

Abstract
Hexameric AAA+ ATPases induce conformational changes in a variety of macromolecules. AAA+ structures contain the nucleotide-binding P-loop with the Walker A sequence motif: GxxGxGK(T/S). A subfamily of AAA+ sequences contains Asn in the Walker A motif instead of Thr or Ser. This noncanonical subfamily includes torsinA, an ER protein linked to human dystonia and DnaC, a bacterial helicase loader. Role of the noncanonical Walker A motif in the functionality of AAA+ ATPases has not been explored yet. To determine functional effects of introduction of Asn into the Walker A sequence, we replaced the Walker-A Thr with Asn in ClpB, a bacterial AAA+ chaperone which reactivates aggregated proteins. We found that the T-to-N mutation in Walker A partially inhibited the ATPase activity of ClpB, but did not affect the ClpB capability to associate into hexamers. Interestingly, the noncanonical Walker A sequence in ClpB induced preferential binding of ADP vs. ATP and uncoupled the linkage between the ATP-bound conformation and the high-affinity binding to protein aggregates. As a consequence, ClpB with the noncanonical Walker A sequence showed a low chaperone activity in vitro and in vivo. Our results demonstrate a novel role of the Walker-A Thr in sensing the nucleotide's γ-phosphate and in maintaining an allosteric linkage between the P-loop and the aggregate binding site of ClpB. We postulate that AAA+ ATPases with the noncanonical Walker A might utilize distinct mechanisms to couple the ATPase cycle with their substrate-remodeling activity.
Keywords: AAA+ ATPase, molecular chaperone, protein aggregation, aggregate reactivation, site-directed mutagenesis, nucleotide binding
Introduction
AAA+ ATPases (ATPases associated with various cellular activities) are molecular machines that use energy from ATP hydrolysis to induce conformational changes in macromolecules or to disassemble macromolecular complexes.1,2 Members of the AAA+ family play essential roles in protein disaggregation and degradation, vesicular transport and membrane fusion, DNA replication and repair, and cytoskeletal regulation. AAA+ ATPases contain one or two sequence modules with several conserved motifs involved in nucleotide binding and ATP hydrolysis. The biologically active form of most characterized AAA+ ATPases is a nucleotide-stabilized hexameric ring.3
Heat-shock protein ClpB, a member of the AAA+ family, is essential for survival of bacteria, yeast, and plants under severe thermal stress.4 ClpB contains two AAA+ sequence modules (D1, D2) and two additional mobile domains: the N-terminal domain and the coiled-coil middle domain.5 ClpB reactivates aggregated proteins in cooperation with the DnaK chaperone system.6–8 The ClpB activity is linked to extraction of single polypeptides from aggregated particles and their forced unfolding by translocation through a narrow channel located at the center of the hexameric ring.9 The substrate translocation by ClpB is driven by ATP hydrolysis and is analogous to the degradation-preceding unfolding/translocation of substrates by the proteasome and other ATP-dependent proteases.10 Aggregated substrates bind to D1 and to the N-terminal domain of ClpB,11,12 become translocated through the channel from D1 to D2, then exit the ClpB oligomer and their further folding in solution may be assisted by other chaperones.
Each AAA+ module of ClpB contains a flexible loop located inside the channel.5 The loops bind polypeptide substrates and are involved in their forced translocation.11,13 ATP stabilizes the position of the D1 loop, whereas the loop retains its flexibility in the presence of ADP.14 This observation is consistent with the ATP-induced high-affinity binding of substrates by ClpB and a low affinity of the ADP state15 and implies a conformational linkage between the channel loop and the ATP-binding site. It is not known, however, how the nucleotide-occupancy information is relayed between the two sites.
Walker A motif is found in the phosphate-binding loop (P-loop) of many ATP-and GTP-binding proteins. The Walker A consensus sequence of AAA+ ATPases: GxxGxGK[T/S], contains an essential lysine which supports binding of nucleotides. The role of the flanking hydroxyl-containing residue (Thr in both AAA+ modules of ClpB) is less understood. Phylogenetic analysis shows that in the highly divergent “evolutionary tree” of AAA+ ATPases, the D2 module of ClpB is specifically related to a family of animal AAA+ proteins called torsins.16 A member of the torsin family, torsinA, has been linked to early onset dystonia, a human neurological disease.17 Interestingly, most torsins, including torsinA, contain a noncanonical Walker A sequence: GxxGxGKN. Besides the torsin family, an Asn in the Walker A motif is found in DnaC, a bacterial AAA+ helicase loader.18 Intriguingly, the biochemical properties of torsinA and DnaC do not fully conform to the AAA+ paradigm: those proteins do not form hexamers and have low ATPase activity in the absence of other macromolecules.18,19
We hypothesized that the last residue of the Walker A motif may participate in coupling the binding of nucleotides with interactions of an AAA+ ATPase with substrates or partner proteins. We replaced the Walker-A Thr in each AAA+ module of ClpB with Asn and discovered that the noncanonical sequence partially inhibited the ATPase and aggregate-reactivation activity, modified the nucleotide-binding affinity, and altered the linkage between nucleotide-occupancy and aggregate binding. Our results suggest a specific role of the Walker-A threonine in controlling the functionality of an AAA+ ATPase.
Results
We asked if a replacement of the canonical Thr in the Walker A motif with Asn would affect the biochemical properties and activity of ClpB. We produced three ClpB variants: two with the mutation in the Walker A motif in one AAA+ module (T213N in D1 or T612N in D2) and the double-mutant with both AAA+ modules modified (T213N/T612N). First, we tested the structural integrity of the mutated ClpB. As shown in Figure 1, ClpB(T213N/T612N) assembled into oligomers in the presence of ATP or ADP. This property of ClpB(T213/T612N) is indistinguishable from the well-described nucleotide-induced oligomerization of wt ClpB.20 The result shown in Figure 1 indicates first that, the T-to-N substitution in the Walker A sequence does not globally destabilize the structure of a AAA+ module and second that, ClpB(T213N/T612N) interacts with and responds to ATP and ADP.
Figure 1.
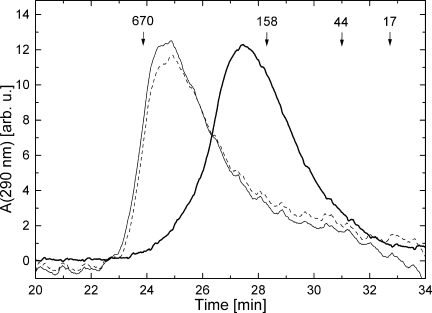
Gel-filtration analysis of ClpB(T213N/T612N). Elution profiles were measured in the absence of nucleotides (thick solid line), with 2 mM ATP (thin solid line), or with 2 mM ADP (broken line). Elution times of the molecular-weight standards (kDa) are indicated.
A replacement of Thr with Asn in the Walker A sequence partially inhibited the rate of ATP hydrolysis by ClpB [Fig. 2(A)]. Single mutations in either D1 or D2 produced a similar ∼50% decrease in the ATPase activity, but the rate of phosphate production from ATP by ClpB(T213N/T612N) was approximately sevenfold lower than that of wt ClpB. This result demonstrates that the T-to-N change in the Walker A sequence may affect the thermodynamics and/or kinetics of the nucleotide binding/dissociation or the catalytic efficiency. Interestingly, the ATPase of all ClpB variants was activated by soluble pseudosubstrates of ClpB: casein and poly-lysine [Fig. 2(B)]. Thus, in spite of being less efficient ATPases, the T-to-N ClpB variants respond to soluble activators. The mechanism of ATPase activation by casein or poly-lysine is not known, but it is accepted that the ATPase acceleration accompanies translocation of the soluble pseudosubstrates through the channel of ClpB.9 Thus, our results suggest that the translocation machinery may remain functional in the ClpB variants with the noncanonical Walker A motif.
Figure 2.
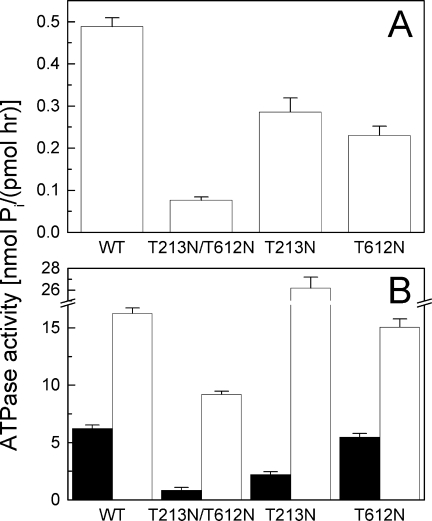
ATPase activity of ClpB. The rate of ATP hydrolysis was measured for wt ClpB or its variants in the absence of other polypeptides (A), in the presence of 0.1 mg/mL κ-casein (B, black bars), or with 0.04 mg/mL poly-l-lysine (B, white bars).
To determine possible effects of the noncanonical Walker A sequence on nucleotide binding, we investigated interactions of ATPγS, a poorly hydrolyzable ATP analog and ADP with wt ClpB and its T213N/T612N variant using titration calorimetry. The experiments were performed under low ionic-strength conditions to induce oligomerization of ClpB before adding nucleotides.21 The nucleotide binding site in D1 of ClpB is different from that in D2.5 Thus, there are two classes of six equivalent nucleotide binding sites in the hexameric ClpB. Studies on a related ATPase ClpX demonstrated complex allosteric linkages between multiple ATP-binding sites resulting in positive as well as negative cooperativity in ATP binding.22 As it is not known which binding mechanism applies to nucleotide interactions with 12 sites in the hexameric ClpB, we restrict our analysis to observing the shape of binding isotherms to obtain information on the effective nucleotide-binding capability of ClpB.
As shown in Figure 3(A), the incremental binding isotherm for ATPγS with wt ClpB parallels that of ADP (with a small difference in the binding enthalpy). This result indicates that wt ClpB displays a similar apparent affinity for ATP and ADP. Under physiological conditions of an ATP excess, the ATP-bound state of wt ClpB is highly populated, which supports efficient ATP-hydrolysis and the ATP-driven chaperone activity. In contrast to wt ClpB, the T213N/T612N variant showed a sigmoidal incremental binding isotherm for ADP, but not for ATPγS [Fig. 3(B)]. A sigmoidal incremental binding curve produces a steeper saturation curve and implies a stronger overall binding affinity for ADP than for ATPγS.23 This result indicates that the noncanonical Walker A sequence in ClpB disturbs the balance between the protein's affinity for ADP and ATP. ClpB(T213N/T612N) may preferentially populate its ADP-bound state which decelerates the ATP-hydrolysis cycle, as was indeed observed in Figure 2(A).
Figure 3.
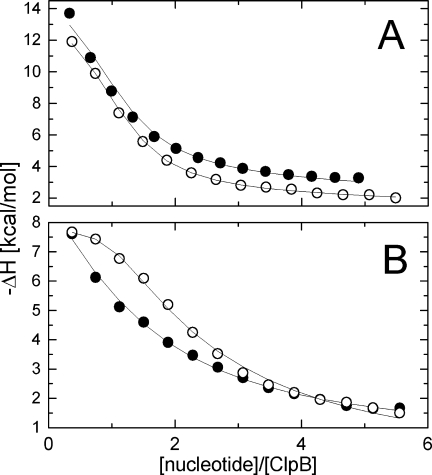
Nucleotide binding to ClpB. Shown are the heat effects of titrating 50 μM wt ClpB (A) and ClpB(T213N/T612N) (B) with ATPγS (•) or ADP (○). Solid lines show fits of an empirical model of multiple ligand-binding sites (MCS ITC software, MicroCal Inc.). The enthalpy of binding is negative which indicates an exothermic reaction.
The biological function of ClpB is recognition and reactivation of aggregated proteins. We produced large aggregates of two substrates: glucose-6-phosphate dehydrogenase (G6PDH) and malate dehydrogenase (MDH) that can be reactivated by ClpB with the DnaK system of cochaperones.8,12 We incubated the aggregates with ClpB in the absence and presence of different nucleotides and then determined the amount of ClpB bound to the aggregated particles (see Fig. 4). Background amounts of ClpB were retained on filters in the absence of aggregates or with aggregates in the presence of ADP or ATP. As expected, binding of wt ClpB to the aggregates was stimulated by ATPγS (which simulates a “frozen” ATP-bound ClpB state), but interestingly, ClpB(T213N) and ClpB(T213N/T612N) showed only background binding to the aggregates in the presence of ATPγS. In contrast, ClpB(T612N) retained its aggregate-binding capability. Thus, introduction of the noncanonical Walker A sequence into D1, but not D2 of ClpB inhibits binding of aggregated substrates under conditions expected to stimulate high-affinity interactions.
Figure 4.
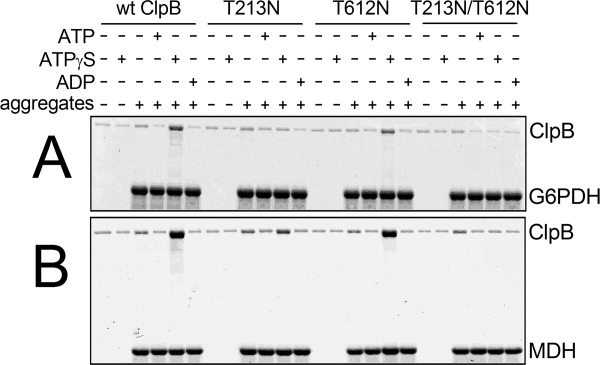
Interactions of ClpB with aggregated substrates. Wt ClpB, ClpB(T213N), ClpB(T612N), or ClpB(T213N/T612N) (1.5 μM protein) were incubated without or with aggregated 3 μM glucose-6-phosphate dehydrogenase (A) or 3 μM malate dehydrogenase (B) without nucleotides or with 5 mM ATP, ATPγS, or ADP. The solutions were passed through a 0.1-μm filter. SDS-PAGE analysis with a Coomassie stain is shown for the fractions retained on the filter and subsequently solubilized with an SDS buffer. Representative results from two independent experiments are shown.
Not surprisingly, ClpB(T213N) and ClpB(T213N/T612N) failed to reactivate aggregated G6PDH in the presence of the DnaK system (see Fig. 5). Interestingly, the rate of reactivation of the aggregated G6PDH by ClpB(T612N) was also significantly lower than that of wt ClpB. These results indicate that the T-to-N substitution in either D1 or D2 Walker A sequence inhibits the chaperone activity of ClpB in vitro. We also found that the modification of the Walker A motif significantly inhibited the activity of ClpB in vivo (see Fig. 6). Compared with wt E. coli, the clpB-null strain poorly survives heat shock due to severe protein aggregation. Survival of that strain at a lethal temperature can be restored after expression of wt ClpB. As shown in Figure 6(inset), the amounts of ClpB(T213N), ClpB(T612N) and ClpB(T213N/T612N) in E. coli cells after 6 h at 50°C were comparable with that of wt ClpB. However, expression of ClpB(T213N), ClpB(T612N), and, most significantly, ClpB(T213N/T612N) did not restore the full viability of cells after heat shock. Thus, introduction of the noncanonical Walker A sequence affects the biological functionality of ClpB.
Figure 5.
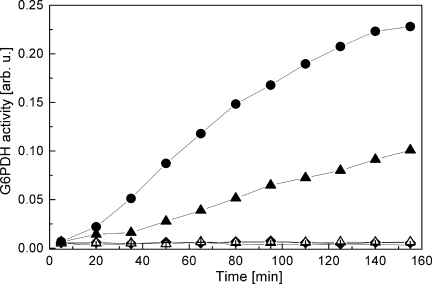
Reactivation of aggregated G6PDH in the presence of ClpB and the DnaK chaperone system. Aggregated 3 μM G6PDH was incubated without ClpB with 1 μM DnaK, 1 μM DnaJ, 0.5 μM GrpE (KJE) (+), KJE with 1.5 μM wt ClpB (•), KJE with 1.5 μM ClpB(T213N) (▵), KJE with 1.5 μM ClpB(T612N) (▴), or KJE with 1.5 μM ClpB(T213N/T612N) (♦). After incubation at 30°C for the indicated time, the G6PDH activity was measured as described in Materials and Methods. Representative results from two independent experiments are shown.
Figure 6.
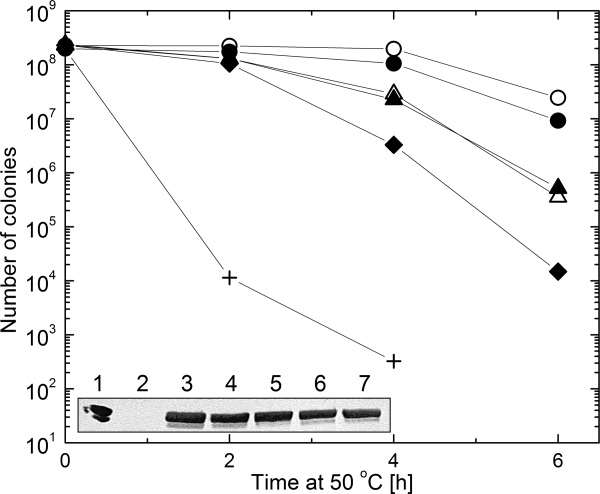
In vivo chaperone activity of ClpB. E. coli cells were grown at 50°C until the indicated time and their survival was determined as described in Materials and Methods. The data are shown for wt MC4100 E. coli (○), ClpB-null E. coli containing a control pGB2 plasmid (+), pGB2 with wt ClpB sequence (•), pGB2 with ClpB(T213N) (▵), pGB2 with ClpB(T612N) (▴), or pGB2 with ClpB(T213N/T612N) (♦). Inset shows immunodetection of ClpB in bacterial cultures after 6 h of heat shock. Purified ClpB is shown in Lane 1, empty pGB2 in Lane 2, wt E. coli in Lane 3, pGB2(wt ClpB) in Lane 4, pGB2(T213N) in Lane 5, pGB2(T612N) in Lane 6, and pGB2(T213N/T612N) in Lane 7.
Discussion
The mechanism of aggregate reactivation by ClpB agrees with the established functional paradigm of the canonical AAA+ ATPases.2 Nucleotides induce association of ClpB into hexamers (see Fig. 1). Wt ClpB displays a high affinity toward aggregated substrates in its ATP-bound conformation (see Fig. 4). The affinities of ClpB for ATP and ADP are similar [see Fig. 3(A)], which supports an efficient nucleotide turnover and energy generation as long as enough ATP is available.
In the nucleotide-free structure of D1 of E. coli ClpB, the Walker-A threonine side chain is exposed inside the nucleotide binding site in the vicinity of Mg2+ which supports binding of the phosphates of ATP.24 In the AMP-PNP-bound ClpB from T. thermophilus, the nucleotide γ-phosphate is sandwiched between the Walker-A Thr and Lys (see Fig. 7).5 A replacement of the Walker-A Thr (or Ser, which is also found in the Walker-A consensus sequence) with a larger amino acid Asn could affect the binding of nucleotides and/or magnesium due to steric conflicts or modified orientation of the hydrogen-bonding groups in the side chain. Indeed, introduction of the noncanonical Walker A into ClpB stabilizes the ADP-bound state and decelerates the ATPase [see Figs. 2(A) and 3(B)]. Our results demonstrate a previously unknown role of the last residue of the Walker A motif in modulating the affinity for nucleotides.
Figure 7.
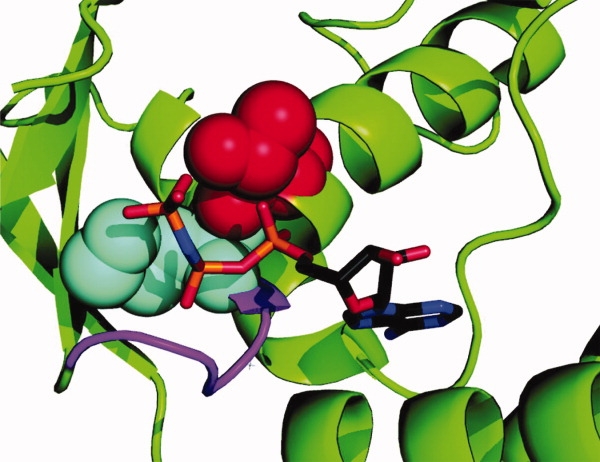
Location of the Walker-A Thr in the nucleotide-binding site of ClpB. Shown is a fragment of the AMP-PNP-bound D1 from T. thermophilus ClpB.5 The nucleotide is shown using a stick representation. The Walker-A Lys (cyan) and Thr (red) are shown in space-fill representation. The rest of P-loop is shown in magenta.
We also discovered that the Walker-A Thr is essential for the high-affinity binding of ClpB to its aggregated substrates in spite of being located at a distance from the substrate binding site (see Fig. 4). The latter result suggests an allosteric linkage between the P-loop in ClpB and the substrate interaction site. Thr213 in D1 of ClpB is positioned 31 Å away from an essential substrate-binding residue, Tyr251 located in an unstructured loop at the channel entrance.5,11 A close proximity of the Walker-A Thr and the nucleotide γ-phosphate (see Fig. 7) suggests that the threonine may participate in detecting the presence of γ-phosphate and relaying that information to the substrate binding loop. A replacement of the Walker A Thr with Asn apparently inhibits the γ-phosphate detection, severs the allosteric linkage, and disables the high-affinity configuration of a distant substrate-binding site even in the presence of saturating ATPγS (see Fig. 4). Interestingly, the T-to-N Walker A mutation inhibits binding of aggregated proteins to ClpB, but does not apparently disable interactions with soluble activators [comp. Figs. 4 and 2(B)].
Importantly, binding of aggregated substrates to ClpB has been inhibited by a T-to-N mutation in D1, but not in D2 (see Fig. 4). This result implies that the D2 loop, which is located inside the channel ∼38 Å from its entrance, does not provide essential contacts during the ATPγS-induced ClpB-substrate interaction. In other words, it is unlikely that in the absence of ATP hydrolysis parts of a substrate become inserted into the channel deep enough to reach the D2 loop. Inhibition of substrate binding in ClpB(T213N) and ClpB(T213N/T612N) is further reflected in the loss of chaperone activity of those ClpB variants (see Fig. 5). Interestingly, the reactivation of G6PDH aggregates by ClpB(T612N) has been also significantly decelerated, as compared with wt ClpB, which indicates that the fully active D1 module needs a cooperation of D2 to achieve efficient translocation of polypeptides through the channel.
All noncanonical ClpB variants were less effective than wt ClpB in supporting the survival of E. coli under aggregation-promoting heat-shock conditions (see Fig. 6). Surprisingly, ClpB(T213N) was equally active in vivo as ClpB(T612N) (compare Figs. 5 and 6). This result suggests that severing of the link between ATP-and substrate binding in ClpB(T213N) may not be as radical in vivo for some thermolabile E. coli proteins as it is in vitro for the large preformed aggregates (see Fig. 4) or that other cellular factors may support aggregate binding to ClpB and may compensate for the loss of affinity in the chaperone.
The postulated role of the last residue of Walker A in regulating the nucleotide-dependent functions is consistent with studies on the oncogene Ras, a GTPase that couples a variety of cellular pathways. The dominant-inhibitory mutant Ras17N, which has been extensively used to suppress Ras signaling, contains Asn in Walker A instead of Ser.25 Ras17N, unlike wt Ras, shows higher affinity for GDP than for GTP, which is consistent with our results on the noncanonical variant of ClpB and demonstrates that the asparagine in Walker A can indeed significantly change the nucleotide-dependent properties of an ATPase or a GTPase. Moreover, our results suggest that the Walker-A T-to-N (or S-to-N) mutation can be added to the toolbox of approaches for studying the function of AAA+ ATPases as the way to disable high-affinity substrate binding even in the presence of ATP.
Finally, we should recall that the Walker A motif with Asn in place of Thr (or Ser) occurs in torsins and DnaC. Bacterial DnaC and metazoan torsins are located in two distant branches of the AAA+ evolutionary tree and their separation can be tracked as far back as the last universal common ancestor of the whole AAA+ superfamily.16 It is intriguing that the introduction of Asn into Walker A apparently occurred twice during evolution in two distant forms of life: bacteria and animals. Like the noncanonical variants of ClpB described in this work, DnaC is not an efficient ATPase, but unlike ClpB, it does not form oligomers.18 Single-stranded DNA and the helicase DnaB (which is a canonical AAA+ ATPase) stimulate the ATP hydrolysis by DnaC. Moreover, DnaC assembles into oligomers on a scaffold of the hexameric DnaB. The properties of DnaC and our results suggest that AAA+ ATPases with Asn in the Walker A motif may differ from their canonical peers in biochemical functionality and may use distinct mechanisms of linkage to other macromolecules.
Perhaps, the noncanonical Walker A sequence in DnaC and torsins evolved to add a new level of control over the ATPase activity and conformation-remodeling function of those AAA+ proteins.
Materials and Methods
Proteins
ClpB and its variants were purified from E. coli as described earlier.20 G6PDH, κ-casein, and poly-l-lysine were obtained from Sigma, MDH from Roche, and GrpE from Assay Designs (Ann Arbor, Michigan). DnaK and DnaJ were purified as described earlier.26,27 Site-directed mutagenesis was performed using the QuickChange procedure (Stratagene). Protein concentrations are given in monomer units.
ATPase activity assay
The rate of ATP hydrolysis at 37°C was determined with a malachite green assay, as described earlier.20 The assays in the absence of other polypeptides contained 2.5 μg ClpB, those in the presence of κ-casein or poly-l-lysine contained 0.25 μg ClpB.
Gel-filtration chromatography
Gel-filtration analysis was performed at room temperature with a Shimadzu HPLC. ClpB samples (20 μL, ∼1 mg/mL protein) were analyzed at 0.06 mL/min on a Superose 6 PC 3.2/30 column (GE Healthcare) equilibrated with 50 mM Tris-HCl pH 7.5, 0.2M KCl, 20 mM MgCl2, 1 mM EDTA, 1 mM DTT without or with 2 mM ATP or ADP. Gel-filtration standards were obtained from Bio-Rad.
Titration calorimetry
Calorimetric titrations were performed at 30°C using a MicroCal MCS-ITC calorimetry system. ClpB was diluted to 50 μM in 50 mM Tris-HCl pH 7.5, 10 mM MgCl2, 1 mM EDTA, 1 mM DTT. ATPγS and ADP were diluted to 1.5 mM in the above buffer. Protein and nucleotide concentrations were verified spectrophotometrically before loading into the calorimeter. The produced heat effects of 15-μL injections of the nucleotide solutions were integrated with Origin software (MicroCal) to produce the values of the incremental enthalpy of binding for each injection (shown in Fig. 3). Control nucleotide injections into the buffer without ClpB produced negligible heat effects.
ClpB-aggregate interaction assay
Large protein aggregates were produced by diluting G6PDH to 220 μM and MDH to 380 μM in 5M urea, 8% glycerol, and 20 mm DTT. After 5-min incubation at 47°C, the sample was diluted 10-fold in the refolding buffer (50 mM triethanolamine/HCl pH 7.5, 20 mM Mg(OAc)2, 30 mM KCl, 10 mM β-mercaptoethanol, 1 mM EDTA) and incubated for 15 min at 47°C and 5 min on ice. Aggregated G6PDH (3 μM) or MDH (3 μM) was then shaken for 5 min at 30°C in the refolding buffer containing 1.5 μM ClpB or its variants without nucleotides or with 5 mM ATP, ATPγS, or ADP. A total of 75-μL aliquots were applied to Amicon Ultrafree-MC centrifugal filter devices with 0.1-μm Durapore membrane (Millipore). Filter units were incubated for 3 min at room temperature and then centrifuged for 4 min at 12,000g. The filters were washed with the refolding buffer containing an appropriate nucleotide. Proteins retained on the membrane were eluted by 10-min incubation at 47°C with 75 μL SDS sample buffer and centrifugation (4 min, 12,000g). The eluates were analyzed by SDS-PAGE and Coomassie staining.
G6PDH reactivation assay
Aggregates of G6PDH (22 μM) were diluted eightfold in the refolding buffer with 6 mM ATP containing 1 μM DnaK, 1 μM DnaJ, and 0.5 μM GrpE (KJE), or KJE with 1.5 μM ClpB or its variants. After incubation at 30°C, aliquots were withdrawn and the G6PDH activity was measured at 30°C in 50 mM Tris-HCl pH 7.8, 5 mM MgCl2 with 2 mM glucose-6-phosphate, and 1 mM NADP+. Absorption at 340 nm was measured after 5 min.
Heat-shock survival assay
The clpB gene together with the heat-shock promoter was subcloned into a low-copy pGB2 plasmid.28 E. coli survival during heat-shock was determined as described earlier.28 Immunodetection of ClpB in E. coli cultures was performed with rabbit polyclonal anti-ClpB antibodies.29
References
- 1.Neuwald AF. Aravind L. Spouge JL. Koonin EV. AAA+: a class of chaperone-like ATPases associated with the assembly, operation, and disassembly of protein complexes. Genome Res. 1999;9:27–43. [PubMed] [Google Scholar]
- 2.Hanson PI. Whiteheart SW. AAA+ proteins: have engine, will work. Nat Rev Mol Cell Biol. 2005;6:519–529. doi: 10.1038/nrm1684. [DOI] [PubMed] [Google Scholar]
- 3.Ogura T. Wilkinson AJ. AAA+ superfamily ATPases: common structure–diverse function. Genes Cells. 2001;6:575–597. doi: 10.1046/j.1365-2443.2001.00447.x. [DOI] [PubMed] [Google Scholar]
- 4.Weibezahn J. Schlieker C. Tessarz P. Mogk A. Bukau B. Novel insights into the mechanism of chaperone-assisted protein disaggregation. Biol Chem. 2005;386:739–744. doi: 10.1515/BC.2005.086. [DOI] [PubMed] [Google Scholar]
- 5.Lee S. Sowa ME. Watanabe YH. Sigler PB. Chiu W. Yoshida M. Tsai FT. The structure of ClpB: a molecular chaperone that rescues proteins from an aggregated state. Cell. 2003;115:229–240. doi: 10.1016/s0092-8674(03)00807-9. [DOI] [PubMed] [Google Scholar]
- 6.Zolkiewski M. ClpB cooperates with DnaK, DnaJ, and GrpE in suppressing protein aggregation. A novel multi-chaperone system from Escherichia coli. J Biol Chem. 1999;274:28083–28086. doi: 10.1074/jbc.274.40.28083. [DOI] [PubMed] [Google Scholar]
- 7.Motohashi K. Watanabe Y. Yohda M. Yoshida M. Heat-inactivated proteins are rescued by the DnaK.J-GrpE set and ClpB chaperones. Proc Natl Acad Sci USA. 1999;96:7184–7189. doi: 10.1073/pnas.96.13.7184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Goloubinoff P. Mogk A. Zvi AP. Tomoyasu T. Bukau B. Sequential mechanism of solubilization and refolding of stable protein aggregates by a bichaperone network. Proc Natl Acad Sci USA. 1999;96:13732–13737. doi: 10.1073/pnas.96.24.13732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Weibezahn J. Tessarz P. Schlieker C. Zahn R. Maglica Z. Lee S. Zentgraf H. Weber-Ban EU. Dougan DA. Tsai FT. Mogk A. Bukau B. Thermotolerance requires refolding of aggregated proteins by substrate translocation through the central pore of ClpB. Cell. 2004;119:653–665. doi: 10.1016/j.cell.2004.11.027. [DOI] [PubMed] [Google Scholar]
- 10.Sauer RT. Bolon DN. Burton BM. Burton RE. Flynn JM. Grant RA. Hersch GL. Joshi SA. Kenniston JA. Levchenko I. Neher SB. Oakes ES. Siddiqui SM. Wah DA. Baker TA. Sculpting the proteome with AAA(+) proteases and disassembly machines. Cell. 2004;119:9–18. doi: 10.1016/j.cell.2004.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schlieker C. Weibezahn J. Patzelt H. Tessarz P. Strub C. Zeth K. Erbse A. Schneider-Mergener J. Chin JW. Schultz PG. Bukau B. Mogk A. Substrate recognition by the AAA+ chaperone ClpB. Nat Struct Mol Biol. 2004;11:607–615. doi: 10.1038/nsmb787. [DOI] [PubMed] [Google Scholar]
- 12.Barnett ME. Nagy M. Kedzierska S. Zolkiewski M. The amino-terminal domain of ClpB supports binding to strongly aggregated proteins. J Biol Chem. 2005;280:34940–34945. doi: 10.1074/jbc.M505653200. [DOI] [PubMed] [Google Scholar]
- 13.Lum R. Tkach JM. Vierling E. Glover JR. Evidence for an unfolding/threading mechanism for protein disaggregation by Saccharomyces cerevisiae Hsp104. J Biol Chem. 2004;279:29139–29146. doi: 10.1074/jbc.M403777200. [DOI] [PubMed] [Google Scholar]
- 14.Lee S. Choi JM. Tsai FT. Visualizing the ATPase cycle in a protein disaggregating machine: structural basis for substrate binding by ClpB. Mol Cell. 2007;25:261–271. doi: 10.1016/j.molcel.2007.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Weibezahn J. Schlieker C. Bukau B. Mogk A. Characterization of a trap mutant of the AAA+ chaperone ClpB. J Biol Chem. 2003;278:32608–32617. doi: 10.1074/jbc.M303653200. [DOI] [PubMed] [Google Scholar]
- 16.Iyer LM. Leipe DD. Koonin EV. Aravind L. Evolutionary history and higher order classification of AAA+ ATPases. J Struct Biol. 2004;146:11–31. doi: 10.1016/j.jsb.2003.10.010. [DOI] [PubMed] [Google Scholar]
- 17.Ozelius LJ. Hewett JW. Page CE. Bressman SB. Kramer PL. Shalish C. de Leon D. Brin MF. Raymond D. Corey DP. Fahn S. Risch NJ. Buckler AJ. Gusella JF. Breakefield XO. The early-onset torsion dystonia gene (DYT1) encodes an ATP-binding protein. Nat Genet. 1997;17:40–48. doi: 10.1038/ng0997-40. [DOI] [PubMed] [Google Scholar]
- 18.Davey MJ. Fang L. McInerney P. Georgescu RE. O'Donnell M. The DnaC helicase loader is a dual ATP/ADP switch protein. EMBO J. 2002;21:3148–3159. doi: 10.1093/emboj/cdf308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kustedjo K. Deechongkit S. Kelly JW. Cravatt BF. Recombinant expression, purification, and comparative characterization of torsinA and its torsion dystonia-associated variant Delta E-torsinA. Biochemistry. 2003;42:15333–15341. doi: 10.1021/bi0349569. [DOI] [PubMed] [Google Scholar]
- 20.Barnett ME. Zolkiewska A. Zolkiewski M. Structure and activity of ClpB from Escherichia coli. Role of the amino-and-carboxyl-terminal domains. J Biol Chem. 2000;275:37565–37571. doi: 10.1074/jbc.M005211200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Akoev V. Gogol EP. Barnett ME. Zolkiewski M. Nucleotide-induced switch in oligomerization of the AAA+ ATPase ClpB. Protein Sci. 2004;13:567–574. doi: 10.1110/ps.03422604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hersch GL. Burton RE. Bolon DN. Baker TA. Sauer RT. Asymmetric interactions of ATP with the AAA+ ClpX6 unfoldase: allosteric control of a protein machine. Cell. 2005;121:1017–1027. doi: 10.1016/j.cell.2005.05.024. [DOI] [PubMed] [Google Scholar]
- 23.Wiseman T. Williston S. Brandts JF. Lin LN. Rapid measurement of binding constants and heats of binding using a new titration calorimeter. Anal Biochem. 1989;179:131–137. doi: 10.1016/0003-2697(89)90213-3. [DOI] [PubMed] [Google Scholar]
- 24.Li J. Sha B. Crystal structure of E. coli Hsp100 ClpB nucleotide-binding domain 1 (NBD1) and mechanistic studies on ClpB ATPase activity. J Mol Biol. 2002;318:1127–1137. doi: 10.1016/S0022-2836(02)00188-2. [DOI] [PubMed] [Google Scholar]
- 25.Feig LA. Tools of the trade: use of dominant-inhibitory mutants of Ras-family GTPases. Nat Cell Biol. 1999;1:E25–E27. doi: 10.1038/10018. [DOI] [PubMed] [Google Scholar]
- 26.Zmijewski MA. Macario AJ. Lipinska B. Functional similarities and differences of an archaeal Hsp70(DnaK) stress protein compared with its homologue from the bacterium Escherichia coli. J Mol Biol. 2004;336:539–549. doi: 10.1016/j.jmb.2003.12.053. [DOI] [PubMed] [Google Scholar]
- 27.Zylicz M. Yamamoto T. McKittrick N. Sell S. Georgopoulos C. Purification and properties of the dnaJ replication protein of Escherichia coli. J Biol Chem. 1985;260:7591–7598. [PubMed] [Google Scholar]
- 28.Kedzierska S. Akoev V. Barnett ME. Zolkiewski M. Structure and function of the middle domain of ClpB from Escherichia coli. Biochemistry. 2003;42:14242–14248. doi: 10.1021/bi035573d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tek V. Zolkiewski M. Stability and interactions of the amino-terminal domain of ClpB from Escherichia coli. Protein Sci. 2002;11:1192–1198. doi: 10.1110/ps.4860102. [DOI] [PMC free article] [PubMed] [Google Scholar]