

Abstract
Disulfide bonds play a critical role in the stabilization of the immunoglobulin β-sandwich sandwich. Under reducing conditions, such as those that prevail in the cytoplasm, disulfide bonds do not normally form and as a result most antibodies expressed in that compartment (intrabodies) accumulate in a misfolded and inactive state. We have developed a simple method for the quantitative isolation of antibody fragments that retain full activity under reducing conditions from large mutant libraries. In E. coli, inactivation of the cysteine oxidoreductase DsbA abolishes protein oxidation in the periplasm, which leads to the accumulation of scFvs and other disulfide-containing proteins in a reduced form. Libraries of mutant scFvs were tethered onto the inner membrane of dsbA cells and mutants that could bind fluorescently labeled antigen in the reducing periplasm were screened by Anchored Periplasmic Expression (APEx; Harvey et al., Proc Natl Acad Sci USA 2004;101:9193–9198.). Using this approach, we isolated scFv antibody variants that are fully active when expressed in the cytoplasm or when the four Cys residues that normally form disulfides are substituted by Ser residues.
Keywords: protein structure/folding, disulfide bonds, directed evolution, intrabodies
Introduction
Disulfide bonds contribute greatly to the stability of antibody immunoglobulin domains.1–3 Typically, in scFv antibody fragments, the disruption of the two conserved disulfide bonds that connect the two β-sheets in each of the VH and VL domains causes a decrease of 4–5 kcal/mol in the ΔG of folding and is accompanied by loss of antigen binding affinity, susceptibility to proteolysis, and aggregation.2,4,5
The expression of antibody fragments within intracellular compartments (intrabodies) constitutes a promising and clinically relevant technology for binding to target proteins relevant to disease progression.5 Intrabodies are being investigated as a potential treatment for human viral infection, cancer therapy, and neurodegenerative diseases.5–8 However, the cytoplasm of eukaryotic and most prokaryotic cells is maintained in a highly reduced state that strongly disfavors the formation of disulfide bonds under physiological conditions. Consequently, most antibodies are not compatible with expression in that compartment and thus cannot be employed as intrabodies. Naturally occurring antibodies exhibiting high thermodynamic stability and antigen binding under conditions where disulfide bonds cannot form are rare.9,10 Therefore, scFvs with desired antigen specificity and sufficiently high stability to be suitable for use as intrabodies need to be generated de novo. Intrabodies have been isolated by screening repertoire libraries using high-throughput screening methods that interrogate antibody function under reducing conditions, such as yeast 2-hybrid, protein complementation assays, and ribosomal display.11–16 In contrast, filamentous phage display necessitates the secretion of antibodies into the oxidizing environment of the bacterial periplasmic space. Therefore, for intrabody applications, phage-derived antibodies to target antigens must be subjected to a second screen to isolate clones compatible with cytoplasmic expression.17 Alternatively, phage display has been employed successfully for the directed evolution of hyperstable antibody frameworks that in some cases can withstand expression in the reducing environment of the cytoplasm.1 In turn, natural or engineered hyperstable antibody frameworks have been used as scaffolds for the creation of large synthetic libraries containing randomized CDRs13,18–20 enabling the isolation of scFvs that are folded in the absence of disulfides. Alternatively, MBP-scFv fusions have been shown to exhibit significant activity when expressed in the cytoplasm of Escherichia coli or mammalian cells.21
The bacterial periplasmic space is a highly oxidizing compartment that strongly favors the formation of protein disulfide bonds. Cysteine oxidation is catalyzed by the highly efficient protein thiol oxidase DsbA.22,23 Upon transferring its disulfide to a substrate protein, DsbA becomes reduced and has to be recycled by the action of the membrane enzyme DsbB, which then transfers the electrons to quinones. In E. coli strains deficient in dsbA (or dsbB), the redox potential of the periplasm is highly reducing, and as a result, proteins accumulate almost exclusively in reduced form.24
dsbA strains are not compatible with phage display because they do not support filamentous phage assembly.25 Earlier, we developed a flow cytometric technique for the screening of antibody fragments, called Anchored Periplasmic Expression (APEx), in which the display of the desired protein is not affected by redox state of the periplasm. In APEx, proteins are anchored onto the periplasmic side of the inner membrane via fusion to either a transmembrane domain of an integral membrane protein or to the signal peptide and the first few N-terminal amino acids of an inner membrane lipoprotein such as NlpA.26,27 The latter anchoring strategy is usually preferable because lipoprotein fusions can be expressed at higher levels. For detection of the displayed protein, the cells are converted to spheroplasts to permeablize the outer membrane, incubated with a fluorescent ligand, and analyzed by flow cytometry. Variations of this technique have been used for the engineering of very high affinity antibody variants, for the isolation of IgG antibodies from hyperimmune libraries, and for selecting mutant antibody fragments with improved expression properties.26,28,29 Recently, we developed a 2-hybrid version of APEx (APEx 2-hybrid) whereby both an antibody fragment and its cognate antigen are expressed in the same cell [Fig. 1(B)]. Briefly, the antibody fragment is anchored on the inner membrane of E. coli, whereas the antigen is expressed as a soluble, epitope-tagged, periplasmic protein. Upon removal of the outer membrane by treatment with Tris-EDTA and lysozyme, most smaller periplasmic proteins (M.W. < 40–50 kDa), including any unbound epitope-tagged antigen, are released quantitatively into the extracellular fluid. However, if the epitope-tagged antigen can bind to the membrane-anchored antibody, it remains associated with the cell. The antibody:antigen complex on spheroplasts can then be detected using a fluorescent anti-epitope tag antibody. Thus, the spheroplasts become fluorescently labeled and can be isolated by flow cytometry. An important advantage of this system compared with the yeast 2-hybrid and other in vivo protein technologies is that binding affinity is directly and quantitatively measured at the single cell level by FACS.28 In this work, we used APEX to screen libraries of scFvs that are able to fold into their native conformation in the reducing periplasm of dsbA cells. As a model system, we isolated variants of the anti-Bacillus anthracis protective antigen (PA) 14B7* scFv that, in contrast to the parental antibody, were able to fold under reducing conditions and could be expressed in fully active form in the bacterial cytoplasm. Thus, the methodology we present here should enable the rapid isolation of antibody fragment variants that can fold into their active conformation under reducing conditions and can be used for intrabody applications.
Figure 1.
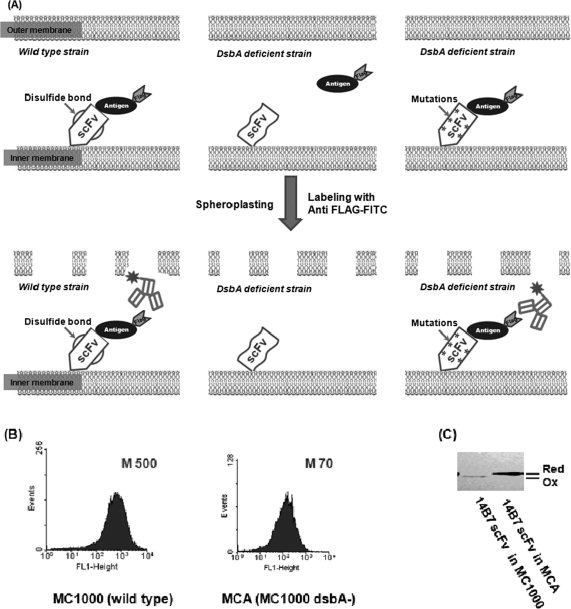
Isolation of active antibody fragments in E. coli dsbA mutants. (A) Schematic showing the screening strategy. Left panel: a correctly folded scFv anchored on the inner membrane of wild-type cells is able to bind antigen. A fluorescent antibody that recognizes an epitope tag on the antigen is used to detect the formation of antibody:antigen complex. Middle panel: in the dsbA mutant, the scFv is reduced and cannot bind antigen. Upon spheroplasting, the antigen diffuses away and hence the cell is not labeled by the fluorescent anti-epitope tag antibody. Right panel: a mutant scFv that is capable of folding in the absence of disulfide bonds can bind antigen in dsbA cells. M, mean fluorescence intensity of the cell population. (B) Fluorescence histograms of cells coexpressing the 14B7* scFv and PA domain IV proteins in either E. coli MC1000 (wild-type) or MCA (MC1000 dsbA). The formation of the antibody:antigen complex was detected by labeling with 200 nM anti-FLAG-FITC. M, mean fluorescent intensity. (C) The redox state of the 14B7* scFv in MC1000 or MCA cells following alkylation with AMS and separation by nonreducing SDS-PAGE.
Results
Assay development
As a model antibody fragment for this study, we used the 14B7* scFv that binds the Protective Antigen (PA) component of the B. anthracis toxin.30 In particular, the 14B7* scFv was anchored onto the inner membrane by fusion to the leader peptide and the first six amino acids of the mature sequence (CDQSSS) of the E. coli lipoprotein NlpA. The 14B7* scFv recognizes a conformational epitope located within PA domain 4 (PA-D4), a 139 amino acid fragment comprising aa 596–735.31 PA-D4 fused to a C-terminal FLAG epitope tag was expressed in soluble form in the periplasmic space.
For simplicity, the scFv and the antigen were expressed as a dicistronic operon downstream from the lac promoter.28 E. coli MC1000 cells expressing the membrane anchored 14B7* scFv together with periplasmic PA-D4 were converted to spheroplasts by treatment with lysozyme and EDTA in the presence of sucrose.26 In spheroplasts, the permeability barrier posed by the outer membrane is severely compromised, allowing the externally added anti-FLAG antibodies conjugated to FITC to interact with the FLAG tag presented by 14B7*:PA-D4 complexes on the surface of the inner membrane, resulting in a high fluorescence signal [Fig. 1(A)]. However, in E. coli MCA (MC1000 dsbA) where the oxidative folding of the scFv protein and thus antigen binding is impaired, the cell fluorescence is 8-fold lower and comparable with that obtained in MC1000 cells expressing the antigen together with an unrelated scFv antibody [Fig. 1(B) and data not shown].
The redox state of the 14B7* scFv in spheroplasted MC1000 and MCA cells was determined by nonreducing SDS-PAGE after trapping of free thiols with 4-acetamido-4′-maleimidostilbene-2,2′-disulfonate (AMS).32 Briefly, proteins were first precipitated with TCA to block thiol oxidation. Then, the precipitated proteins were resuspended in buffer containing the thiol reactive reagent AMS. AMS conjugation increases the molecular weight by 490 Da per thiol. Thus, for a scFv that contains four cysteines this translates to an increase in M.W. of ∼2 kDa. Only a band corresponding to this higher molecular weight species was detected in MCA cells, indicating that, as expected, the cysteine residues in the scFv protein were present in the reduced state. In contrast, in the parental strain MC1000 the scFv was present exclusively in the oxidized state [Fig. 1(C)]. Note that in Fig 1(C), the intensity of the scFv band in MC1000 cells is lower relative to MCA; the lower intensity somehow appears to be a consequence of the sample processing and does not reflect the relative expression levels of the proteins. Western blot analysis of untreated cells revealed that untreated MC1000 cells consistently accumulate a higher amount of scFv protein relative to cells lacking dsbA. Thus, the low FACS signal observed in the dsbA cells correlates with the presence of reduced scFv in the periplasm.
Library screening
The 14B7* scFv gene was subjected to random mutagenesis by error-prone PCR. Following transformation, a library of ∼107 independent colonies was obtained. DNA sequencing of 10 clones selected at random revealed an average of 1.8% nucleotide substitutions per gene. Cells were grown in TB media, protein synthesis was induced with 1 mM IPTG for 4 h at 25°C, and the cells were converted into spheroplasts and, finally, were labeled with 200 nM anti-FLAG-FITC. A total of 5 × 108 cells were sorted in a Cytomation MoFlo droplet deflection flow cytometer and the top 2% of the cells with the highest 530 nm fluorescence (FL-1) were collected and resorted as above. DNA encoding scFv from the sorted population was rescued by PCR amplification and recloned into plasmid p14B7*-D4 After a total of three rounds of sorting, we isolated three distinct clones exhibiting a marked increase in cell fluorescence relative to cells expressing the parental 14B7* scFv. Preliminary experiments indicated that one of these clones likely represented a false positive. The other two clones, named 3-44 and 4-39, were selected for further analysis. DNA sequencing revealed that the two clones contained 23 and 16 nucleotide substitutions resulting in 16 and 13 amino acid substitutions, respectively (see Fig. 2). Variant 3-44 also contained a deletion of one residue in the Gly-Ser linker. The frequency of nucleotide substitutions in the selected clones was 3-to 4-fold higher than that of the presort population (1.8% nt substitutions per gene). The selection of hypermutated clones from libraries generated by error-prone PCR has been observed previously.33 In clone 3-44, mutations were distributed throughout the protein, whereas in clone 4-39, all but one of the amino acid substitutions (VH L4H) were located in the variable light chain (VL). The fluorescence histograms of MCA cells expressing the two variants are shown in Figure 3(A).
Figure 2.

Amino acid sequence alignment of 14B7* scFv and the 3-44 and 4-39 mutants.
Figure 3.
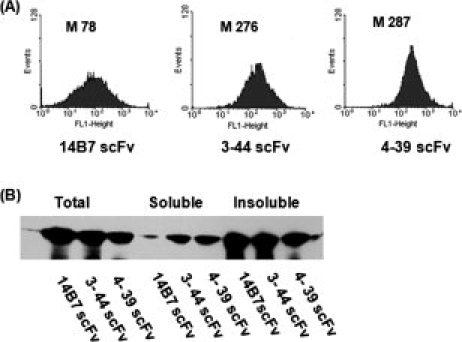
Analysis of isolated scFv clones. (A) FACS histograms of MCA cells expressing 14B7*, 3-44, or 4-39 scFvs. (B) Solubility of the scFvs following expression in the bacterial cytoplasm. Cells were grown at 25°C, and protein synthesis was induced with 1 mM IPTG for 4 h and harvested. Proteins were resolved by SDS-PAGE and scFvs were detected by Western blotting using anti-His-HRP at a 1:10,000 dilution. Samples were normalized by loading aliquots equivalent to OD600 = 2.
Characterization of isolated scFv mutants
To evaluate how well the selected clones are expressed as soluble proteins in the cytoplasm of E. coli, the signal sequence was excised, the genes were placed downstream of the T7 promoter in PET28a, and the resulting plasmids were introduced into Rosetta™ 2(DE3) cells, which is a BL21 strain used to aid in expression of eukaryotic proteins that contain codons which are rarely used in E. coli. Cells were grown in rich media at 25°C, lysed by passing through a French press, and fractionated into soluble and insoluble fractions by high speed centrifugation. The two scFv variants displayed markedly higher accumulation in the soluble fraction [Fig. 3(B)]. Similar results were obtained in cells grown at 37°C (data not shown). Following purification by IMAC chromatography, the yields of the 3-44 and 4-39 scFvs obtained were 0.35 and 0.5 mg/L, respectively. For comparison, the yield of 14B7* expressed and purified under identical conditions was 0.1 mg/L.
The binding of the scFvs to the full length, 83 kDa PA, protein was analyzed by ELISA. Cell lysis results in rapid oxidation of cytoplasmically expressed scFvs, an event that can be prevented by the addition of DTT or by carboxymethylation using iodacetamide [IAA; Fig. 4(A)]. To prevent oxidation, ELISA assays were performed with clarified soluble fractions obtained from cells that had been treated with 100 mM DTT. Under these conditions, the parental 14B7* scFv gave a much lower ELISA signal compared with the 4-39 variant [Fig. 4(B)], indicating that the latter antibody fragment is able to bind antigen under conditions where disulfide bond formation is precluded.
Figure 4.
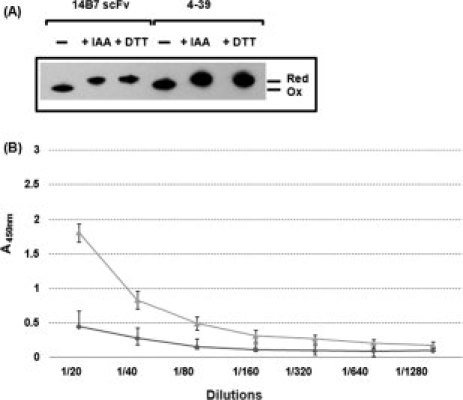
Cysteine redox state and antigen binding activity of scFvs expressed in the E. coli cytoplasm. (A) Cysteine redox state in cytoplasmically expressed scFvs. Upon harvesting, cells were incubated either with 100 mM of prechilled IAA or with the same concentration of DTT for 20 min on ice. The cells were pelleted, lysed in a French press, the cell debris was removed, and the soluble proteins were precipitated by 10% TCA. The precipitated proteins were resuspended in buffer containing AMS. The electrophoretic mobility of scFv proteins was analyzed by nonreducing SDS-PAGE and Western blotting using anti-His-HRP at a 1:10,000 dilution. (B) ELISA: Before lysis, cells were treated with 100 mM DTT, washed with PBS, lysed in a French press, and the soluble proteins were applied to a PA-coated ELISA plate. The bound scFvs were detected by 50 μL of 1:10,000 diluted anti-His-HRP conjugate. ( : 14B7* scFv with 100 mM DTT,
: 14B7* scFv with 100 mM DTT,  : 4-39 scFv with 100 mM DTT).
: 4-39 scFv with 100 mM DTT).
Additionally, the refolding yield of the 4-39 variant following dilution from denaturant solution under reducing conditions was much higher than that of the parental antibody. For these experiments, 1 μM purified scFv protein was first unfolded and reduced in 6M GndHCl and 12.5 mM DTT. Refolding was initiated by diluting the samples 50-fold in buffer containing DTT and 0.01% Tween-20. As a control, the native scFv protein was diluted in buffer as above but also containing 0.12 mM GndHCl corresponding to the final concentration of denaturant in the refolding experiments. The refolding yield of the 4-39 scFv was 80% ± 7.5% compared with 21% ± 11% for 14B7*. Thus, under reducing conditions the mutant antibody is better able to avoid off-pathway reactions during refolding from denaturant solutions.
To further examine the role of disulfide bond formation on the folding and stability of the 14B7* and 4-39 scFvs, the four Cys residues that form the two disulfides in the VH and VL chains were replaced with Ser residues using overlap extension PCR. The van der Waals volume of Ser is similar to Cys, and even though Ser is more hydrophilic, it is tolerated in the interior of scFv antibodies.34 Moreover, Ser substitutions are frequently found in engineered scFvs that can fold in the absence of disulfides.35 Replacement of all the Cys by Ser significantly impaired the ability of 14B7* scFv to bind antigen. In contrast, the antigen binding activity of the 4-39 Cys4→Ser4 scFv variant was identical to that of the wild-type 4-39 [Fig. 5(A)]. The Ser-substituted scFvs showed similar level of cytoplasmic expression and solubility in shake flask cultures as those of the corresponding parental antibodies [Fig. 5(B)]. The 14B7* Cys4→Ser4 and 4-39Cys4→Ser4 scFv variants were purified by Ni chromatography followed by gel filtration FPLC to isolate monomeric protein and the antigen binding kinetics were determined by surface plasmon resonance. Consistent with the ELISA assays, no binding onto immobilized PA83 could be detected for 14B7* Cys4→Ser4. In contrast, the 4-39Cys4→Ser4 exhibited a KD equal to 7.1 × 10−8M. Consistent with the lower FACS signal observed in MCA cells expressing the 4-39 scFv relative to wild-type cells expressing (fully oxidized) 14B7* [compare Figs. 1 and 3(A)], the in vitro determined KD of the 4-39Cys4→Ser4 variant was about 20-fold lower than that of the parental 14B7* scFv (KD = 4 × 10−9M). Interestingly, this reduction in the equilibrium binding constant was almost entirely due to a low association rate constant: kon for 4-39Cys4→Ser4: 3.2 × 104 M−1s−1; for 14B7* 7.1 × 105 M−1s−1: koff for 4-39Cys4→Ser4: 2.3 × 10−3 s−1; for 14B7* 3.0 × 10−3 s−1.
Figure 5.
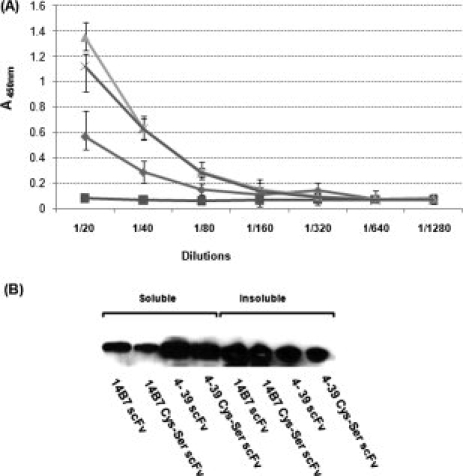
ELISA analysis and expression of Ser-substituted scFvs mutants. (A) ELISA of whole cell lysates incubated with 100 mM DTT and expressing:  : 14B7* scFv,
: 14B7* scFv,  : 14B7* Cys4→Ser4 scFv,
: 14B7* Cys4→Ser4 scFv,  : 4-39 scFv,
: 4-39 scFv,  : 4-39 Cys4→Ser4 scFv. (B) Soluble expression level determined by Western blotting.
: 4-39 Cys4→Ser4 scFv. (B) Soluble expression level determined by Western blotting.
Discussion
With the APEx screening methodology, the physiological environment of the periplasmic space can be exploited for the isolation of scFv variants that not only bind antigen with a desired affinity, but also display other interesting properties. For example, recently Ribnicky et al. combined APEx and export into the periplasm via the Twin Arginine Transporter (Tat) pathway to isolate scFv variants exhibiting faster protein folding kinetics.36 Our laboratory has used APEx and its variations for the isolation of antibody fragments and even full length IgGs from hyperimmune or from synthetic repertoire libraries, for expression optimization, the recognition of antigen immobilized on solid surfaces, and finally for the expression maturation of therapeutically relevant antibodies (Refs. 28, 29, 37, 38; Mazor et al., unpublished data). In this study, we took advantage of the reducing environment in the periplasm in dsbA cells to enable the display of scFvs that lack disulfide bonds. The 14B7* scFv, as most other antibody fragments, fails to fold or bind antigen when disulfide bond formation is disrupted in these cells. Following 14B7* sequence randomization and selection using APEx in dsbA cells, two antibody variants, 3-44 and 4-39, were isolated that displayed enhanced binding to the PA-D4 antigen judging from increased FACS signals. ELISA analysis of the antigen binding activity in lysates from cells expressing scFvs in the cytoplasm and treated with DTT to prevent air-oxidation verified that 4-39, but not 14B7*, was functional under reducing conditions. Even more compelling, the 4-39Cys4→Ser4 scFv construct, in which all four disufide bond forming Cys residues were replaced with Ser was fully active, whereas the 14B7*Cys4→Ser4 scFv was completely inactive. Taken together, these data leave little doubt that 4-39 is capable of binding antigen in the absence of disulfide bond formation, a prerequisite for intrabody applications.
Not surprisingly, the 3-44 and 4-39 variants exhibit higher soluble expression in the E. coli cytoplasm compared with their parental antibody. Similarly, the scFv variants are less prone to aggregation and off-pathway reactions during in vitro refolding from Gnd-HCl under reducing conditions. Although the studies described here were carried out with cells expressing both an antibody fragment library and the antigen (i.e., using the APEx 2-hybrid methodology), it is equally easy to use exogenous, fluorescently labeled antigen for screening. It should be noted, however, that for therapeutic intrabody applications, the antigen is likely to be a polypeptide derived from a cytoplasmic protein and therefore it will be devoid of disulfide bonds. As a result, the expression of therapeutic intrabody targets should be generally compatible with dsbA mutant strains.
Earlier studies had reported the isolation of intrabodies by methods involving cytoplasmically expressed libraries using screening methods such as yeast 2-hybrid or protein complementation assays.5,11,15 However, in these assays readout is not directly correlated with protein affinity, and therefore the isolation of nanomolar affinity antibodies to a desired antigen is a challenge (e.g., Ref.15). Alternatively, antibodies compatible with expression in the cytoplasm have been isolated from libraries based on highly diverse or hyperstable scFvs, which were then screened by conventional phage display.13,18,19,20 However, screening is carried out under conditions where disulfide bonds are formed, so that only a fraction of the antibodies isolated are found to be compatible with cytoplasmic expression (e.g., Ref.20). In the APEx approach described here, screening is carried out using flow cytometry enabling the quantitative isolation of only high affinity clones.37 Moreover, because screening is carried out in the reducing environment of the periplasm of dsbA cells, isolated clones must be able to fold in the absence of disulfide bonds. Consequently, we expect that our strategy might simplify the generation of variants of known, well-characterized scFvs that will be well suited to function as intrabodies or any other application involving the use of antibodies in an environment that prevents disulfide bond formation.
Methods
Bacterial strains and plasmids
Escherichia coli strains Jude 1 [(DH10B F′::Tn10 (Tetr)],39 MC1000 [araD139 (araA-leu)7679 (codB-lac)X74 galE15 galK16],40 MCA [MC1000 dsbA:: Kan5] (laboratory collection), and Rosetta™ 2(DE3) [F− ompT hsdSB(r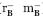 ) gal dcm (DE3) pRARE2 (CamR)] (Novagen) were used in this work.
) gal dcm (DE3) pRARE2 (CamR)] (Novagen) were used in this work.
The plasmids used in this study are listed in supporting information Table S2. The scFv 14B7* is a variant of 14B7 scFv that contains seven amino acid substitutions. These amino acid substitutions residing within the framework regions of the scFv affect expression but not the affinity for the PA antigen (Maynard and Hayhurst, unpublished data); further, X-ray crystallographic analysis revealed that the conformation of the antigen contact residues in 14B7* is virtually identical to that of 14B7 (Leysath and Monzingo, unpublished data).
The PA-D4 gene encoding the B. anthracis protective antigen domain 4 polypeptide was constructed by overlap extension PCR using 15 oligonucleotide primers (PA-D4-F1∼F8 & PA-D4-R1∼R7 in Supp. Info. Table S1). The resulting PCR product was digested with BglI and cloned into SfiI-digested pMoPac16.42 A FLAG epitope tag (DYKDDDDK) was then introduced to the C-terminus of PA-D4 by overlap extension PCR using primers PAD4-Hind-F1,-R1, and-R2 (Table S1). HindIII-digested PCR product was then cloned into pAPEx1 yielding pPAD4-FLAG. For the coexpression of scFvs and PA-D4, the PA-D4 gene was amplified by PCR with PAD4-Hind-F1 and PAD4-Hind-R2 and, after digestion with HindIII, it was cloned into HindIII site of pAPEx1-14B7* scFv yielding p14B7*-D4. For cytoplasmic expression, scFv genes were PCR-amplified from p14B7*-D4, p3-44-D4, and p4-39-D4 using the three forward primers pTrc-14B7*F, pTrc-3-44F, pTrc-4-39F, respectively, and the reverse primer Mopac-R. The PCR product was then digested with NcoI and HindIII and finally ligated into NcoI and HindIII digested pET28a vector resulting in pET28-14B7*scFv, pET28-3-44scFv, and pET28-4-39scFv, respectively.
Serine replacements were performed by multiple overlap PCR reactions using a total of 10 primers (For 4-39: CysSer45F1∼F5 & CysSer45R1∼R5; for 14B7*: CysSer45F1, CysSer14F2∼3, CysSer45F4∼5 & CysSer14R1, CysSer45R2-5). The PCR product was digested with NcoI and HindIII and cloned into pET 28a.
Library construction and FACS screening
The 14B7* scFv was subjected to random mutagenesis by error-prone PCR.37 The DNA product was then ligated into SfiI-digested pD4-FLAG. The ligation product was transformed into E. coli MCA, and the cells were plated on TB (Terrific Broth, Difco) media containing 2% glucose and 40 μg/mL chloramphenicol on large agar plates overnight at 30°C. The plates were scraped and the cells were frozen in TB +15% glycerol aliquots. For screening, the frozen cells were inoculated into fresh TB media as above and grown at 37°C to an OD600 ∼ 0.6. Then, the culture was equilibrated to 25°C for 30 min and 1 mM IPTG was added to induce protein synthesis. After incubation for an additional 4 h at 25°C, cells (equivalent to ∼1 mL of 20 OD600 culture) were pelleted by centrifugation and resuspended in 350 μL of ice-cold 0.1M Tris-HCl, pH 8.0 buffer with 0.75M sucrose, and 100 μg/mL hen egg lysozyme (Sigma-Aldrich, St. Louis, MO). Subsequently, 700 μL of ice-cold 1 mM EDTA (ethylenediaminetetraacetic acid) was gently added and the suspension was left on ice for 10 min followed by the addition of 50 μL of 0.5M MgCl2. After a 10-min incubation on ice, the cells were pelleted by centrifugation at 14,000 rpm and resuspended in phosphate buffered saline containing 200 nM PA conjugated to FITC (List Biological Laboratories, Campbell, CA). The cells were labeled for 1 h at room temperature prior to fluorescence activated cell sorting on a MoFlo (Cytomation, Fort Collins, CO) droplet deflection flow cytometer using a 488-nm Argon laser for excitation. Cells were sorted using an appropriate window for high FITC FL1 emission detected through a 530/40 band pass filter. E. coli cells collected after the first sort were immediately resorted. Subsequently, the scFv genes in the sorted cell suspension were amplified by PCR.28 The amplified scFv DNA was then recloned into the p14B7*-D4 vector, transformed into MCA cells, grown overnight on selective media, and subjected to additional rounds of sorting as above.
Determination of cysteine in vivo redox state
The in vivo redox states of scFv antibodies were determined by derivatization of free thiols by 4-acetamido-4′-maleimidyl-stilbene-2,2′disulfonic acid (AMS, Molecular Probes) in TCA-quenched samples, as described previously.32 Briefly, fresh overnight cultures were diluted to 0.1 OD600 in 5 mL TB media plus appropriate antibiotic and incubated at 37°C for 2–2.5 h. When the OD600 reached 0.5–0.7, the cells were transferred to 25°C with shaking and allowed to equilibrate for 30 min. Subsequently, 1 mM IPTG was added and incubation was continued for an additional 4 h prior to harvesting. At that point, the cells were mixed with 10% trichloroacetic acid (TCA) to block disulfide rearrangement and incubated on ice for 1 h. Precipitated proteins were pelleted by centrifugation for 15 min at 1,400 rpm at 4°C and washed with cold acetone. The pellets were air-dried, resuspended in 100 μL of 10 mM AMS in 50 mM Tris-HCl, pH 8.0, 1% SDS, and 1 mM EDTA (freshly made), then vortexed for 30 min at room temperature, heated for 5 min at 37°C, and reprecipitated with the addition of 1 mL of cold acetone. Following centrifugation, the supernatant was discarded and the pellets were resolubilized in 5× SDS-PAGE loading buffer (without β-mercaptoethanol). After SDS-PAGE, proteins were transferred to nitrocellulose membrane and probed with anti-His-HRP antisera (Sigma-Aldrich, St. Louis, MO). A fully reduced scFv standard was generated by incubating 1 mL of cells with dithiothreitol (DTT) to a final concentration of 100 mM for 20 min on ice, followed by alkylation, as above.
ELISA
Full length (83 kDa) protective antigen (PA83, List Biological Laboratories, Campbell, CA) was coated on ELISA plates (Corning Incorporated, Corning, NY) at a concentration of 4 μg/mL for 16 h at 4°C. The plates were blocked by incubating with 2% nonfat milk in PBS at room temperature. Samples were diluted in PBS with 1% nonfat milk, introduced into the wells and allowed to bind for 1 h at room temperature. Plates were washed three times with PBS containing 0.05% Tween-20 and then anti-His-HRP antiserum diluted 1:10,000 into 2% nonfat milk in PBS was added for another hour. Reactions were developed using Onestep™ Ultra TMB-ELISA (Thermo Scientific, Waltham, MA) and quenched with 4N H2SO4.
Antibody expression and purification
For the purification of the cytoplasmically expressed scFvs, genes were first cloned behind the T7 promoter in pET28a, and the resulting plasmids (Table S2) were transformed into E. coli Rosseta™ 2(DE3) cells. Protein expression and purification were performed by IMAC and size exclusion chromatography as previously described.41 All proteins used in this study were more than 90% pure as judged by Coomassie Brilliant Blue-stained SDS-PAGE.
Cell pellets were resolubilized in 5× SDS-PAGE loading buffer to give total protein fractions. For preparation of soluble and insoluble fractions, cell pellets were resuspended in PBS, disrupted with a French press and centrifuged at 2500 rpm for 10 min to remove cell debris. The supernatant was subjected to ultracentrifugation at 45,000 rpm (134,039g, Beckman L7-55 Ultracentrifuge) for 1 h at 4°C. The pellet was resuspended in 5× SDS-PAGE buffer and boiled for 5 min to give insoluble fraction samples. The supernatant after ultracentrifugation was mixed with 5× SDS-PAGE loading buffer to give soluble fraction samples.
In vitro refolding
Proteins were equilibrated in PBS containing 6M GndHCl with 12.5 mM DTT at a final concentration of 1 μM at room temperature. Refolding was initiated by dilution into 50 volumes of PBS containing 0.01% Tween-20 with 12.5 mM DTT at 4°C. The amount of active protein after refolding was determined by ELISA and was compared with the active protein obtained by diluting 1 μM scFv in PBS with 0.01% Tween-20, 0.12 mM GndHCl (the residual concentration of denaturant following refolding), and 12.5 mM DTT.
Kinetic analysis by surface plasmon resonance
Kinetic studies were performed on a BIAcore 3000 (GE Healthcare). Approximately 750 RU of PA83 were coupled onto a CM5 chip using amine coupling chemistry. Human transferrin was similarly coupled to a reference well for background subtraction. A concentration series of 30–120 nM of scFv was allowed to associate for 5 min and dissociate for 3 min at a constant flow rate of 20 μL/min. Analysis was performed using BIAevaluation software (version 4.1; GE Healthcare).
Acknowledgments
The authors thank Dr. Yariv Mazor for critically reading this manuscript.
Supplemental material
References
- 1.Proba K, Worn A, Honegger A, Pluckthun A. Antibody scFv fragments without disulfide bonds made by molecular evolution. J Mol Biol. 1998;275:245–253. doi: 10.1006/jmbi.1997.1457. [DOI] [PubMed] [Google Scholar]
- 2.Worn A, Pluckthun A. Mutual stabilization of VL and VH in single-chain antibody fragments, investigated with mutants engineered for stability. Biochemistry. 1998;37:13120–13127. doi: 10.1021/bi980712q. [DOI] [PubMed] [Google Scholar]
- 3.Hagihara Y, Mine S, Uegaki K. Stabilization of an immunoglobulin fold domain by an engineered disulfide bond at the buried hydrophobic region. J Biol Chem. 2007;282:36489–36495. doi: 10.1074/jbc.M707078200. [DOI] [PubMed] [Google Scholar]
- 4.Cattaneo A, Biocca S. The selection of intracellular antibodies. Trends Biotechnol. 1999;17:115–121. doi: 10.1016/s0167-7799(98)01268-2. [DOI] [PubMed] [Google Scholar]
- 5.Lo AS, Zhu Q, Marasco WA. Intracellular antibodies (intrabodies) and their therapeutic potential. Handb Exp Pharmacol. 2008;181:343–373. doi: 10.1007/978-3-540-73259-4_15. [DOI] [PubMed] [Google Scholar]
- 6.Messer A, McLear J. The therapeutic potential of intrabodies in neurologic disorders: focus on Huntington and Parkinson diseases. Bio Drugs. 2006;20:327–333. doi: 10.2165/00063030-200620060-00002. [DOI] [PubMed] [Google Scholar]
- 7.Williams BR, Zhu Z. Intrabody-based approaches to cancer therapy: status and prospects. Curr Med Chem. 2006;13:1473–1480. doi: 10.2174/092986706776872899. [DOI] [PubMed] [Google Scholar]
- 8.Cardinale A, Biocca S. Combating protein misfolding and aggregation by intracellular antibodies. Curr Mol Med. 2008;8:2–11. doi: 10.2174/156652408783565595. [DOI] [PubMed] [Google Scholar]
- 9.Proba K, Honegger A, Pluckthun A. A natural antibody missing a cysteine in VH: consequences for thermodynamic stability and folding. J Mol Biol. 1997;265:161–172. doi: 10.1006/jmbi.1996.0726. [DOI] [PubMed] [Google Scholar]
- 10.Tavladoraki P, Girotti A, Donini M, Arias FJ, Mancini C, Morea V, Chiaraluce R, Consalvi V, Benvenuto E. A single-chain antibody fragment is functionally expressed in the cytoplasm of both Escherichia coli and transgenic plants. Eur J Biochem. 1999;262:617–624. doi: 10.1046/j.1432-1327.1999.00443.x. [DOI] [PubMed] [Google Scholar]
- 11.Visintin M, Tse E, Axelson H, Rabbitts TH, Cattaneo A. Selection of antibodies for intracellular function using a two-hybrid in vivo system. Proc Natl Acad Sci USA. 1999;96:11723–11728. doi: 10.1073/pnas.96.21.11723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jermutus L, Honegger A, Schwesinger F, Hanes J, Pluckthun A. Tailoring in vitro evolution for protein affinity or stability. Proc Natl Acad Sci USA. 2001;98:75–80. doi: 10.1073/pnas.011311398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.der Maur AA, Zahnd C, Fischer F, Spinelli S, Honegger A, Cambillau C, Escher D, Pluckthun A, Barberis A. Direct in vivo screening of intrabody libraries constructed on a highly stable single-chain framework. J Biol Chem. 2002;277:45075–45085. doi: 10.1074/jbc.M205264200. [DOI] [PubMed] [Google Scholar]
- 14.Vascotto F, Visintin M, Cattaneo A, Burrone OR. Design and selection of an intrabody library produced de-novo for the non-structural protein NSP5 of rotavirus. J Immunol Methods. 2005;301:31–40. doi: 10.1016/j.jim.2005.03.011. [DOI] [PubMed] [Google Scholar]
- 15.Koch H, Grafe N, Schiess R, Pluckthun A. Direct selection of antibodies from complex libraries with the protein fragment complementation assay. J Mol Biol. 2006;357:427–441. doi: 10.1016/j.jmb.2005.12.043. [DOI] [PubMed] [Google Scholar]
- 16.Visintin M, Melchionna T, Cannistraci I, Cattaneo A. In vivo selection of intrabodies specifically targeting protein-protein interactions: a general platform for an “undruggable” class of disease targets. J Biotechnol. 2008;135:1–15. doi: 10.1016/j.jbiotec.2008.02.012. [DOI] [PubMed] [Google Scholar]
- 17.Gennari F, Mehta S, Wang Y, St Clair Tallarico A, Palu G, Marasco WA. Direct phage to intrabody screening (DPIS): demonstration by isolation of cytosolic intrabodies against the TES1 site of Epstein Barr virus latent membrane protein 1 (LMP1) that block NF-kappaB transactivation. J Mol Biol. 2004;335:193–207. doi: 10.1016/j.jmb.2003.09.073. [DOI] [PubMed] [Google Scholar]
- 18.Desiderio A, Franconi R, Lopez M, Villani ME, Viti F, Chiaraluce R, Consalvi V, Neri D, Benvenuto E. A semi-synthetic repertoire of intrinsically stable antibody fragments derived from a single-framework scaffold. J Mol Biol. 2001;310:603–615. doi: 10.1006/jmbi.2001.4756. [DOI] [PubMed] [Google Scholar]
- 19.Donini M, Morea V, Desiderio A, Pashkoulov D, Villani ME, Tramontano A, Benvenuto E. Engineering stable cytoplasmic intrabodies with designed specificity. J Mol Biol. 2003;330:323–332. doi: 10.1016/s0022-2836(03)00530-8. [DOI] [PubMed] [Google Scholar]
- 20.Philibert P, Stoessel A, Wang W, Sibler AP, Bec N, Larroque C, Saven JG, Courtete J, Weiss E, Martineau P. A focused antibody library for selecting scFvs expressed at high levels in the cytoplasm. BMC Biotechnol. 2007;7:81. doi: 10.1186/1472-6750-7-81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shaki-Loewenstein S, Zfania R, Hyland S, Wels WS, Benhar I. A universal strategy for stable intracellular antibodies. J Immunol Methods. 2005;303:19–39. doi: 10.1016/j.jim.2005.05.004. [DOI] [PubMed] [Google Scholar]
- 22.Ritz D, Beckwith J. Roles of thiol-redox pathways in bacteria. Annu Rev Microbiol. 2001;55:21–48. doi: 10.1146/annurev.micro.55.1.21. [DOI] [PubMed] [Google Scholar]
- 23.Nakamoto H, Bardwell JC. Catalysis of disulfide bond formation and isomerization in the Escherichia coli periplasm. Biochim Biophys Acta. 2004;1694:111–119. doi: 10.1016/j.bbamcr.2004.02.012. [DOI] [PubMed] [Google Scholar]
- 24.Leichert LI, Jakob U. Protein thiol modifications visualized in vivo. PLoS Biol. 2004;2:e333. doi: 10.1371/journal.pbio.0020333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Masip L, Klein-Marcuschamer D, Quan S, Bardwell JC, Georgiou G. Laboratory evolution of Escherichia coli thioredoxin for enhanced catalysis of protein oxidation in the periplasm reveals a phylogenetically conserved substrate specificity determinant. J Biol Chem. 2008;283:840–848. doi: 10.1074/jbc.M705147200. [DOI] [PubMed] [Google Scholar]
- 26.Harvey BR. AustThe University of Texas; 2003. Anchored Periplasmic Expression (APEx): a versatile technology for the flow cytometric selection of high affinity antibodies from Eshcerichia coli expressed libraries; pp. 10–14. Ph.D. Thesis. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ki JJ, Kawarasaki Y, Gam J, Harvey BR, Iverson BL, Georgiou G. A periplasmic fluorescent reporter protein and its application in high-throughput membrane protein topology analysis. J Mol Biol. 2004;341:901–909. doi: 10.1016/j.jmb.2004.05.078. [DOI] [PubMed] [Google Scholar]
- 28.Jeong KJ, Seo MJ, Iverson BL, Georgiou G. APEx 2-hybrid, a quantitative protein-protein interaction assay for antibody discovery and engineering. Proc Natl Acad Sci USA. 2007;104:8247–8252. doi: 10.1073/pnas.0702650104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mazor Y, Van Blarcom T, Mabry R, Iverson BL, Georgiou G. Isolation of engineered, full-length antibodies from libraries expressed in Escherichia coli. Nat Biotechnol. 2007;25:563–565. doi: 10.1038/nbt1296. [DOI] [PubMed] [Google Scholar]
- 30.Little SF, Leppla SH, Cora E. Production and characterization of monoclonal antibodies to the protective antigen component of Bacillus anthracis toxin. Infect Immun. 1988;56:1807–1813. doi: 10.1128/iai.56.7.1807-1813.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Krishnanchettiar S, Sen J, Caffrey M. Expression and purification of the Bacillus anthracis protective antigen domain 4. Protein Expr Purif. 2003;27:325–330. doi: 10.1016/s1046-5928(02)00612-5. [DOI] [PubMed] [Google Scholar]
- 32.Joly JC, Swartz JR. In vitro and in vivo redox states of the Escherichia coli periplasmic oxidoreductases DsbA and DsbC. Biochemistry. 1997;36:10067–10072. doi: 10.1021/bi9707739. [DOI] [PubMed] [Google Scholar]
- 33.Drummond DA, Iverson BL, Georgiou G, Arnold FH. Why high-error-rate random mutagenesis libraries are enriched in functional and improved proteins. J Mol Biol. 2005;350:806–816. doi: 10.1016/j.jmb.2005.05.023. [DOI] [PubMed] [Google Scholar]
- 34.Ladbury JE, Wynn R, Thomson JA, Sturtevant JM. Substitution of charged residues into the hydrophobic core of Escherichia coli thioredoxin results in a change in heat capacity of the native protein. Biochemistry. 1995;34:2148–2152. doi: 10.1021/bi00007a007. [DOI] [PubMed] [Google Scholar]
- 35.Tanaka T, Rabbitts TH. Functional intracellular antibody fragments do not require invariant intra-domain disulfide bonds. J Mol Biol. 2008;376:749–757. doi: 10.1016/j.jmb.2007.11.085. [DOI] [PubMed] [Google Scholar]
- 36.Ribnicky B, Van Blarcom T, Georgiou G. A scFv antibody mutant isolated in a genetic screen for improved export via the twin arginine transporter pathway exhibits faster folding. J Mol Biol. 2007;369:631–639. doi: 10.1016/j.jmb.2007.03.068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Harvey BR, Georgiou G, Hayhurst A, Jeong KJ, Iverson BL, Rogers GK. Anchored periplasmic expression, a versatile technology for the isolation of high-affinity antibodies from Escherichia coli-expressed libraries. Proc Natl Acad Sci USA. 2004;101:9193–9198. doi: 10.1073/pnas.0400187101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jung ST, Jeong KJ, Iverson BL, Georgiou G. Binding and enrichment of Escherichia coli spheroplasts expressing inner membrane tethered scFv antibodies on surface immobilized antigens. Biotechnol Bioeng. 2007;98:39–47. doi: 10.1002/bit.21405. [DOI] [PubMed] [Google Scholar]
- 39.Griswold KE, Kawarasaki Y, Ghoneim N, Benkovic SJ, Iverson BL, Georgiou G. Evolution of highly active enzymes by homology-independent recombination. Proc Natl Acad Sci USA. 2005;102:10082–10087. doi: 10.1073/pnas.0504556102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Calhoun DB, Englander SW. Internal protein motions, concentrated glycerol, and hydrogen exchange studied in myoglobin. Biochemistry. 1985;24:2095–2100. doi: 10.1021/bi00329a043. [DOI] [PubMed] [Google Scholar]
- 41.Segatori L, Paukstelis PJ, Gilbert HF, Georgiou G. Engineered DsbC chimeras catalyze both protein oxidation and disulfide-bond isomerization in Escherichia coli: reconciling two competing pathways. Proc Natl Acad Sci USA. 2004;101:10018–10023. doi: 10.1073/pnas.0403003101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hayhurst A, Happe S, Mabry R, Koch Z, Iverson BL, Georgiou G. Isolation and expression of recombinant antibody fragments to the biological warfare pathogen Brucella melitensis. J Immunol Methods. 2003;276:185–196. doi: 10.1016/s0022-1759(03)00100-5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.