

Abstract
Five mutations in the ENAM gene have been found to cause hypoplastic amelogenesis imperfecta (AI), with phenotypes ranging from localized enamel pitting in carriers to severe hypoplastic AI. To determine the generality of ENAM mutations in hypoplastic AI, we sequenced the ENAM gene in ten Turkish families segregating autosomal hypoplastic AI. In two families, ENAM mutations were found. A novel nonsense mutation (g.12663C>A; p.S246X) was identified in one family segregating local hypoplastic AI as a dominant trait. Affected individuals in a second family segregating autosomal-recessive AI were compound heterozygotes for a novel insertion mutation (g.12946_12947insAGTCAGTACCAGTACTGT GTC) and a previously described insertion (g.13185_13186insAG) mutation. Heterozygous carriers of either insertion had a localized enamel-pitting phenotype. These findings substantiate that enamel phenotypes of ENAM mutations may be dose-dependent, with generalized hypoplastic AI segregating as a recessive trait and localized enamel pitting segregating as a dominant trait.
Keywords: amelogenesis imperfecta, enamel, enamel pitting, gene dosage, enamelin
INTRODUCTION
The amelogenesis imperfecta (AI) disorders are hereditary pathologies of enamel development with autosomal-dominant, autosomal-recessive, and X-linked modes of inheritance (Sundell and Valentin, 1986; Witkop, 1989; Backman, 1997; Aldred et al., 2003). Cardinal clinical findings relate to enamel aberrations, although findings of skeletal and dental malocclusion have been reported (Persson and Sundell, 1982; Cartwright et al., 1999; Ravassipour et al., 2005). Based on the enamel appearance and hypothesized developmental defect, AI can be classified into hypoplastic (secretory defect), hypocalcified (mineralization defect), and hypomaturation (protein processing and crystallite maturation defect) forms. Traditionally, 14 AI subtypes have been recognized based upon clinical phenotype and mode of inheritance (Witkop, 1989), but with continued phenotypic and molecular characterization, it is clear that this system does not account for all AI types (Aldred et al., 2003; Nusier et al., 2004).
To date, mutations in 4 genes have been found to be associated with AI. Hypomaturation AI results from mutations in amelogenin (AMELX) and in the enamel proteases, kallikrein 4 (KLK4), and matrix metalloproteinase 20 (MMP20) (Wright et al., 2003; Hart et al., 2004; Kim et al., 2005a). Mutations in genes encoding the structural proteins amelogenin and enamelin (ENAM) are associated with hypoplastic AI (Rajpar et al., 2001; Hart et al., 2002a,b, 2003; Kida et al., 2002; Mardh et al., 2002; TC Hart et al., 2003; Wright et al., 2003; Kim et al., 2004). Five different ENAM mutations have been reported for AI (Rajpar et al., 2001; Kida et al., 2002; Mardh et al., 2002; PS Hart et al., 2003; TC Hart et al., 2003; Kim et al., 2004, 2005b). Depending on the specific ENAM mutation, the enamel phenotype varies from a generalized thin hypoplastic form that may be transmitted as a dominant (Kida et al., 2002; Rajpar et al., 2001; PS Hart et al., 2003; Kim et al., 2005) or recessive (TC Hart et al., 2003) trait, to a very mild presentation of localized, circumscribed enamel pits transmitted as a dominant trait (TC Hart et al., 2003). A dominant ENAM mutation can also cause an intermediate phenotype, referred to as 'local hypoplastic', in which horizontal rows of enamel pits or grooves circumnavigate the crown (Mardh et al., 2002). To evaluate the generality of ENAM mutations, we performed a mutation screen of the ENAM gene in ten Turkish families segregating autosomal hypoplastic AI.
MATERIALS & METHODS
Identification of Kindreds
Ten families segregating hypoplastic AI were identified when probands presented to the Istanbul University, Department of Pedodontics. Informed consent was obtained according to the Declaration of Helsinki (World Medical Association, 1996), and with institutional review board approval from Istanbul University, The University of Pittsburgh, and the NIDCR/NIH. Dental, medical, and family histories were obtained for available family members. Standard lateral cephalometric radiographs were taken by means of a Cranex-3+Ceph at 10 mA and 81 kV (Cranex Co., Helsinki, Finland). Conventional cephalometric landmarks were identified, and 15 angular and 15 linear measurements were made (Bosch and Athanasiou, 1995).
Mutation and Genotype Analysis
The exons and exon/intron boundaries of the ENAM gene were amplified and sequenced as previously described (PS Hart et al., 2003). Fifty unaffected Turkish control individuals and 50 unaffected controls of European and African descent were also sequenced. For family 2, genotyping of three markers (710M16, AMBL, and 92H22) surrounding the ENAM locus was conducted as previously described (TC Hart et al., 2003).
RESULTS
Clinical Findings
Nine of the families segregated AI as an autosomal-recessive trait, and one family segregated AI as an autosomal-dominant trait. Seven of the ten families were consanguineous.
Family 1
The 16-year-old proband presented with the chief complaint of tooth pain (Fig. 1). Clinical examination revealed that the enamel of unrestored occlusal, lingual, and palatal surfaces of the permanent teeth was hypoplastic, consistent with a diagnosis of local hypoplastic AI (Fig. 1B). Pain resulted from caries of the mandibular molars. The proband's panoramic radiograph revealed localized enamel hypoplasia, absence of the right maxillary third molar, and impaction of the left maxillary and both mandibular third molars (Fig. 1C). Cephalometric and intra-oral examination revealed Angle Class I occlusion with no open bite. Maxillary and mandibular incisors had been previously restored with composite laminates. The proband's 48-year-old father (V-5), diagnosed with AI at the age of 28 yrs, currently had ceramic crowns on most teeth, due to sensitivity and esthetic concerns. The proband's paternal grandfather (IV-4), aunt (V-6), and cousin (VI-4) were also clinically affected with hypoplastic AI. The proband's mother and two siblings were clinically unaffected. The findings are consistent with a diagnosis of autosomal-dominant local hypoplastic AI.
Figure 1.
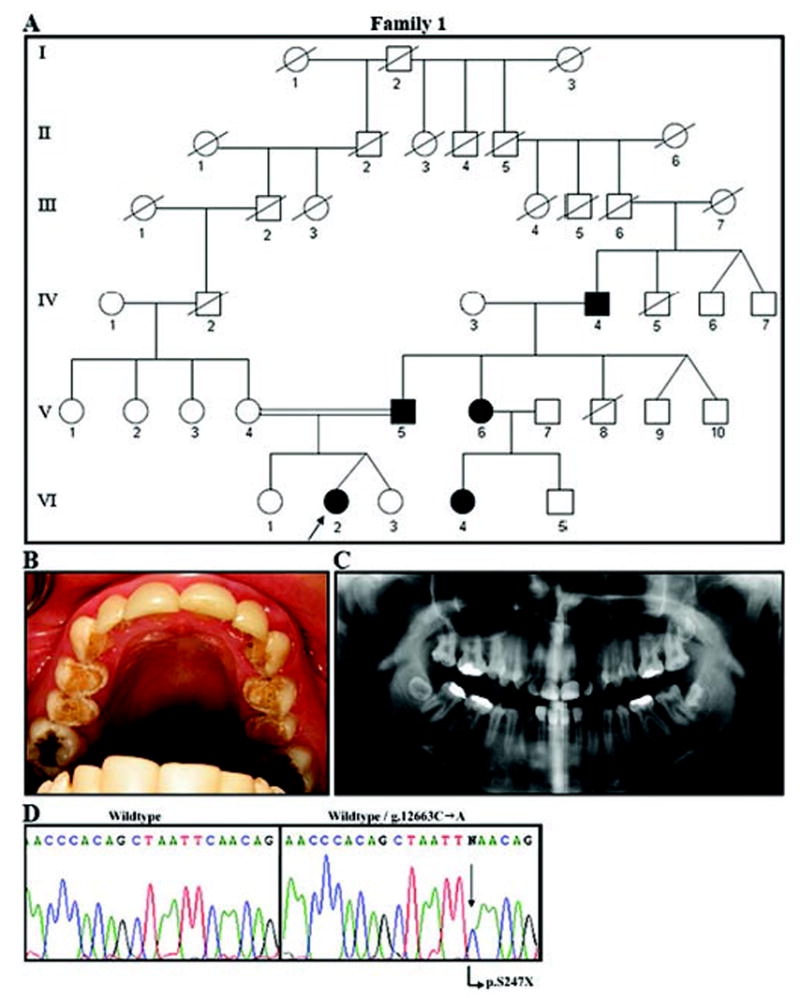
Family segregating autosomal-dominant local hypoplastic amelogenesis imperfecta. (A) Pedigree of Family 1. Blackened symbols represent AI-affected individuals. No information was available on the siblings of IV-4 or on previous generations. (B) Maxillary occlusal view of the proband (VI-2). Hypoplastic enamel is evident on the occlusal and palatal surfaces of the premolars. Laminate restorations are seen on the maxillary incisors and canines. Amalgam restorations are seen on the occlusal sites of molars. (C) The panorex radiograph (from VI-2) shows local hypoplastic enamel. Decreased contrast between enamel and dentin is evident. (D) Molecular analysis of the ENAM gene in this family. The left panel shows the wild-type sequence of a portion of exon 10. On the right is the same part of exon 10 from the proband (VI-2), showing the g.12663C>A mutation (arrow) that results in a premature stop codon (p.S246X).
Family 2
The 10-year-old female proband presented with the chief complaint of a broken maxillary right incisor (Fig. 2A). Based on radiographic and intra-oral findings, she was diagnosed as having generalized thin hypoplastic AI with Angle Class I occlusion and anterior open bite (Figs. 2A–2C). Examination of other family members revealed that the eldest sibling (age 15 yrs; II-1) was affected with generalized thin hypoplastic AI, with Angle Class I occlusion, and no anterior open bite (Fig. 2A), while both parents and the remaining siblings (II-2 and II-3) all had localized, circumscribed enamel pitting (Fig. 2A). These findings are consistent with autosomal-recessive transmission of generalized thin hypoplastic AI (Nusier et al., 2004).
Figure 2.
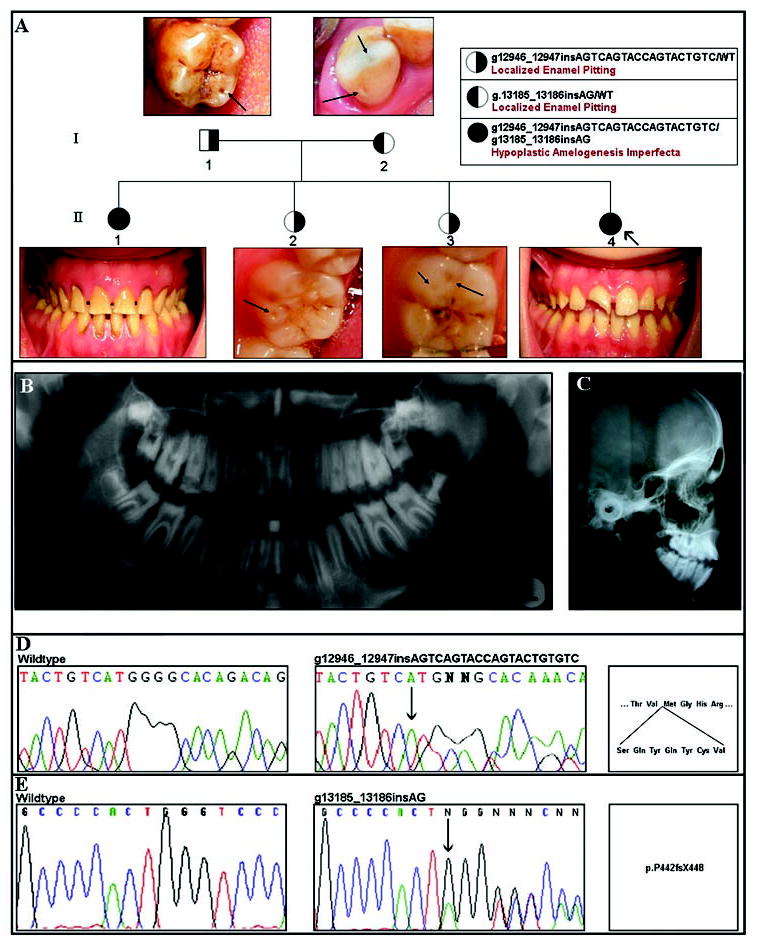
Family segregating autosomal-recessive amelogenesis imperfecta. (A) Pedigree of Family 2. Right-shaded symbols indicate carriers of the 21-bp exon 10 ENAM insertion mutation. Left-shaded symbols indicate carriers of the 2-bp exon 10 ENAM insertion mutation. Individuals with either left- or right-shaded symbols have clinically evident localized enamel pitting. Representative examples of enamel pitting in these individuals are illustrated by the arrows in the photograph above or below their corresponding pedigree symbol. Completely shaded individuals are compound heterozygotes for both exon 10 ENAM mutations and have clinically apparent generalized thin hypoplastic AI. In the proband (II-4), the maxillary right incisor is fractured. All teeth are hypoplastic and yellowish, consistent with generalized hypoplastic amelogenesis imperfecta. In the affected sibling (II-1), all teeth show generalized hypoplastic yellow enamel. (B) Panorex radiograph of proband (II-4) illustrating generalized enamel hypoplasia. (C) Lateral cephalometric radiograph of individual II-4, showing anterior open bite. (D,E) Results of molecular analysis of the ENAM gene in this family. In D, the left panel shows the wild-type sequence of a portion of exon 10. The middle panel shows the same part of exon 10 from the proband (II-4), showing the g.12946_12947insAGTCAGTACCAGTACTGTGTC mutation (arrow) that results in the insertion of 7 amino acids (p.V340_M341insSQYQYCV), as shown in the right panel. In E, the left panel again shows the wild-type sequence for a portion of exon 10. The middle panel shows the same part of exon 10 from the proband (II-4), showing the previously described 2-bp insertion (indicated by the arrow; g.13185_13186insAG) that results in a frameshift and premature truncation (p.P422fsX448).
ENAM Mutation Results
No ENAM mutations were found in eight of the ten families. Families 1 and 2 were found to have an ENAM mutation. In family 1, a novel point mutation in exon 10 segregated with the local hypoplastic AI phenotype as a dominant trait consistent with complete penetrance. According to nomenclature guidelines for ENAM mutations (PS Hart et al., 2003), this mutation is designated as g.12663C>A (c.737C>A) and introduces a premature stop codon (Fig. 1D). Assuming that the mRNA does not undergo nonsense-mediated decay, this mutation would lead to a truncated protein (p.S246X). In family 2, the two AI-affected individuals (II-1 and II-4) were compound heterozygotes for two ENAM mutations, consistent with non-consanguinity. The father was a carrier of a novel 21-base-pair ENAM insertion mutation (g.12946_12947ins AGTCAGTACCAGTACTGTGTC/WT), which he transmitted to all of his children (Fig. 2D). This mutation is predicted to cause an in-frame insertion of 7 amino acids (p.V340_M341insSQYQYCV), resulting in a protein with 1149 amino acids, compared with the wild-type protein of 1142 amino acids (Fig. 2D). The mother (I-2) was a carrier of a two-base-pair insertion mutation (g.13185_13186insAG) in exon 10 (Fig. 2E) that has been previously described in families from another region of Turkey (TC Hart et al., 2003). The families are not known to be related, but genotyping analysis indicated that the haplotype spanning this 2-bp insertion mutation was common to that segregating in the previously identified families, suggesting a common ancestor (data not shown). The mother transmitted this allele to both AI-affected individuals. This mutation causes a frameshift with the introduction of a premature termination codon (p.P422fsX448). Heterozygosity for either of these exon 10 mutations was found to be associated with enamel pitting (Fig. 2A).
None of the 3 ENAM mutations identified in this study was present in the 50 unaffected Turkish control individuals (100 alleles) or in 50 unaffected control individuals (100 alleles) of European and African descent, suggesting that they are not common variants of the ENAM gene.
DISCUSSION
ENAM gene mutations were identified in two of the ten Turkish families segregating hypoplastic AI. While the current study cannot exclude regulatory or intronic mutations of the ENAM gene in the eight families, these families were not consistent with genetic linkage (homozygosity mapping) of the region spanning the ENAM locus. These results suggest genetic heterogeneity. ENAM mutations are responsible for some cases of hypoplastic AI in Turkey, with another gene(s) responsible for the majority of cases. In the two families with identified ENAM mutations, one previously described and two novel mutations were found. This brings the total number of mutations described in ENAM to 7 (Table). Of the described mutations, 4 are single-base substitutions, 2 are insertions, and 1 is a deletion. Although all 7 mutations are predicted to have a profound effect on protein structure, either by premature truncation, or by deletion or insertion of amino acids, it is important to realize that, due to the restricted expression of enamelin (expressed only in developing teeth), these are not verified consequences.
Table.
Mutations Described in ENAM
| Genomic DNAa | cDNAb | Locationc | Proteind | Phenotype | Inheritance | Reference |
|---|---|---|---|---|---|---|
| g.2382A>T | c.157A>T | Exon 5 | p.K53X | Local hypoplastic | AD | Mardh et al., 2002 |
| g.4806A>C | IVS6-2A>C; c.211-2A>C | Intron 6 | p.M71_Q157del | Hypoplastic | AD | Kim et al., 2005b |
| g.6395G>A | IVS8+IG>A; c.534+1G>A | Intron 8 | p.A158_QI78de1 or p. Q 178fsX 191 | Smooth hypoplastic | AD | Rajpar et al., 2001; Hu and Yamakoshi, 2003 |
| g.8344de1G | IV S9+I delG; c.588+1delG | Intron 9 | p.N197fsX277 | Smooth hypoplastic | AD | Kida et al., 2002; PS Hart et al., 2003; Kim et al., 2005b |
| g.12663C>A | c.737C>A | Exon 10 | p.S246X | Local hypoplastic | AD | This report |
| g.13185_13186insAG | c.1258_1259insAG | Exon 10 | p.P422fsX448 | Generalized thin hypoplastic (recessive trait); Localized, circum- scribed pitting (dominant trait) | AR-AI; AD-localized enamel pitting | TC Hart et al., 2003 |
| g.12946_ 12947insAG TCAGTACCAGTAC TGTGTC | c.1020_IO21insAGTC AGTACCAGTACTGT GTC | Exon 10 | p.V340_M341ins SQYQYCV | Generalized thin hypo- plastic (recessive trait); Localized, circum- scribed pitting (dominant trait) | AR-AI AD-localized enamel pitting | This report |
Reference sequence for GenBank accession No. AY167999; the A of the initiator ATG is taken as +1.
Reference sequence for GenBank accession No. AF125373; the A of the initiator ATG is taken as +1.
Exonic numbering assumes 10 exons as described in PS Hart et al. 2003. Mutations reported prior to the publication of these nomenclature guidelines have the corresponding exons listed one less than in this Table, i.e., exon 4 as listed in Mardh et al. 2002 is considered exon 5 according to the nomenclature guidelines.
Initiator methionine as the +1 position.
AD, Autosomal-dominant; AR, Autosomal-recessive.
The type (hypoplastic, hypomineralized, hypomaturation) and extent of enamel defect and the mode of inheritance are important criteria in the AI classification systems (Witkop, 1989; Aldred et al., 2003; Nusier et al., 2004). The novel nonsense mutation (g.12663C>A; p.S247X) identified in AI-affected individuals of Family 1 is etiologic for a local autosomal-dominant hypoplastic form of AI. The enamel defect associated with the novel insertion mutation (g.12946_12947insAGTCAGTCCAGTACTGTGTC) identified in Family 2 is dose-dependent, with a mild, localized enamel-pitting phenotype segregating as a dominant trait, and a more severe, generalized thin hypoplastic AI phenotype segregating as a recessive trait. Of the two individuals who were compound heterozygotes and manifested the generalized thin hypoplastic AI phenotype, one (II-4) had clinical and skeletal open bite, with an ANS-PNS/Go-Me angle of 34 (normative value = 25) (Figs. 2A–2C), while the other (II-1) did not (Fig. 2A). While open bite is more prevalent with AI, the etiologic basis for this is not understood (Ravassipour et al., 2005). This is the second report of gene dosage affecting enamel phenotype in AI. Both cases involve ENAM exon 10 insertion mutations. The AI phenotype in Family 2 occurs when this 21-bp insertion mutation is present in the compound heterozygous state, with a previously described ENAM 2-bp insertion mutation (g.13185_13186insAG) (TC Hart et al., 2003). In the previous report, homozygosity of this 2-bp insertion mutation was associated with generalized hypoplastic AI and anterior open bite in three different consanguineous families. AI-affected individuals from all three families were homozygous for the same mutation. Additionally, 12 individuals from these families were determined to be heterozygous carriers (g.13185-13186insAG/wild-type) of this mutation, and all had localized, circumscribed enamel pits. This pitting phenotype was not found in individuals without the ENAM mutation. This very mild phenotype, which would not be traditionally diagnosed as AI, presents a challenge to the current AI nomenclature. The finding of a second ENAM mutation that segregates the same enamel phenotype, localized enamel pitting as a dominant trait, and generalized thin hypoplastic enamel as a recessive trait, confirms gene dosage effects of ENAM mutations on the enamel phenotype.
The current findings add to the genotype-phenotype correlations reported for ENAM mutations. The phenotype associated with the g.12663C>A mutation primarily involved enamel hypoplasia of the occlusal surfaces and cuspal prominences of the premolar teeth, and the palatal/lingual surfaces of the incisors and canines. There were no regions of horizontal pitting or horizontal grooving of the buccal/facial surfaces. This phenotype appears to be intermediate in severity compared with the local hypoplastic (Mardh et al., 2002) and the more severe smooth hypoplastic enamel phenotypes (Rajpar et al., 2001; Kida et al., 2002; PS Hart et al., 2003; Kim et al., 2005b) previously reported for autosomal-dominant AI mutations. The small differences in phenotype between the g.12663C>A and the g.2382A>T mutations reflect the complexity of enamel formation.
Reported genotype-phenotype correlations for ENAM mutations may provide insight into the molecular basis of enamel hypoplasia. Porcine enamelin is initially secreted as a 186-kDa precursor that rapidly undergoes proteolytic processing to yield several functional cleavage products (Fukae et al., 1996). Sequential cleavage from the C-terminus, presumably by matrix metalloproteinase 20, produces, first, a 155-kDa and then a 142-kDa species (Hu and Yamakoshi, 2003). The 142-kDa species is further cleaved into 89- and 34-kDa products. The 89-kDa product undergoes processing to produce 32- and 25-kDa products. The 32-kDa cleavage product, the predominant form of enamelin found in developing enamel, can be further degraded by kallikrein 4. These various cleavage products appear to have different roles in enamel development, since they are compartmentalized differently in developing enamel (Hu and Yamakoshi, 2003). Based upon known cleavage products of porcine enamelin, we predicted human enamelin cleavage products by aligning amino acid sequences using the LALIGN program (www.ch.EMBnet.org) (Fig. 3).
Figure 3.
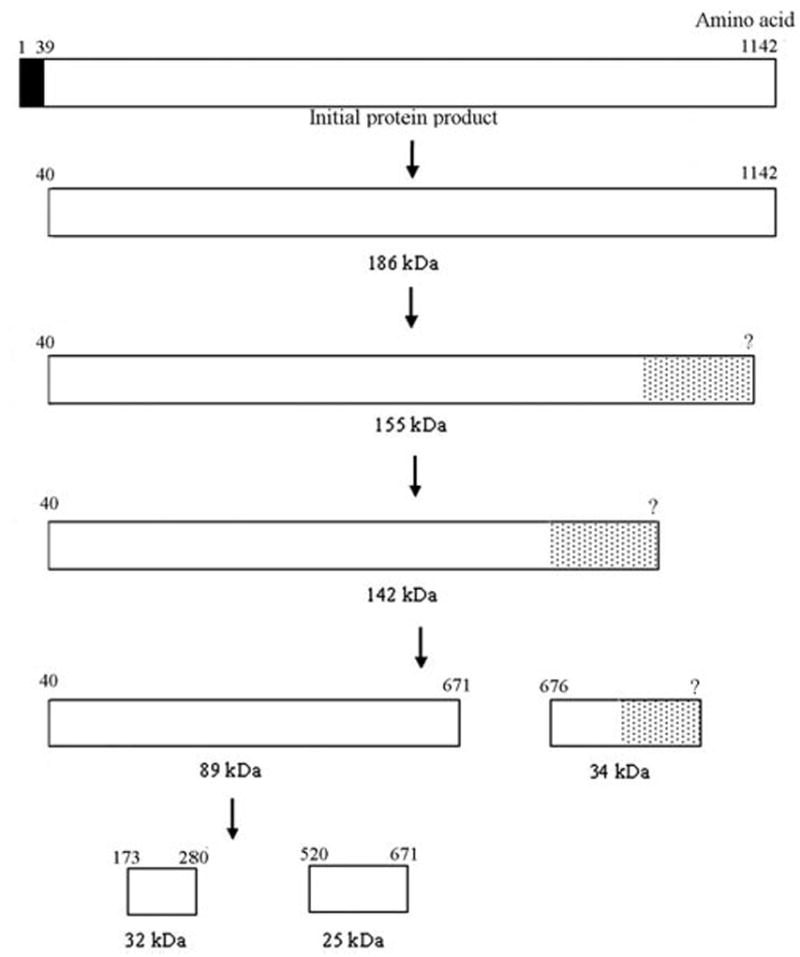
Predicted cleavage of human enamelin, based upon known porcine enamelin cleavage products. The amino acid numbers are shown above the rectangle, which represents the enamelin protein. The black rectangle represents the signal peptide (amino acids 1-39). The dotted rectangles represent cleavage products that have not been characterized at the C-terminus and are therefore shown with a question mark in the amino acid position. Enamelin is initially secreted as a 186-kDa species that undergoes rapid proteolysis to form a variety of cleavage products.
Clinical findings in the two families reported here are consistent with reported genotype-phenotype correlations and emerging understanding of clinical consequences of ENAM mutations. Haploinsufficiency of the enamelin protein results in the milder local hypoplastic autosomal-dominant AI. Mutations which may produce abnormal enamelin protein result in the more severe smooth hypoplastic AI, also inherited as a dominant trait, presumably through a dominant-negative effect. In this report, the dominant mutation in Family 1 (g.12663C>A; p.S246X) should result in haploinsufficiency for the 25-, 32-, and 34-kDa enamelin cleavage products. The 246-amino-acid truncated protein may interfere with enamel formation through a dominant-negative effect, producing a more severe phenotype than the g.2382A>T mutation.
In hypoplastic autosomal-recessive AI, the previously reported affected individuals were predicted to have normal 32-kDa cleavage products, but to lack both the 25- and 34-kDa species (TC Hart et al., 2003). Carriers exhibited localized enamel pitting, presumably due to haploinsufficiency of the 25-and 34-kDa cleavage products. The 21-bp insertion mutation described in this report also results in localized enamel pitting in the heterozygous carrier state. This mutation occurs after the 32-kDa cleavage site. Although the 25- and 34-kDa cleavage sites are still present, the addition of 7 amino acids may change the protein conformation, resulting in either interference with protein cleavage or functional alteration of cleavage products.
All ENAM mutations identified to date result in enamel hypoplasia, but the severity of enamel hypoplasia varies considerably with different mutations. In addition to clinical phenotype, mode of inheritance, and underlying genetic mutation, a robust and clinically useful classification of AI will require a more complete understanding of the molecular pathogenesis that underlies the defective enamel development. The observation of localized enamel pitting in individuals from families segregating autosomal-recessive generalized thin hypoplastic AI should raise suspicion that an ENAM mutation may be etiologic.
Acknowledgments
This work was supported by the National Institutes of Health, NIDCR grant DE12879, and by the NIDCR Intramural Program. The authors thank Michael Gorry for technical assistance.
References
- Aldred MJ, Savarirayan R, Crawford PJM. Amelogenesis imperfecta: a classification and catalogue for the 21st century. Oral Dis. 2003;9:19–23. doi: 10.1034/j.1601-0825.2003.00843.x. [DOI] [PubMed] [Google Scholar]
- Backman B. Inherited enamel defects. Ciba Found Symp. 1997;205:175–182. doi: 10.1002/9780470515303.ch12. [DOI] [PubMed] [Google Scholar]
- Bosch C, Athanasiou AE. Landmarks, variables and norms of various numerical cephalometric analyses—cephalometric morphological and growth data references. In: Athanasiou AE, editor. Orthodontic cephalometry. London: Mosby-Wolfe; 1995. pp. 241–286. [Google Scholar]
- Cartwright AR, Kula K, Wright JT. Craniofacial features associated with amelogenesis imperfecta. J Craniofac Genet Dev Biol. 1999;19:148–156. [PubMed] [Google Scholar]
- Fukae M, Tanabe T, Murakami C, Dohi N, Uchida T, Shimizu M. Primary structure of porcine 89-kDa enamelin. Adv Dent Res. 1996;10:111–118. doi: 10.1177/08959374960100020201. [DOI] [PubMed] [Google Scholar]
- Hart PS, Hart TC, Simmer JP, Wright JT. A nomenclature for X-linked amelogenesis imperfecta. Arch Oral Biol. 2002a;47:255–260. doi: 10.1016/s0003-9969(02)00005-5. [DOI] [PubMed] [Google Scholar]
- Hart PS, Aldred MJ, Crawford PJM, Wright NJ, Hart TC, Wright JT. Amelogenesis imperfecta phenotype-genotype correlations with two amelogenin gene mutations. Arch Oral Biol. 2002b;47:261–265. doi: 10.1016/s0003-9969(02)00003-1. [DOI] [PubMed] [Google Scholar]
- Hart PS, Michalec MD, Seow WK, Hart TC, Wright JT. Identification of the enamelin (g.8344delG) mutation in a new kindred and presentation of a standardized ENAM nomenclature. Arch Oral Biol. 2003;48:589–596. doi: 10.1016/s0003-9969(03)00114-6. [DOI] [PubMed] [Google Scholar]
- Hart PS, Hart TC, Michalec MD, Ryu OH, Simmons D, Hong S, et al. Mutation in kallikrein 4 causes autosomal recessive hypomaturation amelogenesis imperfecta. J Med Genet. 2004;41:545–549. doi: 10.1136/jmg.2003.017657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hart TC, Hart PS, Gorry MC, Michalec MD, Ryu OH, Uygur C, et al. Novel ENAM mutation responsible for autosomal recessive amelogenesis imperfecta and localised enamel defects. J Med Genet. 2003;40:900–906. doi: 10.1136/jmg.40.12.900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu JC, Yamakoshi Y. Enamelin and autosomal-dominant amelogenesis imperfecta. Crit Rev Oral Biol Med. 2003;14:387–398. doi: 10.1177/154411130301400602. [DOI] [PubMed] [Google Scholar]
- Kida M, Ariga T, Shirakawa T, Oguchi H, Sakiyama Y. Autosomal-dominant hypoplastic form of amelogenesis imperfecta caused by an enamelin gene mutation at the exon-intron boundary. J Dent Res. 2002;81:738–742. doi: 10.1177/0810738. [DOI] [PubMed] [Google Scholar]
- Kim JW, Simmer JP, Hu YY, Lin BP, Boyd C, Wright JT, et al. Amelogenin p.M1T and p.W4S mutations underlying hypoplastic X-linked amelogenesis imperfecta. J Dent Res. 2004;83:378–383. doi: 10.1177/154405910408300505. [DOI] [PubMed] [Google Scholar]
- Kim JW, Simmer JP, Hart TC, Hart PS, Ramaswami MD, Bartlett JD, et al. MMP-20 mutation in autosomal recessive pigmented hypomaturation amelogenesis imperfecta. J Med Genet. 2005a;42:271–275. doi: 10.1136/jmg.2004.024505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JW, Seymen F, Lin BPJ, Kiziltan B, Gencay K, Simmer JP, et al. ENAM mutations in autosomal-dominant amelogenesis imperfecta. J Dent Res. 2005b;84:278–282. doi: 10.1177/154405910508400314. [DOI] [PubMed] [Google Scholar]
- Mardh CK, Backman B, Holmgren G, Hu JC-C, Simmer JP, Forsman-Semb K. A nonsense mutation in the enamelin gene causes local hypoplastic autosomal dominant amelogenesis imperfecta (AIH2) Hum Mol Genet. 2002;11:1069–1074. doi: 10.1093/hmg/11.9.1069. [DOI] [PubMed] [Google Scholar]
- Nusier M, Yassin O, Hart TC, Samimi A, Wright JT. Phenotypic diversity and revision of the nomenclature for autosomal recessive amelogenesis imperfecta. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2004;97:220–230. doi: 10.1016/j.tripleo.2003.08.007. [DOI] [PubMed] [Google Scholar]
- Persson M, Sundell S. Facial morphology and open bite deformity in amelogenesis imperfecta. A Roentgenocephalometric study. Acta Odontol Scand. 1982;40:135–144. doi: 10.3109/00016358209012722. [DOI] [PubMed] [Google Scholar]
- Rajpar MH, Harley K, Laing C, Davies RM, Dixon MJ. Mutation of the gene encoding the enamel-specific protein, enamelin, causes autosomal-dominant amelogenesis imperfecta. Hum Mol Genet. 2001;10:1673–1677. doi: 10.1093/hmg/10.16.1673. [DOI] [PubMed] [Google Scholar]
- Ravassipour DB, Powell CM, Phillips CL, Hart PS, Hart TC, Boyd C, et al. Variation in dental and skeletal open bite malocclusion in humans with amelogenesis imperfecta. Arch Oral Biol. 2005;50:611–623. doi: 10.1016/j.archoralbio.2004.12.003. [DOI] [PubMed] [Google Scholar]
- Sundell S, Valentin J. Hereditary aspects and classification of hereditary amelogenesis imperfecta. Community Dent Oral Epidemiol. 1986;14:211–216. doi: 10.1111/j.1600-0528.1986.tb01537.x. [DOI] [PubMed] [Google Scholar]
- Witkop CJJ. Amelogenesis imperfecta, dentinogenesis imperfecta and dentin dysplasia revisited: problems in classification. J Oral Pathol. 1989;17:547–553. doi: 10.1111/j.1600-0714.1988.tb01332.x. [DOI] [PubMed] [Google Scholar]
- World Medical Association. Declaration of Helsinki. Br Med J. 1996;313:1448–1449. [Google Scholar]
- Wright JT, Hart PS, Aldred MJ, Seow K, Crawford PJ, Hong SP, et al. Relationship of phenotype and genotype in X-linked amelogenesis imperfecta. Connect Tissue Res. 2003;44(Suppl 1):72–78. [PubMed] [Google Scholar]