

Abstract
Genetic and biochemical studies suggest that Alzheimer's disease (AD) is caused by a series of events initiated by the production and subsequent aggregation of the Alzheimer's amyloid β peptide (Aβ), the so-called amyloid cascade hypothesis. Thus, a logical approach to treating AD is the development of small molecule inhibitors that either block the proteases that generate Aβ from its precursor (β-and γ-secretases) or interrupt and/or reverse Aβ aggregation. To identify potent inhibitors of Aβ aggregation, we have developed a high-throughput screen based on an earlier selection that effectively paired the folding quality control feature of the Escherichia coli Tat protein export system with aggregation of the 42-residue AD pathogenesis effecter Aβ42. Specifically, a tripartite fusion between the Tat-dependent export signal ssTorA, the Aβ42 peptide and the β-lactamase (Bla) reporter enzyme was found to be export incompetent due to aggregation of the Aβ42 moiety. Here, we reasoned that small, cell-permeable molecules that inhibited Aβ42 aggregation would render the ssTorA-Aβ42-Bla chimera competent for Tat export to the periplasm where Bla is active against β-lactam antibiotics such as ampicillin. Using a fluorescence-based version of our assay, we screened a library of triazine derivatives and isolated four nontoxic, cell-permeable compounds that promoted efficient Tat-dependent export of ssTorA-Aβ42-Bla. Each of these was subsequently shown to be a bona fide inhibitor of Aβ42 aggregation using a standard thioflavin T fibrillization assay, thereby highlighting the utility of our bacterial assay as a useful screen for antiaggregation factors under physiological conditions.
Keywords: Alzheimer's disease, aggregation, chemical biology, chemical chaperone, protein misfolding, twin-arginine translocation
Introduction
Alzheimer's disease (AD) is a progressive neurodegenerative disorder that affects 20–30 million people worldwide.1 Genetic and biochemical studies suggest that AD is caused by a series of events initiated by the production and subsequent aggregation of the Alzheimer's amyloid β peptide (Aβ), the so-called amyloid cascade hypothesis.2,3 Accordingly, a logical approach to treating AD is the development of small molecule inhibitors that block the proteases that generate Aβ from its precursor (β-and γ-secretases) or that interrupt and/or reverse Aβ aggregation. While there are currently no drugs in clinical use that target these mechanisms, many putative disease-modifying inhibitors are under active development and some have shown in vivo efficacy in mouse models and in clinical trials.4–8 One of the most notable examples is Neurochem's Alzhemed (tramiprosate) that was developed to prevent amyloid formation and deposition in the brain, and thus modify the course of AD. Unfortunately, despite Alzhemed's promise in Phase II clinical trials, the recent U.S. Phase III trial is widely considered to have failed and the EU Phase III trial has been discontinued, thus signifying the urgent need for new inhibitors that can be brought to human clinical trials.
Towards the development of new Aβ aggregation blockers, a number of methods for screening putative small-molecule inhibitors of Aβ aggregation have been developed.9–12 The majority of these assays have been performed in vitro using either turbidity or binding of thioflavin T (ThT) to assay the formation of amyloid fibrils from synthetic Aβ peptide. However, based on the high cost of generating pure samples of synthetic or recombinant Aβ peptide, large-scale library screening is cost prohibitive. In addition, there are now numerous reports indicating that small, soluble oligomers of Aβ, rather than large fibrillar plaques, are the principal cause of synaptotoxicity associated with AD.4,13–15 Thus, assays that measure turbidity and binding to ThT, which are inherently based on assembly of Aβ into amyloid, are unlikely to uncover compounds that affect soluble Aβ oligomers. Similarly, the rapid aggregation kinetics of Aβ peptides, especially the 42-residue Aβ42 peptide, make it difficult to prepare samples that are entirely devoid of small aggregates. These preexisting “seeds” are known to promote Aβ aggregation and may therefore prevent the identification of inhibitors that block the earliest stages of aggregation. Finally, inhibitors isolated using these in vitro screens may cross-react with cellular components, especially endogenous β-sheet proteins, and induce cytotoxicity.
To address these shortcomings, we have developed a low-cost, bacterial cell-based assay for the isolation of Aβ aggregation inhibitors. This assay is based on a previously developed genetic selection whereby the folding quality control mechanism of the bacterial twin-arginine translocation (Tat) system was used for the identification of peptides and proteins that fold into a stable and soluble conformation.16 In this earlier study, we found that the efficiency with which Tat-exported β-lactamase (Bla) chimeras localize in the periplasm, and thus confer ampicillin (Amp) resistance to Escherichia coli cells, was dependent on the folding characteristics of the fusion partner polypeptide. By growing cells on increasing concentrations of Amp, we were able to select for protein variants that exhibited higher intracellular stability and were less prone to aggregation. It was observed that the aggregation-prone Aβ42 peptide conferred only low levels of resistance to cells when expressed as an ssTorA-Aβ42-Bla fusion, where the Tat-dependent signal peptide ssTorA was used to specifically target the chimera to the Tat export pathway. Importantly, screening a library of random Aβ42 sequences resulted in the isolation of several solubility-enhanced variants that conferred resistance to cells on Amp,16 indicating that factors affecting the solubility of Aβ42 are sufficient to confer export via the Tat pathway. Here, we hypothesized that small-molecule inhibitors could similarly affect the solubility of Aβ42 in a manner that rescues ssTorA-Aβ42-Bla export to the periplasm. To test this, we successfully developed two high-throughput versions of our genetic folding assay that facilitated isolation of Aβ42-aggregation inhibitors from a combinatorial library of triazine derivatives without the need for synthetic Aβ42. The identification of small molecules that inhibit formation of Aβ42 oligomers highlights the utility of our bacterial assay as a useful screen for antiaggregation factors under biologically relevant conditions.
Results
High-throughput strategies for monitoring Aβ42 aggregation in living cells
Based on our earlier finding that Aβ42 solubility could be effectively discriminated using the Tat quality control mechanism,16 we hypothesized that small, cell-permeable molecules capable of interfering with wildtype (wt) Aβ42 aggregation would promote Tat-dependent export of the ssTorA-Aβ42-Bla reporter to the periplasm (see Fig. 1). Such molecules would represent putative inhibitors of Aβ42 aggregation and could easily be identified in a high-throughput screening (HTS) approach provided that a robust assay was available for reporting the subcellular location of Bla. One strategy for detecting Bla that has localized in the periplasm is to simply monitor cell viability in liquid medium supplemented with Amp.16 Indeed, cells expressing ssTorA-Aβ42-Bla were much less resistant to Amp relative to cells expressing ssTorA-GM6-Bla (see Fig. 2), where GM6 is a solubility-enhanced version of Aβ42 developed previously by Hecht and coworkers17 that we utilized as a positive control in this study. We found that at Amp concentrations of 400 μg/mL and above, in 96-well plates, the growth rate of cells expressing ssTorA-GM6-Bla was ∼16-fold greater than that measured for cells expressing ssTorA-Aβ42-Bla [Fig. 2(a)]. Moreover, a 16-fold difference in minimum inhibitory concentration (MIC), where MIC is defined here as the Amp concentration at which cell growth was <50% of the growth in the absence of Amp, was observed for cells expressing ssTorA-GM6-Bla versus ssTorA-Aβ42-Bla (>800 μg/mL vs. 50 μg/mL, respectively). When the inoculation density was decreased 5-fold, a ∼12-fold difference in growth rate was seen for cells expressing ssTorA-GM6-Bla compared to cells expressing ssTorA-Aβ42-Bla at Amp concentrations of 50 and 100 μg/mL and a 4-fold difference in MIC on Amp was observed (400 μg/mL vs. 50 μg/mL, respectively) [Fig. 2(b)]. Importantly, this data indicates that a dramatic increase in Amp resistance is conferred to cells when the solubility of Aβ42 is enhanced, in this case by genetic mutation. The difference in MIC between soluble and aggregated Aβ42-expressing cells is the basis for the HTS growth assay.
Figure 1.
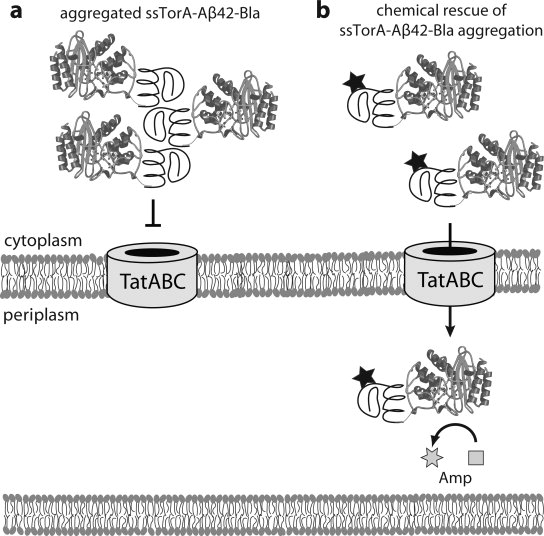
Strategy for isolating Aβ42 aggregation inhibitors using the Tat pathway. (a) ssTorA-Aβ42-Bla expressed in the cytoplasm readily aggregates via Aβ42 self-assembly and thus is not exported by the Tat translocon that recognizes correctly folded proteins. (b) In the presence of small molecules that bind to Aβ42 and prevent aggregation, ssTorA-Aβ42-Bla remains soluble and is efficiently exported to the periplasm where Bla hydrolyzes Amp.
Figure 2.

Amp resistance as an indicator of Aβ42 solubility. Inocula of (a) 1:100 or (b) 1:500 grown in the presence of varying Amp concentrations as indicated. Cells were induced to express ssTorA-Aβ42-Bla (white bars) or ssTorA-GM6-Bla (gray bars). All data was normalized to the growth rate of the 1:100-diluted cells expressing ssTorA-Aβ42-Bla in the absence of Amp. Data represents the average of six replicate experiments and the error bars represent the standard error of the mean (s.e.m.).
As an alternative to using selective pressure to identify aggregation inhibitors, we also explored the development of a fluorescence-based screen that capitalized on our earlier observation that the extent of Aβ42 solubility correlated directly to the quantity and activity of Bla enzyme transported to the periplasm via the Tat system.16 To measure periplasmic Aβ42-Bla activity directly in living cells, we employed the fluorogenic Bla substrate CCF2/AM.18 Intact CCF2/AM is a membrane-permeant ester comprised of two different fluorophores, fluorescein and coumarin, connected via a permissive cephalosporin β-lactam ring. Upon entering cells, host esterases hydrolyze the ester's functional abilities and release the nonpermeant Bla substrate CCF2. Excitation of CCF2 with 409 nm light in the absence of Bla induces FRET within CCF2 and 520 nm light (green) is emitted. After cleavage by Bla, the acceptor is separated from the donor and emission is at 447 nm (blue). The CCF2/AM assay was originally limited to eukaryotic cells because bacteria such as E. coli lacked the esterase activity necessary for activation of the esterified and membrane-permeable CCF2/AM form of the substrate. However, when E. coli cells were supplemented with this activity via periplasmic expression of a fungal lipase known as cutinase, periplasmic Bla could be readily detected using CCF2/AM.19 Indeed, we found that cells expressing Fusarium solani pisi cutinase in the periplasm seemed bright green when incubated with CCF2/AM [Fig. 3(a)], indicating that the substrate had been activated inside cells and remained uncleaved. This result also confirmed that the CCF2/AM substrate was capable of crossing the outer membrane and that after activation by cutinase it was effectively trapped inside cells owing to its negative charge. When cutinase and Bla were both colocalized to the periplasm via the addition of N-terminal Sec signal peptides on each, cells emitted strong blue fluorescence [Fig. 3(b)]. This observation indicated that in these cells the CCF2/AM substrate was activated by periplasmic cutinase and subsequently cleaved by periplasmic Bla, thereby resulting in dominant coumarin donor fluorescence. Thus, heterologous expression of cutinase allowed for facile fluorescent detection of Bla in the periplasm of living E. coli cells. Importantly, when periplasmic cutinase was coexpressed with versions of Bla that were not exported (i.e., due to absence of an export signal), cells emitted green fluorescence (see, for instance, Table I) indicating that a positive signal required colocalization of both cutinase and Bla to the periplasm. This is significant because it shows that there is no measurable false-positive signal arising from a reaction between nonlocalized Bla and CCF2 in the cytoplasm. We speculate that the reason for this is that CCF2 becomes negatively charged in the periplasm due to cutinase activity and thus is incapable of diffusing across the negatively charged inner membrane.
Figure 3.
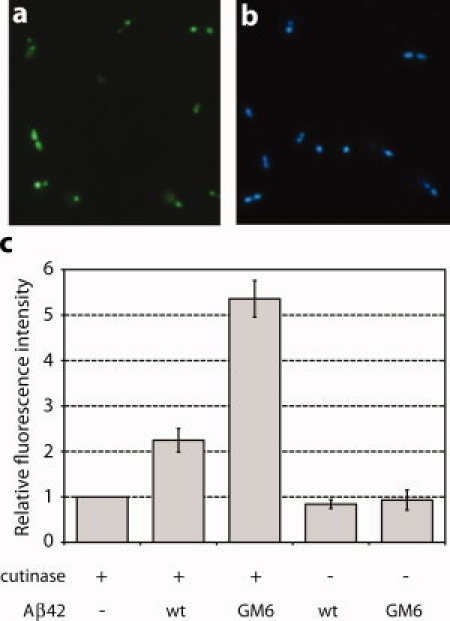
Fluorescence-based detection of Aβ42 aggregation in living E. coli cells using CCF2/AM. (a) Negative control cells expressing Sec-targeted cutinase (ssPhoA-CutA) from pBKPC and (b) positive control cells coexpressing ssPhoA-CutA and TEM-1 Bla with its native Sec signal peptide both from pBPC. Cells were incubated with 1 μM CCF2/AM for 1 h with gentle rocking and visualized by fluorescence microscopy (ex: 409 nm). (c) CCF2 fluorescence for cells expressing ssTorA-Aβ42-Bla or ssTorA-GM6-Bla in the presence (+) or absence (−) of cutinase as indicated. CCF2 fluorescence readings were normalized by dividing fluorescence values measured at 447 nm by those measured at 520 nm, and then normalized to OD600 of the cells. All data was then normalized to the background fluorescence measured in cells expressing cutinase only. Data represents the average of three replicate experiments and the error bars represent the s.e.m.
Table I.
96-Well Plate Assays of Aβ42 Solubility
| Reporter construct | FI/ODa | SDb | CV (%)c | S-to-Bd | SD above meane |
|---|---|---|---|---|---|
| ssTorA-Aβ42-Bla | 1111 | 55 | 5.0 | 1.0 | — |
| ssTorA-GM6-Bla | 2566 | 154 | 6.0 | 2.3 | 9.4 |
| Aβ42-Bla | 1144 | 62 | 5.4 | 1.0 | 0.5 |
| GM6-Bla | 1198 | 67 | 5.6 | 1.1 | 1.3 |
Specific fluorescence activity calculated as the fluorescence intensity (FI) divided by the optical density (OD) (average of 96 readings).
Standard deviation (SD) from measurements across an entire plate.
Coefficient of variance (CV) calculated as the (SD/FI/OD)*100 across an entire plate.
Signal-to-background (S-to-B) calculated by normalizing signals to the FI/OD for ssTorA-Aβ42-Bla negative control.
Number of standard deviations above the average of the ssTorA-Aβ42-Bla negative control.
To determine if CCF2/AM hydrolysis was suitable for detecting Aβ42 solubility and transport, we transformed cells with (i) plasmid pBKPC expressing Sec-targeted cutinase (ssPhoA-CutA) from an arabinose-inducible promoter and (ii) an Aβ42 reporter plasmid encoding either ssTorA-Aβ42-Bla or ssTorA-GM6-Bla. After incubation with CCF2/AM, cells were analyzed in a fluorescence microplate reader (excitation 409 nm; emission 447 nm or 520 nm). Cells expressing cutinase alone emitted a weak blue signal that was just above background [Fig. 3(c)]. In contrast, cells coexpressing cutinase and the efficiently exported ssTorA-GM6-Bla emitted a high level of blue fluorescence that was >12-fold above that observed for cutinase-only control cells and, more importantly, nearly 3-fold more fluorescent than cells expressing the export-incompetent ssTorA-Aβ42-Bla fusion [Fig. 3(c)]. These fluorescent signals were dependent upon cutinase expression as no signal above background was observed for either wt or GM6 versions of Aβ42 expressed in the absence of cutinase [Fig. 3(c)]. Likewise, no signal above background was observed when the cutinase and Aβ42 constructs were coexpressed in ΔtatC cell lacking a functional Tat system (data not shown), indicating that the fluorescent phenotype was entirely dependent on the Tat export system. Collectively, these results indicate that heterologous expression of cutinase allows for facile and quantitative fluorescent discrimination between the soluble ssTorA-GM6-Bla fusion and its aggregation-prone counterpart ssTorA-Aβ42-Bla in living E. coli cells using the cell membrane-permeable substrate CCF2/AM.
Statistical evaluation of Tat-based HTS screen
Next, we sought to demonstrate the robustness and reproducibility of the fluorescent version of our Aβ42 solubility assay in 96-well plate format. For these studies, 96-well plates containing 100 μL of growth medium (M9 minimal medium supplemented with casamino acids and 0.4% arabinose; chosen due to its extremely low fluorescence background) were inoculated with log-phase E. coli cells carrying plasmids for the expression of cutinase and one of the following: (i) ssTorA-Aβ42-Bla; (ii) ssTorA-GM6-Bla; (iii) Aβ42-Bla or (iv) GM6-Bla (the latter two constructs each lacked a Tat signal peptide and thus served as nonexported negative controls). After 18 h of growth and induction of protein expression at 30°C, CCF2/AM was added to each well and incubated for 1 h. Using a fluorescence microplate reader, the fluorescence intensity (FI; ex: 409 nm; em: 447 nm, 520 nm) and cell density (OD; absorbance at 600 nm) in each well were measured. For each of the four Bla fusions, measurements across an entire 96-well plate were made (Table I). Consistent with data in Figure 3, we observed a near 2.5-fold increase in signal for ssTorA-GM6-Bla versus ssTorA-Aβ42-Bla. Importantly, the signal for the positive control ssTorA-GM6-Bla construct (2566 average, 154 standard deviation) was more than nine standard deviations above the average for the ssTorA-Aβ42-Bla negative control (1111 average, 55 standard deviation). Two additional negative controls, Aβ42-Bla and GM6-Bla, yielded signals that were virtually indistinguishable from ssTorA-Aβ42-Bla. It is noteworthy that the coefficient of variation (CV) for 96 measurements made across an entire plate was ≤6.0% for each construct tested. Additionally, the Z-factor,20 which measures the quality or power of an HTS assay, was 0.6 (a value between 0.5 and 1.0 is indicative of an excellent assay). Finally, between-plate and day-to-day variations for each construct were determined to be ≤7.0% and 9.6%, respectively (data not shown).
A search for inhibitors of Aβ42 aggregration
To test whether our assay was able to isolate small molecules that inhibit Aβ42 aggregation by intermolecular effects, we screened a combinatorial library of ∼1000 compounds derived from the triazine scaffold.21,22 Combinatorial diversity in this library was introduced by varying the substituents at positions R1 and R2 on the triazine scaffold [Fig. 4(a)] and the chemical composition of these substituents is described in our earlier work.21,22 We chose this triazine library because: (1) previous studies by Hecht and coworkers showed that aggregation inhibitors already exist in this library23; and (2) compound RS-0466, which was shown by Selkoe and coworkers to block Aβ oligomerization and rescue long-term potentiation, is a triazine derivative.24
Figure 4.

Isolation of triazine derivatives that inhibit Aβ42 aggregation in E. coli. (a) Triazine scaffold where R1 and R2 substituents were varied to generate a combinatorial compound library. (b) R1 and R2 substituents for the four compounds identified as hits using the Aβ42 HTS CCF2/AM assay. (c) Compound 5B2 is structurally similar to positive hit 3B7 at the R1 position but scored as an inactive compound in our cell-based screen.
To implement our screen, we added E. coli cells coexpressing cutinase and ssTorA-Aβ42-Bla to 96-well plates containing growth medium supplemented with inducer and 5 μM of each candidate molecule from the library of triazine derivatives in dimethyl sulfoxide (DMSO). After 18 h of growth and induction of protein expression, CCF2/AM was added to each well and the fluorescence and cell density of each well was measured with a fluorescence microplate reader. In parallel, each compound was screened in control plates containing E. coli cells expressing Aβ42-Bla (lacking the Tat signal sequence). The purpose of this secondary screen was to rule out false positives that might arise due to factors such as: (i) high intrinsic fluorescence of inactive compounds; (ii) nonspecific Bla localization to the periplasm; and/or (iii) release of Bla into the medium caused by compound-induced cell lysis. Importantly, several wells containing triazine derivatives fluoresced at levels that were significantly above background (>three standard deviations above the average signal measured for the ssTorA-Aβ42-Bla negative control with blank DMSO added) for ssTorA-Aβ42-Bla expression but not for Aβ42-Bla. Each of the ∼1000 compounds was tested in quadruplicate and the identification of “hits” was consistent across the four replicates. We consider four of these fluorescent hits, namely compounds 1E4, 3B7, 3G7, and 9D10, to be putative inhibitors of Aβ42 aggregation [Fig. 4(b)]. It is noteworthy that compound 3E2, previously identified by Hecht and coworkers using a bacterial screen based on Aβ42 fused to the green fluorescent protein,23 was scored as a positive hit in some of our plates (three out of four replicates) but was not as strong a hit as the four compounds shown in Figure 4 (each was identified in four out of four replicates). It seems that at the low concentration used here for screening (5 μ)M versus 30–100 μM in,23 3E2 is not as effective as the other lead compounds. It should also be noted that all library compounds were diluted into plates from a 10 mM DMSO stock solution. The use of higher concentrations of our hit molecules (and thus higher DMSO levels in each well) did not seem to have any adverse effects on the cell-based assay. Specifically, we observed a dose-dependent increase in signal up to 30 μM of each compound and the signal remained constant over a concentration range of 30–100 μM for each compound (data not shown). Also interesting is the observation that our HTS assay was capable of effectively discriminating between hits and inactive candidates, for example 5B2, which is identical to 3B7 at position R1 and differs only at position R2 [Fig. 4(c)]. Thus, in the future, it might be possible to screen collections of molecules that only differ at one position as a means to establish the structure/activity relationship between the substituents at each position and the resulting level of aggregation inhibition. Additionally, all of the putative hits were cross-checked for their ability to rescue growth of ssTorA-Aβ42-Bla-expressing cells and each was found to confer a 6–8-fold increase in Amp resistance relative to control cells treated with blank DMSO. Recall that the solubility-enhanced ssTorA-GM6-Bla fusion conferred a ∼12–16-fold increase in Amp resistance relative to control cells (see Fig. 2 above); thus, it seems that both the fluorescence-and viability-based assays are capable of discriminating active and inactive compounds.
Confirmation of hits as Aβ42 aggregation inhibitors
To verify whether our isolated compounds were bona fide inhibitors of Aβ42 aggregation, we monitored the effect of each compound on the aggregation behavior of synthetic Aβ42. It has been established that Aβ42 peptide can be rendered soluble and monomeric After filtration, addition of organic solvents and sonication.25 Dilution of these soluble, monomeric Aβ42 peptides into aqueous buffer results in peptide aggregation and the formation of amyloid fibrils, which can be readily assayed by the binding and subsequent fluorescence of ThT.26 We tested each of our compounds with shaking where the rate of Aβ42 aggregation is faster.23 Specifically, 20 μM of synthetic Aβ42 was incubated in the presence of varying concentrations of each compound (or blank DMSO) for 2.5 h and the resulting fibril formation was measured by the change in ThT fluorescence that results from fibril binding. All four compounds seemed to inhibit Aβ42 in a concentration-dependent manner, similar to the known Aβ42 inhibitor tannic acid (see Fig. 5). Compounds 3B7 and 3G7 exhibited the most potent inhibition with IC50 values between ∼25 and 50 μM. These compounds caused a ∼70–80% reduction in ThT fluorescence at a concentration of 50 μM (see Fig. 5) and 90% reduction at concentrations of 100 μM or greater (data not shown). In contrast, control compound 5B2, which was scored as inactive during the HTS process, did not show any significant inhibition activity (see Fig. 5).
Figure 5.
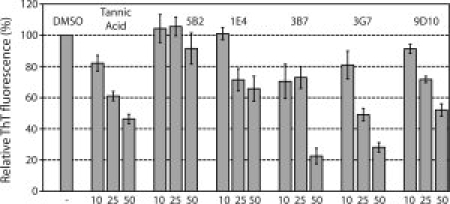
In vitro characterization of aggregation inhibition using synthetic Aβ42. ThT fluorescence of synthetic Aβ42 fibrils after incubation of 20 μM Aβ42 with 10, 25, or 50 μM compounds as indicated. Tannic acid, a known inhibitor of Aβ42 aggregation, served as a positive control. 5B2 was structurally similar to our triazine hits but did not score as a hit in our screen and thus served as a negative control. 1E4, 3B7, 3G7, and 9D10 were compounds identified as Aβ42 aggregation inhibitors in the cell-based CCF2/AM screen of ∼1000 triazine derivatives. ThT fluorescence of Aβ42 incubated with triazine derivatives was expressed as a percentage of the DMSO-only negative control (note that the compounds were all dissolved in DMSO). Data represents the average of three replicate experiments and the error bars represent the s.e.m.
Discussion
We performed a high-throughput screen of a library containing ∼1000 small molecules derived from triazine and identified four lead compounds that promoted efficient Tat-dependent export of ssTorA-Aβ42-Bla, a proven reporter of Aβ42 aggregation.16 This is significant because there are currently only four mildly effective drugs for the treatment of Alzheimer's patients and none of these addresses the underlying molecular cause of AD. Our lead compounds 1E4, 3B7, 3G7, and 9D10 were identified as Aβ42 aggregation inhibitors using a novel fluorescence-based HTS screen enabled by the substrate CCF2/AM. Each compound was cross-checked with the absorbance-based version of our folding assay, then counter-screened in cells that lacked a functional Tat system or with a Aβ42-Bla reporter that lacked an export signal and finally confirmed as bona fide inhibitors of Aβ42 aggregation using an in vitro ThT assay. Compounds 3B7 and 3G7 showed the strongest inhibition (IC50 of ∼25–50 μM) whereas the compound 5B2, which has the same R1 group as 3B7, showed no inhibitory effect on Aβ42 aggregation. The IC50 values found here for 3B7 and 3G7 are comparable to that reported for the triazine derivative 3E2 (∼30 μM) by Hecht and coworkers.23 Our compounds also compared favorably to previously reported inhibitors. In particular, IC50 values for compounds assayed in a manner similar to our analysis (i.e., inhibition of Aβ42 fibril formation using the ThT assay) ranged between 0.1 and 50 μM and included: curcumin (1.1 μM) and its analog rosmarinic acid (1.1 μM),27,28 ferulic acid (5.5 μM), rifampicin (9.1 μM) and tetracycline (10 μM),29 tannic acid (0.1 μM), myricetin (0.43 μM) and nordihydroguaiaretic acid (0.87 μM),30 bis-styrylpyridine and bis-styrylbenzene derivatives (0.1–2.7 μM),31 nicotine (50 μM)32 and N,N'-bis(3-hydroxyphenyl)pyridazine-3,6-diamine (50–100 μM).33 Of all these compounds, tannic acid seems to be one of the best Aβ42 inhibitors with a reported IC50 of ∼0.1 μM.30 It is noteworthy, however, that there appears to be a fair amount of lab-to-lab discrepancy between reported values. For instance, whereas30 report a value of ∼0.1 μM for tannic acid,23 reported an IC50 value for tannic acid (∼25–50 μM) that was similar to the value we measured. This is likely caused by differences in experimental conditions such as incubation time, incubation buffer and source of synthetic Aβ42 between the different reports. Importantly, Kim et al. reported that at concentrations of 25 and 50 μM, the inhibitory effect of their best compound, E2, was similar to or better than tannic acid or another known inhibitor dopamine. Likewise, we observed that our best compounds could inhibit Aβ42 aggregation on par with tannic acid. Thus, while there may be disagreement over the absolute IC50 value for tannic acid, it is clear that the triazine compounds isolated here and by Kim et al. are relatively potent Aβ42 aggregation inhibitors.
It is noteworthy that 5B2 and 3B7 each have the same R1 position side group but differ in their R2 moiety: in 5B2 it is aliphatic whereas in 3B7 it is cyclic. This finding suggests that, like the known Aβ42 aggregation inhibitor tannic acid, steric bulk may be important in blocking Aβ42 aggregation. The other compounds identified in this study all have cyclic moieties in their side groups. We note that compound 3E2,23 shown here to be a slightly less efficient inhibitor in the context of our cell-based screen, contains aliphatic side groups. This would suggest that the binding of Aβ42 by these different triazine derivatives may be site-specific, although at present we do not have any mechanistic data regarding the interactions between Aβ42 and any of the isolated compounds.
A key advantage of our approach is that isolation of these compounds did not require synthetic Aβ42; instead, biosynthesis of Aβ42 was carried out by E. coli cells thereby making this an extremely cost-effective strategy compared to earlier screening studies that were performed in vitro using synthetic Aβ42 peptides.9,10,12 Another advantage of our cell-based approach is its inherent ability to select for membrane-permeant, noncytotoxic compounds. This seems to be rather important in light of emerging evidence that intraneuronal formation of soluble, low-molecular-weight oligomers of Aβ42 is the pathogenically critical process in AD.15,34–39 In fact, one recent report implicates soluble Aβ42 dimers as the smallest synaptotoxic species.40 Thus, compounds that can efficiently cross cell membranes and inhibit the earliest stages of intracellular Aβ42 aggregation may prove to be the most effective therapeutic direction for the treatment of AD. Although we do not currently know the export-incompetent conformation (low-n oligomers (e.g., dimers, trimers, and tetramers), dodecamers, etc.) of Aβ42 in our assay, inhibitors that promote export are highly likely to render ssTorA-Aβ42-Bla monomeric or, at most dimeric, because assemblies larger than this are likely to be incapable of passage through the Tat translocation pore.41 Thus, we suspect that our assay is inherently capable of identifying inhibitors of the earliest stages of Aβ42 aggregation, a feat that is difficult with HTS assays that are dependent upon assembly of Aβ into amyloid (e.g., ThT assay).
To date, only a handful of bacterial HTS assays have been used to screen compound libraries against nonbacterial targets but each of these has successfully uncovered interesting lead molecules including estrogen receptor modulators,42 HIV protease inhibitors43,44 and, akin to our studies here, Aβ42 aggregation inhibitors.23 The use of bacteria for HTS studies is fast, inexpensive and reliable; however, one possible limitation is the potential difficulty of getting diverse compounds into cells. As evidence that permeability is not necessarily a limiting factor, other groups have used E. coli-based screening assays to isolate lead compounds from structurally diverse libraries.42–44 Along similar lines, Hecht and coworkers successfully employed wt E. coli cells expressing an Aβ42-GFP reporter to isolate an aggregation inhibitor from a library of triazine derivatives that was nearly identical to the library screened here.23 It should be noted that their hit compound E2 was also a positive hit in our study, although at the lower screening doses used here it was not as effective as the four inhibitors that we characterized here. Regardless, the ability of their assay and ours to isolate inhibitors would suggest that the triazine scaffold is privileged in its ability to cross biological barriers and/or interfere with intracellular Aβ42 aggregation. Indeed, our own sampling of known Aβ42 aggregation inhibitors that were structurally unrelated to triazine, such as ferulic acid, curcumin, nordihydroguaiaretic acid, tannic acid, nicotine, proline, and tramiposate revealed that none of these promoted export of ssTorA-Aβ42-Bla in E. coli (data not shown). While the reason for this is currently unknown, there are several possible explanations: (i) the outer membrane of may be impermeable to these compounds; (ii) these compounds may be metabolized by E. coli cells or actively effluxed from cells; and/or (iii) the stage at which the compound inhibits Aβ42 aggregation may not be compatible with inhibition of ssTorA-Aβ42-Bla aggregation. To improve screening in the future, we are currently investigating the use of different mutant strains (i.e., with increased outer membrane permeability or decreased efflux activity) or the application of a periplasmic folding assay for Aβ42 aggregation (Mansell, Fisher and DeLisa, manuscript in preparation) since penetration of compounds to the periplasm is reportedly less restricted than to the cytoplasm.45–47 Finally, with the recent development of two eukaryotic cell-based assays for Aβ42 aggregation including an Aβ42-GFP assay in yeast48 and an amyloid precursor protein expressing CHO cell line that secretes SDS-stable Abeta oligomers,24 it will be of great interest to see whether compounds isolated using E. coli-based assays are reliable inhibitors in these other host strains.
Materials and Methods
Bacterial strains and plasmids
E. coli strain BLR(DE3) (Novagen; F-ompT hsdSB (rB- mB-) gal dcm (DE3) Δ(srl-recA)306::Tn10 (TetR)) was routinely used in these studies, although a BW25113 tolC− strain from the Keio collection49 was also used. The plasmids pSALect-Aβ42 and pSALect-GM6 for expressing ssTorA-Aβ42-Bla or ssTorA-GM6-Bla, respectively, have been described previously.16 The deletion of the ssTorA signal peptide from pSALect-Aβ42 and pSALect-GM6 was carried out using QuikChange Site-Directed Mutagenesis Kit (Stratagene), yielding plasmids pSALect(ΔssTorA)-Aβ42 and pSALect(ΔssTorA)-GM6. For the CCF2 assays described below, plasmid pBKPC was constructed by cloning the Fusarium solani pisi cutinase gene (kindly provided by K. Griswold and G. Georgiou) immediately after DNA encoding the Sec-specific signal peptide from E. coli alkaline phosphatase (ssPhoA) in pBAD18-Kan.50 Plasmid maintenance was with 25 μg/mL chlorampenicol (Cam) for pSALect derivatives and 50 μg/mL kanamycin (Kan) for pBKPC. Initial CCF2/AM validation experiments were carried out using pBAD18-ssPhoA-CutA (pBPC) that is AmpR and expressed both cutinase and Bla via the Sec pathway.
CCF2/AM assay
Cells grown overnight in Luria-Bertani broth (LB) supplemented with Cam were diluted 1:100 and grown for 3 h at 37°C. These exponential phase cultures were diluted 1:100 in 96-well black, clear-bottom plates (Corning Costar) containing M9 minimal medium supplemented with 0.5% casamino acids, 0.4% arabinose as sole carbon source and inducer of cutinase expression, 1 mM IPTG, 25 μg/mL Cam, and 50 μg/mL Kan. Cells were incubated at 30°C under quiescent conditions for 18 h. CCF2/AM (Invitrogen) was added to the cultures at 1 μM final concentration and gently mixed by rocking at room temperature for 1 h. CCF2 was excited at 409 nm and emission recorded at 449 nm and 520 nm on a Gemini EM microplate spectrofluorometer (Molecular Devices). Optical density (OD) of cultures was read at 600 nm using a Bio-Tek Synergy HT microplate reader (Bio-Tek Instruments). Fluorescence readings were normalized by dividing fluorescence values measured at 447 nm by those measured at 520 nm, and then normalized to OD600. Five micromolar of each triazine derivative was added when required. Because the compounds were diluted in DMSO, DMSO was added to all cultures at 0.3% final concentration. Cells analyzed by microscopy were cultured identically to the protocol given above for the 96-well plate assay using CCF2/AM. After 1 h incubation with CCF2/AM, 5 μL of cells were removed from the plate and directly applied to a microscope slide for visualization. Fluorescence microscopy was performed on an Axioskop 40 microscope (Zeiss) with Bla filter set (Chroma Technology).
Growth selection assay
Cells grown overnight in LB supplemented with Cam were diluted 1:100 and grown for 3 h at 37°C. These exponential phase cultures were diluted 1:500 in 96-well plates (Corning Costar) containing LB supplemented with 25 μg/mL Cam, 1 mM IPTG, 50 μg/mL Amp selection and, when required, 30 μM triazine-derived compounds. The plates were incubated at 37°C for 16 h under quiescent conditions. Culture OD was read at 600 nm. All cultures contained 0.3% DMSO.
ThT assay
ThT assays were performed with synthetic Aβ42 (Anaspec) as described previously23 with shaking at 37°C for 2.5 h. Compounds were added at 10, 25, and 50 μM. ThT fluorescence was measured at 483 nm (450 nm excitation) on a Gemini EM microplate spectrofluorometer. Readings were normalized to the DMSO-only negative control and expressed as percentage relative fluorescence.
Acknowledgments
The authors thank M. Hecht and A. Fortner (Princeton University) for assistance with the synthetic Aβ42 assays.
References
- 1.Selkoe DJ, Schenk D. Alzheimer's disease: molecular understanding predicts amyloid-based therapeutics. Annu Rev Pharmacol Toxicol. 2003;43:545–584. doi: 10.1146/annurev.pharmtox.43.100901.140248. [DOI] [PubMed] [Google Scholar]
- 2.Hardy JA, Higgins GA. Alzheimer's disease: the amyloid cascade hypothesis. Science. 1992;256:184–185. doi: 10.1126/science.1566067. [DOI] [PubMed] [Google Scholar]
- 3.Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science. 2002;297:353–356. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- 4.Lambert MP, Barlow AK, Chromy BA, Edwards C, Freed R, Liosatos M, Morgan TE, Rozovsky I, Trommer B, Viola KL, Wals P, Zhang C, Finch CE, Krafft GA, Klein WL. Diffusible, nonfibrillar ligands derived from Abeta1-42 are potent central nervous system neurotoxins. Proc Natl Acad Sci USA. 1998;95:6448–6453. doi: 10.1073/pnas.95.11.6448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Aisen PS. The development of anti-amyloid therapy for Alzheimer's disease: from secretase modulators to polymerisation inhibitors. CNS Drugs. 2005;19:989–996. doi: 10.2165/00023210-200519120-00002. [DOI] [PubMed] [Google Scholar]
- 6.Aisen PS, Gauthier S, Vellas B, Briand R, Saumier D, Laurin J, Garceau D. Alzhemed: a potential treatment for Alzheimer's disease. Curr Alzheimer Res. 2007;4:473–478. doi: 10.2174/156720507781788882. [DOI] [PubMed] [Google Scholar]
- 7.Barten DM, Albright CF. Therapeutic strategies for Alzheimer's Disease. Mol Neurobiol. 2008;37:171–186. doi: 10.1007/s12035-008-8031-2. [DOI] [PubMed] [Google Scholar]
- 8.Wolfe MS. Gamma-secretase inhibition and modulation for Alzheimer's disease. Curr Alzheimer Res. 2008;5:158–164. doi: 10.2174/156720508783954767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wood SJ, MacKenzie L, Maleeff B, Hurle MR, Wetzel R. Selective inhibition of Abeta fibril formation. J Biol Chem. 1996;271:4086–4092. doi: 10.1074/jbc.271.8.4086. [DOI] [PubMed] [Google Scholar]
- 10.Esler WP, Stimson ER, Ghilardi JR, Felix AM, Lu YA, Vinters HV, Mantyh PW, Maggio JE. Abeta deposition inhibitor screen using synthetic amyloid. Nat Biotechnol. 1997;15:258–263. doi: 10.1038/nbt0397-258. [DOI] [PubMed] [Google Scholar]
- 11.Lansbury PT., Jr Inhibition of amyloid formation: a strategy to delay the onset of Alzheimer's disease. Curr Opin Chem Biol. 1997;1:260–267. doi: 10.1016/s1367-5931(97)80018-x. [DOI] [PubMed] [Google Scholar]
- 12.Blanchard BJ, Chen A, Rozeboom LM, Stafford KA, Weigele P, Ingram VM. Efficient reversal of Alzheimer's disease fibril formation and elimination of neurotoxicity by a small molecule. Proc Natl Acad Sci USA. 2004;101:14326–14332. doi: 10.1073/pnas.0405941101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hartley DM, Walsh DM, Ye CP, Diehl T, Vasquez S, Vassilev PM, Teplow DB, Selkoe DJ. Protofibrillar intermediates of amyloid beta-protein induce acute electrophysiological changes and progressive neurotoxicity in cortical neurons. J Neurosci. 1999;19:8876–8884. doi: 10.1523/JNEUROSCI.19-20-08876.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Walsh DM, Klyubin I, Fadeeva JV, Cullen WK, Anwyl R, Wolfe MS, Rowan MJ, Selkoe DJ. Naturally secreted oligomers of amyloid beta protein potently inhibit hippocampal long-term potentiation in vivo. Nature. 2002;416:535–539. doi: 10.1038/416535a. [DOI] [PubMed] [Google Scholar]
- 15.Walsh DM, Selkoe DJ. Oligomers on the brathe emerging role of soluble protein aggregates in neurodegeneration. Protein Pept Lett. 2004;11:213–228. doi: 10.2174/0929866043407174. [DOI] [PubMed] [Google Scholar]
- 16.Fisher AC, Kim W, DeLisa MP. Genetic selection for protein solubility enabled by the folding quality control feature of the twin-arginine translocation pathway. Protein Sci. 2006;15:449–458. doi: 10.1110/ps.051902606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wurth C, Guimard NK, Hecht MH. Mutations that reduce aggregation of the Alzheimer's Abeta42 peptide: an unbiased search for the sequence determinants of Abeta amyloidogenesis. J Mol Biol. 2002;319:1279–1290. doi: 10.1016/S0022-2836(02)00399-6. [DOI] [PubMed] [Google Scholar]
- 18.Zlokarnik G, Negulescu PA, Knapp TE, Mere L, Burres N, Feng L, Whitney M, Roemer K, Tsien RY. Quantitation of transcription and clonal selection of single living cells with beta-lactamase as reporter. Science. 1998;279:84–88. doi: 10.1126/science.279.5347.84. [DOI] [PubMed] [Google Scholar]
- 19.Nord O, Gustrin A, Nygren PA. Fluorescent detection of beta-lactamase activity in living Escherichia coli cells via esterase supplementation. FEMS Microbiol Lett. 2005;242:73–79. doi: 10.1016/j.femsle.2004.10.047. [DOI] [PubMed] [Google Scholar]
- 20.Zhang JH, Chung TD, Oldenburg KR. A simple statistical parameter for use in evaluation and validation of high throughput screening assays. J Biomol Screen. 1999;4:67–73. doi: 10.1177/108705719900400206. [DOI] [PubMed] [Google Scholar]
- 21.Bork JT, Lee J, Chang YT. Palladium-catalyzed cross-coupling reaction of resin bound chlorotriazines. Tetrahedron Lett. 2003;44:6141–6144. [Google Scholar]
- 22.Khersonsky SM, Jung DW, Kang TW, Walsh DP, Moon HS, Jo H, Jacobson EM, Shetty V, Neubert TA. Chang YT. Facilitated forward chemical genetics using a tagged triazine library and zebrafish embryo screening. J Am Chem Soc. 2003;125:11804–11805. doi: 10.1021/ja035334d. [DOI] [PubMed] [Google Scholar]
- 23.Kim W, Kim Y, Min J, Kim DJ, Chang YT, Hecht MH. A high-throughput screen for compounds that inhibit aggregation of the Alzheimer's peptide. ACS Chem Biol. 2006;1:461–469. doi: 10.1021/cb600135w. [DOI] [PubMed] [Google Scholar]
- 24.Walsh DM, Townsend M, Podlisny MB, Shankar GM, Fadeeva JV, El Agnaf O, Hartley DM, Selkoe DJ. Certain inhibitors of synthetic amyloid beta-peptide (Abeta) fibrillogenesis block oligomerization of natural Abeta and thereby rescue long-term potentiation. J Neurosci. 2005;25:2455–2462. doi: 10.1523/JNEUROSCI.4391-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bitan G, Teplow DB. Preparation of aggregate-free, low molecular weight amyloid-beta for assembly and toxicity assays. Methods Mol Biol. 2005;299:3–9. doi: 10.1385/1-59259-874-9:003. [DOI] [PubMed] [Google Scholar]
- 26.LeVine H., III Thioflavine T interaction with synthetic Alzheimer's disease beta-amyloid peptides: detection of amyloid aggregation in solution. Protein Sci. 1993;2:404–410. doi: 10.1002/pro.5560020312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ono K, Hasegawa K, Naiki H, Yamada M. Curcumin has potent anti-amyloidogenic effects for Alzheimer's beta-amyloid fibrils in vitro. J Neurosci Res. 2004;75:742–750. doi: 10.1002/jnr.20025. [DOI] [PubMed] [Google Scholar]
- 28.Yang F, Lim GP, Begum AN, Ubeda OJ, Simmons MR, Ambegaokar SS, Chen PP, Kayed R, Glabe CG, Frautschy SA, Cole GM. Curcumin inhibits formation of amyloid beta oligomers and fibrils, binds plaques, and reduces amyloid in vivo. J Biol Chem. 2005;280:5892–5901. doi: 10.1074/jbc.M404751200. [DOI] [PubMed] [Google Scholar]
- 29.Ono K, Hirohata M, Yamada M. Ferulic acid destabilizes preformed beta-amyloid fibrils in vitro. Biochem Biophys Res Commun. 2005;336:444–449. doi: 10.1016/j.bbrc.2005.08.148. [DOI] [PubMed] [Google Scholar]
- 30.Ono K, Hasegawa K, Naiki H, Yamada M. Anti-amyloidogenic activity of tannic acid and its activity to destabilize Alzheimer's beta-amyloid fibrils in vitro. Biochim Biophys Acta. 2004;1690:193–202. doi: 10.1016/j.bbadis.2004.06.008. [DOI] [PubMed] [Google Scholar]
- 31.Byeon SR, Lee JH, Sohn JH, Kim DC, Shin KJ, Yoo KH, Mook-Jung I, Lee WK, Kim DJ. Bis-styrylpyridine and bis-styrylbenzene derivatives as inhibitors for Abeta fibril formation. Bioorg Med Chem Lett. 2007;17:1466–1470. doi: 10.1016/j.bmcl.2006.10.090. [DOI] [PubMed] [Google Scholar]
- 32.Moore SA, Huckerby TN, Gibson GL, Fullwood NJ, Turnbull S, Tabner BJ, El-Agnaf OM, Allsop D. Both the D-(+) and L-(−) enantiomers of nicotine inhibit Abeta aggregation and cytotoxicity. Biochemistry. 2004;43:819–826. doi: 10.1021/bi035728h. [DOI] [PubMed] [Google Scholar]
- 33.Nakagami Y, Nishimura S, Murasugi T, Kaneko I, Meguro M, Marumoto S, Kogen H, Koyama K, Oda T. A novel beta-sheet breaker, RS-0406, reverses amyloid beta-induced cytotoxicity and impairment of long-term potentiation in vitro. Br J Pharmacol. 2002;137:676–682. doi: 10.1038/sj.bjp.0704911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gouras GK, Tsai J, Naslund J, Vincent B, Edgar M, Checler F, Greenfield JP, Haroutunian V, Buxbaum JD, Xu H, Greengard P, Relkin NR. Intraneuronal Abeta42 accumulation in human brain. Am J Pathol. 2000;156:15–20. doi: 10.1016/s0002-9440(10)64700-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Walsh DM, Tseng BP, Rydel RE, Podlisny MB, Selkoe DJ. The oligomerization of amyloid beta-protein begins intracellularly in cells derived from human brain. Biochemistry. 2000;39:10831–10839. doi: 10.1021/bi001048s. [DOI] [PubMed] [Google Scholar]
- 36.D'Andrea MR, Nagele RG, Wang HY, Peterson PA, Lee DH. Evidence that neurones accumulating amyloid can undergo lysis to form amyloid plaques in Alzheimer's disease. Histopathology. 2001;38:120–134. doi: 10.1046/j.1365-2559.2001.01082.x. [DOI] [PubMed] [Google Scholar]
- 37.Echeverria V, Cuello AC. Intracellular A-beta amyloid, a sign for worse things to come? Mol Neurobiol. 2002;26:299–316. doi: 10.1385/MN:26:2-3:299. [DOI] [PubMed] [Google Scholar]
- 38.Tabira T, Chui DH, Kuroda S. Significance of intracellular Abeta42 accumulation in Alzheimer's disease. Front Biosci. 2002;7:a44–a49. doi: 10.2741/tabira. [DOI] [PubMed] [Google Scholar]
- 39.Takahashi RH, Almeida CG, Kearney PF, Yu F, Lin MT, Milner TA, Gouras GK. Oligomerization of Alzheimer's beta-amyloid within processes and synapses of cultured neurons and brain. J Neurosci. 2004;24:3592–3599. doi: 10.1523/JNEUROSCI.5167-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shankar GM, Li S, Mehta TH, Garcia-Munoz A, Shepardson NE, Smith I, Brett FM, Farrell MA, Rowan MJ, Lemere CA, Regan CM, Walsh DM, Sabatini BL, Selkoe DJ. Amyloid-beta protein dimers isolated directly from Alzheimer's brains impair synaptic plasticity and memory. Nat Med. 2008;14:837–842. doi: 10.1038/nm1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gohlke U, Pullan L, McDevitt CA, Porcelli I, de Leeuw E, Palmer T, Saibil HR, Berks BC. The TatA component of the twin-arginine protein transport system forms channel complexes of variable diameter. Proc Natl Acad Sci USA. 2005;102:10482–10486. doi: 10.1073/pnas.0503558102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Skretas G, Meligova AK, Villalonga-Barber C, Mitsiou DJ, Alexis MN, Micha-Screttas M, Steele BR, Screttas CG, Wood DW. Engineered chimeric enzymes as tools for drug discovery: generating reliable bacterial screens for the detection, discovery, and assessment of estrogen receptor modulators. J Am Chem Soc. 2007;129:8443–8457. doi: 10.1021/ja067754j. [DOI] [PubMed] [Google Scholar]
- 43.Melnick L, Yang SS, Rossi R, Zepp C, Heefner D. An Escherichia coli expression assay and screen for human immunodeficiency virus protease variants with decreased susceptibility to indinavir. Antimicrob Agents Chemother. 1998;42:3256–3265. doi: 10.1128/aac.42.12.3256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cheng TJ, Brik A, Wong CH, Kan CC. Model system for high-throughput screening of novel human immunodeficiency virus protease inhibitors in Escherichia coli. Antimicrob Agents Chemother. 2004;48:2437–2447. doi: 10.1128/AAC.48.7.2437-2447.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chen G, Hayhurst A, Thomas JG, Harvey BR, Iverson BL, Georgiou G. Isolation of high-affinity ligand-binding proteins by periplasmic expression with cytometric screening (PECS) Nat Biotechnol. 2001;19:537–542. doi: 10.1038/89281. [DOI] [PubMed] [Google Scholar]
- 46.Sroga GE, Dordick JS. Generation of a broad esterolytic subtilisin using combined molecular evolution and periplasmic expression. Protein Eng. 2001;14:929–937. doi: 10.1093/protein/14.11.929. [DOI] [PubMed] [Google Scholar]
- 47.Sroga GE, Dordick JS. A strategy for in vivo screening of subtilisin E reaction specificity in E. coli periplasm. Biotechnol Bioeng. 2002;78:761–769. doi: 10.1002/bit.10269. [DOI] [PubMed] [Google Scholar]
- 48.Bagriantsev S, Liebman S. Modulation of Abeta42 low-n oligomerization using a novel yeast reporter system. BMC Biol. 2006;4:32. doi: 10.1186/1741-7007-4-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Baba T, Ara T, Hasegawa M, Takai Y, Okumura Y, Baba M, Datsenko KA, Tomita M, Wanner BL, Mori H. Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Mol Syst Biol. 2006;2:2006–0008. doi: 10.1038/msb4100050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Guzman LM, Belin D, Carson MJ, Beckwith J. Tight regulation, modulation, and high-level expression by vectors containing the arabinose PBAD promoter. J Bacteriol. 1995;177:4121–4130. doi: 10.1128/jb.177.14.4121-4130.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]