

Abstract
The fibroblast growth factor receptor 3 (FGFR3) is a member of the FGFR subfamily of the receptor tyrosine kinases (RTKs) involved in signaling across the plasma membrane. Generally, ligand binding leads to receptor dimerization and activation. Dimerization involves the transmembrane (TM) domain, where mutations can lead to constitutive activation in certain cancer types and also in skeletal malformations. Thus, it has been postulated that FGFR homodimerization must be inherently weak to allow regulation, a feature reminiscent of α and β integrin TM interactions. However, we show herein that in FGFR3-TM, four C-terminal residues, CRLR, have a profound destabilizing effect in an otherwise strongly dimerizing TM peptide. In the absence of these four residues, the dimerizing propensity of FGFR3-TM is comparable to glycophorin, as shown using various detergents. In addition, the expected enhanced dimerization induced by the mutation associated to the Crouzon syndrome A391E, was observed only when these four C-terminal residues were present. In the absence of these four residues, A391E was dimer-destabilizing. Finally, using site specific infrared dichroism and convergence with evolutionary conservation data, we have determined the backbone model of the FGFR3-TM homodimer in model lipid bilayers. This model is consistent with, and correlates with the effects of, most known pathological mutations found in FGFR-TM.
Keywords: fibroblast growth factor receptor, receptor tyrosine kinases, equilibrium sedimentation, homodimerization, lipid bilayers, site specific infrared dichroism, molecular dynamics
Introduction
The fibroblast growth factor receptor 3 (FGFR3) is a member of the FGFR subfamily of the receptor tyrosine kinases (RTKs). RTKs are membrane receptors essential for cell proliferation, differentiation, and migration.1 Monomeric RTKs consist of an N-terminal extracellular ligand-binding domain, a single α-helical transmembrane (TM) domain, and a C-terminal cytoplasmic kinase domain. In general, ligand binding induces homodimerization of the receptor, followed by transphosphorylation by the cytoplasmic kinase domain, which leads to the activation of downstream intracellular signaling pathways.2,3
The transmembrane domains of RTKs are likely to be involved in dimerization. By using a TOXCAT system, it has been shown that ErbB1, ErbB2, and ErbB4 TM domains dimerize strongly in bacterial membranes in vivo.4 Also, a peptide corresponding to the transmembrane domain of the ErbB receptor inhibited the autophosphorylation of the receptor,5 presumably by disrupting monomer–monomer interactions at the TM domain.
RTK activation also requires a specific intermonomeric orientation. This is supported by (i) the observed linkage of the kinase domain of the Neu receptor to a chimeric TM domain (rotating the interface of the TM domain led to a periodic oscillation in kinase activation6), (ii) the observed lack of activation by a peptide antagonist that otherwise induced dimerization in the erythropoietin receptor (EPOR),7 and (iii) the selective activation of FGFR3 obtained by a cysteine scan at juxtamembrane residues 370–375.8 In the latter report, although all mutants showed dimerization, only two, G370C and S371C, induced activation. Specific monomer–monomer RTK TM orientation requirements may be the reason for varying disease symptoms and severity in pathological mutations in this part of the molecule.9
Although in some cases, for example, the insulin receptor (IR) or the related insulin growth factor receptor (IGFR), dimerization is constitutive,10,11 tight regulation of signaling events in RTKs is thought to depend on an inherently weak dimerizing propensity, as also suggested for heterodimeric integrins.12 Unregulated receptor activation has been proposed to explain the effects of mutations at the TM domain9 observed in skeletal malformations.13 For example, several mutations have been identified in patients at the transmembrane and juxtamembrane regions, for example, G375C and G380R in achondroplasia, the most common form of dwarfism.14 Mutations G370C, S371C, and Y373C are found in thanatophoric displasia type I (TDI), a lethal form of dwarfism that resembles homozygous achondroplasia,15,16 and A391E is found in both Crouzon syndrome, a complex craniosynostosis disorder, and acanthosis nigricans.17
Mutations at the TM domain of FGFRs have also been shown to be oncogenic. For example, G382D has been found in a multiple myeloma cell line18; A391E, G370C, G380R, and Y373C have been associated with bladder carcinomas.19 In FGFR4, mutation G388R accelerates tumor progression in cancer patients.9,20
Some of these mutations, such as A391E in FGFR3, have been shown to enhance receptor dimerization,21 similar to the oncogenic mutation V664E in the EGFR Neu receptor,22,23 although in the latter case, there was no correlation between receptor-dimerization and TM-dimerization enhancement.4
For other TM mutations, however, increased activation may take place because of either an overexpression of the receptor or a diminished turnover. For example, mutation G380R enhanced the kinase activity of a chimeric Neu/FGFR3 receptor and of FGFR324 because of slow downregulation of the mutant receptor,25,26 leading to receptor accumulation at the cell surface and continuous signaling. Consistent with this interpretation, this mutation was not found to enhance FGFR3-TM dimerization in vitro.27
Although the general mechanism of activation of integrins is opposite to that of RTKs, that is, integrins are activated after the separation of α and β subunits,28 it has been suggested that to allow regulation, the tendency of TM domains to form dimers is weak in both systems.29 Indeed, it has been shown that the synthetic peptide corresponding to native FGFR3-TM, encompassing residues 367DEA to CRLR399, is mostly monomeric in sodium dodecyl sulfate (SDS), with only a minor band corresponding to dimers.21,30 Using a fluorescence resonance energy transfer (FRET) assay in lipid bilayers, the free energy of FGFR-TM dimerization was estimated to be only −3 kcal/mol.21,30 Similarly, the transmembrane domains of epidermal growth factor receptors (ErbBr) do not dimerize strongly in micelles, with reported interaction energies of the order of −1 kcal/mol,31 although this is in contrast with the results obtained using larger constructs in bacterial membranes.4
A model for the homodimer interface in FGFR3-TM has been proposed previously,21 in which Leu377, Val381, Phe384, and Ile387 would be located at the helix–helix interface. However, there are no experimental data supporting that model. Site-specific infrared dichroism (SSID) is especially suited to the study of relatively well-ordered systems, which show a periodicity in the orientations of the carbonyl bonds, as found in tilted α helices that form α-helical transmembrane bundles. The orientation in space of these labeled peptidic carbonyl groups can be obtained using the theory of SSID,32 and we have obtained several backbone structures of transmembrane α-helical bundles using this method that have been corroborated using other techniques.33–36 However, to use SSID, FGFR3-TM must form stable dimers in lipid bilayers. We found that by shortening the two ends of FGFR-TM, we could identify a sequence that is strongly dimerizing, even in SDS. Thus, introducing a labeled carbonyl (13C=18O)37 at two consecutive positions of this peptide, we have collected orientational data using polarized attenuated total reflection Fourier transform infrared spectroscopy (PATIR-FTIR), and we report a backbone model of the FGFR-TM dimer in agreement with this experimental data.
Results
Electrophoresis
The dimerization tendencies of the FGFR-TM synthetic peptides shown in Figure 1(a) were examined by using SDS electrophoresis.
Figure 1.
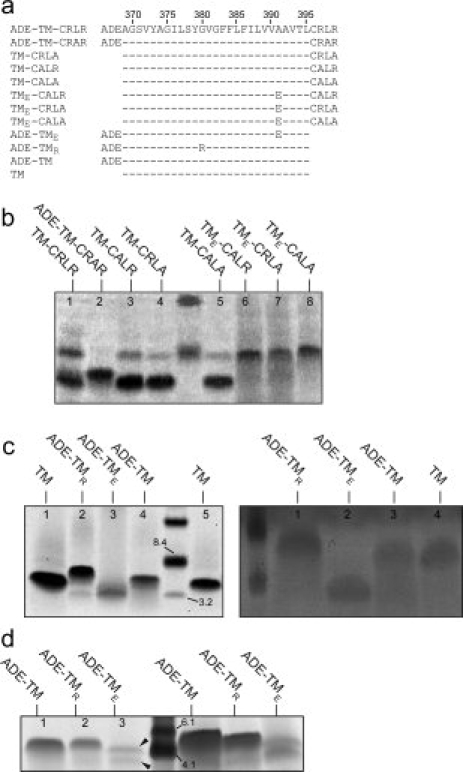
Sequences corresponding to FGFR3-TM and electrophoresis results. (a) Sequences corresponding to synthetic FGFR3-TMs, with the central core of the transmembrane domain (TM) indicated by a broken line, and flanked by N-terminal residues ADE and C-terminal residues CRLR;(b and c, left panel) electrophoreses in SDS, using NuPAGE® Novex 12% Bis-Tris gel; (c, right panel) electrophoresis in SDS, using 15% Tris-Glycine gel; (d) electrophoresis in PFO, using 10–20% Tris-Tricine gel. Peptides are indicated above each line.
Effect of C-terminal residues, CRLR
First, we tested the effect of the C-terminal basic residues (Arg) on dimerization in the absence of the N-terminal acidic residues (fragment ADE). Peptides TM-CRLR [Fig. 1(b)(b), lane 1] and variants that contained one or no C-terminal arginine, TM-CRLA, TM-CALR, and TM-CALA [1(b), lanes 3–5], all formed mostly monomers in SDS, along with a minor proportion of dimers. This behavior is similar to that observed previously for peptide DE-TM-CRLR,21,30 which indicates that neither fragment ADE nor the arginine residues in fragment CRLR have a particularly strong effect on dimer stability.
Mutation A391E is known to induce constitutive activation of FGFR, and in peptide DE-TM-CRLR, it stabilized the dimeric form in SDS, which suggests an interaction between A391E and an unknown residue in the opposite helix.9,21 When we used variants of these peptides, containing the A391E mutation, such as TME-CALR, TME-CRLA, or TME-CALA, we observed mostly dimers [Fig. 1(b), lanes 6–8]. This indicates that the dimer stabilization induced by A391E is not due to interaction to any of the two C-terminal Arg residues in sequence CRLR.
Effect of N-terminal residues, ADE
Then we tested the effect of the absence of fragment CRLR using peptides ADE-TM, ADE-TMR, ADE-TME, and TM [ Fig. 1(a)(a)]. In all cases, a single dimeric form was observed, except for ADE-TME, which was monomeric [ Fig. 1(c)]. This, combined with the dimers observed for TME-CALR, TME-CRLA, or TME-CALA (discussed earlier) shows that (i) the removal of the four C-terminal residues of FGFR3-TM has a strong stabilizing effect on FGFR3-TM dimerization; (ii) the TM core associates strongly in SDS, and fragment CRLR, but not fragment ADE, destabilizes the dimer; and (iii) A391E interacts with some part of the sequence CRLR, but not arginine. It is possible that the interaction of E391 is with leucine or a backbone group. Indeed, peptide ADE-TM-CRA398R, that is, with a L398A mutation, was only monomeric [Fig. 1(b), lane 2], suggesting that this residue is involved in interhelical interactions. In conclusion, these data show that A391 and L398 must be part of the helix–helix interaction in FGFR3-TM.
To confirm the strong dimerizing properties of the core TM peptide [Fig. 1(a)(a)], we performed an electrophoresis in the mild detergent perfluorooctanoic acid (PFO) [ Fig.1(d)]. PFO has been successfully used to determine the correct oligomeric size of α-helical transmembrane bundles in peptides that were monomeric or produced nonspecific oligomers in SDS for example, in the tetrameric phospholemman,38 pentameric small hydrophobic protein in the respiratory syncytial virus,39 or pentameric transmembrane domain of SARS envelope protein36. As expected from the SDS results presented, both the peptide ADE-TM and the peptide-containing mutation G380R ADE-TMR formed only dimers in PFO [Fig. Fig. 1(d), lanes 1 and 2, respectively], whereas mutant A391E, ADE-TME, produced both dimers and monomers [Fig. Fig. 1(d), lane 3, arrows]. The increased amount of dimers in the latter sample versus that found in SDS [Fig. 1(c) (left), lane 3, or 1(c) (right) lane 2] is expected, because PFO is a milder detergent.
Equilibrium sedimentation
To measure the self-association tendency of peptide ADE-TM, we performed a sedimentation equilibrium study in dodecylphosphocholine (DPC) micelles (Fig. 2). DPC is a mimic for DMPC, as it has the same polar headgroups and has been routinely used to determine, for example, the oligomeric size of TM α helical bundles.40 At peptide:detergent molar ratios 1:200 and 1:400, the sedimentation data could be best fitted to a monomer–dimer self-association model, with an association constant (Kaapp) of (1.16 ± 0.6) × 107 M−1. As this value is dependent on the detergent concentration,41 we have calculated it as a mole fraction scale, Kax (Kax = Kaapp × [detergent]),41 where Kax in this case equals 2.8 × 105. From this value, the mole fraction standard-state free energy change due to dimerization, ΔG°X,41 can be estimated at −7.4 kcal/mol. At higher concentrations, the data could not be fitted to a monomer–dimer model. Inclusion of a tetramer improved the fitting, with association constant value K1,4 = 7 × 1010 M−3. Higher-order oligomers have also been observed in tetrameric M2 at high peptide:detergent molar ratios.40 Thus, the high dimerization propensity observed in DPC and PFO, and even SDS, indicates that peptide TM [Fig. 1(a)] must be dimeric in lipid bilayers, because association constants of transmembrane α helices in membranes have a higher value than those in detergents.42,43
Figure 2.
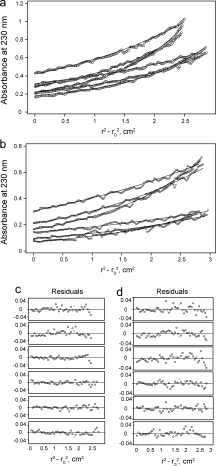
Sedimentation equilibrium results for peptide ADE-TM in DPC micelles. (a) 1:100 and 1:200 protein-to-detergent molar ratio. (b) 1:200 and 1:400 protein-to-detergent molar ratio. Data was obtained at 28k, 34.5k, and 42k rpm (see Materials and Methods). (c) and (d) Residuals for curves in (a) and (b), respectively.
Conformational search for FGFR3 TM homo-dimers
To obtain possible models for the FGFR3-TM homodimer, we used evolutionary conservation data during a global search of homologous sequences of FGFR3-TM (residues 369–395) from various organisms44 [ Fig.3(a)]. Three complete sets, which represent candidates of FGFR3-TM homodimerization, were found [Fig. 3(b)], all right-handed. To compare the helix orientation in these models, the rotational orientation at an arbitrary residue, V381, was used. Clusters ‘1’ and ‘2’ have ωV381 values of ∼70° and −10°, respectively, with a helix tilt of ∼−15° (negative because it is a right-handed dimer). For the cluster labeled ‘3’, the tilt angle was approximately 0°, which means that the ω value in this case is meaningless.32 When the same simulation included juxtamembrane residues (residues 366–399), only cluster ‘1’ was found (not shown).
Figure 3.
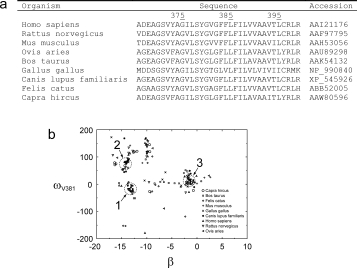
Use of evolutionary conservation data to obtain candidates to FGFR-TM homodimerization. (a) Alignment of FGFR-TM sequences. A systematic search was performed with sequence 369–395 (numbering corresponding to human FGFR3-TM). (b) Evolutionary conserved homodimeric structures obtained after exploring all possible conformational space of FGFR-TM homodimers. Low energy structures are indicated and plotted on a plane (x axis, helix tilt; y axis, rotational orientation for residue V381, ω381). Three “complete sets,” are indicated by circles.
Infrared spectra of FGFR3 TM domain in lipid membranes
The experimental value for ωV381 was determined using SSID32 in DMPC lipid bilayers. Representative infrared spectra of isotopically labeled peptide ADE-TM in DMPC lipid bilayers show a shape of the amide I absorption band, with a maximum at 1657 cm−1 [4(a)], indicative of a predominant α-helical structure.45 Examination of the amide I and II bands in this figure indicates that the percentage of hydrogen/deuterium (H/D) exchange of FGFR-TM amide groups was 20 ± 10%, that is, 24 ± 4 residues are membrane-embedded, which is consistent with the presence of one α-helical transmembrane domain.
Figure 4.
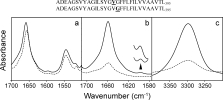
ATR-FTIR spectra of peptide ADE-TM reconstituted in DMPC lipid bilayers. (A) Non polarized infrared spectra of the amide I and II region in H2O, either at low hydration (solid line) or hydrated in D2O (broken line). The small reduction in amide II intensity is due to H/D exchange. (B) amide I region and isotopic 13C=18O label (residue V381), centered at 1590 cm−1 (see insert), obtained at 0° (solid line) or 90° (broken line) polarization. (C) same as in B, but corresponding to the amide A of the spectrum. The synthetic peptide used and the position of the 13C=18O isotopic labels are indicated at the top of the figure.
After confirming that ADE-TM is inserted and forms α helical bundles in lipid bilayers, we investigated the tilt and rotational orientation of the peptide. An example of the infrared dichroic data obtained at two polarizations (see Materials and Methods) is shown for the peptide labeled at V381, for amide I and labeled site [Fig. 4(b)] and for amide A [Fig. 4(c)]. Spectra for the other labeled peptide (G382) were similar and are not shown. The dichroic ratios obtained from each measurement, Rhelix and Rsite are shown in Table I. From the data in this table, we calculated the helix tilt (β) and rotational pitch angle (ω) for residue V38132,46 (β = 16° ± 1° and ωV381 = −14° ± 13°). This value for ω corresponds to cluster '1′ in Figure 3, which was obtained computationally using residues ADE-TM-CRLR in the simulation. The structural model of this homodimer is shown in Figure 5.
Table I.
Dichroic Ratios for Peptide ADE-TM
| Residue | Rhelix (AA) | Rsite |
|---|---|---|
| V381 | 4.5 | 6.3 |
| 4.0 | 4.8 | |
| 4.3 | 5.9 | |
| 4.6 | 6.5 | |
| 4.5 | 6.0 | |
| G382 | 4.7 | 3.0 |
| 5.0 | 3.8 | |
| 4.2 | 3.2 | |
| 4.4 | 2.8 | |
| 4.5 | 4.0 |
Dichroic ratios measured by SSID using the amide A (AA) and the corresponding isotopic label for ADE-TM incorporated in DMPC lipid bilayers (“Materials and Methods”). For each sample, five independent measurements were obtained.
Figure 5.
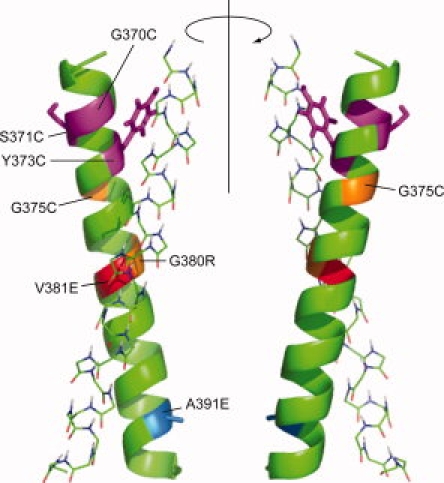
Model of the homodimeric human FGFR3-TM. Homodimeric model for human FGFR3-TM that is consistent with SSID data and with evolutionary conservation, represented by cluster '1′ in Figure 3(b)(b). Residues involved in pathological mutations (see main text) are indicated with different colors on a green helix. For clarity, for one of the α-helices, only the backbone is shown. [Color figure can be viewed in the online issue, which is available at www.interscience.wiley.com.]:
Discussion
It has been shown previously that FGFR3-TM peptide DE-TM-CRLR self-associates weakly.21,30,47 Also, mutation A391E in peptide DE-TM-CRLR stabilized the FGFR-TM homodimer.21 Our results presented here are consistent with these reports; that is, all variants of FGFR-TM containing the four C-terminal residues CRLR were mostly monomeric in SDS, and only dimers were observed in the variants TME-CRLR. The fact that no monomers were observed for TME-CALA, TME-CRLA, or TME-CALR, as compared with DE-TME-CRLR,21 suggests that the N-terminal ADE residues may have some minor destabilizing effect on the dimer.
In the present work, however, we show that after removing the C-terminal CRLR residues, FGFR3-TM self-associates strongly in lipid-like DPC, mild detergent PFO, and even in strong detergent SDS. Therefore, these four residues must be destabilizing in the context of the full receptor molecule, probably because of steric hindrance. This feature may be general to RTKs, in that a conformational change, perhaps removing the steric hindrance at the C-terminal juxtamembrane residues, allows for a strong interaction between TM α helices and activation.
The TM domains of α and β integrins, in contrast, are linked by a salt bridge,28 which stabilizes an otherwise weak interaction; when this salt bridge is broken, α and β chains separate, leading to activation. It is then possible that a fundamentally different form of interaction between TM domains in integrins and RTKs is necessary for an opposite form of activation.
The association constant obtained for the peptide ADE-TM, residues 366–395, is comparable to that of the glycophorin transmembrane (GpA-TM) dimer, where ∼90% of the peptide forms dimers at a 1:1000 peptide:DPC molar ratio.48 The standard free energy of dissociation can be defined in such a way that corresponds to a 1M concentration of detergent.41 Thus, extrapolating experimental data to a 1M detergent concentration yields a GpA-TM dissociation free energy of −3.8 kcal/mol in dodecyldimethylaminobenzoate (C12-DMAB),49 −5.7 kcal/mol in SDS,49 −7.0 kcal/mol in octylpentaoxyethylene (C8E5),41 −7.5 kcal/mol in C12-maltoside,49 and −5.7 kcal/mol in C14 betaine.50 These values are comparable to the value estimated here for FGFR3-TM (fragment TM) of −7.4 kcal/mol. As oligomerization propensities are known to be higher in lipid bilayers,42,43,51 the assumption used in our SSID measurements that peptide TM of FGFR3-TM forms dimers in lipid bilayers is justified.
In the equilibrium sedimentation experiments, the higher-order oligomers observed in DPC at higher peptide:DPC ratio is not unexpected. At peptide to detergent ratios higher than 1/150, it was previously observed51,52 that M2TM begins to form higher-order aggregates, and at peptide/detergent molar ratios more than 0.01, the experimentally measured micelle numbers predict that there would be insufficient detergent to maintain a constant degree of oligomer solvation,52 possibly leading to a higher-order peptide aggregation. However, the abundance of small residues and putative GxxxG-like motifs in the FGFR3-TM also suggests that higher-order oligomer formation may mimic events that could occur in vivo, for example, aggregation of several receptors, as shown for integrins.53 Indeed, FGFR-TM is well conserved at almost all positions around the helix, which suggests interaction of FGFR-TM with itself or with other receptors, as shown for ErbB1 and ErbB2.54
Our results are consistent with TOXCAT experiments that detected high dimerization propensity in TM fragments of members of the ErbB family: ErbB1, ErbB2, ErbB3, and ErbB4.4 In these experiments, the residues equivalent to juxtamembrane CRLR in FGFR, for example, KRRQ in ErbB2, were not included in the TOXCAT fusion construct with staphylococcus nuclease (SN). We speculate therefore that the absence of these residues explains the high affinity observed in bacterial membranes, of about 50% that of GpA-TM. In contrast, only a weak dimerization propensity was observed for ErbB TM fragments fused to SN in detergents.31 As discussed by these authors, the explanation for these discrepancies may lay in the sequence context of the construct. These limitations are clearly not present when using synthetic transmembrane fragments.
It has been proposed that to stabilize the FGFR-TM homodimer, A391E interacts with the other helix.21 The present work shows which interactions are more likely. First, a symmetric interaction between the two side chain Glu carboxylic groups is not possible, as in this case the peptide ADE-TME would show dimerization. Equally, the interaction of the Glu side chain cannot take place with residues N-terminal to A391E because the dimer is destabilized when the C-terminal residues, CRLR, are missing.
Our model for the FGFR3-TM homodimer obtained from SSID is consistent with the mutations found in several diseases and with the severity of their symptoms. For example, G370C and S371C in FGFR3-TM have been linked to thanatophoric dysplasia (TD), always lethal in the neonatal period.55 In contrast, G375C, is found in achondroplasia, a milder form of human dwarfism.24,56 These Cys mutations have been proposed to induce disulfide-mediated stabilization of the dimeric FGFR3 and constitutive activation.8,9 Indeed, it has been shown that G370C and G371C mutants showed high levels of mitogen activated protein kinase phosphorylation, while the activation of the G375C mutant was lower,8 and dimerization in the absence of ligand correlated with the severity of the phenotype: strong for Cys370 and Cys371, intermediate for Cys375, and negligible for the wildtype. Residues 370 and 371 are then expected to be involved in helix-helix interactions in the FGFR3-TM homodimer. The relative homodimerizing tendencies of these Cys mutations in FGFR-TM seem to confirm this,47 as Cys370, Cys371, and Cys375 exhibited the highest, intermediate and lowest propensities for disulfide bond formation, respectively. In our model, G370 is at the interface between the two α helices, whereas G375 is almost opposite to the interacting face, in agreement with the data presented. Further, G380R (achondroplasia), V381E (hyperchondroplasia), G382D (multiple myeloma), and A391E (Crouzon syndrome with acanthosis nigricans, bladder cancer) are located close to interfacial positions.
This structural model of RTKs TM interaction is imperative for understanding disease mechanism caused by mutations found in the TM domain. With this structural model, we are currently designing inhibitory peptides to target the TM domain of FGFR.
Materials and Methods
Preparation of isotopically labeled 13C=18O amino acids and peptide synthesis
Amino acids labeled with carbonyl 13C=18O were obtained as described previously.34,37 The amino acids were then derivatized with 9-fluorenyl-methyloxycarbonyl (FMOC).57 For SSID measurements, two peptides corresponding to the transmembrane domain of FGFR3 (FGFR-TM: ADEAG370SVYAGILSYG380VGFFLFILVV390AAVTL395) (M.W. 3,063.6 d) were synthesized introducing isotopic labels (13C=18O) at consecutive sites, at V381 and G382. The use of only two labels is appropriate to discriminate between possible models that are separated by ω angles of at least 45°, because the error in ω using SSID is usually 10–20°.
All FGFR-TM peptides shown in Figure 1(a) were synthesized using standard solid-phase FMOC chemistry. The peptides were cleaved from the resin with trifluoroacetic acid (TFA) and lyophilized. Before lyophilization, a small amount of HCl was added to the fractions collected, to prevent formation of peptide-TFA adducts (which give rise to a band at ∼1685 cm−1). The lyophilized peptides were then dissolved in a small amount (∼5 μL) of hexafluoroisopropanol (HFIP) (Alfo Aesar, MA) and diluted with acetonitrile (ACN)/water mixture and injected into a Jupiter 5 C4-300 column (Phenomenex, Cheshire, UK) for HPLC purification. The ACN content increased linearly from 30 to 100%, as described previously.58 All solvents contained 0.1% (vol/vol) TFA. Peptide purity was confirmed by matrix-assisted laser desorption ionization-time of flight (MALDI-TOF).
Electrophoresis
Electrophoreses were performed in SDS and PFO.59 Peptides were first dissolved in a minimal amount of HFIP and diluted with NuPAGE® LDS sample buffer (Invitrogen) or PFO sample buffer, containing 4% PFO. Samples were heated at 94°C for 4 min and loaded on a NuPAGE® Novex 12% Bis-Tris gel [Fig. 1(b,c)] with MES SDS running buffer or Tris-Tricine 10–20% gel (Bio-Rad) [Fig. 1(d)] with Tris-tricine PFO running buffer. Electrophoresis were carried out at a constant voltage of 60 V till the dye ran out of the gel and stained with Coomassie blue.
Analytical ultracentrifugation
Sedimentation equilibrium experiments were performed using a Beckman XL-I analytical ultracentrifuge at 25°C.40 The absorbance was measured at 230 nm (ɛ230 = 5000M−1cm−1). The buffer composition was 20 mM MOPS (3-N-morpholino-propanesulfonic acid), 100 mM KCl, 1 mM MgCl2 at pH 7.4, and 10 mM dodecylphosphocholine (DPC). To match the density of DPC, D2O was added to the buffer to a final volume ratio of 50.5%.60 The density-matched buffer was added to the dry peptide–detergent film, obtained by lyophilization.
The samples were centrifuged in three-compartment carbon-epoxy centerpieces with quartz windows for lengths of time sufficient to achieve equilibrium, typically 20 h, and run at 28,000, 345,000, and 42,000 rpm. The equilibrium data sets were analyzed using the program ULTRASCAN.61 At the end of each run, absorbance data at 230 nm was obtained. The monomeric molecular mass of FGFR-TM and its partial specific volume were calculated with the program SEDNTERP.62
The partial specific volume, calculated after correction for partial hydrogen/deuterium exchange,40 was 0.767 cm3/g. Three different concentrations of FGFR-TM were used with peptide-to-detergent ratios were 1:100, 1:200, and 1:400, respectively. The log plot (ln A vs. r2–r) was not linear, indicating that at these peptide-to-detergent ratios more than one species was present. The data was best fitted to a monomer–dimer equilibrium, at the two smallest concentrations. The fit was not good at the highest concentration, and a tetrameric component was added.
Sample preparation for ATR-FTIR
Infrared spectra were recorded on a Nicolet Nexus spectrometer (Madison, USA) purged with N2 and equipped with a MCT/A detector, cooled with liquid nitrogen. Attenuated total reflection (ATR) spectra were measured with a 25-reflections ATR accessory from Graseby Specac (Kent, UK) and a wire grid polarizer (0.25 μm, Graseby Specac). A total of 200 interferograms collected at a resolution of 4 cm−1 were averaged for every sample and processed with 1 point zero filling and Happ-Genzel apodization. The peptide (typically ∼3 mg) was reconstituted in a 30:1 DMPC/peptide molar ratio34 and applied onto a trapezoidal (50 mm × 2 mm × 20 mm) Ge internal reflection element (IRE). A dry, or D2O-saturated, N2 stream flowing through the ATR compartment34 was used to remove bulk water or to achieve D2O exchange, respectively.
H/D exchange
The percentage of isotopic (hydrogen/deuterium, H/D) exchange was calculated from the area ratio amide II/amide I, using non polarized spectra, before and after exchange in D2O, as described previously.63 Non polarized spectra were obtained from the parallel (||) and perpendicular (⊥) ATR polarized spectra, using the expression 1 · (∥) + 1.44 · (⊥), as described previously.64
SSID data analysis
The data were analyzed according to the theory of site-specific dichroism presented in detail elsewhere.32 By measuring the orientation of the amide I transition dipole moment one can determine the helix tilt angle β and the rotational pitch angle ω of a specific dipole moment about the helix axis. The angle α between the transition dipole moment of the vibrational transition and the z axis was taken as 39° for the amide C=O bond and 29° for the N—H bond. The parameters ɛx, ɛy, and ɛz, the electric-field components, were used according to a thick film approximation. Dichroic ratios were calculated as the ratio between the integrated absorptions of the spectra collected with parallel and perpendicular polarized light.
The rotational pitch angle ω is defined arbitrarily as 0° when the transition dipole moment, the helix director, and the z axis all reside in a single plane. The difference of ω between two consecutive residues was assumed to be 100°, as in a canonical α helix. The rotational orientation and tilt for each labeled residue was calculated as described.32,46
Global search molecular dynamics (GSMD) protocol
The simulations were performed using a Compaq Alpha Cluster SC45 that contains 44 nodes. All calculations were carried out using the parallel version of the Crystallography and NMR System (CNS Version 0.3), the Parallel Crystallography and NMR System (PCNS).65 The global search was carried out in vacuo with united atoms, explicitly describing only polar and aromatic hydrogen atoms as described elsewhere66 using CHI 1.1 (CNS Helical Interactions). As the models tested are homooligomers, the interaction between the helices was assumed to be symmetrical.
Trials were carried out starting from either left or right crossing angle configurations. The initial helix tilt, β, was restrained to 0° and the helices were rotated about their long helical axes in 10° increments until the rotation angle reached 350°. Henceforth, the simulation was repeated by increasing the helix tilt in discrete steps of 5°, up to 45°. We must note that the restraint for the helix tilt is not completely strict, that is, at the end of the simulation a drift of up to ±5° from the initial restrained value could be observed in some cases. Three trials were carried out for each starting configuration using different initial random velocities.
Clusters were identified with a minimum number of eight similar structures. Any structure belonging to a certain cluster was within 1.5 Å root mean square deviation (RMSD) from any other structure within the same cluster. Finally, the structures belonging to each cluster were averaged and subjected to energy minimization. These final averaged structures, described by a certain tilt and rotational orientation at a specified arbitrary residue, were taken as the representatives of the respective clusters.
The tilt angle of the models, β, was taken as the average of the angles between each helix axis in the bundle and the bundle axis. The bundle axis, coincident with the normal to the bilayer, was calculated by CHI. The helix axis was calculated as a vector with starting and end points above and below a defined residue, where the points correspond to the geometric mean of the coordinates of the five α carbons N-terminal and the five α carbons C-terminal to the defined residue. The rotational orientation angle ω of a residue is defined by the angle between a vector perpendicular to the helix axis, oriented towards the middle of the peptidic C=O bond of the residue, and a plane that contains both the helical axis and the normal to the bilayer. In this work, to compare the models, a residue was chosen arbitrarily, and the ω angle is always given for residue V381. Intersequence comparisons between low-energy clusters were performed by calculating the RMSD between their α-carbon backbone. Fitting was performed using the program ProFit (http://www.bioinf.org.uk/software/profit). The energies calculated correspond to the total energy of the system, including both bonded, for example, bond, angle, dihedral, improper, and nonbonded, that is, Van der Waals and electrostatic terms.66 The interaction energy for the residues was calculated with the function chi_interaction implemented in CHI.
A total number of homologous 9 sequences corresponding to FGFR-TM were used for the simulations. These sequences were obtained using ncbi homoloGene search (http://www.ncbi.nlm.nih.gov/). Two different sequence lengths were used: residues 366–395 and 369–399.
References
- 1.Schlessinger J. Cell signaling by receptor tyrosine kinases. Cell. 2000;103:211–225. doi: 10.1016/s0092-8674(00)00114-8. [DOI] [PubMed] [Google Scholar]
- 2.Mohammadi M, McMahon G, Sun L, Tang C, Hirth P, Yeh BK, Hubbard SR, Schlessinger J. Structures of the tyrosine kinase domain of fibroblast growth factor receptor in complex with inhibitors. Science. 1997;276:955–960. doi: 10.1126/science.276.5314.955. [DOI] [PubMed] [Google Scholar]
- 3.Hubbard SR, Mohammadi M, Schlessinger J. Autoregulatory mechanisms in protein-tyrosine kinases. J Biol Chem 1998. 1998;273:11987–11990. doi: 10.1074/jbc.273.20.11987. [DOI] [PubMed] [Google Scholar]
- 4.Mendrola JM, Berger MB, King MC, Lemmon MA. The single transmembrane domains of ErbB receptors self-associate in cell membranes. J Biol Chem. 2002;277:4704–4712. doi: 10.1074/jbc.M108681200. [DOI] [PubMed] [Google Scholar]
- 5.Bennasroune A, Fickova M, Gardin A, Dirrig-Grosch S, Aunis D, Cremel G, Hubert P. Transmembrane peptides as inhibitors of ErbB receptor signaling. Mol Biol Cell. 2004;15:3464–3474. doi: 10.1091/mbc.E03-10-0753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bell CA, Tynan JA, Hart KC, Meyer AN, Robertson SC, Donoghue DJ. Rotational coupling of the transmembrane and kinase domains of the Neu receptor tyrosine kinase. Mol Biol Cell. 2000;11:3589–3599. doi: 10.1091/mbc.11.10.3589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Livnah O, Johnson DL, Stura EA, Farrell FX, Barbone FP, You Y, Liu KD, Goldsmith MA, He W, Krause CD, Pestka S, Jolliffe LK, Wilson IA. An antagonist peptide-EPO receptor complex suggests that receptor dimerization is not sufficient for activation. Nat Struct Biol. 1998;5:993–1004. doi: 10.1038/2965. [DOI] [PubMed] [Google Scholar]
- 8.Adar R, Monsonego-Ornan E, David P, Yayon A. Differential activation of cysteine-substitution mutants of fibroblast growth factor receptor 3 is determined by cysteine localization. J Bone Miner Res. 2002;17:860–868. doi: 10.1359/jbmr.2002.17.5.860. [DOI] [PubMed] [Google Scholar]
- 9.Li E, Hristova K. Role of receptor tyrosine kinase transmembrane domains in cell signaling and human pathologies. Biochemistry. 2006;45:6241–6251. doi: 10.1021/bi060609y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Massague J, Pilch PF, Czech MP. A unique proteolytic cleavage site on the beta subunit of the insulin receptor. J Biol Chem. 1981;256:3182–3190. [PubMed] [Google Scholar]
- 11.Luo RZ, Beniac DR, Fernandes A, Yip CC, Ottensmeyer FP. Quaternary structure of the insulin-insulin receptor complex. Science. 1999;285:1077–1080. doi: 10.1126/science.285.5430.1077. [DOI] [PubMed] [Google Scholar]
- 12.Hynes RO. Integrins: bidirectional, allosteric signaling machines. Cell. 2002;110:673–687. doi: 10.1016/s0092-8674(02)00971-6. [DOI] [PubMed] [Google Scholar]
- 13.Muenke M, Schell U. Fibroblast-growth-factor receptor mutations in human skeletal disorders. Trends Genet. 1995;11:308–313. doi: 10.1016/s0168-9525(00)89088-5. [DOI] [PubMed] [Google Scholar]
- 14.Rousseau F, Bonaventure J, Legeai-Mallet L, Pelet A, Rozet JM, Maroteaux P, Le Merrer M, Munnich A. Mutations in the gene encoding fibroblast growth factor receptor-3 in achondroplasia. Nature. 1994;371:252–254. doi: 10.1038/371252a0. [DOI] [PubMed] [Google Scholar]
- 15.Tavormina PL, Rimoin DL, Cohn DH, Zhu YZ, Shiang R, Wasmuth JJ. Another mutation that results in the substitution of an unpaired cysteine residue in the extracellular domain of FGFR3 in thanatophoric dysplasia type I. Hum Mol Genet. 1995;4:2175–2177. doi: 10.1093/hmg/4.11.2175. [DOI] [PubMed] [Google Scholar]
- 16.Rousseau F, el Ghouzzi V, Delezoide AL, Legeai-Mallet L, Le Merrer M, Munnich A, Bonaventure J. Missense FGFR3 mutations create cysteine residues in thanatophoric dwarfism type I (TD1) Hum Mol Genet. 1996;5:509–512. doi: 10.1093/hmg/5.4.509. [DOI] [PubMed] [Google Scholar]
- 17.Meyers GA, Orlow SJ, Munro IR, Przylepa KA, Jabs EW. Fibroblast growth factor receptor 3 (FGFR3) transmembrane mutation in Crouzon syndrome with acanthosis nigricans. Nat Genet. 1995;11:462–464. doi: 10.1038/ng1295-462. [DOI] [PubMed] [Google Scholar]
- 18.Otsuki T, Nakazawa N, Taniwaki M, Yamada O, Sakaguchi H, Wada H, Yawata Y, Ueki A. Establishment of a new human myeloma cell line, KMS-18, having t(4;14)(p16.3;q32.3) derived from a case phenotypically transformed from Ig A-lambda to BJP-lambda, and associated with hyperammonemia. Int J Oncol. 1998;12:545–552. doi: 10.3892/ijo.12.3.545. [DOI] [PubMed] [Google Scholar]
- 19.van Rhijn BW, van Tilborg AA, Lurkin I, Bonaventure J, de Vries A, Thiery JP, van der Kwast TH, Zwarthoff EC, Radvanyi F. Novel fibroblast growth factor receptor 3 (FGFR3) mutations in bladder cancer previously identified in non-lethal skeletal disorders. Eur J Hum Genet. 2002;10:819–824. doi: 10.1038/sj.ejhg.5200883. [DOI] [PubMed] [Google Scholar]
- 20.Bange J, Prechtl D, Cheburkin Y, Specht K, Harbeck N, Schmitt M, Knyazeva T, Muller S, Gartner S, Sures I, Wang H, Imyanitov E, Haring HU, Knayzev P, Iacobelli S, Hofler H, Ullrich A. Cancer progression and tumor cell motility are associated with the FGFR4 Arg(388) allele. Cancer Res. 2002;62:840–847. [PubMed] [Google Scholar]
- 21.Li E, You M, Hristova K. FGFR3 dimer stabilization due to a single amino acid pathogenic mutation. J Mol Biol. 2006;356:600–612. doi: 10.1016/j.jmb.2005.11.077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Smith SO, Smith CS, Bormann BJ. Strong hydrogen bonding interactions involving a buried glutamic acid in the transmembrane sequence of the neu/erbB-2 receptor. Nat Struct Biol. 1996;3:252–258. doi: 10.1038/nsb0396-252. [DOI] [PubMed] [Google Scholar]
- 23.Smith SO, Smith C, Shekar S, Peersen O, Ziliox M, Aimoto S. Transmembrane interactions in the activation of the Neu receptor tyrosine kinase. Biochemistry. 2002;41:9321–9332. doi: 10.1021/bi012117l. [DOI] [PubMed] [Google Scholar]
- 24.Webster MK, Donoghue DJ. Constitutive activation of fibroblast growth factor receptor 3 by the transmembrane domain point mutation found in achondroplasia. Embo J. 1996;15:520–527. [PMC free article] [PubMed] [Google Scholar]
- 25.Monsonego-Ornan E, Adar R, Feferman T, Segev O, Yayon A. The transmembrane mutation G380R in fibroblast growth factor receptor 3 uncouples ligand-mediated receptor activation from down-regulation. Mol Cell Biol. 2000;20:516–522. doi: 10.1128/mcb.20.2.516-522.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cho JY, Guo C, Torello M, Lunstrum GP, Iwata T, Deng C, Horton WA. Defective lysosomal targeting of activated fibroblast growth factor receptor 3 in achondroplasia. Proc Natl Acad Sci USA. 2004;101:609–614. doi: 10.1073/pnas.2237184100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.You M, Li E, Hristova K. The achondroplasia mutation does not alter the dimerization energetics of the fibroblast growth factor receptor 3 transmembrane domain. Biochemistry. 2006;45:5551–5556. doi: 10.1021/bi060113g. [DOI] [PubMed] [Google Scholar]
- 28.Luo BH, Springer TA, Takagi J. A specific interface between integrin transmembrane helices and affinity for ligand. PLoS Biol. 2004;2:776–786. doi: 10.1371/journal.pbio.0020153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schneider D, Engelman DM. Involvement of transmembrane domain interactions in signal transduction by α/β integrins. J Biol Chem. 2004;279:9840–9846. doi: 10.1074/jbc.M312749200. [DOI] [PubMed] [Google Scholar]
- 30.Li E, You M, Hristova K. Sodium dodecyl sulfate-polyacrylamide gel electrophoresis and forster resonance energy transfer suggest weak interactions between fibroblast growth factor receptor 3 (FGFR3) transmembrane domains in the absence of extracellular domains and ligands. Biochemistry. 2005;44:352–360. doi: 10.1021/bi048480k. [DOI] [PubMed] [Google Scholar]
- 31.Stanley AM, Fleming KG. The transmembrane domains of ErbB receptors do not dimerize strongly in micelles. J Mol Biol. 2005;347:759–772. doi: 10.1016/j.jmb.2005.01.059. [DOI] [PubMed] [Google Scholar]
- 32.Arkin IT, MacKenzie KR, Brunger AT. Site-directed dichroism as a method for obtaining rotational and orientational constraints for oriented polymers. J Am Chem Soc. 1997;119:8973–8980. [Google Scholar]
- 33.Kukol A, Adams PD, Rice LM, Brunger AT, Arkin TI. Experimentally based orientational refinement of membrane protein models: a structure for the Influenza A M2 H+ channel. J Mol Biol. 1999;286:951–962. doi: 10.1006/jmbi.1998.2512. [DOI] [PubMed] [Google Scholar]
- 34.Torres J, Adams PD, Arkin IT. Use of a new label, 13C=18O, in the determination of a structural model of phospholamban in a lipid bilayer. Spatial restraints resolve the ambiguity arising from interpretations of mutagenesis data. J Mol Biol. 2000;300:677–685. doi: 10.1006/jmbi.2000.3885. [DOI] [PubMed] [Google Scholar]
- 35.Torres J, Parthasarathy K, Lin X, Saravanan R, Kukol A, Liu DX. Model of a putative pore: the pentameric α-helical bundle of SARS coronavirus E protein in lipid bilayers. Biophys J. 2006;91:938–947. doi: 10.1529/biophysj.105.080119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Parthasarathy K, Ng L, Lin X, Liu DX, Pervushin K, Gong X, Torres J. Structural flexibility of the pentameric SARS coronavirus envelope protein ion channel. Biophys J. 2008;95:L39–L41. doi: 10.1529/biophysj.108.133041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Torres J, Kukol A, Goodman JM, Arkin IT. Site-specific examination of secondary structure and orientation determination in membrane proteins: the peptidic 13C=18O group as a novel infrared probe. Biopolymers. 2001;59:396–401. doi: 10.1002/1097-0282(200111)59:6<396::AID-BIP1044>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- 38.Beevers AJ, Kukol A. Secondary structure, orientation, and oligomerization of phospholemman, a cardiac transmembrane protein. Protein Sci. 2006;15:1127–1132. doi: 10.1110/ps.051899406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gan SW, Ng L, Xin L, Gong X, Torres J. Structure and ion channel activity of the human respiratory syncytial virus (hRSV) small hydrophobic protein transmembrane domain. Protein Sci. 2008;17:813–820. doi: 10.1110/ps.073366208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kochendoerfer GG, Salom D, Lear JD, Wilk-Orescan R, Kent SB, DeGrado WF. Total chemical synthesis of the integral membrane protein influenza A virus M2: role of its C-terminal domain in tetramer assembly 1. Biochemistry. 1999;38:11905–11913. doi: 10.1021/bi990720m. [DOI] [PubMed] [Google Scholar]
- 41.Fleming KG. Standardizing the free energy change of transmembrane helix-helix interactions. J Mol Biol. 2002;323:563–571. doi: 10.1016/s0022-2836(02)00920-8. [DOI] [PubMed] [Google Scholar]
- 42.Cristian L, Lear JD, DeGrado WF. Use of thiol-disulfide equilibria to measure the energetics of assembly of transmembrane helices in phospholipid bilayers. Proc Natl Acad Sci USA. 2003;100:14772–14777. doi: 10.1073/pnas.2536751100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Stouffer AL, Ma C, Cristian L, Ohigashi Y, Lamb RA, Lear JD, Pinto LH, DeGrado WF. The interplay of functional tuning, drug resistance, and thermodynamic stability in the evolution of the M2 proton channel from the influenza A virus. Structure. 2008;16:1067–1076. doi: 10.1016/j.str.2008.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Briggs JAG, Torres J, Arkin IT. A new method to model membrane protein structure based on silent amino acid substitutions. Proteins. 2001;44:370–375. doi: 10.1002/prot.1102. [DOI] [PubMed] [Google Scholar]
- 45.Byler DM, Susi H. Examination of the secondary structure of proteins by deconvolved FTIR spectra. Biopolymers. 1986;25:469–487. doi: 10.1002/bip.360250307. [DOI] [PubMed] [Google Scholar]
- 46.Torres J, Briggs JA, Arkin IT. Multiple site-specific infrared dichroism of CD3-z, a transmembrane helix bundle. J Mol Biol. 2002;316:365–374. doi: 10.1006/jmbi.2001.5267. [DOI] [PubMed] [Google Scholar]
- 47.You M, Spangler J, Li E, Han X, Ghosh P, Hristova K. Effect of pathogenic cysteine mutations on FGFR3 transmembrane domain dimerization in detergents and lipid bilayers. Biochemistry. 2007;46:11039–11046. doi: 10.1021/bi700986n. [DOI] [PubMed] [Google Scholar]
- 48.Doura AK, Kobus FJ, Dubrovsky L, Hibbard E, Fleming KG. Sequence context modulates the stability of a GxxxG-mediated transmembrane helix-helix dimer. J Mol Biol. 2004;341:991–998. doi: 10.1016/j.jmb.2004.06.042. [DOI] [PubMed] [Google Scholar]
- 49.Fisher LE, Engelman DM, Sturgis JN. Effect of detergents on the association of the glycophorin a transmembrane helix. Biophys J. 2003;85:3097–3105. doi: 10.1016/S0006-3495(03)74728-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fleming KG, Ren CC, Doura AK, Eisley ME, Kobus FJ, Stanley AM. Thermodynamics of glycophorin A transmembrane helix dimerization in C14 betaine micelles. Biophys Chem. 2004;108:43–49. doi: 10.1016/j.bpc.2003.10.008. [DOI] [PubMed] [Google Scholar]
- 51.Stouffer AL, Nanda V, Lear JD, DeGrado WF. Sequence determinants of a transmembrane proton channel: an inverse relationship between stability and function. J Mol Biol. 2005;347:169–179. doi: 10.1016/j.jmb.2005.01.023. [DOI] [PubMed] [Google Scholar]
- 52.Stouffer AL, DeGrado WF, Lear JD. Analytical ultracentrifugation Studies of the Influenza M2 homotetramerization equilibrium in detergent solutions. Progr Colloid Polym Sci. 2006;131:108–115. [Google Scholar]
- 53.Li RH, Mitra N, Gratkowski H, Vilaire G, Litvinov R, Nagasami C, Weisel JW, Lear JD, DeGrado WF, Bennett JS. Activation of integrin αIIbβ3 by modulation of transmembrane helix associations. Science. 2003;300:795–798. doi: 10.1126/science.1079441. [DOI] [PubMed] [Google Scholar]
- 54.Gerber D, Sal-Man N, Shai Y. Two motifs within a transmembrane domain, one for homodimerization and the other for heterodimerization. J Biol Chem. 2004;279:21177–21182. doi: 10.1074/jbc.M400847200. [DOI] [PubMed] [Google Scholar]
- 55.Vajo Z, Francomano CA, Wilkin DJ. The molecular and genetic basis of fibroblast growth factor receptor 3 disorders: the achondroplasia family of skeletal dysplasias, Muenke craniosynostosis, and Crouzon syndrome with acanthosis nigricans. Endocr Rev. 2000;21:23–39. doi: 10.1210/edrv.21.1.0387. [DOI] [PubMed] [Google Scholar]
- 56.Shiang R, Thompson LM, Zhu YZ, Church DM, Fielder TJ, Bocian M, Winokur ST, Wasmuth JJ. Mutations in the transmembrane domain of FGFR3 cause the most common genetic form of dwarfism, achondroplasia. Cell. 1994;78:335–342. doi: 10.1016/0092-8674(94)90302-6. [DOI] [PubMed] [Google Scholar]
- 57.Wellings DA, Atherton E. Standard Fmoc protocols. Methods Enzymol. 1997;289:44–67. doi: 10.1016/s0076-6879(97)89043-x. [DOI] [PubMed] [Google Scholar]
- 58.Iwamoto T, You M, Li E, Spangler J, Tomich JM, Hristova K. Synthesis and initial characterization of FGFR3 transmembrane domaconsequences of sequence modifications. Biochim Biophys Acta. 2005;1668:240–247. doi: 10.1016/j.bbamem.2004.12.012. [DOI] [PubMed] [Google Scholar]
- 59.Ramjeesingh M, Huan LJ, Garami E, Bear CE. Novel method for evaluation of the oligomeric structure of membrane proteins. Biochem J. 1999;342(Part 1):119–123. [PMC free article] [PubMed] [Google Scholar]
- 60.Li R, Babu CR, Lear JD, Wand AJ, Bennett JS, DeGrado WF. Oligomerization of the integrin αIIbβ3: roles of the transmembrane and cytoplasmic domains. Proc Nat Sci USA. 2001;98:12462–12467. doi: 10.1073/pnas.221463098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Demeler B. UltraScan, a comprehensive data analysis software package for analytical ultracentrifuge experiments. In: Scott DJ, editor; Harding SE, editor; Rowe AJ, editor. Modern analytical ultracentrifugation: techniques and methods. Royal Society of Chemistry (UK); 2005. pp. 210–229. [Google Scholar]
- 62.Laue TM, Stafford WF., III Modern applications of analytical ultracentrifugation. Annu Rev Biophys Biomol Struct. 1999;28:75–100. doi: 10.1146/annurev.biophys.28.1.75. [DOI] [PubMed] [Google Scholar]
- 63.McGhie EJ, Hume PJ, Hayward RD, Torres J, Koronakis V. Topology of the Salmonella invasion protein SipB in a model bilayer. Mol Microbiol. 2002;44:1309–1321. doi: 10.1046/j.1365-2958.2002.02958.x. [DOI] [PubMed] [Google Scholar]
- 64.Marsh D. Quantitation of secondary structure in ATR infrared spectroscopy. Biophys J. 1999;77:2630–2637. doi: 10.1016/S0006-3495(99)77096-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL. Crystallography & NMR system: a new software suite for macromolecular structure determination. Acta Crystallogr Sect D. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 66.Adams PD, Arkin IT, Engelman DM, Brunger AT. Computational searching and mutagenesis suggest a structure for the pentameric transmembrane domain of phospholamban. Nat Struct Biol. 1995;2:154–162. doi: 10.1038/nsb0295-154. [DOI] [PubMed] [Google Scholar]