

Abstract
A series of resorbable poly(ethylene glycol)-co-poly(glycolic acid) macromonomers have been synthesized with the chemistries from three different photopolymerizable end-groups (acrylates, methacrylates, and urethane methacrylates). The aim of the study is to examine the effects of the chemistry of the cross-linker group on the properties of photocross-linkable hydrogels. PEG-co-PGA (4KG5) hydrogels were prepared by photopolymerization with high vinyl group conversion as confirmed by 1H NMR spectroscopy using DOSY 1D pulse sequence. Our study reveals that the nature of end-groups in a moderately amphiphilic polymer can adjust the distribution and size of the micellar configuration in water leading to changes in the macroscopic structure of hydrogels. By varying the chemistry of the cross-linker group (diacrylates; DA, dimethacrylates; DM, and urethane dimethacrylates; UDM), we determined that the hydrophobocity of a single core polymer consisting of poly(glycolic acid) could be fine-tuned leading to significant variations in the mechanical, swelling, and degradation properties of the gels.
In addition, the effects of cross-linker chemistry on cytotoxicity and proliferation were examined. Cytotoxicity assays showed that all the three types of hydrogels (4KG5 DA, DM, and UDM) were biocompatible and the introduction of RGD ligand enhanced cell adhesion. However, differences in gel properties and stability differentially affected the spreading and proliferation of myoblast C2C12 cells.
Keywords: PEG-co-PGA, end-group chemistry, micelle, biodegradable, hydrogel properties, myoblast cells
1. Introduction
Hydrogels produced by photopolymerization from a variety of polymers have been used extensively for a number of biomedical applications such as scaffolds for tissue engineering and controlled release systems for drug delivery [1-4]. The rapid advances in these fields require not only the development of biocompatible and biodegradable scaffolds but also a versatile system where we can controllably fine-tune their properties with respect to the specific application. To provide a delivery system adaptable to many applications, several groups have reported that hydrogel properties can be adjusted by a number of parameters, such as nature of macromonomers, molecular weight, composition, cross-link density, or external stimuli [5-8]. However, little is known about the effect of polymerizable end-groups on gel structure and properties, such as mechanics, swelling, degradation profiles and cell responses. These end groups, which in this case are cross-linkers, have different chemistries and can affect the microscopic as well as the macroscopic structure of hydrogels. Consequently, the end groups can be used to adjust the amphiphilicity of polymeric networks.
Although many variations of synthetic polymers can form hydrogels via chemical cross-linking, poly(ethylene) glycol (PEG) has been one of the most investigated systems. PEG has been functionalized with diacrylate or dimethacrylate groups and cross-linked to form nondegradable hydrogels via UV photoinitiation [9-11]. Nondegradable hydrogels have been used for diffusion-controlled delivery devices [12,13]. It has been shown that for high molecular weight drugs, such as peptides and proteins, a degradable hydrogel is more suitable for controlled delivery. To obtain degradable hydrogels, PEG has been copolymerized with degradable polymers such as poly(lactic acid), poly(glycolic acid), (PGA), or enzymatically resorbable polysaccharides [14-16]. Erosion of water-soluble polymers occurs by a combination of mechanisms. Linkages in the polymer may be labile to hydrolysis and enzymes, thereby producing oligomeric scission products soluble in water. For hydrophobic polymers, degradation is limited to the surface of the materials. For hydrophilic polymers, on the other hand, water may become entrapped in the polymer and degradation ensues by bulk erosion. Poly(glycolic acid) (PGA) is one of the most thoroughly investigated poly(α-hydroxy esters). This class of polymers is bioerodible as a consequence of hydrolysable bonds along the polymer backbone.
Hydrogels based on PEG-co-poly(α-hydroxy acid) diacrylates and their derivatives contain large amounts of water and have a hydrophilic, nonionic surface. This lowers the driving force for protein absorption on these surfaces from the physiological environment. Non-adhesive hydrogels were modified with a bioactive moiety such as GRGDS peptide sequences to facilitate cell adhesion, spreading, and organization. The RGD sequence is a ubiquitous adhesive peptide, responsible for the integrin-ligand interaction between cells and the surface of RGD-modified hydrogels. Cell adhesion is often a necessary first step in basic cellular processes such as cell proliferation, cellular trafficking, and tissue development.
The primary goal of this research was to elucidate how end-group chemistry might be exploited to fine-tune the properties of degradable hydrogels. We hypothesized that by varying the chemistry of the cross-linking group we could tune the gel microstructure, leading to a significant peculiarity in the hydrogels. The synthesis, characterization, and evaluation of PEG-co-PGA based hydrogels are described. Based on the method of Hubbell and co-workers, the macromonomers of interest in this research were synthesized based on a water-soluble central block of PEG extended with biodegradable oligomers of PGA and terminated with different cross-linkable end-groups [17,11]. The degree of polymerization of the hydrolytically labile extension was relatively small to conserve water solubility of the PEG component. Therefore, the properties of the macromonomer in solution and the gel properties were determined primarily by the water-soluble central domain of PEG. Further, the amphiphilic nature of these macromonomers caused them to assume micellar conformations, enabling gelation. Acrylates, methacrylates, and urethane methacrylates were chosen as terminal end-groups because they enabled photopolymerization, resulting in the formation of a cross-linked network. In addition, varying terminal vinyl group chemistry can influence the physical properties of hydrogels. These water-soluble macromonomers were photopolymerized via a light-sensitive initiation mechanism using long-wave ultraviolet of aqueous solutions (10% and 20% by mass fraction). The gels degraded by hydrolysis of the oligo(α-hydroxy acid) regions into poly(ethylene glycol), α-hydroxy acid, and acidic oligomers [17].
We report that the nature of photopolymerizable end-groups affect the amphiphilicity and micellar configuration of PEG-co-PGA precursors. Such a subtle effect has a significant impact on the physical properties of hydrogels and cell responses. The utility of using various end group chemistry to cross-link synthetic and biocompatible polymers to form versatile hydrogels has the potential to produce multifarious scaffolds appropriate for use in a number of biomedical applications.
2. Materials and methods
2.1. Materials
PEG (MM ≈ 4000 g/mol), acryloyl chloride (AC), methacrylic anhydride (MA), 2-isocyanatoethyl methacrylate (IEM), triethylamine (TEA), stannous octoate, dibutyltin dilaurate (DBTD), and Hoechst dye were purchased from Sigma-Aldrich and used as received. Dichloromethane and ethyl ether were obtained from Fischer, and dichloromethane was dried over activated molecular sieves prior to use. Glycolide was purchased from E. I. du Pont de Nemours. Acryloyl-PEG-N-hydroxysuccinimide (ACRL-PEG-NHS, 3400 g/mol) was purchased from Nektar Therapeutics. GRGDS peptide was purchased from Bachem Bioscience Inc. Photoinitiator Irgacure 2959 (I2959) was obtained from Ciba Specialty Chemicals and used as received. All other chemicals used were of reagent grade and were used without further purification. For in vitro cell culture, murine myofibroblast cells (C2C12) were obtained from American Type Culture Collection (Manassas, VA) and cultured in DMEM purchased from Invitrogen (Carlsbad, CA), supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin, all obtained from Invitrogen. Phosphate buffered saline (PBS) was purchased from Invitrogen. FITC labeled Phalloidin were purchased from Sigma Aldrich (St Louis, MO). Live/Dead® Viability/Cytotoxicity Kit was purchased from Molecular Probes, Inc. (Eugene, OR).
2.2. Methods
2.2.1. Synthesis of degradable macromonomers
4KG5 copolymer was prepared from the reaction of PEG (4 000 g/mol) and glycolide at a ratio of 5 mol of glycolide/mol of PEG. A total of 30 g of dry PEG, 4.35 g of glycolide, and 15 mg of stannous octoate were charged into a 100-mL round-bottomed flask under a nitrogen atmosphere. The reaction mixture was stirred under vacuum at 200 °C for 4 h and at 160 °C for 2 h, and subsequently cooled to room temperature. 4KG5 was dissolved in dichloromethane, precipitated in anhydrous ether, filtered, and dried in a vacuum oven overnight at room temperature.
4KG5 DM and 4KG5 UDM were prepared from the reaction of 4KG5 with MA and IEM, respectively. An example of the synthesis of a 4KG5 DM is as follows. 4KG5 (10 g, ≈ 2.2 mmol), 2.2 equiv of MA (0.74 g, ≈ 4.8 mmol), and TEA (0.4 mL) were reacted in ≈ 30 mL of dichloromethane over freshly activated molecular sieves (≈ 6 g) for 4 d at room temperature. The solution was precipitated twice into ethyl ether. The product was filtered and then dried in a vacuum oven overnight at room temperature. The same route was used to prepare 4KG5 UDM except for the catalyst (6 drops of DBTD were added).
4KG5 DA was prepared from the reaction of 4KG5 with AC. 4KG5 (10 g, ≈ 2.2 mmol) was dissolved in 100 mL of dichloromethane in a 200 mL round bottomed flask and was cooled to 0 °C in a ice bath. A total of (0.44 mL ≈ 4.4 mmol) of TEA and (0.53 mL ≈ 8.8 mmol) of AC were added to the flask, and the reaction mixture was stirred for 12 h at 0 °C and for 12 h at room temperature. The reaction mixture was filtered to remove triethylamine hydrochloride. The solution was precipitated twice into ethyl ether. The product was filtered and then dried in a vacuum oven overnight at room temperature.
2.2.2 Synthesis of ACRL-PEG-Peptide
GRGDS peptide was dissolved in anhydrous DMF containing 4 molar excess of TEA. ACRL-PEG-NHS was also dissolved in anhydrous DMF and, immediately after, mixed with 1.1 molar excess of peptide (Figure 2). After incubating for 3 h at room temperature, ACRL-PEG-GRGDS was precipitated twice in cold anhydrous ether and dried in a vacuum oven overnight at room temperature. The peptide coupling reaction and molecular mass of the product was monitored by MALDI-TOF MS.
Figure 2.

Covalent attachment of the GRGDS bioactive peptide to photopolymerizable ACRL-PEG-NHS macromer.
2.2.3 Macromonomer characterization
1H NMR and MALDI-TOF MS were used to follow and verify each step of the synthesis. High-resolution, 300 MHz proton NMR spectra were taken on a Bruker Avance™ 300 spectrometer. Deuterated chloroform was used as solvent, and the polymer concentrations were varied between 2.5 % and 3 % by mass fraction.
MALDI-TOF MS was performed on a PerSeptive Biosystems Voyager STR in reflectron mode. The MALDI matrix, consisting of dihydrobenzoic acid (DHB) and the macromonomers were dissolved in 1 mL of Tetrahydrofuran (THF). Sodium was used as the cationizing reagent in a 1:1 by volume ratio of THF solution (0.5 mg/mL solution in THF) and macromonomer solutions. ACRL-PEG-Peptide was dissolved in 1 mL of mix solvent methanol/water (50:50) without cationizing agent. All MALDI samples were hand spotted on the target.
2.2.4. Characterization of micelles
The micelles size was measured using dynamic light scattering (DLS) by CONTIN analysis. A Malvern Particle Sizer HPPS 5001 was used with a He-Ne laser (wavelength 633 nm) working at 3.0 mW. The three macromonomers (4KG5 DA, DM, and UDM) were dissolved in de-ionized water at a concentration of 5 mg/mL, filtered, and subsequently introduced into a thermostated scattering cell at 25°C for measurement. The sizes were calculated by volume measurement and expressed as Dav ± S (average diameter ± standard deviation).
2.2.5. Hydrogel preparation
10% and 20% by mass fraction macromonomers were mixed in distilled deionized water. A photoinitiator ((4-(2-hydroxyethoxy)phenyl-(2-propyl)ketone, I-2959) was added to each macromonomer solution to yield a final concentration of 0.05 wt.% I-2959. Cylindrical samples were cured with a long wavelength UV source (365 nm, 300 μW/cm2) for 10 min to obtain chemically cross-linked hydrogels.
2.2.6. Characterization of vinyl group conversion
NMR spectroscopy was used to characterize vinyl conversion of 4KG5 macromonomers after photopolymerization. 1H and 1H 1D DOSY were performed at 300K on a Bruker Avance DMX-500 NMR spectrometer operating at 500.13 MHz and equipped with a Bruker z-gradient probehead. 1H 1D DOSY experiment was performed using a bipolar pulse pair stimulated echo pulse sequence with 2 spoil bipolar gradient pulses of 2% strength with duration of 6 ms and a diffusion time of 0.2 s. The pulse sequence included a longitudinal eddy current delay of 5 ms. Macromonomer solutions were prepared by mixing 10% and 20% macromonomers (4KG5 DA, 4KG5 DM, and 4KG5U DM) solutions (wt/wt) with D2O in the presence of photoinitiator (I2959, 0.05% by mass fraction). 1 mL of each macromonomer solution was transferred and sealed in an NMR tube before being cured under a UV source (365 nm, 300 μW/cm2) to form cross-linked hydrogels. 1H 1D DOSY NMR spectroscopy was used to characterize the efficiency of vinyl group reactivity during photocross-linking. The conversion was evaluated by comparing the relative peaks of uncross-linked and cross-linked methylene protons.
2.2.7. Swelling experiments
Hydrogels disks in triplicate were prepared by photopolymerization in phosphate-buffered saline solution (PBS, pH ∼ 7.4). The equilibrium mass swelling ratio (QM) was calculated by the following equation [18]:
where Ms and Md were fully swollen gel and dried gel weights, respectively.
2.2.8. Mechanical measurements and degradation
Hydrogel disks were prepared by photopolymerization under UV irradiation for 10 min in a Teflon mold of size 0.87 mm thickness and 30 mm diameter. Rheological measurements were performed on a stress-controlled Rheometrics SR5 rheometer in a parallel plate configuration (standard plates, 25 mm diameter). For the degradation studies, the hydrogels were swollen in PBS (pH ≈ 7.4) at room temperature. Hydrogel disks were placed onto the rheometer and covered with a pool of buffered solution during the entire time of the experiment. The elastic modulus (G′) was monitored as a function of time at a fixed frequency (ω = 1 rad/s). The mechanical properties of freshly prepared gels were determined by varying the stress amplitude from 10 – 5000 Pa and determining the linear behavior. The modulus was also used to track degradation but probing the modulus at a fixed frequency and amplitude as a function of time (every hour); some part of the degradation was found to following exponential kinetics and a time constant extracted from the linear portion.
2.2.9. Biocompatibility and cell proliferation
Cell culture studies were carried out using standard aseptic tissue culture techniques. To evaluate the biocompability of the hydrogels, 5×104 C2C12 cells were seeded in vitro onto the surfaces of PEG-co-PGA gels under static conditions. Viability of C2C12 cells was measured by live/dead staining at 24h and 48h post-seeding. For live/dead staining, the cell culture media was aspirated, the wells rinsed with PBS, and live/dead stain added. Cells were incubated for 30 min at 37° C in the dark.
To evaluate the proliferation of cells, C2C12 were cultured on GRGDS-modified 4KG5 DA, DM, and UDM hydrogels after 1, 4, and 7 days of incubation. The cells were fixed with 4% paraformaldehyde and Phalloidin TRITC staining was performed to visualize actin organization and cell anchorage to the polymer surface, and the nucleus was stained with Hoescht blue. Images were captured using a monochrome CCD camera attached to an Axiovert 200 microscope (Zeiss). From the images, the number of pseudocoloured cells and gross percent viability were calculated. Statistical analyses for the proliferation of cells were done using the student's t-test. Pairs were compared for cell counts at different time points (day 1, 4, and 7) and for the three gel types (DA, DM, and UDM).
3. Results
3.1. Preparation of 4KG5DA, DM, and UDM macromonomers and their hydrogels
As shown in Figure 1, oligo(glycolic acid) was polymerized from both ends of the bifunctional PEG block to provide hydrolysable linkages. Finally, each end of the 4KG5 monomer was functionalized with acryloyl chloride, methacrylic anhydride, and 2-isocyanatoethyl methacrylate. These multi-functional vinyl groups provide rapid gelation by containing four sites for cross-linking on each macromonomer (4KG5 DA, DM, and UDM). The gels degrade upon hydrolysis of the oligo(α-hydroxy acid) regions into water-soluble components: PEG, α-hydroxy acid, and acidic oligomers. Previous studies have shown that mechanical properties, cross-linking densities, and the degradation rates of these gels can be tailored by the molecular mass of PEG, appropriate choice and number of degradable linkages present on the cross-linking molecule [19]. After functionalization with vinyl moieties, the macromonomers were cross-linked via photopolymerization process using a UV light initiating system under mild conditions. To investigate end-group effect on macroscopic hydrogel properties, a series of macromonomers containing three different polymerizable group chemistries was used.
Figure 1.

Reaction scheme for the synthesis of photolymerizable 4KG5DA, 4KG5DM, and 4KG5UDM. Poly (ethylene glycol)-co-poly-(glycolic acid) (4KG5) was synthesized by ring-opening polymerization of glycolide/poly(ethylene glycol) using stannous octoate as a catalyst. The molar ratio of glycolide to PEG is 5:1, which means 2.5 of glycolide per chain end. However, since 2 glycolic acid moieties are generated from one glycolide, 5 glycolic acid units are extended per chain end. 4KG5 copolymer, which has α- and ω-terminated hydroxyl groups, was subsequently end-capped with acrylate, methacrylate, and 2-isocyanatoethyl methacrylate.
3.2. Characterization of macromonomers
A series of controlled molecular mass PEG-co-poly(α-hydroxy acid) diacrylate 4KG5 DA, PEG-co-poly(α-hydroxy acid) dimethacrylate 4KG5 DM, and PEG-co-poly(α-hydroxy acid) urethane dimethacrylate 4KG5 UDM of high purity and low dispersity was prepared. The products were characterized by proton nuclear magnetic resonance (1H NMR) and matrix-assisted laser desorption ionization time-of-flight mass spectrometry (MALDI-TOF MS). Proton NMR and MALDI-TOF MS together provide comprehensive information regarding the degree of acrylate, methacrylate, and isocyanatoethyl methacrylate conversions, molecular mass, and product purity. Figure 3 shows typical 1H NMR spectra for 4KG5 DA, DM, and UDM. PEG shows two main chains chemical shift at δPEG ≈ 3.64 and 4.22. PGA extensions show three chemical shifts at δPGA ≈ 4.61, 4.70, and 4.80. The chemical shifts of methylene protons for DA, DM, and UDM are δDA ≈ 6.19, 6.34, and 6.64; δDM ≈ 5.55 and 6.10; and δ UDM ≈ 5.80 and 6.21, respectively. Upon reaction with 4KG5, these protons shift at δDA ≈ 5.96, 6.23, and 6.49; δDM≈ 5.64 and 6.20; and δUDM ≈ 5.57 and 6.13, respectively, and the chemical shift for the proton from the urethane linker appears at δUDM ≈ 5.25. The 1H NMR spectra for the three types of macromonomers show expected peaks, and the lack of additional peaks suggests that unreacted compounds, catalysts and solvents have all been quantitatively removed.
Figure 3.

1H NMR for 4KG5 DA, DM, and UDM with their characteristic vinylic peaks (∼ 5.5-6.5 ppm).
MALDI-TOF MS is a powerful technique from which the molecular mass, molecular mass distribution, and end group functionalities can be determined. The MALDI-TOF MS spectra of PEG, 4KG5, and 4KG5 UDM are shown in Figure 4. Intrinsic to MALDI analysis, the relative signal intensities decrease and the breadth of the peak appears to increase as the molecular mass increases. The number average molecular masses (Mn) of the materials obtained from proton NMR and MALDI-TOF MS spectra for each step of the synthesis increase, confirming that the reactions are achieved with high conversion and low impurities. The conversions and molecular mass results for 4KG5 DA, DM, and UDM are listed in Table 1. Figure 4 shows a shift of the molecular masses from the starting product PEG (Mn= 3923 ± 39 g.mol-1), to the intermediate macromer 4KG5 (Mn= 4393 ± 44 g.mol-1), and to the polymerizable macromer 4KG5 UDM (Mn= 4621 ± 46 g.mol-1). Combined analyses of 1H NMR and MALDI-TOF MS confirmed the formation of prepolymers of high purity and narrow polydispersity (PD ≤ 1.03). Also, ACRL-PEG-Peptide has been characterized by MALDI-TOF MS. Figure 5 shows a molecular mass shift of 649 ± 13 g.mol-1, which corresponds to the peptide molecular mass gained after reaction.
Figure 4.

MALDI-TOF of a series of 4k PEG, 4KG5 copolymer, and functionalized 4KG5UDM.
Table 1.
Molecular Mass (MM) results for 4KG5 and its derivative macromonomers. Percentage conversion corresponds to conversion of glycolide monomer to PGA at the end of PEG chains (4KG5) and end-group modification of 4KG5 monomers with methacrylate end groups (4KG5 DA, DM, and UDM).
| MM (g/mol) | Conversion (%) | Mn (NMR) | Mn (MALDI) | Mw (MALDI) | PDI |
|---|---|---|---|---|---|
| 4KG5 | 72 | 3944 | 4393 | 4437 | 1.01 |
| 4KG5 DA | 81 | 4284 | 4567 | 4711 | 1.02 |
| 4KG5 DM | 92 | 4302 | 4583 | 4720 | 1.03 |
| 4KG5 UDM | 97 | 4400 | 4621 | 4760 | 1.03 |
Figure 5.

MALDI-TOF MS of ACRL-PEG-NHS and ACRL-PEG-Peptide. MS spectrum of ACRL-PEG-NHS is shifted to the right (higher molecular mass) after covalent attachment of the peptide.
The micellar sizes for the three macromonomers in water were measured using DLS. The 4KG5 DA, UDM, and DM micelles were found to have diameters of 33.5 ± 1.4 nm, 40.3 ± 1.6 nm, and 56.3 ± 2.3 nm, respectively (Figure 6). This indicates that at constant molecular weight, composition, and concentration, the nature of the end-group does significantly alter the micellar structure of the block copolymers.
Figure 6.

Characterization of the micellar sizes by DLS for the three macromonomers in deionized water at a concentration of 5 mg/mL. The 4KG5 DA, UDM, and DM micelles were found to have diameters of 33.5 ± 1.4 nm, 40.3 ± 1.6 nm, and 56.3 ± 2.3 nm, respectively. The sizes were calculated by volume measurement and expressed as Dav ± S (average diameter ± standard deviation).
3.3. Characterization of vinyl group conversion
In order to characterize vinyl group conversion after photopolymerization, 1H 1D DOSY NMR spectroscopy was used to improve the resolution of peaks. The gradient spin-echo pulse sequence effectively suppressed the slow-diffusing components of the broad macromonomer resonance allowing for the vinyl group signals to be clearly visible. As shown in Figure 7, full disappearance of methylene protons (between 5.5 - 6.5 ppm) for 4KG5 DA, DM, and UDM macromonomers was reached after 10 min irradiation under UV in the presence of photoinitiator I2959 at both macromonomer mass fractions (10% and 20%). Characterization of methylene protons also indicated high reactivity during the photopolymerization process. This suggests that high vinyl conversions can be achieved for all three types of hydrogels regardless of the macromonomer concentration or end group chemistry.
Figure 7.

1H NMR of uncross-linked (top) and 1H DOSY 1D of photocross-linked (bottom) macromonomers (4KG5DA, 4KG5DM, and 4KG5UDM) in D2O at two mass fractions (10% and 20%). Photopolymerization is induced directly in an NMR tube. 1mL of macromonomer solution containing the photoinitiator (I2959) was transferred into the NMR tube before curing with a long wavelength UV source (365 nm, 300 μW/cm2) to obtain hydrogel. The vinylic peaks (between 5.5-6.5 ppm) disappeared after 10 min irradiation.
3.4. Mechanical and swelling measurements
To characterize the mechanical strength and degree of cross-linking of the three types of hydrogels, the elastic modulus (G′) and viscous modulus (G″) were measured. The shear modulus (G*) is shown in figure 8, measured at 1 rad/sec and found to be linear for applied stresses between 10 – 5000 Pa. The shear moduli of hydrogels prepared using various end group chemistry and at various mass fraction, exhibit a greater mechanical strength for 20% weight hydrogels compared to 10% weight hydrogels due to a higher degree of cross-linking. Furthermore, the shear modulus is also affected by the nature of the end-group chemistries. We noticed significant differences between 4KG5 DA, DM, and UDM hydrogels for both mass fractions.
Figure 8.

Shear modulus (G*) of hydrogels prepared using various 4KG5 end groups (DA, UDM, and DM) and at various mass fractions (10% and 20%).
The degradation behavior of swollen, covalently cross-linked hydrogels was characterized by monitoring changes in their elastic and storage moduli with time. As reported in Table 2, at the chosen conditions (pH 7.4 and room temperature), total degradation time for these gels into water-soluble products varied from 1 d up to 27 d, depending on polymer concentration and nature of the cross-linker used. While the gels with urethane methacrylate cross-linker groups degrade relatively quickly, the acrylate and methacrylate cross-linked gels seem more stable against hydrolysis and have significantly longer degradation times.
Table 2.
Degradation time of hydrogels prepared using various 4KG5 end-groups and at various mass fractions. The characterization of the hydrogels was performed in triplicate.
| Hydrogel | Weight % | Degradation Time Constant τ (hr) |
Degradation Time (days) |
|---|---|---|---|
| 4KG5 DA | 10% | 13.7 ± 0.3 | 4 ± 0.2 |
| 20% | 57.1 ± 1.2 | 8 ± 0.4 | |
| 4KG5 DM | 10% | 33.4 ± 0.7 | 14 ± 0.7 |
| 20% | 105.3 ± 2.1 | 27 ± 1.3 | |
| 4KG5 UDM | 10% | 7.5 ± 0.2 | 1 ± 0.1 |
| 20% | 12.6 ± 0.3 | 5 ± 0.2 |
Hydrogel swelling coupled with the analysis of the elastic shear modulus of the hydrogel is a good indicator of mechanical strength and, proportionally, the cross-link density of entangled hydrogel networks. From the data collected based upon weight of the hydrogel disks, the hydrogel swelling ratio based on mass (QM) was determined by dividing mass of the swelled disks (MS) by the mass of the dry disks (Md). Using an approximation, the volumetric swelling ratio (Qv) is determined from the equation [18]:
Where ρp is the density of the polymer (∼1.2 g/mL), ρs is the density of the solvent (water 1 g/mL), and QM is the mass swelling ratio of the hydrogel (Ms/Md). As shown in Figure 9, the hydrogels reached their equilibrium swelling size within 60 min when incubated in PBS at 37°C before reaching a plateau. The water content for the three types of hydrogels was a function of the macromonomer mass fraction. Overall, as expected, 10% hydrogels having a lower concentration of cross-links swelled to a greater extent when compared to 20% hydrogels. Surprisingly, end-group chemistry also seems to have a significant impact on the amount of water absorbed by the gels. A similar trend was observed for both macromonomer mass fractions; 4KG5 DA gels had the highest swelling ratio (Qv = 8.21 and 5.44) followed by 4KG5 DM (Qv = 6.95 and 4.77) and 4KG5 UDM (Qv = 5.74 and 4.03) hydrogels for 10% and 20% hydrogels, respectively.
Figure 9.

Volumetric swelling ratio (Qv) versus time of a series of 4KG5 DA, DM, and UDM hydrogels in PBS at two mass fractions (10% and 20%). The Qv value is reported for each type of gels and mass fraction.
3.5. Biocompatibility and cell proliferation
4KG5 DA, DM, and UDM, were used to perform a preliminary assessment of cellular responses to the hydrogels. C2C12 cells were examined after 4 d post-seeding using phase contrast and fluorescent microscopy. Live/dead cell staining (Figure 10) show that the hydrogels were biocompatible, non-toxic and more than 90% viable cells were scored. The polymerized hydrogels possess relatively nonadherent surface to C2C12 cells, presumably due to poor protein adsorption to the hydrophilic polymer surface [20]. The incorporation of the GRGDS peptide allowed modulation of hydrogel properties from cell non-adhesive to adhesive. GRGDS promoted cell interactions causing adhesion, spreading, and proliferation of cells.
Figure 10.
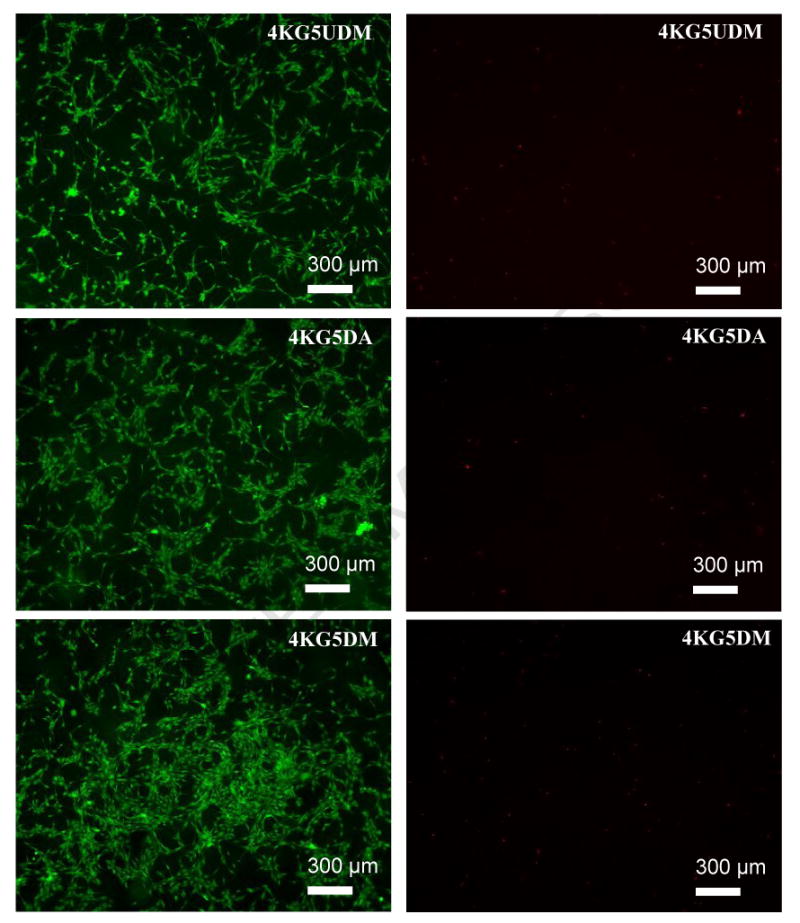
Live stain (left) and dead stain (right) of 4KG5 DA, DM, and UDM hydrogels seeded with C2C12 cells after 4 d of incubation.
Cell proliferation was assessed by counting the number of Hoechst blue stained nucleus (Figure 11). As reported in Figure 12, the cells proliferated on the gels with time. Cell proliferation for the three types of gel was similar at 24 h of culture but the cell numbers increased significantly by day 4 on 4KG5 DM hydrogels when compared to 4KG5 DA and UDM hydrogels, respectively, and by day 7 the number of cells cultured on 4KG5 DM hydrogels was nearly twice as high as the number of cells cultured on 4KG5 UDM hydrogels, 2015 cells/5mm2 vs. 1063 cells/5mm2, respectively.
Figure 11.
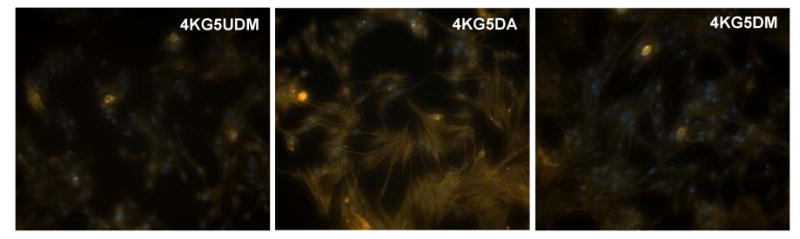
Nuclear stain of C2C12 cells cultured on 20% 4KG5 DA, DM, and UDM hydrogels after 4 d of incubation.
Figure 12.

Proliferation of C2C12 cells cultured on GRGDS-modified 4KG5 DA, DM, and UDM hydrogels after 1, 4, and 7 d of incubation. Cells were stained with Hoechst dye. Data represent the mean of three experiments ± standard deviation of cell number/normalized section (5mm2). A statistical significance of p ≤ 0.005 was obtained for all the comparisons (except for the pair comparison with the symbol *). Statistical significance was calculated using the student's t-test for multiple comparisons.
4. Discussion
The PEG-co-PGA macromonomers used to form hydrogels are multifunctional, with three types of cross-linkable groups having a double bond located at each end of the macromonomer chain. Acrylates, methacrylates, and urethane methacrylates were chosen as terminal end groups for three primary reasons: first, their photopolymerization results in the formation of cross-linked network; second, the unreacted components have relatively low cytotoxicity; and third, the physical properties of hydrogels can be fine-tuned by varying the terminal vinylic group chemistry.
A three-dimensional network is formed through chain polymerization of the functional end-groups. As mentioned previously, the amphiphilic nature of the macromonomers causes them to assume a micellar conformation, which in turn enables photopolymerization with nearly full vinyl conversion after 10 min exposure under UV.
Hydrogel properties are readily controlled through the copolymer composition, molecular weight, and percent of macromonomer in solution prior to polymerization. However, in this study, we are introducing another substantial parameter, the chemistry of cross-linkers, which also plays a key role in the physical properties of the gels. It is, therefore, clear that the nature of these cross-linker end-groups has the ability to adjust the three-dimensional network microstructure through variation of hydrophilicity/hydrophobicity of PGA segments. PEG-co-PGA macromonomers may form aggregates in aqueous solution; further, their size is a function of hydrophobicity of the PGA segments and presumably to the nature of cross-linker end group chemistry. The amphiphilic block copolymers based on PEG extended with PGA self-assembled into nanoscale micelle-like structures in aqueous solutions. These micelle-like structures consist of a corona/core structure similar to that of conventional amphiphilic surfactants, where the association of the hydrophobic block forms the core. The hydrophilic corona forms the shell in the aqueous environment and these shells are interconnected due to the bifunctionality of the block copolymers. This occurs by the hydrophilic PEG chains stretching out and forming a “link” between the micelles. As shown by DLS characterization, the disparity in their micellar sizes can be attributed to differences in the degree of hydrophobicity of cross-linkers and intermolecular hydrogen bonding (urethane groups from UDM functionalities), altering the conformation of the micellar core.
Aggregate formation is essentially a phase-separation process, and as hypothesized previously, the aggregate size was a function of cross-linker chemistry. Consequently, by changing aggregate size we expected there would be a commensurate change in the number, density, or size of the cross-linking sites. Therefore, mechanical properties, water absorption, and the degradation rates would be impacted, since larger aggregates would lead to a higher cross-link density. Given the range of properties observed simply by changing cross-linker chemistry, these effects can be quite significant.
By varying the end-group chemistry and cross-link density in the network, one has control over the physical properties of the PEG-co-PGA hydrogels. The underlying hypothesis of this work is that by varying the chemistry of the cross-linker group and the gel concentration, it is possible to change the gel microstructure, mechanical properties, swelling behavior, and degradation kinetics.
Figure 8 shows the shear moduli of PEG-co-PGA hydrogels prepared from different photoreactive end-groups (DA, DM, and UDM) and as a function of mass fraction (10% and 20%). As expected, the shear moduli monotonically increased as the oligomers mass fraction increased for the three gel types; this was caused by an increase in cross-link density. As mentioned previously, gel microstructure is expected to affect the mechanical properties. 4KG5 DM vs. 4KG5 DA and DM hydrogels with structures that contain larger cross-linked clusters, which reinforce the networks, exhibit higher shear moduli at both macromonomer concentrations.
Furthermore, network structure clearly affects hydrogel swelling ratio. Hydrogels with lower aggregate size, which imply a weaker cross-link density, e.g. 4KG5 DA hydrogels, exhibit higher swelling as more water can be absorbed. However, surprisingly 4KG5 DM, having the highest cross-link density with respect to the network microstructure, did not exhibit the lowest swelling ratio. This was a result of a shrinking process observed on the 4KG5 UDM hydrogels while swelling in PBS. This unexpected behavior can be attributed to possible hydrogen bonding between urethane groups and ethylene oxide from the backbone leading to a more compact structure. Moreover, as shown in Figure 9, 10% gels have a bigger volumetric swelling ratio than 20% gels, presumably because they possess a larger amount of free water, which in turn may promote oligomeric scission via a bulk erosion process along the polymer backbone.
PEG-co-PGA segments contain hydrolytically labile ester bonds that provide the network with its degradable characteristic. The gels degrade upon hydrolysis of the oligo(α-hydroxy acid) regions into PEG, α-hydroxy acid, and oligo(acrylic acid), oligo(methacrylic acid) or oligo(urethane methacrylic acid) depending on the nature of the end-group chemistry. These components are all water-soluble and are either naturally existent in the body or are rapidly eliminated by excretion through the kidneys. When exposed to water, these linkages within the PGA segments are cleaved at a rate given by hydrolysis kinetics. The degradation rates of the hydrogels were predetermined by the macromonomer mass fraction. As expected, the degradation for the 20% hydrogels was slower when compared to 10% hydrogels since a larger number of cross-links have to be broken before reaching a point where the remaining polymer chains are no longer combined to form a gel with an infinite weight-average molecular weight. As reported previously and confirmed with the current investigation, at a given gel concentration, the overall degradation timescale is influenced by the chemistry of the end-group precursors [21]. The order of hydrophilicity of these cross-linker groups was UDM > DA > DM, and more hydrophilic networks resulted in decreased degradation time. UDM is expected to be the more hydrophilic end-group due to possible hydrogen bonding between the urethane group and molecules of water. DM appears to be the more hydrophobic end-group due to methyl fragment of the methacrylate group resulting in a more stable cross-linked network during the process of hydrolysis.
Myoblast C2C12 cells seeded in 4KG5 DA, DM, and UDM hydrogels (Figure 10) were used as preliminary assessment for determining the biocompatibility of these materials. Given the high purity of the product and assuming networks with a relatively high degree of conversion can be achieved, we expect the cells to be viable in the hydrogels. Live cells are distinguished by their intracellular esterase activity, which is determined by the enzymatic conversion of nonfluorescent cell-permeable Calcein AM to an intensely green fluorescent calcein. Live cells enzymatically activated the fluorescent calcein. The dead stain Ethidium homodimer-1 is excluded by intact membranes of live cells. Therefore, Ethidium homodimer-1 only enters cells with damaged membranes and attaches to nucleic acids within dead cells to produce red fluorescence. The cell viability is thus measured through these physical biochemical properties. Figure 10 shows the live/dead cell stain for the three types of hydrogels. Live and dead cell stains show that cells are completely (or nearly completely) viable after 4 d of incubation.
While the addition of cell adhesion peptides to hydrogels has been shown to increase cell attachment and spreading, we have found that hydrogel chemistry also affects attachment and proliferation of cells grown in a peptide-grafted scaffold. These results reflect the fact that cells require cell adhesion ligands, but also indicate that hydrogel microstructure may affect the peptide concentration and distribution. Proliferation of C2C12 cells growing on peptide-modified 4KG5 DA, DM, and UDM hydrogels was examined by staining cells for proliferating cell nuclei. As shown in Figure 12, there was a slight difference in the number of proliferating cells in hydrogels containing DA and DM end-group functionalities at the three time-points of incubation (308 cells/5mm2 vs. 346 cells/5mm2 after 1 d, 880 cells/5mm2 vs. 1057 cells/5mm2 after 4 d, 2015 cells/5mm2 vs. 1660 cells/5mm2 after 7 d of incubation, respectively). Strikingly, for the same concentration of GRGDS (4 mM), the percentage of proliferating cells was significantly lower for 4KG5 UDM hydrogels. This can be attributed to rapid degradation of this type of hydrogel before being replaced by a new extracellular matrix, lowering surface area available for cells to migrate and proliferate. Furthermore, the hydrophilicity of such hydrogels may intervene in cell attachment efficiency, which is known to be essential for cell growth. These results indicate that changes in gel properties (mechanics, hydrophobicity, and stability against degradation) are attributed to the nature of end-group chemistry, which impacted cell function. These observations also demonstrate that the nanoscale organization and distribution of adhesion peptide may be affected by the size of aggregates of hydrophobic PGA fragments. As described previously, the aggregates are sensitive to the nature of end-groups, suggesting that the chemistry of cross-linkers may be an important variable in controlling cell attachment and therefore proliferation.
5. Conclusions
In summary, a series of resorbable hydrogels were prepared by cross-linking polymerization of PEG-co-PGA macromonomers containing three types of end-group functionalities: diacrylates, dimethacrylates, and urethane dimethacrylates. These end-group chemistries seem to have a significant impact on the micellar configuration and their characteristics. Significant variations on the macroscopic physical properties of the gels were observed for the three types of hydrogels at both macromonomer mass fractions (10% and 20%). It is shown that by varying the chemistry of the cross-linker, it is possible to tune the physical properties of PEG-co-PGA hydrogels by more than an order of magnitude. Quantitative differences in the measurements of shear moduli, volumetric swelling ratios, and degradation times were attributed to differences in size and distribution of aggregates. The nature of these cross-linker end-groups has the ability to adjust the three-dimensional network microstructure through variation of hydrophilicity/hydrophobicity of PGA segments. This outcome suggests that a slight variation in gel chemistry has the potential to greatly affect gel structure, properties, and degradation behavior. Preliminary cell experiments showed that these hydrogels were biocompatible, and that the proliferation of MC2C12 cells was strongly dependent on the nature of cross-linker end-group functionalities. By tuning scaffold chemistry, and subsequently, gel structure and degradation behavior, a cell-carrier with unique properties can be engineered, providing an elastic framework for cell attachment, migration, and proliferation. The ability to adjust network structure and gel properties by controlling the cross-linker chemistry provides an additional strategy in the design of degradable scaffolds for tissue engineering.
Acknowledgments
Financial support was provided from NIDCR DE R01-15392-4 (JOH) and from U.S. Army DAMD 17-02-1-0717 (NRW). NRW also acknowledges support from a 3M Non-Tenured Faculty Grant.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Graham NB. Hydrogels: their future, Part I. Med Device Technol. 1998;9:18–22. [PubMed] [Google Scholar]
- 2.Graham NB. Hydrogels: their future, Part II. Med Device Technol. 1998;9(3):22–5. [PubMed] [Google Scholar]
- 3.Nuttelman CR, Rice MA, Rydholm AE, Salinas CN, Shah DN, Anseth KS. Macromolecular monomers for the synthesis of hydrogel niches and their application in cell encapsulation and tissue engineering. Prog Polym Sci. 2008;33:167–179. doi: 10.1016/j.progpolymsci.2007.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nair LS, Laurencin CT. Biodegradable polymers as biomaterials. Prog Polym Sci. 2007;32:762–798. [Google Scholar]
- 5.Zhang YL, Chu CC. The effect of molecular weight of biodegradable hydrogel components on indomethacin release from dextran and poly(DL)lactic acid based hydrogels. J Bio Comp Polym. 2002;17:65–85. [Google Scholar]
- 6.Huglin MB, Rego JM. Influence of temperature on swelling and mechanical-properties of a sulfobetaine hydrogel. Polymer. 1991;32:3354–3358. [Google Scholar]
- 7.Liu Y, Huglin MB. Effective cross-linking densities and elastic-moduli of some physically cross-linked hydrogels. Polymer. 1995;36:1715–1718. [Google Scholar]
- 8.Sen M, Kantoglu O, Guven O. The effect of external stimuli on the equilibrium swelling properties of poly(N-vinyl 2-pyrrolidone itaconic acid) poly-electrolyte hydrogels. Polymer. 1999;40:913–917. [Google Scholar]
- 9.Bryant SJ, Anseth KS. The effects of scaffold thickness on tissue engineered cartilage in photocrosslinked poly(ethylene oxide) hydrogels. Biomaterials. 2001;22:619–626. doi: 10.1016/s0142-9612(00)00225-8. [DOI] [PubMed] [Google Scholar]
- 10.West JL, Hubbell JA. Polymeric biomaterials with degradation sites for proteases involved in cell migration. Macromolecules. 1999;32:241–244. [Google Scholar]
- 11.Lin-Gibson S, Bencherif S, Cooper JA, Wetzel SJ, Antonucci JM, Vogel BM, Horkay F, Washburn NR. Synthesis and characterization of PEG dimethacrylates and their hydrogels. Biomacromolecules. 2004;5:1280–1287. doi: 10.1021/bm0498777. [DOI] [PubMed] [Google Scholar]
- 12.Peppas NA, editor. Hydrogels in medecine and pharmacy. I, II, and III. Boca Raton, FL: CRC Press; 1988. [Google Scholar]
- 13.Illum L, Davis SS, editors. Polymers in controlled drug delivery. Bristol: Wright; 1987. [Google Scholar]
- 14.Behravesh E, Sikavitsas VI, Mikos AG. Quantification of ligand surface concentration of bulk-modified biomimetic hydrogels. Biomaterials. 2003;24:4365–4374. doi: 10.1016/s0142-9612(03)00338-7. [DOI] [PubMed] [Google Scholar]
- 15.Park YD, Tirelli N, Hubbell JA. Photopolymerized hyaluronic acid-based hydrogels and interpenetrating networks. Biomaterials. 2003;24:893–900. doi: 10.1016/s0142-9612(02)00420-9. [DOI] [PubMed] [Google Scholar]
- 16.Metters AT, Anseth KS, Bowman CN. Fundamental studies of a novel, biodegradable PEG-b-PLA hydrogel. Polymer. 2000;41:3993–4004. [Google Scholar]
- 17.Sawhney AS, Pathak CP, Hubbell JA. Bioerodible hydrogels based on photopolymerized poly(ethylene glycol)-co-poly(alpha-hydroxy acid) diacrylate macromers. Macromolecules. 1993;26:581–587. [Google Scholar]
- 18.Leach JB, Bivens KA, Patrick CW, Jr, Schmidt CE. Photocrosslinked hyaluronic acid hydrogels: natural, biodegradable tissue engineering scaffolds. Biotechnology & Bioengineering. 2003;82:578–58. doi: 10.1002/bit.10605. [DOI] [PubMed] [Google Scholar]
- 19.Davis KA, Burdick JA, Anseth KS. Photoinitiated crosslinked degradable copolymer networks for tissue engineering applications. Biomaterials. 2003;24:2485–2495. doi: 10.1016/s0142-9612(02)00582-3. [DOI] [PubMed] [Google Scholar]
- 20.Hern DL, Hubbell JA. Incorporation of adhesion peptides into nonadhesive hydrogels useful for tissue resurfacing. J Biomed Mater Res. 1998;39:266–276. doi: 10.1002/(sici)1097-4636(199802)39:2<266::aid-jbm14>3.0.co;2-b. [DOI] [PubMed] [Google Scholar]
- 21.Bencherif SA, Sheehan JA, Hollinger JO, Walker LM, Matyjaszewski K, Washburn NR. Influence of cross-linker chemistry on release kinetics of PEG-co-PGA hydrogels. J Biomed Mater Res Part A. doi: 10.1002/jbm.a.32069. in press. [DOI] [PubMed] [Google Scholar]