

Abstract
New technologies for imaging molecules, particularly optical technologies, are increasingly being used to understand the complexity, diversity and in vivo behaviour of cancers. ‘Omic’ approaches are providing comprehensive ‘snapshots’ of biological indicators, or biomarkers, of cancer, but imaging can take this information a step further, showing the activity of these markers in vivo and how their location changes over time. Advances in experimental and clinical imaging are likely to improve how cancer is understood at a systems level and, ultimately, should enable doctors not only to locate tumours but also to assess the activity of the biological processes within these tumours and to provide ‘on the spot’ treatment.
Imaging has become an indispensable tool in cancer research, clinical trials and medical practice. In the past three decades, there has been a huge increase in the number of imaging technologies and their applications (Fig. 1), but several challenges remain. At present, molecular imaging systems enable doctors to see where a tumour is located in the body. Ultimately, it is hoped that some of these systems will also help doctors to visualize the expression and activity of particular molecules, cells and biological processes that influence the behaviour of tumours and/or responsiveness to therapeutic drugs.
Figure 1. Imaging technologies used in oncology.

Many macroscopic imaging technologies (shown above the timeline) are in routine clinical use, and there have been huge advances in their capabilities to obtain anatomical and physiological information since the beginning of the twentieth century. Shown are some examples of bones (X-rays), soft tissue (ultrasound, MRI and CT rows), three-dimensional organs (CT and MRI rows) and physiological imaging (MRI and PET rows). Microscopic and other intravital optical techniques (shown below the timeline) have developed over the past decade and now allow studies of genetic, molecular and cellular events in vivo. Shown are surface-weighted, whole-mouse, two-dimensional techniques (macroscopic reflectance row); tomographic three-dimensional techniques, often in combination with other anatomical modalities (tomography row); and intravital microscopy techniques (microscopy row). The timeline is approximate and is not to scale. BLI, bioluminescence imaging; CT, computed tomography; DOT, diffuse optical tomography; FMT, fluorescence-mediated tomography; FPT, fluorescence protein tomography; FRI, fluorescence reflectance imaging; HR-FRI, high-resolution FRI; LN-MRI, lymphotropic nanoparticle-enhanced MRI; MPM, multiphoton microscopy; MRI, magnetic resonance imaging; MSCT, multislice CT; OCT, optical coherence tomography; OFDI, optical frequency-domain imaging; PET, positron-emission tomography. (PET image reproduced, with permission, from ref. 4. Diffusion MRI image courtesy of B. Ross and A. Rehemtulla, Univ. Michigan Medical School, Ann Arbor. X-ray image (left) reproduced, with permission, from ref. 86. Diaphanoscopy image reproduced, with permission, from ref. 87. Fibre-optic image reproduced, with permission, from ref. 88. BLI image courtesy of K. Shah, Massachusetts General Hospital, Harvard Medical School, Boston, Massachusetts. Lifetime FRI image courtesy of U. Mahmood and C. Salthouse, Massachusetts General Hospital. DOT image reproduced, with permission, from ref. 89. FPT image courtesy of G. Zacharakis and V. Ntziachristos, Massachusetts General Hospital. FMT-MRI image courtesy of J. Chen, Massachusetts General Hospital. Bright-field image (left) reproduced, with permission, from ref. 14. Bright-field image (right) courtesy of T. Mempel, Massachusetts General Hospital. Epifluorescence image courtesy of F. Jaffer, Massachusetts General Hospital. OCT, OFDI image reproduced, with permission, from ref. 22. MPM image reproduced, with permission, from ref. 51. Microendoscopy image reproduced, with permission, from ref. 17.)
Perhaps the biggest growth area is fluorescence imaging, with various technologies being adapted for in vivo analysis. Indeed, because of developments in fluorescent imaging, researchers are now on the verge of being able to address some of the big questions in molecular oncology. How do the components of intracellular signalling pathways interact in real time? What are the kinetics and flux rates of such networks? What are the differences between networks in malignant cells and normal cells? Can we exploit these differences to make drugs that are less toxic and more efficacious? And what are the ‘hubs’ (the crucial signalling nodes) that will translate into the most efficient read-outs of how a cancer is progressing and whether a therapy is effective? In this article, we highlight recent advances in molecular imaging, with an emphasis on fluorescence imaging. We first provide an overview of the imaging technologies and then focus on specific applications in cancer biology and oncology. Cellular nanoimaging1,2 and clinical imaging technologies3-5 have been reviewed recently and so will not be covered in depth.
Overview of imaging technologies
Imaging systems can be grouped by the energy used to derive visual information (X-rays, positrons, photons or sound waves), the spatial resolution that is attained (macroscopic, mesoscopic or microscopic) or the type of information that is obtained (anatomical, physiological, cellular or molecular). Macroscopic imaging systems that provide anatomical and physiological information are now in widespread clinical and preclinical use: these systems include computed tomography (CT), magnetic resonance imaging (MRI) and ultrasound. By contrast, systems that obtain molecular information are just emerging, and only some are in clinical and preclinical use: these systems include positron-emission tomography (PET), single-photon-emission CT (SPECT), fluorescence reflectance imaging, fluorescence-mediated tomography (FMT), fibre-optic microscopy, optical frequency-domain imaging, bioluminescence imaging, laser-scanning confocal microscopy and multiphoton microscopy.
For imaging technologies to be adapted more widely and to be complementary to other types of molecular measurement, the read-outs need to meet certain criteria: they need to be quantitative, high resolution, longitudinal (that is, allow imaging over time), comprehensive, standardized, digital and sensitive to molecular perturbations in the system. In terms of quantification, imaging technologies can provide data that are absolute or relative. Absolute quantification is possible in techniques where signals are independent of position in the sample. CT, FMT, MRI and PET inherently provide quantifiable information. Relative quantification is obtained from image data sets whose signals are depth and sample-type dependent but can be validated by rigorous experimental design. Bioluminescence imaging, fluorescence reflectance imaging and multiphoton microscopy technologies belong in this category. Table 1 summarizes the spatial resolution, depth penetration, imaging time and cost of available systems.
Table 1. Overview of imaging systems.
| Technique | Resolution* | Depth | Time† | Quantitative‡ | Multi- channel |
Imaging agents |
Target | Cost§ | Main small-animal use |
Clinical use |
|---|---|---|---|---|---|---|---|---|---|---|
| MRI | 10-100 μm | No limit | Minutes to hours | Yes | No | Paramagnetic chelates, magnetic particles | Anatomical, physiological, molecular | $$$ | Versatile imaging modality with high soft-tissue contrast | Yes |
| CT | 50 μm | No limit | Minutes | Yes | No | Iodinated molecules | Anatomical, physiological | $$ | Imaging lungs and bone | Yes |
| Ultrasound | 50 μm | cm | Seconds to minutes | Yes | No | Microbubbles | Anatomical, physiological | $$ | Vascular and interventional imaging∥ | Yes |
| PET | 1-2 mm | No limit | Minutes to hours | Yes | No | 18F-, 64Cu- or 11C-labelled compounds | Physiological, molecular | $$$ | Versatile imaging modality with many tracers | Yes |
| SPECT | 1-2 mm | No limit | Minutes to hours | Yes | No | 99mTc- or 111In-labelled compounds | Physiological, molecular | $$ | Imaging labelled antibodies, proteins and peptides | Yes |
| Fluorescence reflectance imaging | 2-3 mm | <1 cm | Seconds to minutes | No | Yes | Photoproteins, fluorochromes | Physiological, molecular | $ | Rapid screening of molecular events in surface-based disease | Yes |
| FMT | 1 mm | <10 cm | Minutes to hours | Yes | Yes | Near-infrared fluorochromes | Physiological, molecular | $$ | Quantitative imaging of fluorochrome reporters | In development |
| Bioluminescence imaging | Several mm | cm | Minutes | No | Yes | Luciferins | Molecular | $$ | Gene expression, cell and bacterium tracking | No |
| Intravital microscopy¶ | 1 μm | <400-800 μm | Seconds to hours | No | Yes | Photoproteins, fluorochromes | Anatomical, physiological, molecular | $$$ | All of the above at higher resolutions but limited depths and coverage | In development# |
For high-resolution, small-animal imaging systems. (Clinical imaging systems differ.)
Time for image acquisition.
Quantitative here means inherently quantitative. All approaches allow relative quantification.
Cost is based on purchase price of imaging systems in the United States: $, <US$100,000; $$,US$100,000-300,000; $$$, >US$300,000.
Interventional means used for interventional procedures such as biopsies or injection of cells under ultrasound guidance.
Laser-scanning confocal or multiphoton microscopy.
For microendoscopy and skin imaging. (Table adapted, with permission, from ref. 85.)
For routine clinical practice and for testing the efficacy of drugs in clinical trials, CT, MRI, PET and SPECT are useful. Adaptations of these systems with much higher spatial resolutions have become available for use in experimental mouse models, allowing the development of new imaging probes for use in the clinic. By contrast, fluorescence reflectance imaging, FMT, fibre-optic microscopy and optical frequency-domain imaging are still mainly used experimentally, but they have clear potential for translation into the clinic. Because each technology has unique strengths and limitations, platforms that combine several technologies — such as PET-CT, FMT-CT, FMT-MRI and PET-MRI — are emerging, and these multimodal platforms have improved the reconstruction and visualization of data.
Complementary approaches to imaging include the use of portable in vivo flow cytometers and molecular ‘nanolabs’ to track circulating tumour cells (Box 1), as well as implantable, miniaturized fibre-optic multiphoton microscopy systems and implantable sensors for imaging molecular information in tumour environments. Other imaging technologies have recently been described, including thermal, electromagnetic and terahertz imaging, but their use in vivo or in oncology is not as established as the techniques described here.
Box 1 | Quantification of circulating tumour cells.
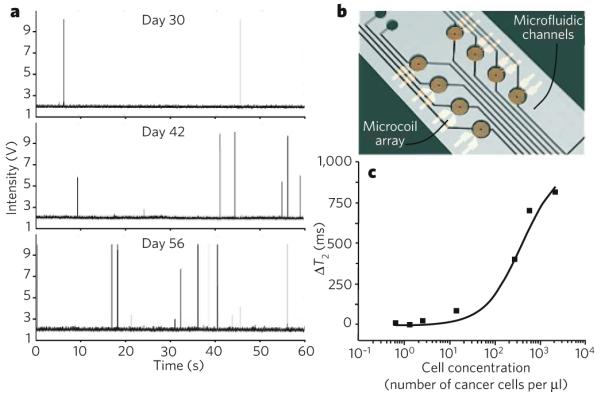
The number of circulating tumour cells (CTCs) is a sensitive biomarker for tumour progression and metastasis94. Therefore, the quantification of CTCs is emerging as useful for diagnosing and ‘staging’ cancer, for assessing responses to treatment, and for evaluating whether there is residual disease. To be clinically useful, emerging technologies need to be highly sensitive (with detection limits around 1 cell per ml of blood) and highly specific for CTCs. Several such methods have recently been described and are in use experimentally.
Imaging CTCs non-invasively in microvessels of the skin could improve the sensitivity of detection by allowing large volumes of blood to be analysed. Confocal detection of CTCs with a dedicated in vivo flow cytometer was first demonstrated in 2004 (ref. 95), and portable systems have been described more recently96. In the figure, part a shows circulating green fluorescent protein (GFP)-positive multiple myeloma cells detected by in vivo flow cytometry in ear arterioles. Measurements were taken over 60 s at various intervals after tumour cells were injected (three separate days are shown). Each signal spike represents a single CTC. Therefore, the data show an increase in the numbers of CTCs over time, and this occurred together with tumour progression. Another technique that has been advocated is using multiphoton microscopy to image the peripheral vasculature after intravenous injection of tumour-cell-specific fluorescent ligands, such as fluorescent folates, which are thought to be internalized by tumour cells97.
An alternative blood-screening method recently reported uses a highly sensitive microfluidic platform. The platform consists of an array of microposts that are made chemically functional with antibodies specific for epithelial cell-adhesion molecule (EpCAM) and therefore captures CTCs of epithelial origin 98 (not shown).
Finally, a chip-based diagnostic MRI (DMR) platform with multiple channels allows rapid and quantitative detection of biological targets99, as shown in parts b and c of the figure. Using functionalized magnetic nanoparticles as proximity sensors to amplify molecular interactions100, the DMR system can carry out highly sensitive and selective measurements on small volumes of unprocessed biological samples, including the profiling of circulating cells and the multiplexed identification of cancer biomarkers99. Part c of the figure shows detectable DMR changes (change in the time of magnetic relaxation, ΔT2) of whole blood as a function of added cancer cells. (Panel a courtesy of C. Lin, Massachusetts General Hospital, Boston, Massachusetts, and A. Kung and I. Ghobrial, Dana-Farber Cancer Institute, Boston, Massachusetts. Panel b adapted, with permission, from ref. 99.)
Key developments in optical imaging
Photons travelling through tissue and interacting with tissue components form the basis of optical imaging techniques. In this section, we cover systems for the observation of molecular targets at the microscopic and macroscopic levels, as well as non-fluorescence-based optical imaging systems.
Fluorescence imaging
Fluorescence illumination and observation has been one of the most rapidly adapted imaging technologies, in both medicine and biological sciences. Fluorescence has spurred the development of sophisticated microscopic, mesoscopic and macroscopic imaging systems, as well as miniaturized fibre-optic approaches, imaging probes and genetic reporter systems. Fluorescence refers to the property of certain molecules to absorb light at a particular wavelength and to emit light of a longer wavelength after a brief interval known as the fluorescence lifetime. The phenomenon of fluorescence was known by the middle of the nineteenth century and was then adapted to fluorescence microscopy in the early twentieth century. Medical applications of fluorescence imaging date back to 1924, when the autofluorescence of endogenous porphyrins was observed in tumours illuminated with ultraviolet light6. In 1942, red fluorescence by tumours was observed after intravenous administration of porphyrins7, and the first use of fluorescein (which emits green light) to improve the detection of brain tumours was reported in 1948 (ref. 8). Britton Chance and others pioneered the reconstruction of light through tissue in the 1980s (ref. 9), on the basis of the observation that near-infrared photons (650-900 nm) travel through tissue much more efficiently than those in the visible range10. Concomitantly, fluorescence imaging with targeted molecular probes was developed11-13. Today, tomographic reconstruction of near-infrared photons, spectral unmixing, image fusion and multichannel imaging have become routine, and several preclinical and clinical imaging systems are commercially available.
Microscopic fluorescence imaging
Microscopic imaging has been used to study the activity of cells in many biological settings, including tumours, by a variety of illumination techniques. In 1839, Rudolph Wagner visualized leukocytes rolling in blood vessels within membranous translucent tissues by using bright-field transillumination14. With the advent of fluorescence microscopy, it then became possible to analyse multiple cell types simultaneously and in deep positions in solid tissues. Several imaging approaches based on fluorescence microscopy that were established for visualizing cells in vitro have recently been adapted for in vivo imaging: multiphoton microscopy, laser-scanning confocal microscopy, fibre-optic approaches and spectrally encoded endoscopy.
Multiphoton microscopy imaging systems achieve depth resolutions of up to 800 μm, yield three-dimensional information from light emitted by differentially labelled fluorescent objects (detected through multiple channels) and from second harmonic signals, and can provide hours of imaging at high spatial resolutions. Intravital (in vivo) multiphoton microscopy provides relative quantification of fluorescence signal (as described earlier), but it can be used to derive truly quantitative parameters of intravascular and interstitial cell migration (such as velocity, displacement, persistence time, and chemotactic and motility indices)15. In the frequently used animal models, cells are investigated in the popliteal and inguinal lymph nodes, the cranial bone marrow, tumour window chambers surgically implanted in the dorsum of the animal, and some exteriorized organs harbouring orthotopic cancers. Ancillary techniques such as second harmonic generation and fluorescence resonance energy transfer (FRET) are also increasingly being adopted for in vivo imaging, because they can add structural or molecular detail. Techniques to quantify the FRET level include imaging after photobleaching or fluorescence lifetime.
Confocal microscopy set-ups have also become more widespread, because they are user-friendly and less costly than multiphoton microscopy. But cell death as a result of phototoxicity can be a limitation if observation times are lengthy, and optical penetration into tissues is lower.
Fibre-optic approaches have been adapted for intravital microscopy recently, potentially allowing imaging at orthotopic sites. Miniaturized systems are available for experimental work, and several other systems are being adapted for endoscopic clinical imaging16,17.
Finally, spectrally encoded endoscopy is another new three-dimensional endoscopic technique that allows miniaturization of endoscopes, thus overcoming many of the limitations of the fibre-optic bundles of conventional endoscopes18.
Macroscopic fluorescence imaging
Macroscopic fluorescence imaging systems rely on photographic principles to collect images in low light. There are two main types of imaging approach: fluorescence reflectance and tomographic fluorescence.
Fluorescence reflectance imaging systems consist of an excitation source, filters and a charge-coupled-device camera to obtain two-dimensional (planar) images. They are useful for imaging events in surface tumours (such as xenografts) and surgically exposed organs, and for intra-operative use. But they have a limited depth resolution beyond 3-5 mm and are not inherently quantitative.
Tomographic fluorescence systems19 (FMT, FMT-CT, FMT-MRI and fluorescence protein tomography) reconstruct three-dimensional maps of fluorochromes on the basis of sophisticated algorithms, and these systems are quantitative. The early tomographic fluorescence systems used mice immersed in index-matching fluids to simplify the theoretical constraints associated with boundary modelling and to calculate light propagation. More recent ‘free-space’ systems eliminate the need for index-matching fluid. FMT can now also be combined with CT or MRI for improved photon reconstruction and image visualization20. Tomographic fluorescence systems have also been adapted to reconstruct visible fluorescence (for example, fluorescence protein tomography), thus enabling fluorescent proteins or genetically modified cells to be tracked irrespective of cell division in vivo21. New fluorescence protein tomography systems can image organisms (such as fruitflies and nematodes) at mesoscopic resolution.
Non-fluorescence-based optical imaging
Several optical imaging systems that are not based on fluorescence have also been introduced: bioluminescence imaging, optical coherence tomography, photoacoustic microscopy and tissue spectroscopy.
Bioluminescence imaging has emerged as a useful technique for imaging small experimental animals. The imaging signal depends on the expression levels of a luciferase enzyme, the presence of exogenously administered enzyme substrate (a luciferin), ATP, oxygen and depth. Numerous luciferase-luciferin pairs have been harnessed for in vivo imaging. Particularly useful pairs are firefly (Photinus pyralis) luciferase-luciferin, because of the high wavelength of emitted light and quantum yields, and Renilla reniformis luciferase-coelenterazine and Gaussia princeps luciferase-coelenterazine, because of their flash kinetics and ability to generate photons outside cells. Typical doses of luciferin are in large excess (>100 mg per kg body weight administered intraperitoneally) and are injected immediately before data acquisition. In contrast to fluorescence techniques, there is no inherent background noise, which makes this technique highly sensitive. However, at present, this method does not allow absolute quantification of target signal; instead, its primary uses are in binary mode (that is, yes or no for luciferase expression) or as an imaging tool to follow the same animal over time in identical conditions, including positioning of the body.
Optical coherence tomography is based on light scattering and can be used to image microscopic structures in vivo (at a resolution of 2-15 μm and to a depth of 1-3 mm). Studies in humans have shown that this method identifies dysplasia and colonic adenomas. Until now, however, it has been too slow to provide comprehensive microscopic imaging and has therefore been used as a point-sampling technique with a field of view comparable to that in a conventional biopsy. Optical frequency-domain imaging leverages the high sensitivity of optical coherence tomography but is several orders of magnitude faster22 and can thus be used to screen larger organ samples for the presence of malignancies.
Photoacoustic microscopy uses short laser pulses to irradiate tissue and temporarily raise its temperature (by millikelvins). Thermo-elastic expansion then causes the emission of photoacoustic waves that can be measured by wide-band ultrasonic transducers, offering improved depth resolution in the 3-20 mm range23. The technique combines ultrasonic-scale spatial resolution with high sensitivity to tissue light absorption and can yield information on physiology or on exogenously administered light absorbers. Newer generations of imaging systems provide macroscopic tomographic capabilities (a process known as photoacoustic tomography) or use radio-frequency pulses to generate acoustic waves (a process known as thermal acoustic tomography).
Finally, tissue spectroscopy detects relative changes in the way in which light interacts with tissue and has been used extensively to improve early detection of gastrointestinal malignancies24. The vibrational spectra of biological specimens can be used to identify the biochemical constituents of tissue, but there are practical limitations of this technique for in vivo use, owing to its relatively low sensitivity and limited spatial and temporal resolutions. Much stronger vibrational signals can be obtained with coherent anti-Stokes Raman scattering (CARS), a nonlinear Raman technique25. CARS microscopy is useful for mapping lipid compartments, protein clusters and water distribution. Other optical contrast mechanisms such as absorption, polarization, coherence, interference and elastic (or Mie) scattering can also be exploited to generate images, some of which might be particularly useful for oncological applications.
Labelling
With few exceptions, cells, whether they are normal cells or cancer cells, cannot easily be distinguished from each other by in vivo imaging. Consequently, the molecules and cells of interest need to be labelled to become visible. Labelling techniques can be broadly divided into three categories: genetic reporters, injectable imaging agents and exogenous cell trackers.
Genetic reporters
Genetic reporter strategies have largely been limited to mouse models of cancer, but their use is widespread in basic biological sciences26,27. The types of protein expressed by the reporter genes used in optical imaging are fluorescent proteins (green, far-red and photoswitchable fluorescent proteins), bioluminescent proteins (luciferases from several organisms) or other proteins (such as herpes simplex virus 1 thymidine kinase, transferrin receptor, somatostatin receptor type 2, dopamine receptor D2 and human Na+/I- symporter). The optical reporter genes are cloned into the promoter or enhancer region of a gene of interest or engineered to be expressed as a fusion with the protein of interest, allowing longitudinal studies of biological processes. Such reporters are particularly valuable in genetically engineered mouse models in which the activation of oncogenes and/or tumour-suppressor genes is under temporal and tissue-specific control, because the reporters enable not only oncogenic transformation to be studied but also drug action, toxicity and resistance28,29.
Three genetic reporter strategies are available to study protein-protein interactions in vivo: modified mammalian two-hybrid systems30; bioluminescence resonance energy transfer (BRET)31 and FRET32; and split reporter33. Emerging two-hybrid systems are fairly simple and rapid, but they still need to be optimized. The reaction occurs in the nucleus, so the system can mainly be used to interrogate interactions between nuclear proteins or proteins that are modified to translocate to the nucleus. BRET, unlike FRET, usually does not cause photodamage to the cells and does not require excitation of the donor with an external light source, so it has less background noise, which might ultimately translate into being able to detect protein-protein interactions at lower concentrations. Split-reporter strategies, in which a reporter gene is split into components that become active when reconstituted, generate specific signals and can be used to image interacting proteins anywhere in the cell, but they need to be adapted for reversible interactions. These three genetic reporter strategies also allow monitoring of the concentrations of ions, sugars, lipids and other active molecules in vivo, so they could further broaden the application of intravital microscopy. Using a combination of the above genetic-reporter strategies for labelling should increase our knowledge of entire biological pathways and accelerate a systems-wide understanding of biological complexity.
Injectable imaging agents
Injectable imaging agents that are specific for molecular targets have the advantage of being usable in both experimental animals and humans. But, at present, such agents exist for <5% of the molecular targets of interest in cancer. This is largely because in vivo imaging agents face more stringent design criteria than in vitro reagents. The basic underlying difficulty is to design agents with high target-to-background ratios. The ideal agent should contribute minimally to the background signal (for example, there should be minimal non-specific tissue extravasation, internalization by macrophages, or renal or hepatic elimination, which obscures adjacent organs) but yield high local concentrations at the intended sites. For these reasons, in vivo agents are often designed using chemical or biological amplification strategies. Chemical amplification strategies include using multiple copies of a label in a single molecule (multivalency), pairing of deactivators and fluorochromes (quenching), using covalent target binders (active-site binding), inducing magnetic changes on target interaction (relaxivity changes and magnetic switches), releasing caged compounds by photolysis (uncaging) and using distance-dependent interaction between excited states of dyes (FRET). Biological amplification strategies include intracellular sequestration of the imaging agent (cellular trapping), generation of signal by endogenous enzymes in target cells (enzymatic conversion), and a two-step activation process in which molecular agents are tagged first by a multivalent agent and then by a fluorescent conjugate (pre-targeting).
Some generic types of imaging probe are summarized in Fig. 2. Of particular interest in the past few years have been high-throughput methods such as phage display34; systematic evolution of ligands by exponential enrichment (SELEX); fragment-based, template-based and nanoparticle-based35 synthesis for library design; and fast in vivo screening techniques36. Spectrally resolved imaging agents should foster systems approaches that allow the evaluation of multiple cellular and/or molecular activities in situ.
Figure 2. High-affinity imaging agents with appropriate pharmacokinetics are essential for imaging at the molecular level.

Different strategies have been pursued to develop agents for molecular imaging, and the various types of agent available and examples of these are indicated. Small molecules are shown in the top row, and macromolecular agents and nanotechnology-derived agents are shown in the bottom row. Four main types of small molecule are used. Small ligands refers to imageable small molecules (for example, 18F-labelled drugs and fluorescent peptides), whereas active-site binders (green) attach to specific protein pockets in enzymes (blue) either covalently or non-covalently. Site-specific protein tags (pink) achieve a similar function but at sites of interest in engineered proteins (white). Environmentally sensitive probes (for example, 4-N,N,-dimethylaminophthalimidoalanine (4-DAPA); yellow) change their physicochemical properties on interaction with the target (in this case, Tyr and T90β). Two main types of macromolecule are used. Supramolecular structures are synthetic agents that have been useful as enzyme substrates for the imaging of cathepsins and proteases or for delineating new microvasculature: shown is part of a poly-l-lysine backbone (blue) derivatized with protease-cleavable side chains (red) and polyethylene glycol (grey). Engineered proteins (with optimized pharmacokinetics) refers to other macromolecules that have been used for some targets. Finally, a host of nanomaterials, including inorganic nanoparticles (grey) and bionanoparticles, that can be used for imaging phagocytic cells or cell-surface targets (green) is emerging. (Images adapted, with permission, from ref. 90 (active-site binders), ref. 91 (site-specific protein tags), ref. 92 (environmentally sensitive probes), Mimetibody.com (engineered proteins) and ref. 93 (inorganic nanoparticles.)
Exogenous cell trackers
Exogenous cell trackers are being used increasingly to study adoptive cell-transfer therapies. Recent translational approaches for cell labelling include an [111In]oxine tracer37 (which is approved by the US Food and Drug Administration), derivatized magnetic nanoparticles38-40, and novel amine-reactive esters of benzindolium-derived fluorochromes for macroscopic and microscopic imaging41.
Applications in oncology
In this section, we review the roles in tumorigenesis that have been ascribed to various cells and molecules on the basis of in vivo studies. We also highlight some of the main applications of, and recent advances in, tumour imaging. Several other review articles have discussed the use of established clinical imaging technologies3-5.
Tumours and the host response
Stimuli from tumours affect components of the extracellular matrix (ECM) (such as collagen, fibronectin and glycoproteins) and cells present in the environment of the tumour (such as T cells, macrophages, dendritic cells, mast cells, neutrophils, fibroblasts and endothelial cells). These ECM components and cells can have complex regulatory functions mediated by proteolytic enzymes, cytokines, growth factors and angiogenesis-promoting factors. Recent advances in intravital imaging have made it possible to quantify and catalogue the behaviour and functions of some of the cellular and molecular components of the immune system that control tumour growth (Fig. 3). Importantly, intravital microscopy studies indicate that the behaviour of immune cells in tissues is dictated by local factors in the organ that often cannot be reproduced in vitro (at least for the foreseeable future).
Figure 3. Behaviour of tumour cells and cells in the tumour stroma in situ, as elucidated by in vivo imaging.

Intravital microscopy can be used to derive quantitative parameters on intravascular and interstitial cell migration, interaction and viability in the tumour environment and the lymphatic organs. A, In vivo microscopic recordings in anaesthetized mice can be used to obtain data at cellular resolution. Some of the types of process that can be observed are illustrated. a, Leukocytes (green) trafficking in blood microvessels (red). b, B cells (pink) and T cells (green) inside and outside high-endothelial venules (diffuse red), respectively, in a tumour-draining lymph node. Collagen fibres are also visualized (blue). c, A cytotoxic T cell (CTL) (green) recognizing and killing a target cell (pink). Yellow dots track the position of the centre of the CTL at 15 s intervals. Collagen fibres are also visualized (blue). d, Simultaneous tracking of multiple cell types: a CTL (green) is firmly bound to an antigen-presenting cell (blue), and another CTL is transiently interacting with a FOXP3+ regulatory T (Treg) cell (red). (Panels b-d reproduced, with permission, from ref. 51.) B, A typical analysis of cellular behaviour from data retrieved from time-lapse intravital imaging can assess four properties: the motility of cell populations (based on mean displacement over the square root of time); the instantaneous velocity of a single cell over time; the average cell-cell interaction time (in this case, A shows CTL-Treg-cell interactions, and B shows CTL-target cell); and the viability of a single cell over time (as measured by the ratio of cytoplasmic fluorescence to nuclear fluorescence, Fc:Fn)15,51. C, A model for the in vivo role of tumour stromal cells in tumour growth and metastasis, based on intravital imaging studies, is shown. Immune cells such as monocytes (which differentiate into tumour-associated macrophages, TAMs), CTLs and Treg cells are recruited into the tumour stroma from the blood vessels (dashed lines indicate direction) and can be visualized by intravital microscopy. Imaging studies can be used to investigate how these and other cells (such as carcinoma-associated fibroblasts, CAFs) interact with each other or with tumour cells and how they participate in tumorigenesis. In particular, in vivo studies have examined how CTLs recognize tumour-associated antigens at the surface of tumour cells41,48,49 and exert cytotoxic activity51 (a), how Treg cells suppress the function of CTLs51 (b), how TAMs and CAFs remodel the extracellular matrix (ECM) and promote invasion (c) and angiogenesis54,56 (d), and how TAMs also participate in tumour-cell intravasation54 (e). Macroscopic imaging techniques are complementary and allow bulk cell migration to be measured. Objects that can be imaged in vivo include the cell types described here, as well as molecular targets (or parameters) for which specific probes have been designed (indicated in red). bFGF, basic fibroblast growth factor; CSF1, colony-stimulating factor 1 (also known as M-CSF); CXC-L12, CXCchemokine ligand 12; EGF, epidermal growth factor; EGFR, EGF receptor; IFN-γ, interferon-γ; IL, interleukin; MMP, matrix metalloproteinase; mTOR, mammalian target of rapamycin; PI(3)K, phosphatidylinositol-3-OH kinase; ROS, reactive oxygen species; TGF-β, transforming growth factor-β; TIE2, endothelial tyrosine kinase (also known as TEK); TNF, tumour-necrosis factor; UPA, urokinase-type plasminogen activator; VCAM1, vascular cell-adhesion molecule 1; VEGF, vascular endothelial growth factor; VVF, vascular volume fraction.
Antitumour cytotoxic T cells (CTLs) that recognize short antigenic peptides presented by tumour cells can kill their targets in an antigen-dependent and contact-dependent manner by releasing preformed cytotoxic granules or by generating potent inflammatory responses through the secretion of cytokines42,43. Other immune cells suppress the function of these CTLs, thus promoting tumour growth: these include FOXP3+ regulatory T (Treg) cells44,45 and tumour-associated macrophages (TAMs)46,47.
The immunological functions mediated by effector and suppressor cells have mostly been characterized in vitro. In vivo studies at cellular resolution have been carried out only recently. A recent finding from two intravital multiphoton microscopy studies is that CTLs are recruited from blood vessels in the tumour periphery and then infiltrate towards the centre of the tumour48,49. Sustained locomotion of tumour-infiltrating CTLs requires the presence of cognate antigen (that is, the particular antigen recognized by each CTL); thus, only CTLs specific for tumour antigens can infiltrate tumours deeply. In conditions in which CTLs efficiently reject tumours, the CTLs migrate randomly throughout the tumour environment at high instantaneous velocities and pause to form antigen-dependent interactions with tumour cells. The dynamics of CTL-tumour cell interactions are diverse: some tumour-specific CTLs engage in long-lasting interactions, whereas others establish sequential, short-term contacts. The reasons for these different behaviours are unknown, but during this interaction time, CTLs could be delivering their cytotoxic granules and/or integrating signals for the production of pro-inflammatory cytokines. The progeny of CTLs that have come into contact with antigen typically include cells with distinct effector functions50, which might have diverse modes of interaction with tumour cells in vivo. When CTLs detach from their tumour-cell targets, they become highly motile again, presumably in search of intact tumour cells.
The presence of Treg cells can compromise killing by CTLs and prevent efficient tumour rejection. Indeed, another intravital multiphoton microscopy study has shown that CTLs have a constrained window of opportunity to induce target-cell death before the cell-cell conjugates dissociate (typically about 15 min), and Treg cells suppress the function of tumour-specific CTLs by delaying granule exocytosis beyond this time frame (typically >30 min)51. In the presence of these Treg cells, the CTLs behave otherwise ‘normally’ in situ: they proliferate extensively, are highly motile, contain cytotoxic granules, and form antigen-dependent conjugates with target cells. Interestingly, Treg-cell-mediated suppression is reversible, because in vivo depletion of Treg cells quickly restores CTL function51. These findings have clinical significance because patients with cancer also accumulate Treg cells in tumour stroma44, where CTLs fail to exert a cytotoxic function52. Intravital imaging studies in mouse models indicate that Treg-cell-mediated suppression requires cytokines such as transforming growth factor-β but not prolonged interactions with CTLs51. Because Treg cells are relatively poor producers of transforming growth factor-β, they might function by mobilizing cellular intermediates, such as TAMs. Indeed, CTLs in the tumour stroma form long-lasting interactions with TAMs49, which might supply a variety of immunosuppressive signals. Clearly, more intravital microscopy studies are needed to identify precisely the protumorigenic and antitumorigenic activities of the various cells in the tumour stroma.
Tumour invasion and metastasis
The motility of tumour cells is an essential feature for intravasation at primary sites and extravasation at distant sites to form metastases. For cells to migrate, they need to adhere to selected substrates, to reorganize their cytoskeleton, and to acquire the spatial asymmetry that enables them to turn intracellularly generated forces into locomotion. A core set of proteins, including those of the cofilin pathway, coordinates actin polymerization and is essential for the formation of cellular protrusions and crawling. Multiphoton microscopy imaging and gene-expression profiling in a mouse model of breast cancer have shown that the expression of genes in the cofilin pathway is altered in tumour cells and that these cells show dysregulated chemotaxis towards environmental stimuli, such as epidermal growth factor (EGF) and the chemokine CXCL12 (ref. 53).
Imaging studies have also confirmed that tumour-cell migration and invasion depend on stromal cells such as TAMs and carcinoma-associated fibroblasts (CAFs)54-56 (Fig. 3). By visualizing differentially labelled tumour cells and TAMs in situ, it has been shown that motile tumour cells preferentially migrate towards perivascular TAMs that reside deep within breast tumours. The tumour cells move efficiently along linear paths in association with proteoglycans that are present on ECM fibres, which might be guidance cues for movement towards blood vessels.
Tumour cells eventually exit into the blood vessels in proximity to TAMs; the efficiency of this egress depends on the number of perivascular TAMs but not on the number of blood vessels. It is possible that the EGF receptor (EGFR) at the surface of tumour cells responds directly to chemoattractants such as EGF that are secreted by perivascular TAMs. Indeed, inhibition of the EGFR in this model markedly decreases the number of tumour cells exiting into the blood54. Tumour cells can also secrete colony-stimulating factor 1 (CSF1; also known as M-CSF), which stimulates TAMs to secrete EGF, thereby further promoting tumour progression (through promoting intravasation, which can lead to metastasis). Together with TAMs, CAFs are present in large numbers in the stromal compartment of most invasive human breast cancers. CAFs secrete more CXCL12 than do fibroblasts in non-cancerous regions. The increased amount of CXCL12 has several effects: it promotes carcinoma cell growth by direct paracrine stimulation of the chemokine receptor CXCR4 on tumour cells; it recruits endothelial progenitor cells from the blood vessels, giving rise to highly vascularized tumours; and it supports the metastasis of tumour cells towards lymphatic vessels55,57. Other cells present in the tumour stroma, such as mast cells58, neutrophils59 and mesenchymal stem cells60, can also promote cancer invasion and metastasis, but their activity in situ has not been investigated.
Matrix remodelling and angiogenesis
TAMs and tumour cells overproduce matrix metalloproteinases, plasminogen activators, cathepsins and other hydrolases. These enzymes degrade structural components of the ECM and thereby facilitate migration of the tumour cells, as well as that of endothelial cells and immune cells. The enzymes also change the nature of the interaction between cells and the ECM, promote cell growth, and promote the ‘angiogenic switch’ by liberating various ECM-bound cytokines. Angiogenesis — the formation of new blood vessels — provides oxygen and nutrients to tumours and promotes invasion and metastasis. There are several agents in development to image the key cells and effector molecules involved in these processes. Functional nanomaterials specific for macrophage subtypes have been reported35, and several imaging agents also allow the visualization of proteases involved in ECM remodelling and the measurement of angiogenesis and associated parameters.
Three-dimensional molecular sensing of protease activity in vivo, for example, is now feasible by combining activatable imaging probes based on biologically compatible near-infrared fluorochromes61 and FMT for positional accuracy and detection in deep tissues62. These imaging probes can accumulate in tumours, because the probes have long half-lives in the blood and because the new blood vessels in the tumour are leaky; the proteases in the tumour stroma then cleave the probe, which consists of a labelled protein, thereby releasing the previously quenched fluorochromes locally. Using a combination of FMT and CT further improves signal localization and shows tumour burden; the procedure can be repeated serially (over time) to follow tumour progression20. The role of specific proteases in ECM degradation or other activities has not been investigated in vivo at subcellular resolution, although the protease sensors mentioned above will probably be considered for future multiphoton microscopy investigations.
Longitudinal intravital microscopy studies have also uncovered unique features of tumour blood vessels and angiogenesis63,64. For example, blood flow in tumours is highly variable, can reverse direction over time and does not deliver nutrients effectively. In many cancers, tumour blood vessels have defective endothelial barriers with relatively large pores, and pericytes, which normally provide support to endothelial cells, are often absent. These abnormalities, together with pressure from proliferating cells, contribute to high interstitial pressure and hypoxia in tumours. In addition, intravital microscopy studies65-67 and tomographic fluorescence mapping68 are being used to compare the efficacy of angiogenesis inhibitors and to investigate the efficacy of therapeutic drug delivery. Interestingly, studies of tumour blood-vessel responses have shown that certain antiangiogenic agents transiently normalize the structure and function of tumour vasculature, instead of ‘pruning’ the vessels as was expected69. This finding indicates that, at least in mouse models, there is a window of opportunity for improving drug delivery and for increasing sensitivity to radiation treatment.
Cell death
Real-time imaging of cell death would be a coveted application with which to assess the efficacy of cytotoxic drugs and, potentially, to monitor the toxicity of these drugs in normal tissues. Successful new-generation drugs are likely to be designed to exploit the differences (rather than the similarities) in activation of the apoptotic machinery between malignant and normal cells. The ability to image such effects in vivo could thus have far-reaching implications for assessing therapeutic efficacy and toxicity.
Bioluminescence imaging has been used to evaluate tumour growth and regression (for example, in response to therapy) in experimental animals with tumours engineered to express a luciferase70, although this technique allows only relative quantification (as mentioned earlier). Strategies to image cell death directly include the use of recombinant photoprotein reporters activatable by caspase-3 cleavage71,72, and fluorescent73 or radio-labelled74 ligands, such as annexin V, with high affinity for apoptotic cells. In fact, pharmaceutical companies are increasingly using whole-animal optical imaging systems, with tumour size or molecule-specific imaging signal as the primary read-outs for drug efficacy. Complementing the macroscopic information provided by bioluminescence imaging, recent approaches to image apoptosis at the single-cell level in vivo include labelling components in the cytoplasm and the nucleus (DNA) with different fluorochromes and then monitoring the cytoplasm:nucleus fluorescence ratio (Fc:Fn) over time; dead cells typically show a decrease in this ratio, owing to DNA fragmentation and loss of plasma-membrane integrity 51. An alternative strategy is to visualize cellular morphology, because condensation of the cell body and blebbing of the plasma membrane are characteristic of cells entering apoptosis. These approaches have been used successfully to elucidate the in vivo dynamics of CTL-mediated killing and the mechanisms used by Treg cells to blunt the cytotoxic functions of CTLs49,51. In the clinic, analysing glucose metabolism by using PET (after administering the probe [18F]fluorodeoxyglucose, which is preferentially taken up by metabolically active cells) is progressively being used as a surrogate for treatment efficacy75,76.
Clinical detection of epithelial neoplasias
New fibre-optic imaging technologies are being explored to detect early-stage epithelial cancers in the gastrointestinal tract77 (Fig. 4), the lungs78 and other easily accessible surfaces. These developments are important for several reasons. First, traditional endoscopy can fail to detect up to 20% of lesions during a procedure. Second, treatments for early-stage cancers are often curative, in contrast to later-stage treatments. Third, advanced imaging technologies can assist therapeutic endoscopic procedures (including laser-based surgery, cryotherapy, electrocautery and stenting). Last, emerging newer procedures could be used to differentiate premalignant or malignant lesions from inflammatory lesions in individuals who are at high risk of developing cancer.
Figure 4. Clinical imaging.

With a common computing platform, data obtained by different imaging techniques can be seamlessly assembled (and fused) for the screening, detection, characterization (in vivo pathology) and real-time treatment of early-stage cancers. In the example shown, a multislice CT image of the abdomen is acquired, and the slices are assembled in three dimensions. A suspicious colonic lesion is identified (at the intersection of the dashed lines) and reconstructed in three dimensions to reveal a polyp-like growth (arrow). Post-processing algorithms that explore differences in tissue and air-barium attenuation (insets) can be used to suggest, but not confirm, the presence of a malignancy. Near-infrared endoscopy using protease beacons then allows the detection of small and otherwise difficult-to-identify lesions. The bright signal in the near-infrared fluorescence channel indicates large amounts of proteases in a tumour (lower right) but not in a normal colon (upper right). White-light views of the same images are shown on the left. Microendoscopy shows a malignant signature in vivo, which can be used to make treatment decisions on the spot in real time. The microscopy image shows a polyp-like lesion, which contains mucosal cells (green), proteases (red) and new microvasculature (blue). Real-time local treatment can be achieved by lesion removal or light-activated therapy. 3D, three-dimensional.
Two clinical advances are particularly noteworthy given their high sensitivity and specificity: near-infrared fluorescence endoscopy, and fibre-optic confocal laser-scanning microendoscopy. Near-infrared fluorescence endoscopy, in combination with new imaging agents, is an emerging technology that has strong potential for increasing sensitivity79. The agents that are closest to clinical trials are fluorescent substrates that are activated by tumour-associated cathepsins20,80. Second, fibre-optic confocal laser-scanning microendoscopy has emerged as a tool for identifying cellular and subcellular structures on the basis of reflected light, autofluorescence or exogenous imaging agents. This endoscopic technique allows diagnosis of colorectal cancer, helps to target intervention to relevant areas, and decreases the number of biopsies required for cancer surveillance17. It is expected that this combination of a macroscopic strategy and a microscopic strategy — that is, using near-infrared fluorescence endoscopy to identify suspicious lesions and then fibre-optic multichannel microendoscopy to analyse them microscopically — will ultimately allow ‘virtual histology’.
These approaches herald the beginning of a new era that will give researchers and doctors a unique look at cellular structures and functions at and below epithelial surfaces. There is a clear need and opportunity for developing sets of biologically compatible, diagnostic near-infrared fluorescence imaging agents that are specific for different cell types and tissue structures and that enable multichannel measurements to be made, similar to the reagents available for in vitro fluorescence microscopy34. Finally, the capability of multichannel macroscopic and microscopic systems will allow immediate treatment decisions. Several agents that have combined diagnostic and therapeutic (‘theranostic’) properties are in development81.
Intra-operative imaging
Fixed-geometry and hand-held fluorescence imaging systems should improve oncological surgical procedures. There are several emerging applications: defining tumour margins and identifying small metastases to improve the accuracy of surgical removal, mapping sentinel lymph nodes, and defining anatomy during surgical intervention. Most systems are based on reflectance imaging at video rate, combined with the use of near-infrared fluorochromes. Tumour margins have been visualized by using several fluorescence strategies, including targeting cancer cells82, and targeting the proteolytic or phagocytic activities of tumour cells or host cells in the periphery79,83. Sentinel lymph-node mapping has been improved through the use of near-infrared fluorochrome-labelled albumin, fluorescent nanoparticles and red-shifted quantum dots84.
Challenges for the future
In the past decade, enormous strides have been made in the imaging sciences, and many new technologies, agents and reporters have been applied to oncology research, clinical trials and patient care. What has facilitated these advances, and how do we ensure that they will continue in the future? Advances have been fuelled by the convergence of biology, chemistry, physics and engineering in interdisciplinary centres across the world. Miniaturization, nanotechnology and computation have also been driving forces behind innovation. Commercial availability of new imaging systems and imaging probes has also contributed to the adoption of these techniques. One area that has lagged behind, however, is the creation of information technology infrastructures that serve macroscopic and microscopic imaging communities alike. Although the Digital Imaging and Communications in Medicine standards are common in medical imaging and are the backbone of picture archiving and communication systems and electronic patient records, such standards have not commonly been adopted by the in vivo microscopy community. There are additional opportunities in image archival, distribution, analysis, visualization and creation of standardized databases to compare and analyse data sets.
Imaging is one of the few technologies that can generate longitudinal data sets in intact host environments. As the research community starts to adopt a more systems-wide analysis, it is clear that the data will need to fulfil certain criteria: they need to be truly quantitative (that is, absolute not only relative), comprehensive and able to be integrated with other molecular data sets. Quantification and integration are some of the more immediate needs.
The translation of imaging agents and technologies into the clinic has been much slower than was initially hoped. The reasons for this are complex and include considerable regulatory hurdles, market forces, lower profit margins for imaging than for therapeutic drugs, and the lack of reimbursement strategies for newer imaging agents. Despite these hurdles, several new technologies are poised to enter clinical trials. With the deeper understanding of the molecular basis of disease that has been gained by imaging studies — and the recent advances in detection technology, imaging systems, reconstruction algorithms and visualization tools — the transformative promise of imaging is likely to be fulfilled soon. Applying the new molecular imaging tools to humans will make a fundamental improvement in how cancer is understood in vivo and should allow earlier detection, stratification of patients for treatment, and objective evaluation of new therapies in a given patient. The outcome will be considerably better management and care of those with cancer.
Acknowledgements
The authors acknowledge financial support from the National Institutes of Health.
References
- 1.Soon L, Braet F, Condeelis J. Moving in the right direction — nanoimaging in cancer cell motility and metastasis. Microsc. Res. Tech. 2007;70:252–257. doi: 10.1002/jemt.20411. [DOI] [PubMed] [Google Scholar]
- 2.Deisseroth K, et al. Next-generation optical technologies for illuminating genetically targeted brain circuits. J. Neurosci. 2006;26:10380–10386. doi: 10.1523/JNEUROSCI.3863-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brindle KM. New approaches for imaging tumour responses to treatment. Nature Rev. Cancer. 2008;8:94–107. doi: 10.1038/nrc2289. [DOI] [PubMed] [Google Scholar]
- 4.Quon A, Gambhir SS. FDG-PET and beyond: molecular breast cancer imaging. J. Clin. Oncol. 2005;23:1664–1673. doi: 10.1200/JCO.2005.11.024. [DOI] [PubMed] [Google Scholar]
- 5.Neves AA, Brindle KM. Assessing responses to cancer therapy using molecular imaging. Biochim. Biophys. Acta. 2006;1766:242–261. doi: 10.1016/j.bbcan.2006.10.002. [DOI] [PubMed] [Google Scholar]
- 6.Policard A. Étude sur les aspects offerts par des tumeurs expérimentales examinées à la lumière de Wood. C. R. Séances Soc. Biol. Fil. 1924;91:1423–1424. [Google Scholar]
- 7.Auler H, Banzer G. Untersuchungen ueber die Rolle der Porphyrine bei geschwulstranken Menschen und Tieren. Z. Krebsforsch. 1942;53:65–68. [Google Scholar]
- 8.Moore GE, Peyton WT, French LA. The clinical use of fluorescein in neurosurgery. The localization of brain tumors. J. Neurosurg. 1948;5:392–398. doi: 10.3171/jns.1948.5.4.0392. [DOI] [PubMed] [Google Scholar]
- 9.Chance B. Optical method. Annu. Rev. Biophys. Biophys. Chem. 1991;20:1–28. doi: 10.1146/annurev.bb.20.060191.000245. [DOI] [PubMed] [Google Scholar]
- 10.Jobsis FF. Noninvasive, infrared monitoring of cerebral and myocardial oxygen sufficiency and circulatory parameters. Science. 1977;198:1264–1267. doi: 10.1126/science.929199. [DOI] [PubMed] [Google Scholar]
- 11.Tatsuta M, et al. Diagnosis of gastric cancers with fluorescein-labeled monoclonal antibodies to carcinoembryonic antigen. Lasers Surg. Med. 1989;9:422–426. doi: 10.1002/lsm.1900090415. [DOI] [PubMed] [Google Scholar]
- 12.Folli S, et al. Immunophotodiagnosis of colon carcinomas in patients injected with fluoresceinated chimeric antibodies against carcinoembryonic antigen. Proc. Natl Acad. Sci. USA. 1992;89:7973–7977. doi: 10.1073/pnas.89.17.7973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pelegrin A, et al. Antibody-fluorescein conjugates for photoimmunodiagnosis of human colon carcinoma in nude mice. Cancer. 1991;67:2529–2537. doi: 10.1002/1097-0142(19910515)67:10<2529::aid-cncr2820671024>3.0.co;2-b. [DOI] [PubMed] [Google Scholar]
- 14.Wagner R. Erläuterungstafeln zur Physiologie und Entwicklungsgeschichte. Voss; Leipzig: 1839. [Google Scholar]
- 15.Halin C, Rodrigo Mora J, Sumen C, von Andrian UH. In vivo imaging of lymphocyte trafficking. Annu. Rev. Cell Dev. Biol. 2005;21:581–603. doi: 10.1146/annurev.cellbio.21.122303.133159. [DOI] [PubMed] [Google Scholar]
- 16.Flusberg BA, et al. Fiber-optic fluorescence imaging. Nature Methods. 2005;2:941–950. doi: 10.1038/nmeth820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kiesslich R, Goetz M, Vieth M, Galle PR, Neurath MF. Confocal laser endoscopy for in vivo diagnosis of colorectal cancer. Nature Clin. Pract. Oncol. 2007;4:480–490. doi: 10.1038/ncponc0881. [DOI] [PubMed] [Google Scholar]
- 18.Yelin D, et al. Three-dimensional miniature endoscopy. Nature. 2006;443:765. doi: 10.1038/443765a. [DOI] [PubMed] [Google Scholar]
- 19.Ntziachristos V, Ripoll J, Wang LV, Weissleder R. Looking and listening to light: the evolution of whole-body photonic imaging. Nature Biotechnol. 2005;23:313–320. doi: 10.1038/nbt1074. [DOI] [PubMed] [Google Scholar]
- 20.Grimm J, et al. Use of gene expression profiling to direct in vivo molecular imaging of lung cancer. Proc. Natl Acad. Sci. USA. 2005;102:14404–14409. doi: 10.1073/pnas.0503920102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zacharakis G, et al. Volumetric tomography of fluorescent proteins through small animals in vivo. Proc. Natl Acad. Sci. USA. 2005;102:18252–18257. doi: 10.1073/pnas.0504628102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yun SH, et al. Comprehensive volumetric optical microscopy in vivo. Nature Med. 2006;12:1429–1433. doi: 10.1038/nm1450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- This paper describes the development of a fibre-optic imaging technique that is useful for diagnostic imaging of epithelial disease.
- 23.Zhang HF, Maslov K, Stoica G, Wang LV. Functional photoacoustic microscopy for high-resolution and noninvasive in vivo imaging. Nature Biotechnol. 2006;24:848–851. doi: 10.1038/nbt1220. [DOI] [PubMed] [Google Scholar]
- 24.Liu Y, et al. Optical markers in duodenal mucosa predict the presence of pancreatic cancer. Clin. Cancer Res. 2007;13:4392–4399. doi: 10.1158/1078-0432.CCR-06-1648. [DOI] [PubMed] [Google Scholar]
- 25.Evans CL, et al. Chemical imaging of tissue in vivo with video-rate coherent anti-Stokes Raman scattering microscopy. Proc. Natl Acad. Sci. USA. 2005;102:16807–16812. doi: 10.1073/pnas.0508282102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gross S, Piwnica-Worms D. Spying on cancer: molecular imaging in vivo with genetically encoded reporters. Cancer Cell. 2005;7:5–15. doi: 10.1016/j.ccr.2004.12.011. [DOI] [PubMed] [Google Scholar]
- 27.Giepmans BN, Adams SR, Ellisman MH, Tsien RY. The fluorescent toolbox for assessing protein location and function. Science. 2006;312:217–224. doi: 10.1126/science.1124618. [DOI] [PubMed] [Google Scholar]
- 28.Ventura A, et al. Restoration of p53 function leads to tumour regression in vivo. Nature. 2007;445:661–665. doi: 10.1038/nature05541. [DOI] [PubMed] [Google Scholar]
- 29.Sharpless NE, Depinho RA. The mighty mouse: genetically engineered mouse models in cancer drug development. Nature Rev. Drug Discov. 2006;5:741–754. doi: 10.1038/nrd2110. [DOI] [PubMed] [Google Scholar]
- 30.Ray P, et al. Noninvasive quantitative imaging of protein-protein interactions in living subjects. Proc. Natl Acad. Sci. USA. 2002;99:3105–3110. doi: 10.1073/pnas.052710999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Perroy J, Pontier S, Charest PG, Aubry M, Bouvier M. Real-time monitoring of ubiquitination in living cells by BRET. Nature Methods. 2004;1:203–208. doi: 10.1038/nmeth722. [DOI] [PubMed] [Google Scholar]
- 32.Jares-Erijman EA, Jovin TM. FRET imaging. Nature Biotechnol. 2003;21:1387–1395. doi: 10.1038/nbt896. [DOI] [PubMed] [Google Scholar]
- 33.Paulmurugan R, Massoud TF, Huang J, Gambhir SS. Molecular imaging of drug-modulated protein-protein interactions in living subjects. Cancer Res. 2004;64:2113–2119. doi: 10.1158/0008-5472.can-03-2972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kelly K, et al. Targeted nanoparticles for imaging incipient pancreatic ductal adenocarcinoma. PLoS Med. doi: 10.1371/journal.pmed.0050085. in the press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Weissleder R, Kelly K, Sun EY, Shtatland T, Josephson L. Cell-specific targeting of nanoparticles by multivalent attachment of small molecules. Nature Biotechnol. 2005;23:1418–1423. doi: 10.1038/nbt1159. [DOI] [PubMed] [Google Scholar]
- This study used libraries of nanoparticles to identify cell-specific targeting agents that can be used for in vivo labelling.
- 36.Kelly KA, Waterman P, Weissleder R. In vivo imaging of molecularly targeted phage. Neoplasia. 2006;8:1011–1018. doi: 10.1593/neo.06610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pittet MJ, et al. In vivo imaging of T cell delivery to tumors after adoptive transfer therapy. Proc. Natl Acad. Sci. USA. 2007;104:12457–12461. doi: 10.1073/pnas.0704460104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bulte JW. Intracellular endosomal magnetic labeling of cells. Methods Mol. Med. 2006;124:419–439. doi: 10.1385/1-59745-010-3:419. [DOI] [PubMed] [Google Scholar]
- 39.Arbab AS, Liu W, Frank JA. Cellular magnetic resonance imaging: current status and future prospects. Expert Rev. Med. Devices. 2006;3:427–439. doi: 10.1586/17434440.3.4.427. [DOI] [PubMed] [Google Scholar]
- 40.Pittet MJ, Swirski FK, Reynolds F, Josephson L, Weissleder R. Labeling of immune cells for in vivo imaging using magnetofluorescent nanoparticles. Nature Protocols. 2006 doi: 10.1038/nprot.2006.11. doi:10.1038/nprot.2006.11. [DOI] [PubMed] [Google Scholar]
- 41.Swirski FK, et al. A near-infrared cell tracker reagent for multiscopic in vivo imaging and quantification of leukocyte immune responses. PLoS ONE. 2007;2:e1075. doi: 10.1371/journal.pone.0001075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Boon T, Coulie PG, Van den Eynde BJ, van der Bruggen P. Human T cell responses against melanoma. Annu. Rev. Immunol. 2006;24:175–208. doi: 10.1146/annurev.immunol.24.021605.090733. [DOI] [PubMed] [Google Scholar]
- 43.Romero P, et al. Ex vivo staining of metastatic lymph nodes by class I major histocompatibility complex tetramers reveals high numbers of antigen-experienced tumor-specific cytolytic T lymphocytes. J. Exp. Med. 1998;188:1641–1650. doi: 10.1084/jem.188.9.1641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Curiel TJ, et al. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nature Med. 2004;10:942–949. doi: 10.1038/nm1093. [DOI] [PubMed] [Google Scholar]
- 45.Chen ML, et al. Regulatory T cells suppress tumor-specific CD8 T cell cytotoxicity through TGF-β signals in vivo. Proc. Natl Acad. Sci. USA. 2005;102:419–424. doi: 10.1073/pnas.0408197102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nagaraj S, et al. Altered recognition of antigen is a mechanism of CD8+ T cell tolerance in cancer. Nature Med. 2007;13:828–835. doi: 10.1038/nm1609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yang L, et al. Abrogation of TGFβ signaling in mammary carcinomas recruits Gr-1+CD11b+ myeloid cells that promote metastasis. Cancer Cell. 2008;13:23–35. doi: 10.1016/j.ccr.2007.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Boissonnas A, Fetler L, Zeelenberg IS, Hugues S, Amigorena S. In vivo imaging of cytotoxic T cell infiltration and elimination of a solid tumor. J. Exp. Med. 2007;204:345–356. doi: 10.1084/jem.20061890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mrass P, et al. Random migration precedes stable target cell interactions of tumor-infiltrating T cells. J. Exp. Med. 2006;203:2749–2761. doi: 10.1084/jem.20060710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- References 48 and 49 were the first in vivo studies of T-cell infiltration and motility in tumour stroma and T-cell formation of cognate-antigen-dependent contacts with tumour cells.
- 50.Veiga-Fernandes H, Walter U, Bourgeois C, McLean A, Rocha B. Response of naive and memory CD8+ T cells to antigen stimulation in vivo. Nature Immunol. 2000;1:47–53. doi: 10.1038/76907. [DOI] [PubMed] [Google Scholar]
- 51.Mempel TR, et al. Regulatory T cells reversibly suppress cytotoxic T cell function independent of effector differentiation. Immunity. 2006;25:129–141. doi: 10.1016/j.immuni.2006.04.015. [DOI] [PubMed] [Google Scholar]
- This study found that Treg cells reversibly suppress CTL-mediated antitumour immunity by allowing CTLs to acquire full effector potential but withholding their ‘license to kill’.
- 52.Zippelius A, et al. Effector function of human tumor-specific CD8 T cells in melanoma lesions: a state of local functional tolerance. Cancer Res. 2004;64:2865–2873. doi: 10.1158/0008-5472.can-03-3066. [DOI] [PubMed] [Google Scholar]
- 53.Wang W, Eddy R, Condeelis J. The cofilin pathway in breast cancer invasion and metastasis. Nature Rev. Cancer. 2007;7:429–440. doi: 10.1038/nrc2148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wyckoff JB, et al. Direct visualization of macrophage-assisted tumor cell intravasation in mammary tumors. Cancer Res. 2007;67:2649–2656. doi: 10.1158/0008-5472.CAN-06-1823. [DOI] [PubMed] [Google Scholar]
- This paper reports that the intravasation of breast tumour cells in vivo occurs in association with perivascular TAMs.
- 55.Orimo A, et al. Stromal fibroblasts present in invasive human breast carcinomas promote tumor growth and angiogenesis through elevated SDF-1/CXCL12 secretion. Cell. 2005;121:335–348. doi: 10.1016/j.cell.2005.02.034. [DOI] [PubMed] [Google Scholar]
- 56.Granot D, et al. In vivo imaging of the systemic recruitment of fibroblasts to the angiogenic rim of ovarian carcinoma tumors. Cancer Res. 2007;67:9180–9189. doi: 10.1158/0008-5472.CAN-07-0684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Muller A, et al. Involvement of chemokine receptors in breast cancer metastasis. Nature. 2001;410:50–56. doi: 10.1038/35065016. [DOI] [PubMed] [Google Scholar]
- 58.Soucek L, et al. Mast cells are required for angiogenesis and macroscopic expansion of Myc-induced pancreatic islet tumors. Nature Med. 2007;13:1211–1218. doi: 10.1038/nm1649. [DOI] [PubMed] [Google Scholar]
- 59.Shojaei F, et al. Bv8 regulates myeloid-cell-dependent tumour angiogenesis. Nature. 2007;450:825–831. doi: 10.1038/nature06348. [DOI] [PubMed] [Google Scholar]
- 60.Karnoub AE, et al. Mesenchymal stem cells within tumour stroma promote breast cancer metastasis. Nature. 2007;449:557–563. doi: 10.1038/nature06188. [DOI] [PubMed] [Google Scholar]
- 61.Weissleder R, Tung CH, Mahmood U, Bogdanov A., Jr. In vivo imaging of tumors with protease-activated near-infrared fluorescent probes. Nature Biotechnol. 1999;17:375–378. doi: 10.1038/7933. [DOI] [PubMed] [Google Scholar]
- 62.Ntziachristos V, Tung CH, Bremer C, Weissleder R. Fluorescence molecular tomography resolves protease activity in vivo. Nature Med. 2002;8:757–760. doi: 10.1038/nm729. [DOI] [PubMed] [Google Scholar]
- 63.Carmeliet P, Jain RK. Angiogenesis in cancer and other diseases. Nature. 2000;407:249–257. doi: 10.1038/35025220. [DOI] [PubMed] [Google Scholar]
- 64.McDonald DM, Choyke PL. Imaging of angiogenesis: from microscope to clinic. Nature Med. 2003;9:713–725. doi: 10.1038/nm0603-713. [DOI] [PubMed] [Google Scholar]
- 65.Ferrara N, Kerbel RS. Angiogenesis as a therapeutic target. Nature. 2005;438:967–974. doi: 10.1038/nature04483. [DOI] [PubMed] [Google Scholar]
- 66.Brown EB, et al. In vivo measurement of gene expression, angiogenesis and physiological function in tumors using multiphoton laser scanning microscopy. Nature Med. 2001;7:864–868. doi: 10.1038/89997. [DOI] [PubMed] [Google Scholar]
- This study used intravital microscopy and tumour-window-chamber models to monitor gene expression, cell adhesion, delivery of therapeutics, angiogenesis and blood-vessel permeability in deep regions of tumours.
- 67.Willett CG, et al. Direct evidence that the VEGF-specific antibody bevacizumab has antivascular effects in human rectal cancer. Nature Med. 2004;10:145–147. doi: 10.1038/nm988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Montet X, Ntziachristos V, Grimm J, Weissleder R. Tomographic fluorescence mapping of tumor targets. Cancer Res. 2005;65:6330–6336. doi: 10.1158/0008-5472.CAN-05-0382. [DOI] [PubMed] [Google Scholar]
- 69.Jain RK. Normalization of tumor vasculature: an emerging concept in antiangiogenic therapy. Science. 2005;307:58–62. doi: 10.1126/science.1104819. [DOI] [PubMed] [Google Scholar]
- 70.Contag CH, Jenkins D, Contag PR, Negrin RS. Use of reporter genes for optical measurements of neoplastic disease in vivo. Neoplasia. 2000;2:41–52. doi: 10.1038/sj.neo.7900079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lee KC, et al. Noninvasive molecular imaging sheds light on the synergy between 5-fluorouracil and TRAIL/Apo2L for cancer therapy. Clin. Cancer Res. 2007;13:1839–1846. doi: 10.1158/1078-0432.CCR-06-1657. [DOI] [PubMed] [Google Scholar]
- 72.Laxman B, et al. Noninvasive real-time imaging of apoptosis. Proc. Natl Acad. Sci. USA. 2002;99:16551–16555. doi: 10.1073/pnas.252644499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ntziachristos V, et al. Visualization of antitumor treatment by means of fluorescence molecular tomography with an annexin V-Cy5.5 conjugate. Proc. Natl Acad. Sci. USA. 2004;101:12294–12299. doi: 10.1073/pnas.0401137101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Blankenberg FG, Vanderheyden JL, Strauss HW, Tait JF. Radiolabeling of HYNIC-annexin V with technetium-99m for in vivo imaging of apoptosis. Nature Protocols. 2006 doi: 10.1038/nprot.2006.17. doi:10.1038/nprot.2006.17. [DOI] [PubMed] [Google Scholar]
- 75.Avril N, et al. Prediction of response to neoadjuvant chemotherapy by sequential F-18-fluorodeoxyglucose positron emission tomography in patients with advanced-stage ovarian cancer. J. Clin. Oncol. 2005;23:7445–7453. doi: 10.1200/JCO.2005.06.965. [DOI] [PubMed] [Google Scholar]
- 76.Lordick F, et al. PET to assess early metabolic response and to guide treatment of adenocarcinoma of the oesophagogastric junction: the MUNICON phase II trial. Lancet Oncol. 2007;8:797–805. doi: 10.1016/S1470-2045(07)70244-9. [DOI] [PubMed] [Google Scholar]
- 77.Dekker E, Fockens P. New imaging techniques at colonoscopy: tissue spectroscopy and narrow band imaging. Gastrointest. Endosc. Clin. N. Am. 2005;15:703–714. doi: 10.1016/j.giec.2005.08.006. [DOI] [PubMed] [Google Scholar]
- 78.Herth FJ, Eberhardt R, Ernst A. The future of bronchoscopy in diagnosing, staging and treatment of lung cancer. Respiration. 2006;73:399–409. doi: 10.1159/000093369. [DOI] [PubMed] [Google Scholar]
- 79.Marten K, et al. Detection of dysplastic intestinal adenomas using enzyme-sensing molecular beacons in mice. Gastroenterology. 2002;122:406–414. doi: 10.1053/gast.2002.30990. [DOI] [PubMed] [Google Scholar]
- 80.Joyce JA, et al. Cathepsin cysteine proteases are effectors of invasive growth and angiogenesis during multistage tumorigenesis. Cancer Cell. 2004;5:443–453. doi: 10.1016/s1535-6108(04)00111-4. [DOI] [PubMed] [Google Scholar]
- This paper shows that cathepsins promote invasive growth and angiogenesis in pancreatic islet tumours.
- 81.McCarthy JR, Jaffer FA, Weissleder R. A macrophage-targeted theranostic nanoparticle for biomedical applications. Small. 2006;2:983–987. doi: 10.1002/smll.200600139. [DOI] [PubMed] [Google Scholar]
- 82.Koyama Y, et al. Spectral fluorescence molecular imaging of lung metastases targeting HER2/neu. Clin. Cancer Res. 2007;13:2936–2945. doi: 10.1158/1078-0432.CCR-06-2240. [DOI] [PubMed] [Google Scholar]
- 83.Kirsch DG, et al. A spatially and temporally restricted mouse model of soft tissue sarcoma. Nature Med. 2007;13:992–997. doi: 10.1038/nm1602. [DOI] [PubMed] [Google Scholar]
- This study combined a new mouse model of soft-tissue sarcoma that mimics human sarcomas with a hand-held imaging device that identifies residual tumour tissue during intra-operative molecular imaging.
- 84.Kim S, et al. Near-infrared fluorescent type II quantum dots for sentinel lymph node mapping. Nature Biotechnol. 2004;22:93–97. doi: 10.1038/nbt920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Rudin M, Weissleder R. Molecular imaging in drug discovery and development. Nature Rev. Drug Discov. 2003;2:123–131. doi: 10.1038/nrd1007. [DOI] [PubMed] [Google Scholar]
- 86.Rontgen WC. On a new kind of rays. Nature. 1896;53:274–276. doi: 10.1126/science.3.59.227. [DOI] [PubMed] [Google Scholar]
- 87.Drexler B, Davis JL, Schofield G. Diaphanography in the diagnosis of breast cancer. Radiology. 1985;157:41–44. doi: 10.1148/radiology.157.1.4034975. [DOI] [PubMed] [Google Scholar]
- 88.Figueiredo JL, Alencar H, Weissleder R, Mahmood U. Near infrared thoracoscopy of tumoral protease activity for improved detection of peripheral lung cancer. Int. J. Cancer. 2006;118:2672–2677. doi: 10.1002/ijc.21713. [DOI] [PubMed] [Google Scholar]
- 89.Ntziachristos V, Yodh AG, Schnall M, Chance B. Concurrent MRI and diffuse optical tomography of breast after indocyanine green enhancement. Proc. Natl Acad. Sci. USA. 2000;97:2767–2772. doi: 10.1073/pnas.040570597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Drahl C, Cravatt BF, Sorensen EJ. Protein-reactive natural products. Angew. Chem. Int. Ed. Engl. 2005;44:5788–5809. doi: 10.1002/anie.200500900. [DOI] [PubMed] [Google Scholar]
- 91.Roberti MJ, Bertoncini CW, Klement R, Jares-Erijman EA, Jovin TM. Fluorescence imaging of amyloid formation in living cells by a functional, tetracysteine-tagged α-synuclein. Nature Methods. 2007;4:345–351. doi: 10.1038/nmeth1026. [DOI] [PubMed] [Google Scholar]
- 92.Venkatraman P, et al. Fluorogenic probes for monitoring peptide binding to class II MHC proteins in living cells. Nature Chem. Biol. 2007;3:222–228. doi: 10.1038/nchembio868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Harisinghani MG, et al. Noninvasive detection of clinically occult lymph-node metastases in prostate cancer. N. Engl. J. Med. 2003;348:2491–2499. doi: 10.1056/NEJMoa022749. [DOI] [PubMed] [Google Scholar]
- 94.Cristofanilli M, et al. Circulating tumor cells, disease progression, and survival in metastatic breast cancer. N. Engl. J. Med. 2004;351:781–791. doi: 10.1056/NEJMoa040766. [DOI] [PubMed] [Google Scholar]
- 95.Georgakoudi I, et al. In vivo flow cytometry: a new method for enumerating circulating cancer cells. Cancer Res. 2004;64:5044–5047. doi: 10.1158/0008-5472.CAN-04-1058. [DOI] [PubMed] [Google Scholar]
- This paper reports the first in vivo flow-cytometry-based method for quantifying circulating tumour cells.
- 96.Boutrus S, et al. Portable two-color in vivo flow cytometer for real-time detection of fluorescently-labeled circulating cells. J. Biomed. Opt. 2007;12:020507. doi: 10.1117/1.2722733. doi:10.1117/1.2722733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.He W, Wang H, Hartmann LC, Cheng JX, Low PS. In vivo quantitation of rare circulating tumor cells by multiphoton intravital flow cytometry. Proc. Natl Acad. Sci. USA. 2007;104:11760–11765. doi: 10.1073/pnas.0703875104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Nagrath S, Haber DA, Toner M. Isolation of rare circulating epithelial cells in cancer patients by microchip technology. Nature. 2007;450:1235–1239. doi: 10.1038/nature06385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Lee H, Sun EY, Ham D, Weissleder R. Chip-NMR biosensor for detection and molecular analysis of cells. Nature Med. doi: 10.1038/nm.1711. in the press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- This study developed a low-cost diagnostic MRI platform for rapid, quantitative, multichannel detection of biological targets in unprocessed samples.
- 100.Perez JM, Josephson L, O’Loughlin T, Hogemann D, Weissleder R. Magnetic relaxation switches capable of sensing molecular interactions. Nature Biotechnol. 2002;20:816–820. doi: 10.1038/nbt720. [DOI] [PubMed] [Google Scholar]