more representative of its general performance. The training and test set proteins are designated in Table I.
Multichain proteins
At present, StoneHinge only predicts on single-chain proteins. The StoneHingeP methodology is applicable to oligomeric proteins, but interpretation of the results is not straightforward. For example, the two largest rigid subdomains of the protein may fall on different chains. Assigning a hinge between these two domains would not make sense, as they are not covalently connected. It is also possible that one chain contains two domains with an intervening hinge, whereas another chain contains a single large domain not involved in the hinge motion. In this case, selecting the two largest domains of the complex would not necessarily select the domains bounding the hinge. Because of these difficulties, StoneHinge will generate a StoneHingeP hydrogen-bond dilution plot (e.g., Supp. Info. Fig. 1) for oligomeric proteins without making a prediction. The dilution plot can then be manually interpreted to locate potential hinges.
StoneHingeD predictions
StoneHingeD predictions utilize DomDecomp,12 which is freely available at http://stonehinge.molmovdb.org or by request to Prof. George Phillips (phillips@biochem.wisc.edu). StoneHingeD makes predictions based on DomDecomp GNM normal mode analysis, which identifies domains as groups of residues undergoing anticorrelated motion at the lowest nonzero frequency. The last residue assigned to one domain is consecutive with the first residue of the next. Thus, hinges are identified as pairs of residues at anticorrelated domain boundaries.
StoneHinge consensus prediction
To combine StoneHingeP and StoneHingeD results, the residues in a StoneHingeD prediction are assigned as a hinge if they fall within five residues of a StoneHingeP prediction. (N.B. For cases presented, the same results are obtained if a more stringent criterion, falling within two residues, is used.) For proteins in which hinges are only predicted by one method, the predictions of that method are used alone, as in the cases of glutamine binding protein and the closed state of LAO binding protein. However, these predictions have lower confidence, as both component methods tend to overpredict the number of hinge residues.
FlexProt predictions
StoneHinge results were compared with hinges identified by the FlexProt server of Shatsky, Nussinov, and Wolfson (http://bioinfo3d.cs.tau.ac.il/FlexProt/), which detects hinges by superimposing domains from open and closed structures.11,32 FlexProt requires both the open and closed conformations of the protein as input, and the hinges are identified as the pair of residues bordering adjacent rigid fragments. The hinge assignments were made using default settings: a maximum of 3.0 Å RMSD between matched fragments in the two structures and a minimum size of 15 amino acids for matched fragments. Hinges reported here are based on the alignment with lowest RMSD between all matched fragments.
Assessing prediction sensitivity
There can be variability when assessing hinge motion from experimental or simulation data, as evidenced by the differences observed between the literature and visually-defined hinges. Therefore, we assessed the sensitivity of the predictors using the intersection of the hinges from the literature and visual hinge sets. Hinges from these sets were said to intersect, or represent consensus, if they included amino acids within five residues of each other. These hinges are referred to as the gold standard set.
StoneHinge availability
StoneHinge is available online at http://stonehinge.molmovdb.org, as shown in Figure 4. Proteins can be submitted for prediction online, and the predicted hinge locations will be mapped onto the three-dimensional structure and can be rotated in Jmol. Hinge predictions are also given in tabular form for StoneHingeP, StoneHingeD, and the StoneHinge consensus. Additionally, the hydrogen-bond dilution plot used for StoneHingeP predictions can be displayed. For those preferring to run the software locally, the StoneHinge Perl code is also available for download under the GNU Public License at the aforementioned web site. It can be run locally on Linux or Unix systems and modified as desired.
Figure 4.
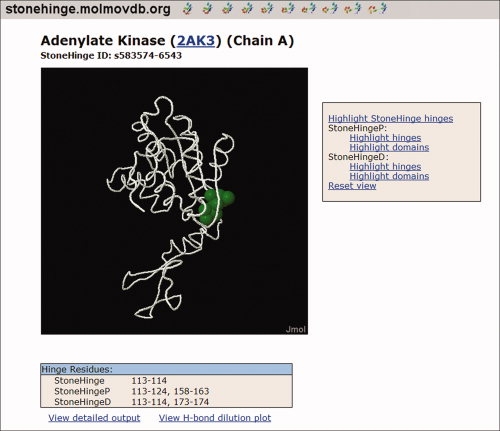
Proteins can be submitted online for a StoneHinge analysis at http://stonehinge.molmovdb.org. This view of the user interface shows predictions made on adenylate kinase in the open form. The StoneHinge hinges are highlighted in the Jmol viewer (http://www.jmol.org), which can also display the StoneHingeP and StoneHingeD hinges and domains. Predicted hinge residues are listed in the table below the molecular graphics. [Color figure can be viewed in the online issue, which is available at www.interscience.wiley.com.]:
Protein structures
All protein structures were retrieved from the Protein Data Bank.53 Protein Data Bank entries are as follows: 1CTP,54 1ATP,55 4BJL,56 2LAO, 1LST,57 2AK3,58 1AKE,59 1GGG,34 1WDN,60 2BPG,61 1BPD,62 1CFD,63 1CLL,64 1K23, 1K20,14 1URP,15 2DRI,65 1PGB,66 1HKA,67 1BCK,68 2APR,69 3APR,70 2CLA,71 3CLA,72 2SGA,73 and 5SGA.74
Acknowledgments
The authors thank Sameer Arora for his early contributions to the StoneHinge code, as well as Sibsankar Kundu and George Phillips for providing the DomDecomp software.
References
- 1.Hayward S, Kitao A, Berendsen HJ. Model-free methods of analyzing domain motions in proteins from simulation: a comparison of normal mode analysis and molecular dynamics simulation of lysozyme. Proteins. 1997;27:425–437. doi: 10.1002/(sici)1097-0134(199703)27:3<425::aid-prot10>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]
- 2.Scheibel T, Lindquist SL. The role of conformational flexibility in prion propagation and maintenance for Sup35p. Nat Struct Biol. 2001;8:958–962. doi: 10.1038/nsb1101-958. [DOI] [PubMed] [Google Scholar]
- 3.Schneidman-Duhovny D, Inbar Y, Nussinov R, Wolfson HJ. Geometry-based flexible and symmetric protein docking. Proteins. 2005;60:224–231. doi: 10.1002/prot.20562. [DOI] [PubMed] [Google Scholar]
- 4.Cozzini P, Kellogg GE, Spyrakis F, Abraham DJ, Costantino G, Emerson A, Fanelli F, Gohlke H, Kuhn LA, Morris GM, Orozco M, Pertinez TA, Rizzi M, Sotriffer CA. Target flexibility: an emerging consideration in drug discovery and design. J Med Chem. 2008;51:6237–6255. doi: 10.1021/jm800562d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gerstein M, Lesk AM, Chothia C. Structural mechanisms for domain movements in proteins. Biochemistry. 1994;33:6739–6749. doi: 10.1021/bi00188a001. [DOI] [PubMed] [Google Scholar]
- 6.Gerstein M, Jansen R. Studying macromolecular motions in a database framework: from structure to sequence. In: Thorpe M, Johnson T, Tsai J, Krebs W, editors; Duxbury P, editor. Rigidity theory and applications. New York, NY: Kluwer Academic/Plenum Publishers; 1999. [Google Scholar]
- 7.Krebs W. Molecular Biophysics and Biochemistry. New Haven: Yale University; 2001. The database of macromolecular motions: a standardized system for analyzing and visualizing macromolecular motions in a database framework; p. 288. Dissertation. [Google Scholar]
- 8.Maiorov V, Abagyan R. A new method for modeling large-scale rearrangements of protein domains. Proteins. 1997;27:410–424. doi: 10.1002/(sici)1097-0134(199703)27:3<410::aid-prot9>3.0.co;2-g. [DOI] [PubMed] [Google Scholar]
- 9.Zhang XJ, Wozniak JA, Matthews BW. Protein flexibility and adaptability seen in 25 crystal forms of T4 lysozyme. J Mol Biol. 1995;250:527–552. doi: 10.1006/jmbi.1995.0396. [DOI] [PubMed] [Google Scholar]
- 10.Hayward S, Berendsen HJ. Systematic analysis of domain motions in proteins from conformational change: new results on citrate synthase and T4 lysozyme. Proteins. 1998;30:144–154. [PubMed] [Google Scholar]
- 11.Shatsky M, Nussinov R, Wolfson HJ. Flexible protein alignment and hinge detection. Proteins. 2002;48:242–256. doi: 10.1002/prot.10100. [DOI] [PubMed] [Google Scholar]
- 12.Kundu S, Sorensen DC, Phillips GN., Jr Automatic domain decomposition of proteins by a Gaussian network model. Proteins. 2004;57:725–733. doi: 10.1002/prot.20268. [DOI] [PubMed] [Google Scholar]
- 13.Gerstein M, Schulz G, Chothia C. Domain closure in adenylate kinase. Joints on either side of two helices close like neighboring fingers. J Mol Biol. 1993;229:494–501. doi: 10.1006/jmbi.1993.1048. [DOI] [PubMed] [Google Scholar]
- 14.Ahn S, Milner AJ, Futterer K, Konopka M, Ilias M, Young TW, White SA. The “open” and “closed” structures of the type-C inorganic pyrophosphatases from Bacillus subtilis and Streptococcus gordonii. J Mol Biol. 2001;313:797–811. doi: 10.1006/jmbi.2001.5070. [DOI] [PubMed] [Google Scholar]
- 15.Bjorkman AJ, Mowbray SL. Multiple open forms of ribose-binding protein trace the path of its conformational change. J Mol Biol. 1998;279:651–664. doi: 10.1006/jmbi.1998.1785. [DOI] [PubMed] [Google Scholar]
- 16.Zehfus MH. Binary discontinuous compact protein domains. Protein Eng. 1994;7:335–340. doi: 10.1093/protein/7.3.335. [DOI] [PubMed] [Google Scholar]
- 17.Holm L, Sander C. Dictionary of recurrent domains in protein structures. Proteins. 1998;33:88–96. doi: 10.1002/(sici)1097-0134(19981001)33:1<88::aid-prot8>3.0.co;2-h. [DOI] [PubMed] [Google Scholar]
- 18.Holm L, Sander C. Parser for protein folding units. Proteins. 1994;19:256–268. doi: 10.1002/prot.340190309. [DOI] [PubMed] [Google Scholar]
- 19.Islam SA, Luo J, Sternberg MJ. Identification and analysis of domains in proteins. Protein Eng. 1995;8:513–525. doi: 10.1093/protein/8.6.513. [DOI] [PubMed] [Google Scholar]
- 20.Siddiqui AS, Barton GJ. Continuous and discontinuous domains: an algorithm for the automatic generation of reliable protein domain definitions. Protein Sci. 1995;4:872–884. doi: 10.1002/pro.5560040507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Swindells MB. A procedure for detecting structural domains in proteins. Protein Sci. 1995;4:103–112. doi: 10.1002/pro.5560040113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Swindells MB. A procedure for the automatic determination of hydrophobic cores in protein structures. Protein Sci. 1995;4:93–102. doi: 10.1002/pro.5560040112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Painter J, Merritt EA. Optimal description of a protein structure in terms of multiple groups undergoing TLS motion. Acta Crystallogr. 2006;D62:439–450. doi: 10.1107/S0907444906005270. [DOI] [PubMed] [Google Scholar]
- 24.Flores SC, Gerstein MB. FlexOracle: predicting flexible hinges by identification of stable domains. BMC Bioinformatics. 2007;8:215. doi: 10.1186/1471-2105-8-215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Flores SC, Lu LJ, Yang J, Carriero N, Gerstein MB. Hinge Atlas: relating protein sequence to sites of structural flexibility. BMC Bioinformatics. 2007;8:167. doi: 10.1186/1471-2105-8-167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hinsen K. Analysis of domain motions by approximate normal mode calculations. Proteins. 1998;33:417–429. doi: 10.1002/(sici)1097-0134(19981115)33:3<417::aid-prot10>3.0.co;2-8. [DOI] [PubMed] [Google Scholar]
- 27.Jacobs DJ, Rader AJ, Kuhn LA, Thorpe MF. Protein flexibility predictions using graph theory. Proteins. 2001;44:150–165. doi: 10.1002/prot.1081. [DOI] [PubMed] [Google Scholar]
- 28.Bahar I, Atilgan AR, Erman B. Direct evaluation of thermal fluctuations in proteins using a single-parameter harmonic potential. Fold Des. 1997;2:173–181. doi: 10.1016/S1359-0278(97)00024-2. [DOI] [PubMed] [Google Scholar]
- 29.Flores SC, Keating KS, Painter J, Morcos FG, Nguyen K, Merritt E, Kuhn LA, Gerstein M. HingeMaster: normal mode hinge prediction approach and integration of complementary predictors. Proteins. 2008;73:299–319. doi: 10.1002/prot.22060. [DOI] [PubMed] [Google Scholar]
- 30.Krebs WG, Gerstein M. The morph server: a standardized system for analyzing and visualizing macromolecular motions in a database framework. Nucleic Acids Res. 2000;28:1665–1675. doi: 10.1093/nar/28.8.1665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Flores S, Echols N, Milburn D, Hespenheide B, Keating K, Lu J, Wells S, Yu EZ, Thorpe M, Gerstein M. The database of macromolecular motions: new features added at the decade mark. Nucleic Acids Res. 2006;34:D296–D301. doi: 10.1093/nar/gkj046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shatsky M, Nussinov R, Wolfson HJ. FlexProt: alignment of flexible protein structures without a predefinition of hinge regions. J Comput Biol. 2004;11:83–106. doi: 10.1089/106652704773416902. [DOI] [PubMed] [Google Scholar]
- 33.Tsigelny I, Greenberg JP, Cox S, Nichols WL, Taylor SS, Ten Eyck LF. 600 ps molecular dynamics reveals stable substructures and flexible hinge points in cAMP dependent protein kinase. Biopolymers. 1999;50:513–524. doi: 10.1002/(SICI)1097-0282(19991015)50:5<513::AID-BIP5>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- 34.Hsiao CD, Sun YJ, Rose J, Wang BC. The crystal structure of glutamine-binding protein from Escherichia coli. J Mol Biol. 1996;262:225–242. doi: 10.1006/jmbi.1996.0509. [DOI] [PubMed] [Google Scholar]
- 35.Kumar A, Widen SG, Williams KR, Kedar P, Karpel RL, Wilson SH. Studies of the domain structure of mammalian DNA polymerase beta. Identification of a discrete template binding domain. J Biol Chem. 1990;265:2124–2131. [PubMed] [Google Scholar]
- 36.Ikura M, Clore GM, Gronenborn AM, Zhu G, Klee CB, Bax A. Solution structure of a calmodulin-target peptide complex by multidimensional NMR. Science. 1992;256:632–638. doi: 10.1126/science.1585175. [DOI] [PubMed] [Google Scholar]
- 37.Meador WE, Means AR, Quiocho FA. Target enzyme recognition by calmodul2.4 A structure of a calmodulin-peptide complex. Science. 1992;257:1251–1255. doi: 10.1126/science.1519061. [DOI] [PubMed] [Google Scholar]
- 38.van der Spoel D, de Groot BL, Hayward S, Berendsen HJ, Vogel HJ. Bending of the calmodulin central helix: a theoretical study. Protein Sci. 1996;5:2044–2053. doi: 10.1002/pro.5560051011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.DeLano WL. The PyMOL molecular graphics system. San Carlos, CA: DeLano Scientific; 2002. [Google Scholar]
- 40.Raymer ML, Sanschagrin PC, Punch WF, Venkataraman S, Goodman ED, Kuhn LA. Predicting conserved water-mediated and polar ligand interactions in proteins using a K-nearest-neighbors genetic algorithm. J Mol Biol. 1997;265:445–464. doi: 10.1006/jmbi.1996.0746. [DOI] [PubMed] [Google Scholar]
- 41.Mayer KL, Earley MR, Gupta S, Pichumani K, Regan L, Stone MJ. Covariation of backbone motion throughout a small protein domain. Nat Struct Biol. 2003;10:962–965. doi: 10.1038/nsb991. [DOI] [PubMed] [Google Scholar]
- 42.Lei M, Zavodszky MI, Kuhn LA, Thorpe MF. Sampling protein conformations and pathways. J Comput Chem. 2004;25:1133–1148. doi: 10.1002/jcc.20041. [DOI] [PubMed] [Google Scholar]
- 43.Zavodszky MI, Sanschagrin PC, Korde RS, Kuhn LA. Distilling the essential features of a protein surface for improving protein-ligand docking, scoring, and virtual screening. J Comput Aided Mol Des. 2002;16:883–902. doi: 10.1023/a:1023866311551. [DOI] [PubMed] [Google Scholar]
- 44.Zavodszky MI, Lei M, Thorpe MF, Day AR, Kuhn LA. Modeling correlated main-chain motions in proteins for flexible molecular recognition. Proteins. 2004;57:243–261. doi: 10.1002/prot.20179. [DOI] [PubMed] [Google Scholar]
- 45.Ahmed A, Gohlke H. Multiscale modeling of macromolecular conformational changes combining concepts from rigidity and elastic network theory. Proteins. 2006;63:1038–1051. doi: 10.1002/prot.20907. [DOI] [PubMed] [Google Scholar]
- 46.Chun HM, Padilla CE, Chin DN, Watanabe M, Karlov VI, Alper HE, Soosaar K, Blair KB, Becker OM, Caves LSD, Nagle R, Haney D, Farmer B. MBO(N)D: a multibody method for long-time molecular dynamics simulations. J Comput Chem. 2000;21:159–184. [Google Scholar]
- 47.Williams MA, Goodfellow JM, Thornton JM. Buried waters and internal cavities in monomeric proteins. Protein Sci. 1994;3:1224–1235. doi: 10.1002/pro.5560030808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gohlke H, Kuhn LA, Case DA. Change in protein flexibility upon complex formation: analysis of Ras-Raf using molecular dynamics and a molecular framework approach. Proteins. 2004;56:322–337. doi: 10.1002/prot.20116. [DOI] [PubMed] [Google Scholar]
- 49.Lindahl E, Hess B, van der Spoel D. GROMACS 3.0: a package for molecular simulation and trajectory analysis. J Mol Model. 2001;7:306–317. [Google Scholar]
- 50.Hespenheide BM, Rader AJ, Thorpe MF, Kuhn LA. Identifying protein folding cores from the evolution of flexible regions during unfolding. J Mol Graph Model. 2002;21:195–207. doi: 10.1016/s1093-3263(02)00146-8. [DOI] [PubMed] [Google Scholar]
- 51.Rader AJ, Hespenheide BM, Kuhn LA, Thorpe MF. Protein unfolding: rigidity lost. Proc Natl Acad Sci USA. 2002;99:3540–3545. doi: 10.1073/pnas.062492699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tanford C. The hydrophobic effect: formation of micelles and biological membranes. 2nd Edn. New York: Wiley; 1980. [Google Scholar]
- 53.Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. The protein data bank. Nucleic Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Karlsson R, Zheng J, Xuong N, Taylor SS, Sowadski JM. Structure of the mammalian catalytic subunit of cAMP-dependent protein kinase and an inhibitor peptide displays an open conformation. Acta Crystallogr D Biol Crystallogr. 1993;49:381–388. doi: 10.1107/S0907444993002306. [DOI] [PubMed] [Google Scholar]
- 55.Zheng J, Trafny EA, Knighton DR, Xuong NH, Taylor SS, Ten Eyck LF, Sowadski JM. 2.2 A refined crystal structure of the catalytic subunit of cAMP-dependent protein kinase complexed with MnATP and a peptide inhibitor. Acta Crystallogr D Biol Crystallogr. 1993;49:362–365. doi: 10.1107/S0907444993000423. [DOI] [PubMed] [Google Scholar]
- 56.Huang DB, Ainsworth CF, Stevens FJ, Schiffer M. Three quaternary structures for a single protein. Proc Natl Acad Sci USA. 1996;93:7017–7021. doi: 10.1073/pnas.93.14.7017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Oh BH, Pandit J, Kang CH, Nikaido K, Gokcen S, Ames GF, Kim SH. Three-dimensional structures of the periplasmic lysine/arginine/ornithine-binding protein with and without a ligand. J Biol Chem. 1993;268:11348–11355. [PubMed] [Google Scholar]
- 58.Diederichs K, Schulz GE. The refined structure of the complex between adenylate kinase from beef heart mitochondrial matrix and its substrate AMP at 1.85 A resolution. J Mol Biol. 1991;217:541–549. doi: 10.1016/0022-2836(91)90756-v. [DOI] [PubMed] [Google Scholar]
- 59.Muller CW, Schulz GE. Structure of the complex between adenylate kinase from Escherichia coli and the inhibitor Ap5A refined at 1.9 A resolution. A model for a catalytic transition state. J Mol Biol. 1992;224:159–177. doi: 10.1016/0022-2836(92)90582-5. [DOI] [PubMed] [Google Scholar]
- 60.Sun YJ, Rose J, Wang BC, Hsiao CD. The structure of glutamine-binding protein complexed with glutamine at 1.94 A resolution: comparisons with other amino acid binding proteins. J Mol Biol. 1998;278:219–229. doi: 10.1006/jmbi.1998.1675. [DOI] [PubMed] [Google Scholar]
- 61.Pelletier H, Sawaya MR, Kumar A, Wilson SH, Kraut J. Structures of ternary complexes of rat DNA polymerase beta, a DNA template-primer, and ddCTP. Science. 1994;264:1891–1903. [PubMed] [Google Scholar]
- 62.Sawaya MR, Pelletier H, Kumar A, Wilson SH, Kraut J. Crystal structure of rat DNA polymerase beta: evidence for a common polymerase mechanism. Science. 1994;264:1930–1935. doi: 10.1126/science.7516581. [DOI] [PubMed] [Google Scholar]
- 63.Kuboniwa H, Tjandra N, Grzesiek S, Ren H, Klee CB, Bax A. Solution structure of calcium-free calmodulin. Nat Struct Biol. 1995;2:768–776. doi: 10.1038/nsb0995-768. [DOI] [PubMed] [Google Scholar]
- 64.Chattopadhyaya R, Meador WE, Means AR, Quiocho FA. Calmodulin structure refined at 1.7 A resolution. J Mol Biol. 1992;228:1177–1192. doi: 10.1016/0022-2836(92)90324-d. [DOI] [PubMed] [Google Scholar]
- 65.Bjorkman AJ, Binnie RA, Zhang H, Cole LB, Hermodson MA, Mowbray SL. Probing protein-protein interactions. The ribose-binding protein in bacterial transport and chemotaxis. J Biol Chem. 1994;269:30206–30211. [PubMed] [Google Scholar]
- 66.Gallagher T, Alexander P, Bryan P, Gilliland GL. Two crystal structures of the B1 immunoglobulin-binding domain of streptococcal protein G and comparison with NMR. Biochemistry. 1994;33:4721–4729. [PubMed] [Google Scholar]
- 67.Xiao B, Shi G, Chen X, Yan H, Ji X. Crystal structure of 6-hydroxymethyl-7,8-dihydropterin pyrophosphokinase, a potential target for the development of novel antimicrobial agents. Structure. 1999;7:489–496. doi: 10.1016/s0969-2126(99)80065-3. [DOI] [PubMed] [Google Scholar]
- 68.Kallen J, Mikol V, Taylor P, Walkinshaw MD. X-ray structures and analysis of 11 cyclosporin derivatives complexed with cyclophilin A. J Mol Biol. 1998;283:435–449. doi: 10.1006/jmbi.1998.2108. [DOI] [PubMed] [Google Scholar]
- 69.Suguna K, Bott RR, Padlan EA, Subramanian E, Sheriff S, Cohen GH, Davies DR. Structure and refinement at 1.8 A resolution of the aspartic proteinase from Rhizopus chinensis. J Mol Biol. 1987;196:877–900. doi: 10.1016/0022-2836(87)90411-6. [DOI] [PubMed] [Google Scholar]
- 70.Suguna K, Padlan EA, Smith CW, Carlson WD, Davies DR. Binding of a reduced peptide inhibitor to the aspartic proteinase from Rhizopus chinensis: implications for a mechanism of action. Proc Natl Acad Sci USA. 1987;84:7009–7013. doi: 10.1073/pnas.84.20.7009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Gibbs MR, Moody PC, Leslie AG. Crystal structure of the aspartic acid-199 –> asparagine mutant of chloramphenicol acetyltransferase to 2.35-A resolution: structural consequences of disruption of a buried salt bridge. Biochemistry. 1990;29:11261–11265. doi: 10.1021/bi00503a015. [DOI] [PubMed] [Google Scholar]
- 72.Leslie AG. Refined crystal structure of type III chloramphenicol acetyltransferase at 1.75 A resolution. J Mol Biol. 1990;213:167–186. doi: 10.1016/S0022-2836(05)80129-9. [DOI] [PubMed] [Google Scholar]
- 73.Moult J, Sussman F, James MN. Electron density calculations as an extension of protein structure refinement. Streptomyces griseus protease A at 1.5 A resolution. J Mol Biol. 1985;182:555–566. doi: 10.1016/0022-2836(85)90241-4. [DOI] [PubMed] [Google Scholar]
- 74.James MN, Sielecki AR, Brayer GD, Delbaere LT, Bauer CA. Structures of product and inhibitor complexes of Streptomyces griseus protease A at 1.8 A resolution. A model for serine protease catalysis. J Mol Biol. 1980;144:43–88. doi: 10.1016/0022-2836(80)90214-4. [DOI] [PubMed] [Google Scholar]