

Abstract
Mammalian sterile 20 (MST1) kinase, a member of the sterile 20 (Ste-20) family of proteins, is a proapoptotic cytosolic kinase that plays an important role in the cellular response to oxidative stress. In this study, we report on the development of a potent and selective MST1 kinase inhibitor based on a ruthenium half-sandwich scaffold. We show that the enantiopure organoruthenium inhibitor, 9E1, has an IC50 value of 45 nM for MST1 and a greater than 25-fold inhibitor selectivity over the related Ste-20 kinases, p21 activated kinase 1 (PAK1), and p21 activated kinase 4 (PAK4) and an almost 10-fold selectivity over the related Thousand and one amino acids kinase 2 (TAO2). Compound 9E1 also displays a promising selectivity profile against unrelated protein kinases, however, the proto-oncogene serine/threonine protein kinase PIM1 (PIM-1) and glycogen synthase kinase 3 (GSK-3β) are inhibited with IC50 values in the low nanomolar range. We also show that 9E1 can inhibit MST1 function in cells. A cocrystal structure of a related compound with PIM-1 and a homology model with MST1 reveals the binding mode of this scaffold to MST1 and provides a starting point for the development of improved MST1 kinase inhibitors for possible therapeutic application.
INTRODUCTION
The MST1 (mammalian sterile 20) kinase is a proapoptotic cytosolic kinase that plays an important role in diverse biological processes including the cellular response to oxidative stress.1, 2 Activation of MST1 via the caspase-3 pathway leads to apoptosis and overexpression of MST1 results in the induction of apoptosis in a variety of cell backgrounds through a pathway that involves activation of SAPK (stress-activated protein kinase.1, 3 Upon activation of the apoptotic pathway, MST1 is cleaved by caspase-3, resulting in its translocation to the nucleus.
Physiologically relevant MST1 substrates have not been identified until recently. Studies by Azad and coworkers demonstrate that cytosolic MST1 kinase phosphorylates the Forkhead box protein O (FOXO) transcription factor.2 Specifically, under conditions of oxidative stress, MST1 kinase phosphorylates FOXO at conserved serines within the DNA binding domain resulting in the disruption of FOX complexes with 14-3-3 protein in the cytosol and the subsequent translocation of FOXO to the nucleus to activate FOXO-regulated genes.4, 5 Once nuclear, FOXO proteins regulate the expression of genes involved in apoptosis, cell cycle transitions, DNA repair, oxidative stress, muscle growth, cell differentiation and glucose metabolism.5, 6 The MST1-FOXO signaling pathway is also feedback regulated since FOXO activates the pro-apoptotic gene Fas ligand (FasL), which encodes a protein that is responsible for the activation of MST1 kinase.7, 8 The MST1 kinase also regulates the FOXO/DAF16 signaling pathway and the correlation between the misregulation of FOXO with several pathological states including cancer and diabetes mellitus implicates the MST1-FOXO signaling pathway as an important target for inhibition for possible therapeutic application.
The caspase cleaved form of MST1 kinase has also been reported to be responsible for phosphorylation of histone H2B at serine fourteen.9 This phosphorylation event has been linked to chromatin condensation, which is a hallmark of apoptosis. Phosphorylation of H2B in conjunction with other modifications promotes DNA fragmentation and cell death. The currently available caspase inhibitors have not been effective in decreasing cell death after the initial stress (such as ischemia) has occurred,10 perhaps because caspases are no longer needed after effector caspases initiate the cell death pathway. It remains an intriguing possibility that the cell death response may be more effectively blunted through the inhibition of downstream activators of apoptosis such as MST1. To date, however, MST1-selective inhibitors have not yet been reported.
One challenge in developing a MST1-selective kinase inhibitor, is that it is a member of a large family of related Ste-20 kinases. The Ste-20 kinases are further divided into the p21-activated kinase (PAK) and germinal center kinase (GCK) families.11 They are characterized by the presence of a highly conserved kinase domain and a noncatalytic region of significant sequence divergence that enables the kinases to interact with various signaling molecules and regulatory proteins. Other well known members of the family include PAK1, PAK4 and TAO2 kinases.12, 13 Many of Ste-20 proteins are part of the MAP (mitogen activated protein) kinase pathway and the deregulation of these kinases has been associated with disease.14, 15
Chemically inert metal complexes can serve as promising scaffolds for the design of enzyme inhibitors.16 For example, organometallic compounds containing a chelating pyridocarbazole moiety have been successfully exploited for the development of potent and selective inhibitors for the kinases GSK-3, PIM-1, phosphatidyl-inositol-3-kinase alpha (PI3Kα) and others.17, 18
In this study, we use a fragment-based drug design approach and initially screened libraries of ruthenium and platinum-containing organometallic kinase inhibitors against MST1 to identify cyclopentadienyl ruthenium half-sandwich complexes as sub-micromolar inhibitors for MST1. A subsequent functionalization of the cyclopentadienyl moiety according to a recently established procedure resulted in a further improvement by about an order of magnitude, yielding a mid-nanomolar MST1 inhibitor.19 Additionally, we also show that this compound is cell permeable and can inhibit MST1 in vitro and in cells and can block downstream phosphorylation of endogenous histone H2B, a marker associated with the onset of apoptosis.
RESULTS
Identification of a lead structure by screening of an organometallic library
An initial compound library containing 58 compounds (Figure S1) with various substituents around both the ruthenium or platinum metal and the pyridocarbazole ring system were screened against MST1.18-22 The MST1 kinase assay was initially performed using 50 nM of enzyme, 250 μM axltide peptide derived from the mouse insulin receptor 1 as a substrate, 100 μM ATP and 5 μM of the respective inhibitor. Figure 1A summarizes the results of this assay in the form of a histogram plotting the percentage of remaining enzyme activity as a function of added compound. The results of this initial screen demonstrated that among the various metal fragment combinations tested, compounds containing the half-sandwich cyclopentadienyl (Cp) moiety together with a CO ligand were the most potent MST1 inhibitors. In particular, the screen yielded five potent hits one of which contained an isoquinoline hetereocycle that was distinct from inhibitors that were previously described for PIM-1 and GSK-3β, so was used for further studies.17,18 The structures of three representative inhibitors are shown in Figure 1B and the IC50 value for the isoquinoline compound 2 was calculated using a dose response curve (Figure 1C). The IC50 value of compound 2 for MST1 falls within the sub-micromolar range providing an excellent starting point for further inhibitor optimization. Therefore, compound 2 was subsequently used as a lead structure for further modifications.
Figure 1.
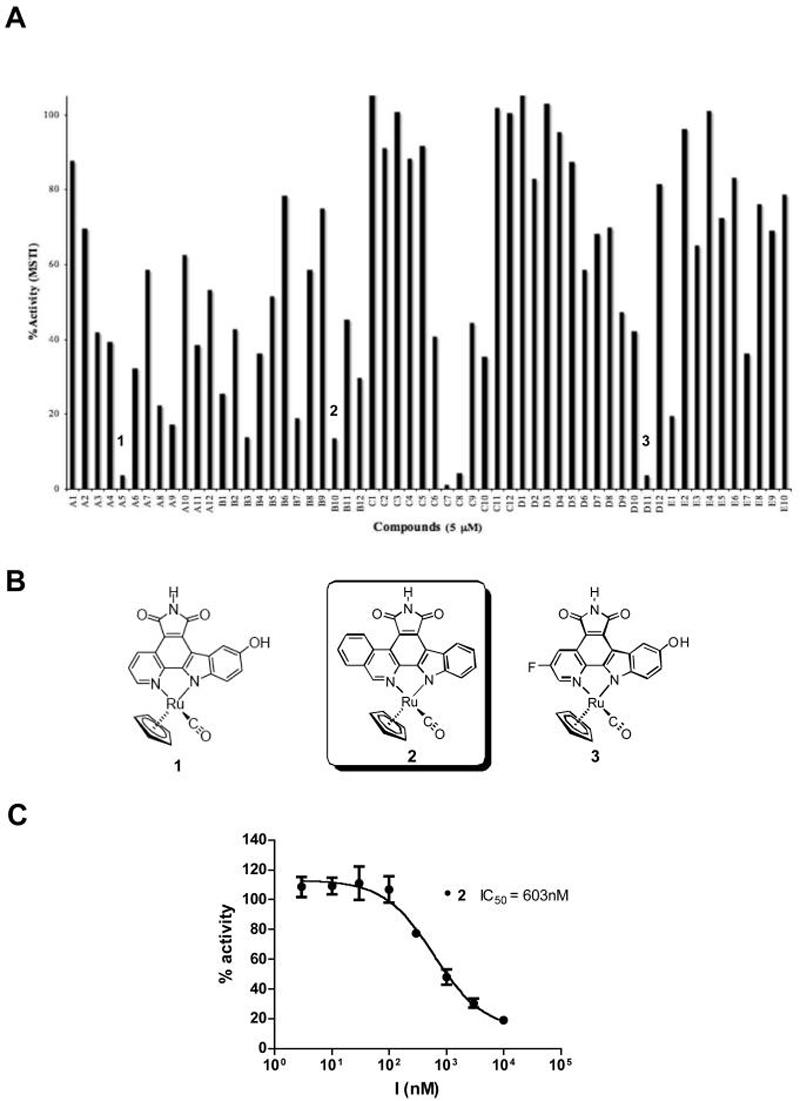
A) Screening of an 58-member organometallic pyridocarbazole library against MST1. Histogram with % activity represented on the y-axis plotted against the compound identifier on the x-axis. See supporting information for the identity of the compounds other than 1-3. The screen was carried out at a compound concentration of 5 μM, an MST1 concentration of 50 nM and an ATP concentration of 100 μM. B) Chemical structures of the three identified hits. C) The IC50 curve for compound 2. Final MST1 concentration used was 1 nM in the presence of 100 μM ATP. The GraphPad Prism software was used to make the graphs.
Improving the lead structure by derivatization of the Cp moiety
We next functionalized the Cp ligand by a recently developed rapid amide formation protocol.19 For this, we used compound 8, bearing an N-hydroxysuccinimide (NHS) ester functionality at the Cp ring (Figure 2). NHS-ester 8 was synthesized starting from TBS-protected isoquinoline analogue 4 by reacting with ruthenium complex 5 in the presence of K2CO3 to afford the ruthenium-pyridocarbazole complex 6 and subsequently the TBS- deprotected complex 7 after treatment with TBAF (38% over two steps). The reaction of 7 with NHS in the presence of the coupling reagent EDCI yielded the activated ester 8 in 83% yield. This activated ester enabled us to rapidly synthesize amide libraries, simply by quenching of the NHS-ester 8 with a library of primary amines.19 Following this approach we synthesized a small library of 44 cyclopentadienyl-amides (compounds 9-13 in Figure 3B and Figure S2) and carried out a screening without purification at an inhibitor concentration corresponding to the IC50 of the first generation hit, compound 2, of 615 nM and an enzyme concentration of 1 nM. The results revealed the compounds 9, 10, 11, 12 and 13 as the most potent MST1 inhibitors with less than 10% of remaining enzyme activity at an inhibitor concentration of 615 nM (Figures 3A and 3B). Substituents with a secondary or tertiary amine in the β-position to the amide group greatly enhanced the MST1 inhibitor potency. However, changing the position of the basic amine or introducing bulky substituents into the side chain significantly decreased the potency for MST1 inhibition (Figure 3A and Figure S2). We observed similar structure-activity relationships with the protein kinase PIM-1.19 In order to further characterize one of these hits, we resynthesized and purified compound 9 and determined the IC50 values of the single enantiomers 9E1 (R-configuration) and 9E2 (S-configuration) (Figure S3). The promising effect on optimization of the scaffold can be seen in the IC50 values that show a decrease from the first generation compound 2 with an IC50 value of 603 nM to the third generation compound 9E1 with an IC50 value of about 10-fold lower. Specifically, dose response curves employing the two enantiomers of compound 9E reveals IC50 values of 45 nM and 212 nM for compounds 9E1 and 9E2, respectively for MST1.
Figure 2.

Modification of the cyclopentadienyl moiety by a rapid amide formation procedure. TBAF = tetrabutylammonium fluoride, TBS = tert-butyldimethylsilyl. Compounds 6, 7, 8 and 9-53 are formed as racemic mixtures.
Figure 3.
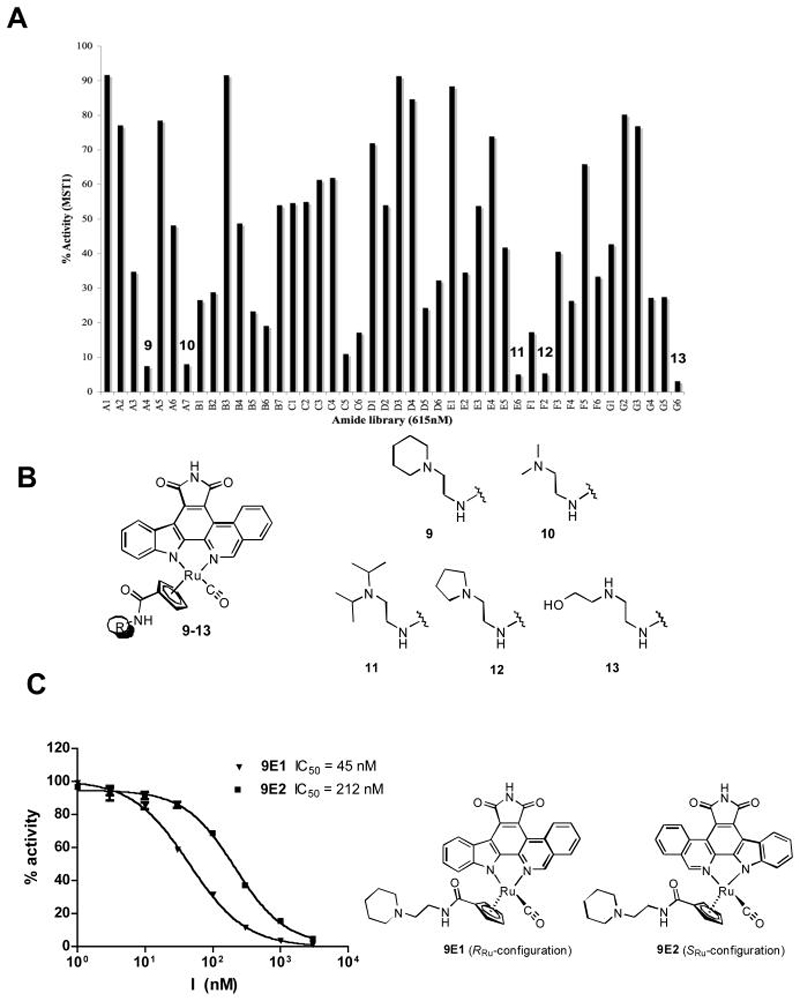
Screening of the amide library for MST1 inhibitors. A) Screening of 44 amide based ligands was performed at an MST1 concentration of 1 nM and an inhibitor concentration of 615 nM. The data are shown as a histogram with % activity represented on the y-axis plotted against the compound identifier on the x-axis. B) Structural scaffold and the structures of the most potent MST1 compounds from this series. See also supporting information for additional amide side chains. C) Compound 9 enantiomer selectivity for MST1. An enzyme concentration of 1 nM was used for the calculation at an ATP concentration of 100 μM. GraphPad Prism was used for IC50 determination and curve fitting. Experiments were carried out in triplicate and error bars represent standard deviation from the mean. A comparison of IC50 values between the two enantiomers shows that MST1 has a strong preference for the 9E1 enantiomer. For the assignment of absolute configurations, see the supporting information.
Selectivity against other Ste-20 kinases
To determine the selectivity of the most potent MST1 inhibitor, compound 9E1 was tested against related Ste-20 kinases including PAK1, PAK4 and TAO2. We used myelin basic protein (MBP) as a common in vitro substrate for all four kinases. The inhibition profiles and IC50 values are shown in Figure 4A and they reveal that compound 9E1 has at least an 8-fold discrimination for MST1 over the other related kinases. Interestingly, the 9E2 enantiomer shows greater potency for TAO2 over MST1 (Figure 4B), revealing that the stereochemistry around the Ru metal is an important component of the kinase inhibitor selectivity of compound 9. Taken together, we have shown that compound 9E1 and related compounds have sub-micromolar potency for MST1 and an almost 10-fold preference for inhibition of MST1 over TAO2 and more than 20 fold preference over PAK1 and PAK4.
Figure 4.
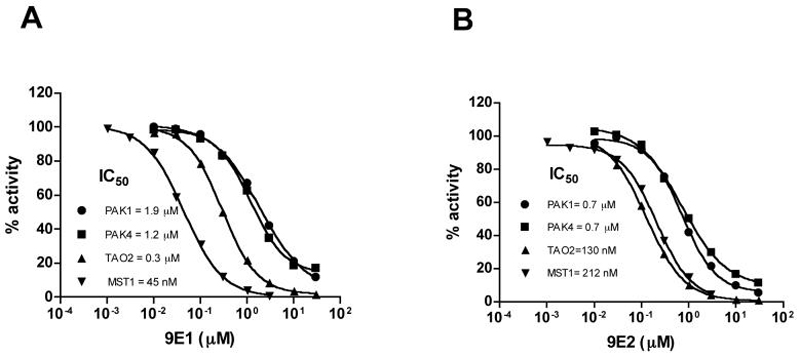
Selectivity profile of MST1 inhibitor for Ste-20 proteins. IC50 curves of compound 9E1 (A) and 9E2 (B) against four Ste-20 proteins. MBP was used as a common substrate for PAK1, PAK4 and TAO2.
Selectivity against unrelated kinases
Ruthenium based organometallic half-sandwich compounds related to compound 2 and 9E1 have been exploited for the design of inhibitors for GSK-3β,23 PIM-1,24 and PI3K.18 We were therefore interested in determining the selectivity profile of 9E1 against these kinases. As shown in Figures 5A and 5B, the inhibitor 9E1 had strong discrimination for MST1 over the proto-oncogene serine/threonine-protein kinase B-raf (BRAF) and PI3K showed no inhibition of these other kinases at an inhibitor concentration of 100 nM, while showing 70% inhibition of MST1 at this inhibitor concentration. Both BRAF and PI3K had IC50 values in the micromolar range and in comparison to MST1 are poorly inhibited by both enantiomers of compound 9. In contrast, PIM-1 and GSK-3β showed inhibition profiles with IC50 values in the low nano-molar range, demonstrating that 9E1 had some but not complete selectivity for MST1.
Figure 5.
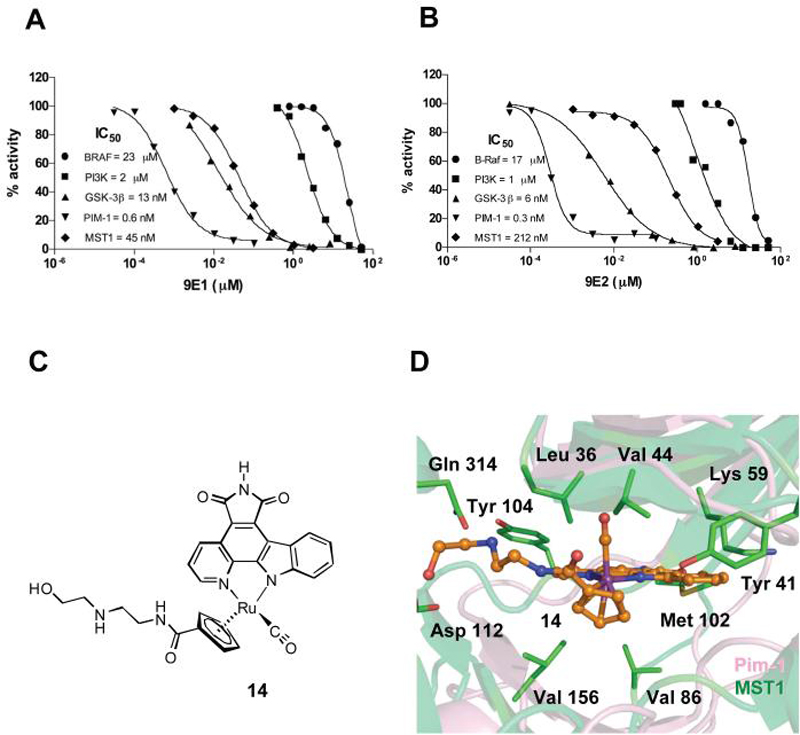
Selectivity profile of MST1 inhibitors for unrelated protein kinases. A) Comparison of IC50 values of compound 9E1 against four unrelated protein kinases. GraphPad Prism was used to obtain the IC50 value for each kinase. The IC50 values for each of the kinases are: MST1, 45 nM; PIM-1, 0.6 nM; GSK-3β, 13 nM; PI3K, 2 μM; BRAF, 23 μM. B) Comparison of IC50 values of compound 9E2 against four unrelated protein kinases: MST1, 212 nM; PIM-1, 0.3 nM; GSK-3β, 6 nM; PI3K, 1 μM; BRAF, 17 μM. C) Compound 14 cocrystallized with PIM-1. D) Superposition of the MST1 homology model (green) onto the crystal structure of PIM-1 (violet) co-crystallized with compound 14 highlighting active site interactions of amine moiety of the inhibitor.
To further probe the selectivity of the 9E1 inhibitor, we submitted the 9E1 inhibitor and staurosporine (as a control) to Millipore's kinase profiler service for screening against 50 kinases using inhibitor concentrations of 10 nM. The results of this study are shown in figure S4 and reveal that only 4 of the 50 kinases assayed are inhibited by the 9E1 compound to below 85% activity. Not surprisingly PIM-1 (4% activity) and GSK-3β (44% activity) are potently inhibited by the 9E1 inhibitor, confirming our earlier results (Figure 5A). In addition, MST1, spleen tyrosine kinase (Syk) and maternal embryonic leucine zipper kinase (MELK) are each inhibited to 81% activity by the 9E1 inhibitor while the other kinases are more poorly inhibited if at all. Of note, in contrast to the 9E1 inhibitor, that shows greater than 15% inhibition for MST1 and 4 other of 50 of the kinases tested, staurosporine shows greater than 15% inhibition for more than half of the kinases profiled, showing that the 9E1 MST1 inhibitor is considerably more selective than staurosporine. Taken together, these studies support the conclusion that the 9E1 inhibitor shows significant, albeit not complete, selectivity for MST1 over several other kinases.
Crystallography and homology modeling
Given that the MST1 9E1 compound is a potent inhibitor against PIM-1, we exploited the ability to cocrystallize PIM-1 complexes with organometallic kinase inhibitors.24 Specifically, we successfully cocrystallized PIM-1 with an 9E1 analogue, compound 14, containing a derivatized side chain with a basic amine in the beta-position, (Figures 5C, S5 and Table S1). We then used the program modeller to build a homology model of MST1 based on high sequence identity within the Ste20 family members and superimposed the model onto the PIM-1 X-ray structure. As expected, the structure reveals that the inhibitor occupies the ATP binding site of PIM-1 (Figure 5D and supporting information). The model suggests that the binding of the pyridocarbazole ring into the MST1 active site is stablized by the surrounding hydrophobic residues Tyr 104, Met 102, Leu 36, Leu 157 and Val 86. Experimental data shows that the IC50 of the isoquinoline backbone inhibitor is further improved by about 10-fold by the addition of the aliphatic amine moiety. The homology model shows that this could be mediated by the amine moiety of the inhibitor that may form hydrogen bonds within the active site of MST1, which could result in further stablization of the ligand bound to MST1 (Figure 5D). A protonated state of the amine moeity might also be stablized by proximal negatively charged amino acids. In the case of PIM-1, Glu 171, Asp128 and Asp 131 encompass the binding pocket. In the case of MST1 the amide side chain is modeled to be stabilized by Asp112, Glu311 and Glu317. MST1 kinase can potentially further anchor the ligand by hydrogen bonding or charge interactions of the Gln 314 residue which is present in an extra C-terminal helix-turn helix motif at the end of the kinase domain that is unique to the Ste-20 family and absent in PIM-1 (Figures 5D and S4). Indeed, it appears that this region shows low sequence conservation within the Ste-20 kinase family members, suggesting that it may be exploited for the further optimization of inhibitor selectivity against other members of the Ste-20 kinases. Together, the modeling of the MST1 kinase domain based on the structure of PIM-1 bound to compound 14 sugggests a mode of interaction of compound 9E1 with MST1, although the details of such interaction requires further studies, preferably involving a MST1/9E1 crystal structure.
Cell permeability of compound 9E1 and inhibition of endogenous MST1 kinase activity
To establish if the 9E1 inhibitor is able to permeate cells at a concentration range where MST1 inhibition can occur and where cells are able to tolerate the compound without significant cell death, we performed a MTT cell viability assay using HeLa cells. The assay was performed for both enantiomers of compound 9 and the results indicates that significant cell death occurs at a compound concentrations of about 5 μM and a plot of cell viability against inhibitor concentration shows that 50% of the cells die at a compound concentration of about 1.2 ΜM (Figure 6A).
Figure 6.
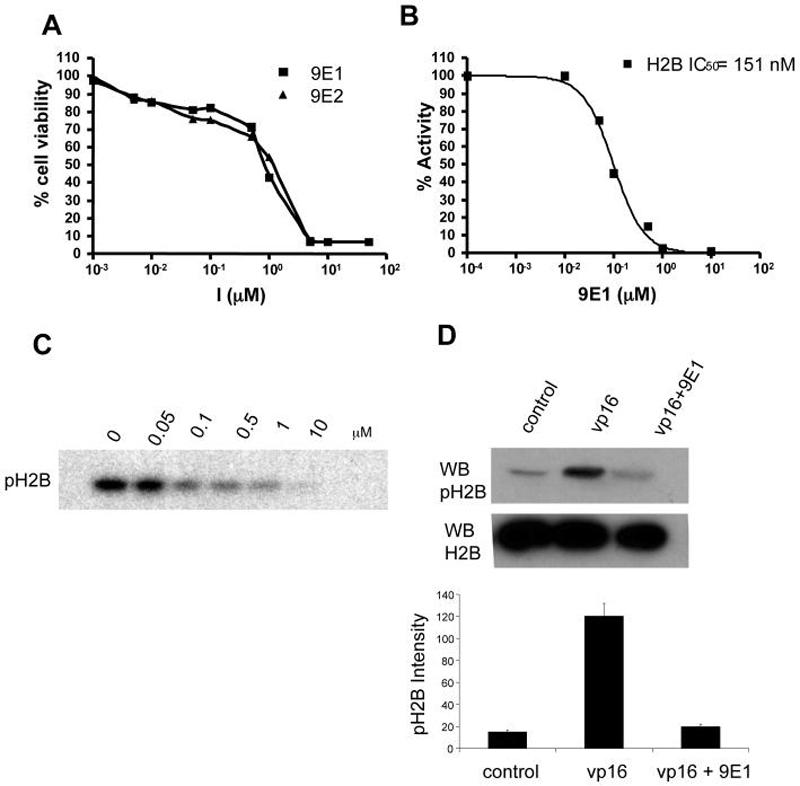
Characterization of MST1 inhibition in cells. A) Cell proliferation assay. The assay was performed for both the enantiomers of compound 9. The IC50 for cell survival is 1.2μM. B) Ex-vivo assay with the endogenously expressed MST1 immunoprecipitated from HeLa cells using MST1 specific antibody (Millipore Inc.). The IC50 curve for the inhibition profile of H2B is measured at an ATP concentration of 100 μM. C) in vitro kinase assay performed on histone H2B using endogenous MST1. The radiography experiment shows γP32 incorporation into histone H2B at indicated concentrations of inhibitor. D) Inhibition by MST1 in cells: The downstream inhibition of MST1 was monitored by probing histone H2B serine 14 phosphorylation levels in HeLa cells as a function of induction of apoptosis in the presence or absence of 1μM 9E1 inhibitor. The phosphorylation status was probed using phospho-S14 histone H2B antibody. Total histone H2B levels were also monitored using histone H2B antibody.
To test inhibition of endogenous MST1 in cells, we also used HeLa cells as a model system. Endogenous MST1 kinase was immunoprecipitated using an anti-MST1 antibody that recognizes the regulatory region of MST1 kinase and hence does not interfere with the kinase activity of MST1. The immunoprecipitated MST1 was used to perform an in vitro kinase assay against the histone H2B substrate. Cells were incubated with various concentrations of inhibitor ranging from 50 nM to10 μM for 48 hours and MST1 kinase was immunoprecipitated and subsequently assayed in vitro for histone H2B phosphorylation. The results of these studies are shown in Figures 6B and 6C and indicate that the 9E1 inhibitor can enter into cells and can potently inhibit endogenous MST1 kinase activity. The inhibition profile of MST1 kinase reveals an IC50 value of about 151 nM of compound 9E1 for MST1 inhibition of MST1-mediated H2B phsophorylation in cells.
To determine if compound 9E1 is able to inhibit the phosphorylation of serine-14 on histone H2B, an endogenous target of MST1 kinase, we incubated HeLa cells with compound 9E1 at a concentration of 1 μM and initiated an apoptotic signal leading to S14 phosphorylation of histone H2B by the addition of an apoptotic inducing agent etoposide (vp16). We monitored the levels of histone S14 phosphorylation in normal cells, cells induced with vp16 and cells incubated with vp16 followed by incubation with the compound 9E1 (Figure 6D) and the results show that in the presence of compound 9E1, S14 phosphorylation of histone H2B is inhibited by more than 90% at an inhibitor concentration of 1 μM. This result demonstrates that compound 9E1 can inhibit a downstream target of MST1 in cells. Taking our in vitro and cellular results together demonstrates that compound 9E1 is a potent and specific MST1 inhibitor that functions to inhibit MST1-mediated phosphoryaltion both in vitro and in cells.
DISCUSSION
In this study, we use organoruthenium compounds as small molecule protein kinase inhibitors. A constant challenge in the field of kinase inhibitor design is to develop selective and potent inhibitors that target the kinase of interest without inhibiting other essential kinases. Staurosporine is an example of an ATP competitive kinase inhibitor that inhibits multiple kinases with moderate to high efficacy.25 The rational underlying the organoruthenium compounds described here is to use the staurosporine scaffold but to combinatorially alter chemical space around it so as to achieve size and shape complementarily for a particular kinase active site pocket. The appeal of this approach lies in the fact that the same scaffold can be easily varied through small modifications in the ring system and ligands attached to the ruthenium metal to create compounds with high potency, in the low nanomolar or sub nanomolar range, and a high degree of kinase selectivity. We used this strategy in this study to develop highly selective and potent MST1 kinase inhibitors that function both in vitro and in cells.
Analysis of several structurally characterized kinases in the context of the known activity of compound 9E1 against these kinases, provides clues into additional avenues that might be exploited to develop organoruthenium inhibitors with greater MST1 specificity. A comparision of the crystal structures of the Ste-20 kinases, PAK1, PAK4 and TAO2 reveals that the active site for PAK1 is more open than PAK4 and TAO2, possibly preventing intimate inhibitor-kinase contacts (Figure S6). This may explain why compound 9E1 inhibits PAK1 more weakly than PAK4 and TAO2. The more compact sizes of the PAK4 and TAO2 active sites might allow for more intimate binding of the the isoquinilone inhibitor (Figure S6). The TAO2 active site is also flanked on one side by an additional C-terminal helix that is not present in other kinases and that is predicted from modeling studies to come in close proximity to the amide tail of the isoquinoline inhibitor (Figure S6). This proximity might allow for additional protein-inhibitor contacts that may account for the better inhibitor binding of 9E1 to TAO2 relative to PAK1 and PAK4. The BRAF and PI3K kinases have the most restrictive active sites, possibly correlating with the poor inhibitory properties of the most potent MST1 kinase inhibitors against these other unrelated kinases (Figure S6). Taken together, these comparisons suggests that additional R group substitutions of the isoquinilone ring of these potent organometallic MST1 inhibitors might further increase MST1 specificity.
The PIM-1 and GSK-3β kinases have the largest active sites of the kinases described above and these relatively large active site might easily accommodate the MST1 inhibitors, thus correlating with the related inhibitor profiles of these two kinases. However, we do show that MST1 and PIM-1 have preference for different enantiomers of compound 9. Therefore, the nature of the ligands and stereochemistry around the ruthenium metal can further be exploited to increase the specificty of organoruthenium MST1 inhibitors.
The MST1 kinase is activated during conditions of oxidative stress and it plays a prominant role in the stress induced apoptosis response, in part, through the phosphorylation of nuclear histone H2B.9 MST1 also phosphorylates the FOXO transcription factor in the cytosol, resulting in its nuclear translocation and activation of genes involved in stress tolerance, metabolism and cellular proliferation.5 MST1 therefore has roles in both cell proliferation and death. Because of this distinction, it is no surprise that the fine-tuning of MST1 activity has been found to be important for health and disease. Indeed, the misregulation of the MST1 pathway has been implicated in diseases such as cancer, diabetes and age related disorders.15, 26 Additionally, based on expression patterns and signaling pathways that MST1 kinase is associated with further suggests roles in neurodegenerative and cardiac disorders where premature apoptosis causes cell death.15, 27 Therefore, MST1 is an attractive target for small molecule inhibition for possible therapeutic applications. Here we show that the development of organoruthenium MST1-specific inhibitors provides an attractive avenue of investigation along this path.
EXPERIMENTAL SECTION
Commercial reagents
Recombinant histone H2B (New England Biolabs) from human was used as a MST1 substrate at a concentration of 1mg/mL. Radiolabeled γ-P32 ATP (3000 Ci/mmol) was purchased from Perkin Elmer Life Sciences. TAO2 kinase was purchased from Cell Signaling. All other reagents were purchased from Sigma Pharmaceuticals.
Protein cloning, expression and purification
MST1 kinase
Human full length MST1 kinase was amplified from an Open Biosystem plasmid (ID # 7939613) and PCR amplified into a Pachis-Tev baculoviral transfer vector. Recombinant viruses were selected and amplified and harvested in Sf9 cells. Cells were lysed by sonication in wash buffer (50 mM Tris pH 8.0, 400 mM NaCl, 2 mM imidazole, 1 mM β-mercaptoethanol) supplemented with protease inhibitors. The clarified supernatant was applied to Ni2+ nitrilotriacetic acid- agarose (Qiagen) and washed thoroughly with wash buffer supplemented with 10 mM imidazole. The resulting protein was eluted with 25 mM Hepes, pH 7.5, 100 mM NaCl, 200 mM imidazole. The eluted protein was concentrated and loaded onto a Superdex 200 (Pharmacia) gel filtration column pre-equilibrated with 25 mM Hepes pH 7.5, 100 mM NaCl. The eluted fractions were pooled, concentrated and flash frozen.
PAK4 kinase
Human PAK4 plasmid was a gift from Dr. S Knapp (Oxford University, Center for Structural Genomics Botnar Research Centre, UK). Kinase domain of PAK4 (291-591) was subcloned into a pET151-TOPO his tag vector. The protein was expressed in BL21DE3 bacterial cells and induced with 1 mM IPTG overnight at 18°C. The cells were collected by centrifugation and resuspended in 50 mM Hepes pH 7.5, 500 mM NaCl, 5 mM DTT. The cells were lysed and after centrifugation the clarified supernatant was purified by Ni affinity purification as described above for the MST1 protein and the protein was treated overnight with TEV protease to cleave off the 6xHis tag. Further purification was achieved by using SP-Sepharose ion exchange and Superdex 200 gel filtration steps. The protein was concentrated to 9 mg/mL and stored in 50 mM Tris pH 8.0, 150 mM NaCl.
PAK1 kinase
Human PAK1 plasmid was a gift from Dr. J. Kissil (The Wistar Institute). The kinase domain of PAK1 (249-545) was sub cloned into a pFastbac baculoviral system with an N-terminal 6xHis-tag. The protein production and Ni affinity purification was carried out as described above. The protein was finally concentrated to 1.2 mg/ml and stored in a buffer containing 20 mM Tris pH 8.0, 125 mM NaCl, 5 mM DTT.
Protein kinase assay
Kinase assays were performed using labeled γP32 ATP and the incorporation of labeled phosphate onto the axltide peptide substrate was monitored. Kinase assays were performed using kinase buffer containing 20 mM MOPS, 30 mM MgCl2, 0.8 μg/μl BSA, pH 7.0, 5 μCu of labeled γP32 ATP and a final concentration of 100 μM of ATP was used for each reaction. Reaction was performed in a final volume of 25 μL. To determine IC50 values, MST1 was pre-incubated for 30 minutes with various concentrations of inhibitor and an equivalent amount of DMSO was used as a negative control. The reaction was initiated by addition of the substrate and the requisite amount of ATP. All reactions were incubated at room temperature for 30 minutes and then stopped by spoting 17.5 μL of the reaction mixture on a circular P81 phosphocellulose paper (diameter 2.1 cm, Whatman) followed by washing four times (five minutes each wash) with 0.75% phosphoric acid and once with acetone. The dried P81 papers were transferred to a scintillation vial and 4 ml of scintillation cocktail was added and the counts per minute (CPM) determined with a Packard 1500 Tri-Carb Liquid Scintillation Analyzer. IC50 values were defined to be the concentration of inhibitor at which the CPM was 50% of the control sample, corrected for by the background counts. IC50 curves were plotted using Graphpad Prism software.
For all IC50 studies within the Ste-20 family members, myelin basic protein (MBP) was used as a substrate. PAK1, PAK4 and TAO2 kinases were used at a final concentration of 1 nM, 2 nM and 10 nM respectively. The kinases were preincubated with various concentrations of inhibitor in the presence of the substrate for 15 minutes and reactions were initiated by addition of ATP. After 45 minutes, reactions were terminated and measured as described previously for MST1.
The kinase assays for BRAF, PIM-1, PI3K and GSK-3β are described in the supplemental text.
Cell culture
Hela cells were maintained in DMEM media supplemented with 10% FBS. The cells were isolated and cultured as described previously.28
Adherent Cell Proliferation Analysis
Cells were plated into a 96-well plate at a density of 2.5 × 104/mL and left to grow overnight. Cells were treated with increasing concentrations of enantiomers 9E1 and 9E2 at concentrations from 50-0.01 μM in triplicate. In each instance, cells were left to grow for 72 hours before being treated with 20 μL of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazoliumbromide (MTT) for 3 hours (Sigma, St. Louis, MO). After this time, the medium was rapidly removed and the MTT crystals were solubilized using DMSO. The resulting absorbance was read in a plate reader at 560 nm. Absorbance readings were calculated by subtracting the absorbance from blank wells and the reduction in cell growth was calculated as a percentage of control absorbance in the absence of any drug. The presented data shows the mean of at least three independent experiments.
Immunoprecipitation IC50 Assay
Mid log phase Hela cells were treated with indicated concentrations of inhibitor compound 9E1 for 48 hrs. The cells were then collected and washed twice with PBS. The resulting pellet was resuspended in lysate buffer and incubated on ice for 30 minutes. The lysate was clarified by centrifugation for 20 minutes at 13,000 rpm and the supernatant was quantified for total protein content. To extract endogenous MST1 kinase, 600 μg of the clarified lysate was incubated with 6 μL of anti-MST1 antibody (Millipore) for 30 minutes and then 25 μL of Protein G beads (GE healthcare) were added and the mixture was further incubated overnight at 4°C. The immuno-complexes were washed three times in PBS and used for in vitro kinase assays as described above.
Acid extraction of histones
Histones were isolated from Hela cells by an acid extraction procedure. In short, HeLa cells were pelleted and washed with PBS. The cell pellet was dissolved in 0.4 N H2SO4 followed by brief sonication. The supernatant was clarified by centrifugation at 14,000 rpm. Histones were subsequently precipitated by adding 10% tricholoacetic acid to the clarified supernantant. The pellet was washed with acetone and dissolved in water. The total protein content was quantified and equal amounts of samples were loaded onto an SDS gel for further analysis by western blot.
Synthesis of organoruthenium MST1 inhibitors
All reactions were carried out using oven-dried glassware and conducted under a positive pressure of nitrogen unless otherwise specified. NMR spectra were recorded on a DMX-300 (300 MHz), DMX-360 (360 MHz), DMX-400 (400 MHz), DRX-500 (500 MHz) or Bruker AM-500 (500 MHz) spectrometer. Infrared spectra were recorded on either Perkin-Elmer 1600 series FTIR or Nicolet 510 FTIR spectrometer. Low-resolution mass spectra were obtained on an LC platform from Micromass using ESI technique. ES-TOF spectra were measured by Waters Micromass MS Technologies. High-resolution mass spectra were obtained with a Micromass AutoSpec or Thermo LTQ-FT instrument using either CI or ES ionization. Compounds 429 and 519 were prepared according to reported literature procedures. Reagents and solvents were used as received from standard suppliers. Chromatographic separations were performed using Silia-P Flash silica gel from Silicycle Inc or MN Kiesegel 60M from Macherey-Nagel GmbH.
Compound 6
A suspension of ligand 4 (75 mg, 0.166 mmol), 5 (122 mg, 0.216 mmol), and K2CO3 (25 mg, 0.183 mmol) in CH3CN/CH2Cl2/2-(trimethylsilyl)ethanol (7:2:1) was purged with nitrogen and stirred at room temperature overnight. The following day, the reaction remained incomplete and was heated to 35°C for 6 hours. The resulting dark red reaction mixture was cooled to room temperature and concentrated to dryness in vacuo. The crude material was adsorbed onto silica gel and subjected to silica gel chromatography using hexanes:EtOAc (3:1). Unable to completely purify 6 from unreacted ligand 4, the impure material was carried to the next step. An analytically pure sample was obtained by subjecting 6 to silica gel chromatography using toluene:acetone (10:1). 1H NMR (500 MHz, CDCl3): δ (ppm) 10.61 (d, J = 8.5 Hz, 1H), 9.42 (s, 1H), 9.10 (d, J = 7.9 Hz, 1H), 8.10-8.07 (m, 2H), 7.83 (ddd, J = 8.0, 7.0, 0.9 Hz, 1H), 7.62 (ddd, J = 8.2, 7.0, 1.2 Hz, 1H), 7.48 (d, J = 8.1 Hz, 1H), 7.41 (ddd, J = 7.9, 7.0, 0.9 Hz, 1H), 6.07 (s, 1H), 5.87 (s, 1H), 5.39 (s, 1H), 5.26 (s, 1H), 4.08-4.05 (m, 2H), 1.11 (s, 9H), 0.70 (s, 6H), 0.62-0.53 (m, 2H), -0.06 (s, 9H). 13C NMR (125 MHz, CDCl3): δ(ppm) 199.0, 175.4, 174.7, 166.1, 159.2, 156.1, 153.8, 140.2, 133.7, 133.2, 133.0, 130.3, 128.6, 128.3, 128.0, 127.0, 126.2, 124.2, 120.9, 119.7, 117.3, 116.4, 114.6, 93.7, 85.6, 82.2, 80.6, 76.7, 63.7, 26.9, 19.4, 17.3, -1.4, -3.3. IR (film): ν(cm-1) 3407, 3119, 2928, 2859, 1969, 1688, 1620, 1582, 1537, 1472, 1375, 1333, 1294, 1242, 1142, 1078, 1042, 924, 837, 764. HRMS calculated for C39H42N3O5RuSi2 (M+H)+ 790.1707, and observed (M+H)+ 790.1696.
Compound 7
A solution of 6 (carried from previous step) in THF (7.5 mL) was purged with nitrogen and tetrabutylammonium fluoride (1 M solution in THF) (664 μL, 0.664 mmol) was added. The reaction mixture was then heated to 45 °C for 2 hours. The resulting red reaction mixture was cooled to 0 °C, quenched with dropwise addition of saturated NH4Cl and extracted into EtOAc. The organic layer was washed with 1 M HCl followed by brine, dried using Na2SO4, filtered and evaporated. The crude material was subjected to silica gel chromatography with CH2Cl2:CH3OH (10:1 to 3:1). The combined product eluents were dried to provide carboxylic acid 7 (36 mg, 38%, over two steps) as a purple solid. 1H NMR (500 MHz, DMSO-d6): δ (ppm) 11.32 (s, 1H), 10.52 (d, J = 8.6 Hz, 1H), 9.94 (s, 1H), 8.85 (d, J = 7.9 Hz, 1H), 8.34 (d, J = 7.8 Hz, 1H), 8.13 (t, J = 8.1 Hz, 1H), 7.97 (t, J = 7.4 Hz, 1H), 7.62 (d, J = 8.1 Hz, 1H), 7.57 (t, J = 7.5 Hz, 1H), 7.34 (t, J = 7.4 Hz, 1H), 6.23 (s, 1H), 6.18 (s, 1H), 5.65 (s, 1H), 5.60 (s, 1H). 13C NMR (125 MHz, DMSO-d6): δ (ppm) 200.4, 170.9, 170.0, 166.5, 161.0, 155.1, 153.3, 139.0, 132.9, 131.5, 131.3, 129.4, 128.9, 128.6, 127.6, 126.9, 124.7, 123.1, 120.1, 119.1, 116.3, 115.2, 114.3, 90.3, 89.7, 81.5, 81.3, 79.4. IR (film): ν(cm-1) 3408, 3063, 2927, 2859, 1957, 1742, 1698, 1626, 1581, 1537, 1467, 1438, 1380, 1349, 1296, 1229, 1158, 757, 692, 642. HRMS calcd for C28H16N3O5Ru (M+H)+ 576.0128, and observed (M+H)+ 576.0124.
Compound 8
A solution of carboxylic acid 7 (36 mg, 0.063 mg) in DMF (1.8 mL) was prepared and cooled to 0 °C. N-hydroxysuccinimide (11 mg, 0.095 mmol) and N-(3-dimethylaminopropyl)-N'-ethylcarbodiimide hydrochloride (18 mg, 0.095 mmol) were added respectively and the reaction was stirred at room temperature overnight. The reaction mixture was then diluted with saturated NH4Cl and extracted into CH2Cl2. The combined organic layers were washed with brine, dried using Na2SO4, filtered and evaporated. The crude material was subjected to silica gel chromatography with CH2Cl2:CH3OH (50:1 to 15:1). The combined product eluents were dried to provide activated ester 8 (35 mg, 83%) as a light purple solid. 1H NMR (500 MHz, DMSO-d6): δ (ppm) 10.55 (d, J = 8.5 Hz, 1H), 9.93 (s, 1H), 8.84 (d, J = 7.7 Hz, 1H), 8.34 (d, J = 8.0 Hz, 1H), 8.16 (t, J = 7.8 Hz, 1H), 7.97 (t, J = 7.4 Hz, 1H), 7.61-7.56 (m, 2H), 7.34 (t, J = 7.2 Hz, 1H), 6.59 (s, 1H), 6.50 (s, 1H), 5.93 (s, 1H), 5.81 (s, 1H), 2.71 (s, 4H). 13C NMR (125 MHz, DMSO-d6): δ (ppm). 199.1, 171.0, 170.0, 170.0, 162.1, 160.8, 155.2, 153.1, 139.1, 133.1, 131.6, 131.5, 129.4, 129.4, 128.9, 128.6, 127.8, 126.9, 124.8, 123.2, 120.4, 119.2, 116.4, 115.0, 114.6, 94.3, 88.9, 84.2, 79.5, 72.1, 25.3. IR (film): ν(cm-1) 3380, 3128, 2925, 2854, 1964, 1732, 1705, 1582, 1534, 1451, 1379, 1272, 1209, 1074, 1022, 989, 747, 688, 645. HRMS calcd for C32H19N4O7Ru (M+H)+ 673.0292, found (M+H)+ 673.0279.
Compound 9 as a racemate
To a solution of ester 8 (15 mg, 0.022 mmol) in DMF (1 mL) was added N-(2-aminoethyl)piperidine (6.2 μL, 0.044 mmol) and the reaction mixture was stirred at room temperature for 2 hours. The DMF was then removed using high vacuum and the crude material co-evaporated once with MeOH. The crude material was subjected to silica gel chromatography with CH2Cl2:CH3OH (10:1 to 3:1). The combined product eluents were dried to provide amide 9 (13 mg, 87%) as a purple solid. 1H NMR (300 MHz, CD3OD-d4): δ(ppm) 10.41 (d, J = 8.5 Hz, 1H), 9.76 (s, 1H), 8.87 (d, J = 7.9 Hz, 1H), 8.23 (dd, J = 8.1, 1.2 Hz, 1H), 7.90 (ddd, J = 8.5, 7.0, 1.0 Hz, 1H), 7.77 (ddd, J = 8.0, 7.0, 1.0 Hz, 1H), 7.62-7.57 (m, 2H), 7.32 (ddd, J = 8.0, 6.0, 2.0 Hz, 1H), 6.29 (s, 1H), 6.20 (s, 1H), 5.47 (t, J = 2.1, 2H), 3.19-3.10 (m, 1H), 2.98-2.89 (m, 1H), 2.33 (s, 4H), 1.76-1.69 (m, 2H), 1.50-1.39 (m, 6H). 13C NMR (100 MHz, DMSO-d6): δ(ppm). 200.9, 171.0, 170.0, 162.8, 161.2, 155.1, 153.7, 139.0, 132.8, 131.4, 131.3, 129.5, 128.9, 128.6, 127.7, 126.8, 124.7, 123.1, 120.0, 119.1, 116.3, 115.4, 114.1, 89.8-88.8, 84.5, 80.7, 56.5-55.9, 53.3-52.6, 24.4-22.1. IR (film): ν(cm-1) 3293, 3094, 2920, 2733, 1960, 1744, 1703, 1599, 1541, 1435, 1377, 1225, 1123, 1022, 862, 747, 685, 636. HRMS calcd for C35H29N5O4Ru (M+H)+ 686.1336, and observed (M+H)+ 686.1324.
Single enantiomers of 9: 9E1 and 9E2
The racemic NHS-ester 4 was resolved to the individual enantiomers with a Chiral Pak 1B analytical HPLC column (Daicel/Chiral Technologies). Each injection was conducted isocratic using hexanes:EtOH (1:24) with a flow rate of 0.8 mL/min. Each enantiomer was then reacted separately with N-(2-aminoethyl)piperidine to provide both 9E1 (R-configuration) and 9E2 (S-configuration). The absolute configuration with assigned based on the CD-spectra and a comparison with known reference compounds (see supporting information).
Library Synthesis
All reactions were conducted in 96-well polypropylene plates in a total volume of 40 μL. A 32.2 mM stock solution of 8 in DMF was prepared. 100 mM stock solutions of each amine in DMF were individually prepared. To each reaction well was added DMF (14.0 μL), activated ester 8 (10.0 μL of 32.2 mM stock) and amine (16.0 uL of 100 mM stock). After mixing the reactions, the well plate was heat-sealed and allowed to stand at room temperature overnight. The reactions were terminated with addition of acetic acid (40 μL of 16.4 mM stock in DMSO) and stored at -78 °C until assayed for activity.
PIM-1/inhibitor structure determination
PIM-1 kinase was expressed and purified as described previously.22 Dephosphorylated PIM-1 was concentrated to 5mg/ml in 50 mM HEPES pH 7.5, 250 mM NaCl, 5% glycerol, 10 mM DTT. The compound 14 was added from a 10mM DMSO stock solution to give a final concentration of 1mM. Crystals were grown at 4°C in a 4 μL sitting drops where 2 μL of protein solution was mixed with 2 μL of the precipitate stock containing 0.2 M magnesium chloride,100 mM BisTrisPropane (pH 6.5), 20% PEG3350, 10% ethylene glycol and 0.3% DMSO. Cryoprotected crystals yielded X-ray diffraction to 2.05 A° using a RigakuRU-200HB rotating anode X-ray generator equipped with R-axis IV++ image plate detector. Diffraction data were indexed and merged using the program Crystal Clear (d*Trek).30 The structure was determined by molecular replacement using a structure of PIM-1 (PDB code the 2BZJ, inhibitor, water molecules, and ions removed) as a search model for rotation and translation functions in AmoRe.31 Iterative cycles of refinement and manual rebuilding of the model were performed using the program REFMAC532 and O,33 respectively. Data collection and refinement statistics are listed in Table S1.
Supplementary Material
ACKNOWLEDGEMENT
We would like to thank Peng Xie for his contribution in providing inhibition data for BRAF and PI3K; Michael Brent for providing purified FoxO protein and Mercedes Lioni for help with the cellular experiments. We also thank the Wistar Protein Expression Facility for baculovirus protein production and the David Christianson lab for use of their X-ray data collection facilities. This work was supported by NIH grants to R.M. (CA114046-) and E.M. (GM071695).
Abbreviations
- BRAF
Proto-oncogene serine/threonine-protein kinase B-Raf
- FOXO
Forkhead box protein O
- GSK-3
Glycogen synthase kinase 3
- MELK
Maternal embryonic leucine zipper kinase
- MST1
Mammalian Sterile 20
- PAK1
p21 activated kinase 1
- PAK4
p21 activated kinase 4
- PIM1
Proto-oncogene serine/threonine protein kinase PIM1
- PI3K
Phosphatidyl-inositol-3-kinase
- Syk
Spleen tyrosine kinase
- TAO2
Thousand and one amino acids kinase 2
Footnotes
Additional supporting information is available on the web.
REFERENCES
- 1.Lee KK, Ohyama T, Yajima N, Tsubuki S, Yonehara S. MST, a physiological caspase substrate, highly sensitizes apoptosis both upstream and downstream of caspase activation. J. Biol. Chem. 2001;276(22):19276–19285. doi: 10.1074/jbc.M005109200. [DOI] [PubMed] [Google Scholar]
- 2.Lehtinen MK, Yuan Z, Boag PR, Yang Y, Villen J, Becker EB, DiBacco S, de la Iglesia N, Gygi S, Blackwell TK, Bonni A. A conserved MST- FOXO signaling pathway mediates oxidative-stress responses and extends life span. Cell. 2006;125(5):987–1001. doi: 10.1016/j.cell.2006.03.046. [DOI] [PubMed] [Google Scholar]
- 3.Ura S, Masuyama N, Graves JD, Gotoh Y. MST1-JNK promotes apoptosis via caspase-dependent and independent pathways. Genes Cells. 2001;6(6):519–530. doi: 10.1046/j.1365-2443.2001.00439.x. [DOI] [PubMed] [Google Scholar]
- 4.Barthel A, Schmoll D, Unterman TG. FoxO proteins in insulin action and metabolism. Trends Endocrinol. Metab. 2005;16(4):183–189. doi: 10.1016/j.tem.2005.03.010. [DOI] [PubMed] [Google Scholar]
- 5.Lam EW, Francis RE, Petkovic M. FOXO transcription factors: key regulators of cell fate. Biochem. Soc. Trans. 2006;34(Pt 5):722–726. doi: 10.1042/BST0340722. [DOI] [PubMed] [Google Scholar]
- 6.Burgering BM, Kops GJ. Cell cycle and death control: long live Forkheads. Trends Biochem. Sci. 2002;27(7):352–360. doi: 10.1016/s0968-0004(02)02113-8. [DOI] [PubMed] [Google Scholar]
- 7.Graves JD, Draves KE, Gotoh Y, Krebs EG, Clark EA. Both phosphorylation and caspase-mediated cleavage contribute to regulation of the Ste20-like protein kinase Mst1 during CD95/Fas-induced apoptosis. J. Biol. Chem. 2001;276(18):14909–14915. doi: 10.1074/jbc.M010905200. [DOI] [PubMed] [Google Scholar]
- 8.Barthelemy C, Henderson CE, Pettmann B. Foxo3a induces motoneuron death through the Fas pathway in cooperation with JNK. BMC Neurosci. 2004;5:48–57. doi: 10.1186/1471-2202-5-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cheung WL, Ajiro K, Samejima K, Kloc M, Cheung P, Mizzen CA, Beeser A, Etkin LD, Chernoff J, Earnshaw WC, Allis CD. Apoptotic phosphorylation of histone H2B is mediated by mammalian sterile twenty kinase. Cell. 2003;113(4):507–517. doi: 10.1016/s0092-8674(03)00355-6. [DOI] [PubMed] [Google Scholar]
- 10.Loetscher H, Niederhauser O, Kemp J, Gill R. Is caspase-3 inhibition a valid therapeutic strategy in cerebral ischemia? Drug Discov. Today. 2001;6(13):671–680. doi: 10.1016/s1359-6446(01)01826-8. [DOI] [PubMed] [Google Scholar]
- 11.Pombo CM, Force T, Kyriakis J, Nogueira E, Fidalgo M, Zalvide J. The GCK II and III subfamilies of the STE20 group kinases. Front. Biosci. 2007;12:850–859. doi: 10.2741/2107. [DOI] [PubMed] [Google Scholar]
- 12.Eswaran J, Lee WH, Debreczeni JE, Filippakopoulos P, Turnbull A, Fedorov O, Deacon SW, Peterson JR, Knapp S. Crystal Structures of the p21-activated kinases PAK4, PAK5, and PAK6 reveal catalytic domain plasticity of active group II PAKs. Structure. 2007;15(2):201–213. doi: 10.1016/j.str.2007.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhou T, Raman M, Gao Y, Earnest S, Chen Z, Machius M, Cobb MH, Goldsmith EJ. Crystal structure of the TAO2 kinase domain: activation and specificity of a Ste20p MAP3K. Structure. 2004;12(10):1891–1900. doi: 10.1016/j.str.2004.07.021. [DOI] [PubMed] [Google Scholar]
- 14.Dan I, Watanabe NM, Kusumi A. The Ste20 group kinases as regulators of MAP kinase cascades. Trends Cell Biol. 2001;11(5):220–230. doi: 10.1016/s0962-8924(01)01980-8. [DOI] [PubMed] [Google Scholar]
- 15.Ling P, Lu TJ, Yuan CJ, Lai MD. Biosignaling of mammalian Ste20-related kinases. Cell Signal. 2008;20(7):1237–1247. doi: 10.1016/j.cellsig.2007.12.019. [DOI] [PubMed] [Google Scholar]
- 16.Meggers E. Exploring biologically relevant chemical space with metal complexes. Curr. Opin. Chem. Biol. 2007;11(3):287–292. doi: 10.1016/j.cbpa.2007.05.013. [DOI] [PubMed] [Google Scholar]
- 17.Meggers E, A.-G. GE, Bregman H, Maksimoska J, Mulcahy SP, Pagano N, William DS. Pyrido[2,3-a]pyrrolo[3,4-c]carbazole-5,7(6H)-diones: Synthesis, Cyclometalation, and Protein Kinase Inhibition. Synlett. 2007;8:1177–1189. [Google Scholar]
- 18.Xie P, Williams DS, Atilla-Gokcumen GE, Milk L, Xiao M, Smalley KSM, Herlyn M, Meggers E, Marmorstein R. Structure-based design of an organoruthenium Phosphatidyl-Inositol-3-Kinase inhibitor reveals a switch governing lipid kinase potency and selectivity. ACS Chemical Biology. 2008;3(5):305–316. doi: 10.1021/cb800039y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bregman H, Meggers E. Ruthenium half-sandwich complexes as protein kinase inhibitors: an N-succinimidyl ester for rapid derivatizations of the cyclopentadienyl moiety. Org. Lett. 2006;8(24):5465–5468. doi: 10.1021/ol0620646. [DOI] [PubMed] [Google Scholar]
- 20.Williams DS, Carroll PJ, Meggers E. Platinum complex as a nanomolar protein kinase inhibitor. Inorg. Chem. 2007;46(8):2944–2946. doi: 10.1021/ic062055t. [DOI] [PubMed] [Google Scholar]
- 21.Bregman H, Carroll PJ, Meggers E. Rapid access to unexplored chemical space by ligand scanning around a ruthenium center: discovery of potent and selective protein kinase inhibitors. J. Am. Chem. Soc. 2006;128(3):877–884. doi: 10.1021/ja055523r. [DOI] [PubMed] [Google Scholar]
- 22.Pagano N, Maksimoska J, Bregman H, Williams DS, Webster RD, Xue F, Meggers E. Ruthenium half-sandwich complexes as protein kinase inhibitors: derivatization of the pyridocarbazole pharmacophore ligand. Org. Biomol. Chem. 2007;5(8):1218–1227. doi: 10.1039/b700433h. [DOI] [PubMed] [Google Scholar]
- 23.Atilla-Gokcumen GE, Williams DS, Bregman H, Pagano N, Meggers E. Organometallic compounds with biological activity: a very selective and highly potent cellular inhibitor for glycogen synthase kinase 3. Chembiochem. 2006;7(9):1443–1450. doi: 10.1002/cbic.200600117. [DOI] [PubMed] [Google Scholar]
- 24.Debreczeni JE, Bullock AN, Atilla GE, Williams DS, Bregman H, Knapp S, Meggers E. Ruthenium half-sandwich complexes bound to protein kinase PIM-1. Angew. Chem. Int. Ed. Engl. 2006;45(10):1580–1585. doi: 10.1002/anie.200503468. [DOI] [PubMed] [Google Scholar]
- 25.Prudhomme M. Biological targets of antitumor indolocarbazoles bearing a sugar moiety. Curr. Med. Chem. Anticancer. Agents. 2004;4(6):509–521. doi: 10.2174/1568011043352650. [DOI] [PubMed] [Google Scholar]
- 26.Huang H, Tindall DJ. Dynamic FoxO transcription factors. J. Cell. Sci. 2007;120(Pt 15):2479–2487. doi: 10.1242/jcs.001222. [DOI] [PubMed] [Google Scholar]
- 27.Yamamoto S, Yang G, Zablocki D, Liu J, Hong C, Kim SJ, Soler S, Odashima M, Thaisz J, Yehia G, Molina CA, Yatani A, Vatner DE, Vatner SF, Sadoshima J. Activation of Mst1 causes dilated cardiomyopathy by stimulating apoptosis without compensatory ventricular myocyte hypertrophy. J. Clin. Invest. 2003;111(10):1463–1474. doi: 10.1172/JCI17459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Smalley KS, Brafford P, Haass NK, Brandner JM, Brown E, Herlyn M. Up-regulated expression of zonula occludens protein-1 in human melanoma associates with N-cadherin and contributes to invasion and adhesion. Am. J. Pathol. 2005;166(5):1541–1554. doi: 10.1016/S0002-9440(10)62370-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bregman H, Williams DS, Meggers E. Pyrido[2,3-a]pyrrolo[3,4-c]carbazole-5,7(6H)-diones: Synthesis, Cyclometalation, and Protein Kinase Inhibition. Synthesis. 2005:1521–1527. [Google Scholar]
- 30.Pflugrath JW. The finer things in X-ray diffraction data collection. Acta Crystallogr. D. 1999;55(Pt 10):1718–1725. doi: 10.1107/s090744499900935x. [DOI] [PubMed] [Google Scholar]
- 31.Navaza J. AMoRe: an Automated Package for Molecular replacement. Acta Crystallogr. A. 1994;50:157–163. [Google Scholar]
- 32.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D. 1997;53(Pt 3):240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 33.Jones TA, Zou JY, Cowan SW, Kjeldgaard M. Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr. A. 1991;47(Pt 2):110–119. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.