

Abstract
mPlum is a far-red fluorescent protein with emission maximum at ∼650 nm and was derived by directed evolution from DsRed. Two residues near the chromophore, Glu16 and Ile65, were previously revealed to be indispensable for the far-red emission. Ultrafast time-resolved fluorescence emission studies revealed a time dependent shift in the emission maximum, initially about 625 nm, to about 650 nm over a period of 500 ps. This observation was attributed to rapid reorganization of the residues solvating the chromophore within mPlum. Here, the crystal structure of mPlum is described and compared with those of two blue shifted mutants mPlum-E16Q and-I65L. The results suggest that both the identity and precise orientation of residue 16, which forms a unique hydrogen bond with the chromophore, are required for far-red emission. Both the far-red emission and the time dependent shift in emission maximum are proposed to result from the interaction between the chromophore and Glu16. Our findings suggest that significant red shifts might be achieved in other fluorescent proteins using the strategy that led to the discovery of mPlum.
Keywords: far-red fluorescent proteins, crystallography, hydrogen bond, structural rearrangement, dynamic stokes shift
Introduction
Far-red fluorescent proteins (FPs) hold great promise in whole-body imaging since the principle light absorbers in tissue, that is hemoglobin, water and lipids, have the lowest combined absorption coefficient in the near-infrared region around 650–900 nm (the so-called NIR window).1 So far, three far-red fluorescent proteins have been discovered, all with emission maximum (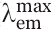 ) of ∼650 nm: HcRed ( = 645 nm),2 aeFP595 AQ143 ( = 655 nm)3 and mPlum ( = 649 nm).4 Of these, only mPlum is monomeric and it also exhibits the highest quantum yield, about 0.10.
) of ∼650 nm: HcRed ( = 645 nm),2 aeFP595 AQ143 ( = 655 nm)3 and mPlum ( = 649 nm).4 Of these, only mPlum is monomeric and it also exhibits the highest quantum yield, about 0.10.
It is generally accepted that the diversity of emission maxima in fluorescent proteins results from a combination of two different mechanisms: chemical modifications to the chromophore structure and influences due to the immediate environment of the chromophore. On the basis of the chemical structures, the existing fluorescent proteins can be divided into two groups. Cyan and green fluorescent proteins contain a chromophore chemically identical to that of GFP5 while yellow, orange, and red emission results from additional modifications to the polypeptide backbone adjacent to a GFP-like chromophore.6–14 In DsRed, the peptide bond immediately preceding the chromophore is oxidized to an acylimine; consequently the conjugation of the chromophore extends over the polypeptide backbone, increasing both the excitation and emission maxima relative to GFP.6,7 zRFP57414 and eqFP611 contain a DsRed-like chromophore although in the latter case, the chromophore adopts a trans conformation instead of the more typical cis conformation.9 In Kaede and EosFP, light-driven backbone cleavage results in extension of the conjugation of a GFP-like chromophore to the imidazole ring of the histidine in the chromophore-forming tripeptide, leading to a green to red shift in emission.8,10,13
Further modifications to the acylimine of a DsRed-like chromophore also occur in several fluorescent proteins. In the “kindling fluorescent protein” (KFP), hydrolysis of the acylimine linkage was proposed, resulting in backbone cleavage and the introduction of a new carbonyl group conjugated with the chromophore.11 In mOrange, the attack of γ-hydroxyl group of Thr66 upon the carbonyl carbon of Phe65 results in a novel five-membered 2-hydroxy dihydrooxazole ring, forming a three-ring chromophore.12 This has been suggested to reduce the conjugation of the Phe65 carbonyl with the rest of the chromophore, thereby accounting for the orange emission. Similar modifications also occur in a natural orange fluorescent protein named mKO (-CYG-chromophore),15 and possibly to CcalOFP1 (-TYG-chromophore).16 Likewise, the yellow fluorescent protein zFP538 ( = 538 nm) undergoes a similar rearrangement to form a tetrahydropyridine ring, due to attack of the Nɛ of Lys 66 on the acylimine α-carbon.17
Changes in the chromophore environment can perturb emission maxima by up to ∼20 nm. An important example is the yellow fluorescent protein YFP (GFP-T203Y variant) in which the side chain of Tyr203 stacks against the phenol moiety of the chromophore, red-shifting both the excitation and emission maxima.18,19 Another example is mCherry, in which the protonated Glu215 carboxyl group forms a hydrogen bond with the imidazolinone ring nitrogen. At pH >10.5, deprotonation of Glu215 is proposed to account for the blue shift in the emission maximum from 609 nm to 594 nm.12
mPlum is a red fluorescent protein (QY = 0.10), created through directed evolution of a monomeric DsRed variant (mRFP1.2) using somatic hypermutation (SHM).4 Saturation mutagenesis of mPlum previously revealed that Glu16 and Ile65 are both important for the far-red emission, as a blue shift of 20–40 nm in the emission maximum results when either side chain is modified.4 It has not been reported whether similar residues are important for the far-red emission in HcRed and aeFP595 AQ143.
Abbyad et al.20 conducted ultrafast time-resolved fluorescence emission studies on mPlum and its progenitors, which revealed a time dependent red shift in the emission maximum, initially about 625 nm, to about 650 nm over a period of 500 ps. As the temperature is lowered from RT both the initial and final emission maxima shift to the blue but the dynamic component is observed at all temperatures above the glass transition. This dynamic Stokes shift, which is absent from the parents DsRed and mRFP, was attributed to rapid reorganization of the residues solvating the chromophore within mPlum. Thus, the red emission of mPlum seems to depend in part on dynamic processes that have so far not been observed or analyzed in previously studied fluorescent proteins.
We initiated crystallographic studies of mPlum in the hopes of determining the structural basis for both the time-independent and time-dependent components of the far-red emission. Here, we report crystal structures of mPlum and its blue shifted variants E16Q and I65L. The results suggest that a unique interaction between the chromophore and Glu16 is responsible for the dynamic Stokes shift.
Results
Temperature-dependent spectroscopy
The spectroscopic properties of mPlum and its variants E16Q, E16L, and I65L were measured at temperatures between room and 77 K. All variants exhibit similar excitation maxima at about 590 nm. However, their emission maxima are substantially different (Table I). At room temperature, mPlum has an emission peak at ∼650 nm, E16Q at 630 nm, I65L at 625 nm, and E16L at 620 nm. Therefore, substitution of Glu16 or Ile65 has a significant effect on the emission, consistent with earlier results.4
Table I.
Spectroscopic Properties of mPlum and Selected Variants
| Protein | Excitation maximum (nm) | Emission maxima (298 K/77 K) |
|---|---|---|
| mPlum | 590 | 649/621 |
| E16Q | 590 | 630/608 |
| E16H | 588 | 619/600 |
| E16L | 588 | 618/598 |
| E16I/H17La | N.A. | 615/N.A. |
| E16V/H17Ta | N.A. | 617/N.A. |
| E16G/H17Ya | N.A. | 626/N.A. |
| E16S/H17Va | N.A. | 626/N.A. |
These values are from Ref. 4. The dependence of emission peak on residue 17 is negligible. N.A., not available.
Upon temperature decrease to 77 K, all variants exhibit a blue shift of ∼20–30 nm in emission maximum, an effect that is significantly larger than seen in GFP (<10 nm blue shift).21 In more detail, mPlum gives the largest blue shift and its emission spectrum at 77 K is unstructured and rather broad, comparable with that observed at room temperature [Fig. 1(A)]. On the other hand, the emission spectrum of E16L is significantly narrowed and appears structured at 77 K [Fig. 1(B)]. The E16Q and I65L spectra at 77 K are intermediate in structure to those of mPlum and E16L at 77 K (data not shown).
Figure 1.
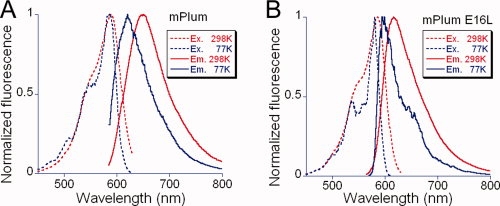
Fluorescence spectra of mPlum and E16L variant. (A) Excitation (dashed lines) and emission (solid lines) spectra of mPlum at room (298 K, red) and low (77 K, blue) temperature. (B) Excitation (dashed) and emission (solid) spectra of mPlum-E16L variant at room (298 K, red) and low (77 K, blue) temperature.
Structural studies
mPlum (a mixture of mature and immature species, separated mature and immature species)20 and two blue shifted mPlum variants E16Q and I65L (purified mature form for each) were crystallized and the structures determined by molecular replacement at various resolutions in the range 1.34–1.9 Å using radiation from synchrotron and home sources. The space group is P212121 in all cases and there are two molecules in the asymmetric unit. The final atomic models have excellent statistics (See supp. info. Supp. Table S1). The final electron density map of mPlum suggests that chain A and B contain an immature and mature chromophore, respectively. When an attempt is made to restrain the geometry of the chromophore in chain A as appropriate for the mature species containing an acylimine, large peaks are revealed adjacent to the chromophore in the (Fo-Fc) difference electron density map (data not shown). We concluded that the asymmetric unit of the mPlum crystal contains a heterodimer of a mature and immature chain. This is consistent with the fact that the physical properties of the mature and immature species are sufficiently different that they can be separated by ion exchange chromatography.20
The final (2Fo-Fc) electron density maps for mPlum (mature species in chain B), E16Q and I65L variants (2) clearly indicate that Met66 Cα adopts sp2 geometry. Therefore, the peptide bond between Ile65 and Met66 is oxidized to an acylimine as found for the parent DsRed6,7,22 and the closely related proteins mCherry and mStrawberry.12 The peptide bond between Ile65 and Met66 is in the cis configuration for all mature species (Fig. 2), with the carbonyl carbon inclined at ∼55° from the approximate plane of the remainder of the chromophore.
Figure 2.
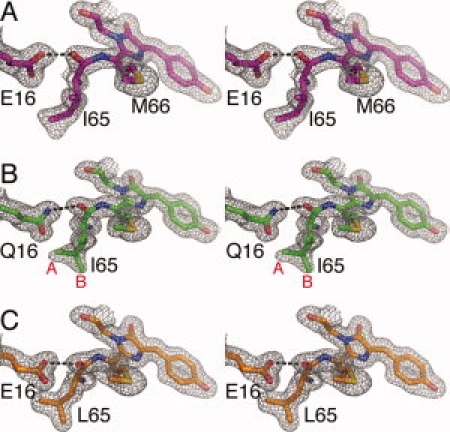
Stereoview of portions of the final (2Fo-Fc) electron density maps. (A) mPlum, (B) E16Q, and (C) I65L variants. Maps are contoured at 1σ level. Atoms of the chromophore are shown in stick representation with nitrogen blue, sulfur yellow, and oxygen red. Note that I65 in panel B has two conformations (A and B).
In mPlum, the chromophore environment (Supp. info. Fig. S1) is substantially more hydrophobic than that of DsRed and is similar to that found in other mFruits.12 The increased hydrophobicity results from replacement of the internal polar residues Lys83 and Lys163 with bulky nonpolar groups, and the possible protonation of Glu215 (see later). Compared with DsRed, the K163M mutation leads to the loss of the hydrogen bond and electrostatic interaction between Lys163 and chromophore phenol oxygen, which presumably results in the charge redistribution of the chromophore.
A substantial shift in the position of Lys70 (3.5 Å) takes place in mPlum compared with DsRed [Fig. 3(A)], which results in the loss of a salt bridge between Lys70 and Glu215 observed in DsRed. This shift can be ascribed to the K83L mutation in mPlum, which eliminates the potential repulsive electrostatic interaction between Lys70 and Lys83 found in DsRed. The new conformation of Lys70 is stabilized through hydrogen bonding and attractive electrostatic interactions with Glu148 (Supp. info. Fig. S1). A similar conformational change of Lys70 is apparent in the E16Q and I65L variants (data not shown) and other mFruits, as well as in the parent DsRed variant K83M.12
Figure 3.
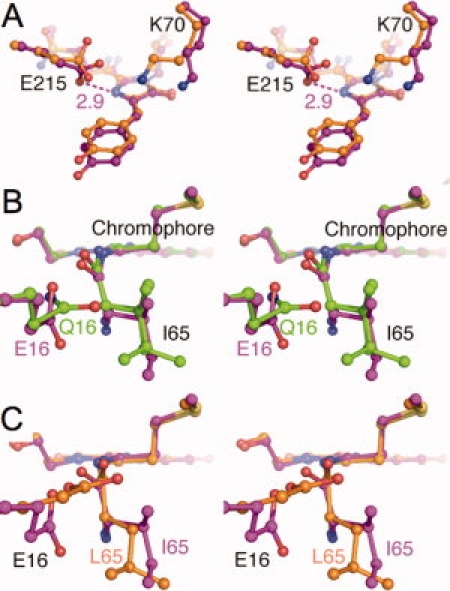
Stereo images of chromophore environment comparison. The mPlum model (purple) is superposed onto (A) DsRed (orange), (B) mPlum E16Q (green), (C) mPlum I65L variant (orange), respectively. Note that in panel (B) Ile65 of E16Q variant shows two conformations. Hydrogen bonds are shown in dashed lines, labeled with approximate lengths in angstroms.
The universally conserved Glu215 in mPlum rotates ∼90° in χ3 and forms a hydrogen bond with the imidazolinone ring nitrogen [N2, 3(A)], compared with DsRed. This conformational change of Glu215 is also observed in the E16Q and I65L mutants and other red-shifted mFruits.12 In mPlum and its variants, Glu215 is, thus, believed to be protonated and to donate a proton to the chromophore N2.
Similar chromophore environmental changes, including conformational rearrangement of Lys70 and Glu215, have previously been argued and to some extent demonstrated to account for red shifts in mCherry and closely related proteins, compared with the progenitor DsRed.12 Thus, we propose that that the time-independent emission red shift of mPlum can also be ascribed to these modifications.
However, other details of the chromophore environment are unique to mPlum. As identified by mutagenesis, Glu16 is a key residue and replaces Val16 found in DsRed and all other mFruits. The side chain of Glu16 is buried inside the protein and its carboxyl group forms a hydrogen bond with the acylimine oxygen of Ile65 [2(A)]. This is the first instance in which an interaction with the acylimine oxygen has been reported for any red fluorescent protein. The unique hydrogen bond between the carboxylate of Glu16 and the acylimine oxygen in mature mPlum (chain B) was refined to have length 2.5 Å. A similar but somewhat longer hydrogen bond (2.7 Å) also exists in chain A of mPlum containing the immature chromophore.
The crystal structure of mature E16Q reveals that the NH2 group of Gln16 also forms a hydrogen bond with the acylimine carbonyl of Ile65 [Fig. 2(B)]. This hydrogen bond was refined to have length 2.8 Å. The side chain of Gln16 rotates ∼70° in χ3, compared with Glu16 in mPlum (in both chain A and B) [Fig. 3(B)], so that the carboxylate and amino groups are arranged at roughly right angles to each other. This rearrangement may be related to disorder in the side chain conformation of Ile65 in the mature E16Q variant [Fig. 2(B)], which is observed in the electron density map to have at least two conformations. In mPlum, Ile65 is apparently restricted to a single conformation, the same in each independent chain.
Mutagenesis demonstrated that Ile65 is also a key residue for the far-red emission of mPlum. In the blue shifted mPlum I65L variant, the carboxyl group of Glu16 still forms a hydrogen bond (2.7 Å) with the carbonyl of Ile65 [Fig. 2(C)]. However, in I65L Glu16 rotates about 150° in χ1 and 30° in χ3, compared with mPlum [Fig. 3(C)]. Interestingly, as shown in Figure 3(B,C), the carboxyl group of Glu16 in I65L, as well as the Gln16 in E16Q is roughly parallel to the chromophore plane, whereas in mPlum the carboxyl moiety is perpendicular to the chromophore plane.
Discussion
Saturation mutagenesis of mPlum had demonstrated that the combination of Glu16 and Ile65 is indispensable for far-red emission at about 650 nm.4 Replacement of either group results in a blue shift in emission of 25–35 nm. The derived variants can be divided into two groups based on their emission maxima: ∼615 and ∼625 nm (for convenience, the emission properties are summarized in Table 1). In the Glu16 derived variants, the 615 nm group contains variants with residues incapable of forming a hydrogen bond with the acylimine oxygen, for example, the E16I and E16V variants. On the other hand, the 625 nm group includes mutants with residues capable of forming a hydrogen bond with the acylimine oxygen directly (e.g. the E16Q variant) or possibly indirectly through a buried water molecule (e.g. the E16G and E16S variants). In the Ile65-derived variants, the I65H mutant has emission maximum less than 610 nm while the others emit at about 625 nm. We chose E16Q and I65L variants as representatives for the 625 nm group and E16L for representative of the 615 nm group, to study the interactions responsible for the far-red emission of mPlum. Excitation spectra of mPlum and these representative mutants indicate that they share the same excitation maximum. In the following, we will argue that reorientation of the unique hydrogen bond between Glu16 and the chromophore is largely responsible for the dynamic Stokes shift.
Temperature dependence
Although all FPs studied show blue shifts in emission at low T, at 77 K mPlum exhibits a much larger Stokes shift (∼30 nm) than observed for E16L (∼10 nm). The E16Q variant shows an intermediate Stokes shift of ∼20 nm at 77 K. Furthermore, at 77 K the excitation and emission spectra of E16L show considerable structure, indicative of a distribution of reasonably well-defined states, that is largely absent from the mPlum spectrum. These data suggest that even at very low temperature, significant solvent reorganization takes place during the excited state in mPlum, but not in the E16L variant. However, as evidenced by the E16Q mutant, the nature of the hydrogen bond (i.e. whether the proton could in principle be transferred), is clearly important for far-red emission. In the following, we will argue that both the type and precise orientation of the Glu16 hydrogen bond is critical for emission at 650 nm.
Unique hydrogen bond
The crystal structure of mPlum reveals a unique hydrogen bond between Glu16 and the acylimine oxygen of the chromophore. No similar hydrogen bond has previously been observed or reported in the other red fluorescent proteins such as eqFP611,9 HcRed,23 DsRed,6,7 zRFP574,14 and other mFruits.12 Indeed, the residue corresponding to Glu16 in mPlum is hydrophobic in all known red fluorescent proteins, that is valine, leucine, isoleucine, and phenolalanine. This is also the case for the more recently derived far-red fluorescent proteins mKate (λem = 635 nm)24 and RFP639 (λem = 639 nm).24 As the proton acceptor is conjugated with the rest of the chromophore, it is reasonable to assume that the interaction is capable of significantly affecting the electronic configuration of the chromophore. Furthermore, it has long been established that hydrogen bonds are directional interactions so that the details of distance and orientation could have important consequences for excitation and emission spectra.
In the Leu16 variant, formation of a similar hydrogen bond is not possible, and model building suggests that a water molecule would neither be readily accommodated nor have its hydrogen bond requirements satisfied in the available volume. Indeed, none is seen in other structures (such as DsRed) where yet smaller valine is found in the 16 position. However, such a bound water molecule could possibly be found in certain mPlum variants for example, if Glu16 were replaced by a smaller polar residue (e.g. glycine, serine).
Structural rearrangements in mPlum/I65L
Mere formation of a hydrogen bond between the side chain at position 16 and the acylimine can only partly explain the far-red emission of mPlum, as glutamine is unable to substitute completely for glutamatic acid. A structural comparison between mPlum and the I65L variant revealed that although the hydrogen bond is maintained, the carboxyl group of Glu16 is reoriented by about 90° due to a steric clash between the two side chains. Interestingly, the new conformation of Glu16 closely resembles that of Gln16 in the E16Q mutant. This strongly suggests that Ile65 is important for the far-red emission in that it permits or helps maintain a specific, strong interaction between of Glu16 and the acylimine oxygen in the excited state. The proposed role of Ile65 is consistent with the fact that all Ile65-derived mutants but I65H have approximately the same emission maximum. Interpretation of the I65H result must necessarily await structural studies.
On the basis of these observations, we propose that the dynamic Stokes shift of mPlum is largely caused by the conformational relaxation of Glu16 after the chromophore is excited, which lowers the excited state energy of the chromophore. Furthermore, we propose that the observed ∼500 ps time constant for the emission shift in mPlum is primarily due to the structural rearrangement of Glu16, rather than a change in the hydrogen-bonding interaction (e.g. proton transfer), which would likely take place on a much faster time scale.
Of course, these data do not rule out the possibility that the new conformation might facilitate the rapid formation of a different type of hydrogen bond in the excited state, such as a “strong” or “low barrier” hydrogen bond.25 It is generally accepted that upon excitation, n–π* charge transfer takes place from the phenol moiety toward the imidazolinone moiety,26,27 which increases the polarity of the carbonyl at position 65 and would thus strengthen the hydrogen-bonding interaction with Glu16. It has been shown experimentally that hydrogen-bonding interactions between a solute and solvent can induce a red shift of the solute. For example, a positive solvatochromic shift was observed for an aminobenzodifuranone derivative (ABF) in hexamethylphosphoramide (HMPA), which was explained by a hydrogen bond between the solvent HMPA and the solute ABF, stabilizing the excited state of the solute relative to the ground state.28
In summary, we propose that the large Stokes shift in mPlum results from the presence of and strengthening of the Glu16-chromophore hydrogen bond interaction in the excited state, which stabilizes the excited state of the chromophore relative to the ground state. As glutamine cannot fully substitute for glutamic acid, the possibility of proton transfer between Glu16 and the chromophore in the excited state remains an open question. Finally, since other far-red fluorescent proteins studied to date do not seem to exhibit an interaction comparable with the Glu16-chromophore hydrogen bond in mPlum, introduction of such an interaction in those proteins might lead to further red shifts in the emission maxima.
Materials and Methods
Mutagenesis, protein expression, and crystallization
Protein was expressed in E. coli (Top10) by use of the pBAD His tagged expression system (Invitrogen).4 Mutagenesis was performed using the QuikChange method (Stratagene). Protein was purified by Ni2+ affinity chromatography over Ni-NTA agarose (Qiagen, Chatsworth, CA) and then buffer exchanged with PD-10 Sephadex columns (Amersham Pharmacia) into 50 mM HEPES (pH 7.9). Mature and immature mPlum mutants were separated by ion exchange chromatography using cationic column (Hi Load 16/10 SP Sepharose) and pH-gradient method with 50 mM citric acid at pH 5.0 and 6.0 (7.0 for mPlum E16L). Crystals of mPlum, mature E16Q, and I65L mutants, were all obtained by hanging drop vapor diffusion: 2 μL protein solution (∼25 mg/mL) with 2 μL well solution (200 mM NaCl, 100 mM Tris pH 8.5, 30% PEG 3400) after one week.
Spectroscopy
Protein samples were diluted to 44 μg/mL in 2 mL 100 mM HEPES pH 8.1. Temperature-dependent fluorescence spectra were taken with a Fluorolog spectrofluorimeter (Spex Industries, Edison, NJ). For low temperature (77 K) measurements, protein samples were diluted in 1 mL phosphate-buffered saline containing 50% glycerol, transferred to a quartz tube, and immersed in a Dewar filled with liquid nitrogen. Absorbance spectra (400 μg/mL protein in the same buffers) were recorded with a HP 8453 UV–vis spectrophotometer.
Data collection and structure solution
Diffraction data sets were collected from single flash-frozen crystals of mature and immature mPlum and mature E16Q mutant (100 K) using ADSC-Q315 detector on beam line 8.2.2 at the Advanced Light Source (Berkeley, CA). Data were reduced using HKL2000 (HKL Research) or d*TREK (Rigaku). Molecular replacement was performed with EPMR29 using the DsRed A chain (PDB entry 1G7K) as a search model, providing unambiguous identification of the space group and unique solutions to the rotation and translation problems. Rigid body refinement was performed using TNT30 and model building was conducted with Coot31 in several stages of increasing resolution. The chromophore entries for the TNT geometry library was derived using the program AM4 as described.7 Shelx-9732 was used in the final stages of refinement. The linkages among backbone nitrogen and alpha carbon of Met66 and carbonyl carbon of Ile65, were unrestrained from the beginning of refinement with Shelx-97.
Statistics of data collection and refinement for the atomic models are presented in the Table S1 (Supp. Info.). No electron density is apparent for residues 1–5 and 223–236 for all proteins, indicating disorder. PROCHECK33 reveals that there are no residues in either the disallowed or “generously allowed” regions of the Ramachandran diagram. Upon superposition of α-carbons with the A subunit of the parent protein DsRed rms deviations for either of two chains of all three proteins is 0.4 Å. The models of all proteins are thus remarkably similar to that of DsRed although there is dramatic change in quaternary structure: from tetrameric to monomeric, consistent with results of other mFruits.12
Acknowledgments
The authors thank Dr. Roger Tsien at University of California, San Diego for generous donation of mPlum cDNA.
Glossary
Abbreviations
- FP
fluorescent protein
- HB
hydrogen bond
- RT
room temperature
Supplemental material
References
- 1.Weissleder R, Ntziachristos V. Shedding light onto live molecular targets. Nat Med. 2003;9:123–128. doi: 10.1038/nm0103-123. [DOI] [PubMed] [Google Scholar]
- 2.Gurskaya N, Fradkov A, Terskikh A, Matz M, Labas Y, Martynov V, Yanushevich Y, Lukyanov K, Lukyanov S. GFP-like chromoproteins as a source of far-red fluorescent proteins. FEBS lett. 2001;507:16–20. doi: 10.1016/s0014-5793(01)02930-1. [DOI] [PubMed] [Google Scholar]
- 3.Shkrob M, Yanushevich Y, Chudakov D, Gurskaya N, Labas Y, Poponov S, Mudrik N, Lukyanov S, Lukyanov K. Far-red fluorescent proteins evolved from a blue chromoprotein from Actinia equina. Biochem J. 2005;392:649–654. doi: 10.1042/BJ20051314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wang L, Jackson W, Steinbach P, Tsien R. Evolution of new nonantibody proteins via iterative somatic hypermutation. Proc Natl Acad Sci USA. 2004;101:16745–16749. doi: 10.1073/pnas.0407752101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Henderson JN, Remington SJ. Crystal structures and mutational analysis of amFP486, a cyan fluorescent protein from Anemonia majano. Proc Natl Acad Sci USA. 2005;102:12712–12717. doi: 10.1073/pnas.0502250102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wall M, Socolich M, Ranganathan R. The structural basis for red fluorescence in the tetrameric GFP homolog DsRed. Nat Struct Biol. 2000;7:1133–1138. doi: 10.1038/81992. [DOI] [PubMed] [Google Scholar]
- 7.Yarbrough D, Wachter R, Kallio K, Matz M, Remington S. Refined crystal structure of DsRed, a red fluorescent protein from coral, at 2.0-angstrom resolution. Proc Natl Acad Sci USA. 2001;98:462–467. doi: 10.1073/pnas.98.2.462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mizuno H, Mal TK, Tong KI, Ando R, Furuta T, Ikura M, Miyawaki A. Photo-induced peptide cleavage in the green-to-red conversion of a fluorescent protein. Mol Cell. 2003;12:1051–1058. doi: 10.1016/s1097-2765(03)00393-9. [DOI] [PubMed] [Google Scholar]
- 9.Petersen J, Wilmann PG, Beddoe T, Oakley AJ, Devenish RJ, Prescott M, Rossjohn J. The 2.0-A crystal structure of eqFP611, a far red fluorescent protein from the sea anemone Entacmaea quadricolor. J Biol Chem. 2003;278:44626–44631. doi: 10.1074/jbc.M307896200. [DOI] [PubMed] [Google Scholar]
- 10.Nienhaus K, Nienhaus GU, Wiedenmann J, Nar H. Structural basis for photo-induced protein cleavage and green-to-red conversion of fluorescent protein EosFP. Proc Natl Acad Sci USA. 2005;102:9156–9159. doi: 10.1073/pnas.0501874102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Quillin ML, Anstrom DA, Shu XK, O'Leary S, Kallio K, Chudakov DA, Remington SJ. Kindling fluorescent protein from Anemonia sulcata: dark-state structure at 1.38 angstrom resolution. Biochemistry. 2005;44:5774–5787. doi: 10.1021/bi047644u. [DOI] [PubMed] [Google Scholar]
- 12.Shu X, Shaner NC, Yarbrough CA, Tsien RY, Remington SJ. Novel chromophores and buried charges control color in mFruits. Biochemistry. 2006;45:9639–9647. doi: 10.1021/bi060773l. [DOI] [PubMed] [Google Scholar]
- 13.Hayashi I, Mizuno H, Tong KI, Furuta T, Tanaka F, Yoshimura M, Miyawaki A, Ikura M. Crystallographic evidence for water-assisted photo-induced peptide cleavage in the stony coral fluorescent protein Kaede. J Mol Biol. 2007;372:918–926. doi: 10.1016/j.jmb.2007.06.037. [DOI] [PubMed] [Google Scholar]
- 14.Pletneva N, Pletnev V, Tikhonova T, Pakhomov AA, Popov V, Martynov VI, Wlodawer A, Dauter Z, Pletnev S. Refined crystal structures of red and green fluorescent proteins from the button polyp Zoanthus. Acta Crystallogr. 2007;63:1082–1093. doi: 10.1107/S0907444907042461. [DOI] [PubMed] [Google Scholar]
- 15.Kikuchi A, Fukumura E, Karasawa S, Mizuno H, Miyawaki A, Shiro Y. Structural characterization of a thiazoline-containing chromophore in an orange fluorescent protein, monomeric Kusabira orange. Biochemistry. 2008;47:11573–11580. doi: 10.1021/bi800727v. [DOI] [PubMed] [Google Scholar]
- 16.Schnitzler CE, Keenan RJ, McCord R, Matysik A, Christianson LM, Haddock SH. Spectral diversity of fluorescent proteins from the Anthozoan Corynactis californica. Mar Biotechnol (NY) 2008;10:328–342. doi: 10.1007/s10126-007-9072-7. [DOI] [PubMed] [Google Scholar]
- 17.Remington S, Wachter R, Yarbrough D, Branchaud B, Anderson D, Kallio K, Lukyanov K. zFP538, a yellow-fluorescent protein from Zoanthus, contains a novel three-ring chromophore. Biochemistry. 2005;44:202–212. doi: 10.1021/bi048383r. [DOI] [PubMed] [Google Scholar]
- 18.Ormo M, Cubitt A, Kallio K, Gross L, Tsien R, Remington S. Crystal structure of the Aequorea victoria green fluorescent protein. Science. 1996;273:1392–1395. doi: 10.1126/science.273.5280.1392. [DOI] [PubMed] [Google Scholar]
- 19.Wachter R, Elsliger M, Kallio K, Hanson G, Remington S. Structural basis of spectral shifts in the yellow-emission variants of green fluorescent protein. Struct Folding Des. 1998;6:1267–1277. doi: 10.1016/s0969-2126(98)00127-0. [DOI] [PubMed] [Google Scholar]
- 20.Abbyad P, Childs W, Shi X, Boxer SG. Dynamic stokes shift in green fluorescent protein variants. Proc Natl Acad Sci USA. 2007;104:20189–20194. doi: 10.1073/pnas.0706185104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Creemers TM, Lock AJ, Subramaniam V, Jovin TM, Volker S. Three photoconvertible forms of green fluorescent protein identified by spectral hole-burning. Nat Struct Biol. 1999;6:557–560. doi: 10.1038/9335. [DOI] [PubMed] [Google Scholar]
- 22.Tubbs JL, Tainer JA, Getzoff ED. Crystallographic structures of discosoma red fluorescent protein with immature and mature chromophores: linking peptide bond trans-cis isomerization and acylimine formation in chromophore maturation. Biochemistry. 2005;44:9833–9840. doi: 10.1021/bi0472907. [DOI] [PubMed] [Google Scholar]
- 23.Wilmann PG, Petersen J, Pettikiriarachchi A, Buckle AM, Smith SC, Olsen S, Perugini MA, Devenish RJ, Prescott M, Rossjohn J. The 2.1A crystal structure of the far-red fluorescent protein HcRed: inherent conformational flexibility of the chromophore. J Mol Biol. 2005;349:223–237. doi: 10.1016/j.jmb.2005.03.020. [DOI] [PubMed] [Google Scholar]
- 24.Kredel S, Nienhaus K, Oswald F, Wolff M, Ivanchenko S, Cymer F, Jeromin A, Michels FJ, Spindler KD, Heilker R, Nienhaus GU, Wiedenmann J. Optimized and far-red-emitting variants of fluorescent protein eqFP611. Chem Biol. 2008;15:224–233. doi: 10.1016/j.chembiol.2008.02.008. [DOI] [PubMed] [Google Scholar]
- 25.Perrin CL, Nielson JB. “Strong” hydrogen bonds in chemistry and biology. Annu Rev Phys Chem. 1997;48:511–544. doi: 10.1146/annurev.physchem.48.1.511. [DOI] [PubMed] [Google Scholar]
- 26.Agmon N, Rettig W, Groth C. Electronic determinants of photoacidity in cyanonaphthols. J Am Chem Soc. 2002;124:1089–1096. doi: 10.1021/ja003875m. [DOI] [PubMed] [Google Scholar]
- 27.Marques MAL, Lopez X, Varsano D, Castro A, Rubio A. Time-dependent density-functional approach for biological chromophores: the case of the green fluorescent protein. Phys Rev Lett. 2003;90:258101. doi: 10.1103/PhysRevLett.90.258101. [DOI] [PubMed] [Google Scholar]
- 28.Gorman AA, Hutchings MG, Wood PD. Solvatochromism of an aminobenzodifuranone: an unprecedented positive wavelength shift. J Am Chem Soc. 1996;118:8497–8498. [Google Scholar]
- 29.Kissinger CR, Gehlhaar DK, Fogel DB. Rapid automated molecular replacement by evolutionary search. Acta Crystallogr D Biol Crystallogr. 1999;55:484–491. doi: 10.1107/s0907444998012517. [DOI] [PubMed] [Google Scholar]
- 30.Tronrud DE, Teneyck LF, Matthews BW. An efficient general-purpose least-squares refinement program for macromolecular structures. Acta Crystallogr A. 1987;43:489–501. [Google Scholar]
- 31.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 32.Sheldrick GM, Schneider TR. SHELXL: high-resolution refinement. Macromol Crystallogr B. 1997;277:319–343. [PubMed] [Google Scholar]
- 33.Laskowski RA, Macarthur MW, Moss DS, Thornton JM. Procheck—a program to check the stereochemical quality of protein structures. J Appl Crystallogr. 1993;26:283–291. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.