

Abstract
The matrix (MA) domain of the HIV-1 structural precursor Gag (PrGag) protein targets PrGag proteins to membrane assembly sites, and facilitates incorporation of envelope proteins into virions. To evaluate the specific requirements for the MA membrane-binding domain (MBD) in HIV-1 assembly and replication, we examined viruses in which MA was replaced by alternative MBDs. Results demonstrated that the pleckstrin homology domains of AKT protein kinase and phospholipase C δ1 efficiently directed the assembly and release of virus-like particles (VLPs) from cells expressing chimeric proteins. VLP assembly and release also was mediated in a phorbol ester-dependent fashion by the cysteine-rich binding domain of phosphokinase C γ. Although alternative MBDs promoted VLP assembly and release, the viruses were not infectious. Notably, PrGag processing was reduced, while cleavage of GagPol precursors resulted in the accumulation of Pol-derived intermediates within virions. Our results indicate that the HIV-1 assembly machinery is flexible with regard to its means of membrane association, but that alternative MBDs can interfere with the elaboration of infectious virus cores.
Keywords: assembly, Gag, HIV, matrix, membrane, retrovirus
Introduction
The matrix (MA) domain of the HIV-1 precursor Gag (PrGag) protein serves at least two assembly functions: it targets PrGag proteins to membrane assembly sites, and it facilitates the incorporation of the SU/TM envelope (Env) glycoprotein complex into virions (Mammano et al., 1995; Ono and Freed, 1999, 2004; Ono et al., 1997, 2000a, b; Pal et al., 1990; Wang and Barklis, 1993; Wang et al., 1993; Wyma et al., 2000; Yu et al., 1992; Zhou et al., 1994). In terms of membrane targeting, there is some debate as to the pathways by which PrGag proteins travel to arrive at productive assembly sites. One possible itinerary involves PrGag association with intracellular membranes such as multivesicular bodies (MVBs), followed by vesicular delivery to the plasma membrane (PM) (Bryant and Ratner, 1990; Davis et al., 2006; Dorfman et al., 1994; Freed and Martin, 1995, 1996). This model is compatible with the transport of preassembled virions to the PM as vesicle cargoes, or with the delivery of PrGag proteins on vesicle cytoplasmic faces to final PM assembly sites (Jones et al., 1990). An alternative travel plan for PrGag proteins is a direct route to the PM (Jouvenet et al., 2006; Ono and Freed, 2004; Saad et al. 2006). In this case, the accumulation of PrGag proteins on intracellular membranes could be viewed as representing dead end assembly products, or internalized PM components, enroute to lysosomal degradation. An intermediate hypothesis is that direct PM targeting and vesicle-mediated transport both can yield infectious virions, depending on the cell type or context (Ono and Freed, 2004).
A number of biochemical and genetic studies have identified different features of HIV-1 MA that influence PrGag localization, membrane binding, and assembly (Bryant and Ratner, 1990; Facke et al., 1993; Lindwasser and Resh, 2002; Murray et al., 2005; Ono and Freed, 1999; Ono et al., 2000a, 2004; Pal et al, 1990; Saad et al., 2006; Tang et al., 2004; Wang and Barklis, 1993; Wang et al., 1993; Zhang et al. 1994). With regard to localization, the amino-terminal α-helical segment of HIV-1 MA recently was shown to associate with the δ subunit of the AP-3, and this interaction appears important for PrGag MVB localization (Dong et al., 2005). Evidence also has shown that residues at the C-terminus of MA mediate PrGag association with the μ subunit of the clathrin-associated adapter complex AP-2, but that PrGag processing eliminates this interaction (Batonick et al., 2005). Consistent with the role of AP-2 in PM cargo internalization, inhibition of MA-AP-2 binding increased virus particle release from cells (Batonick et al., 2005). In addition to adapter protein binding, HIV-1 MA also contributes to the binding of the cellular motor protein KIF-4, an association that plausibly regulates the distribution of PrGag (Tang et al., 1999). Recent investigations also demonstrated that MA residues 5-16 are necessary for PrGag binding to the tail interacting protein TIP47, which mediates a functional association of MA with Env (Lopez-Varges et al., 2006).
While the above protein-protein interactions affect virus infectivity and PrGag localization, membrane binding is impacted greatly through MA-lipid interactions. The MA N-terminus is critical in this process. Like other mammalian retrovirus matrix proteins, HIV-1 MA is myristoylated at its N-terminus, and myristoylation is essential for efficient membrane binding, virus assembly, and infectivity (Bryant and Ratner, 1990; Pal et al., 1990; Saad et al., 2006; Tang et al., 2004; Wang and Barklis, 1993). Studies support a myristoyl switch model for MA membrane association, in which the myristate group of PrGag inserts into membranes to promote binding, but that this fatty acid is retracted on proteolytic processing of PrGag, conferring a reduced membrane affinity for mature MA proteins (Hermuda-Matsumato and Resh, 1999; Paillart and Gottlinger, 1999; Spearman et al., 1997; Tang et al., 2004). Although the MA myristate is necessary for tight PrGag-membrane binding, it is not the only contributor. Analysis indicates that basic amino acids between residues 15 and 40 facilitate electrostatic interactions with negatively charged phospholipid headgroups on the cytoplasmic faces of membranes (Murray et al., 2005; Ono and Freed, 1999; Zhou et al., 1994). However, HIV-1 MA also specifically associates with phosphatidylinositol (4,5) bisphosphate [PI(4,5)P2] (Ono et al., 2004; Saad et al., 2006). In particular, the PI(4,5)P2 headgroup and 2′-acyl chain are accommodated by a cleft in the protein, and binding results both in the membrane-anchoring by the PI(4,5)P2 2′-acyl chain, and in triggering the exposure of the MA myristate (Saad et al., 2006). Given the affinity of MA for PI(4,5)P2, it is plausible that PI(4,5)P2 helps direct PrGag to cholesterol- and dihydrosphingomyelin-rich membrane domains, where HIV-1 assembly appears to occur (Booth et al., 2006; Brugger et al., 2006; Holm et al., 2003; Nguyen and Hildreth, 2000; Ono and Freed, 2001).
Despite the many documented roles for HIV-1 MA, it is not absolutely required for virus infectivity. We originally demonstrated the infectivity of a 106 residue MA deletion mutant that retained only the myristoylation signal and matrix-capsid (CA) cleavage site (Wang et al., 1993), an observation that subsequently was confirmed by others (Reil et al., 1998). In such cases, infection required either an Env protein pseudotype, or truncation of the HIV-1 Env cytoplasmic tail (Reil et al., 1998; Wang et al., 1993). These deletion mutant results imply that wild type (wt) HIV-1 Env function requires MA, but that intact MA is not strictly required for viral replication.
The studies above suggest that any membrane targeting and anchor protein might suffice for directing the assembly of infectious HIV-1 particles. However, a caveat to this interpretation is that the aforementioned MA deletion proteins retained myristoylation signals (Reil et al., 1998; Wang et al., 1993). Thus, a more narrow hypothesis is that membrane interactions mediated by myristate are compatible with HIV-1 assembly and infectivity, while other membrane interaction motifs may or may not serve this role. To analyze this hypothesis further, we have examined HIV-1 MA protein substitution variants for their abilities to replace MA. For our investigations, we chose three alternative membrane binding domains (MBDs). One of these was the AKT protein kinase pleckstrin homology (PH) domain, which preferentially binds to 3′ phosphatidyl inositol (PI) lipids such as phosphatidyl inositol (3,4) bisphosphate [PI(3,4)P2], and the PM-enriched phosphatidyl inositol (3,4,5) trisphosphate [PI(3,4,5)P3] (Haugh et al., 2000; Hurley and Meyer, 2001; Lemmon, 2003, Marshall et al., 2001). The second MBD was the PH of phospholipase C δ1 (PLC), which has affinity for PM-localized PI(4,5)P2 (Hurley and Meyer, 2001; Lemmon, 2003; Stauffer et al., 1998). Finally, we examined the phosphokinase Cγ cysteine-rich C1a plus C1b binding domain (CBD), which binds PM diacylglycerol (DAG), and can be activated by phorbol esters to move from intracellular stores to the PM (Colon-Gonzalez and Kazanietz, 2006; Edwards and Newton, 1997; Hurley and Meyer, 2001; Oancea and Meyer, 1998; Oancea et al., 1998). When the AKTPH, PLCPH, and PKCCBD were employed as substitutes for MA, we found that the two PH domains readily directed PrGag proteins to assemble and release virus-like particles (VLPs) from cells, while PKCCBD-Gag proteins could be induced to release VLPs with phorbol esters. However, the VLPs that used alternative MBDs were noninfectious, either with HIV-1 Env, or with other Env pseudotypes. Assembly-competent but non-infectious AKTPH- and PLCPH-derived VLPs showed nearly normal viral RNA (vRNA), Env protein pseudotype, and entry levels, but had reduced reverse transcriptase (RT) activities, and were defective for DNA synthesis in target cells. Moreover, all MA substitution viruses showed moderate protease (PR) processing defects of the PrGag proteins. Additionally, precursor GagPol (PrGagPol) processing resulted in the accumulation of Pol-derived intermediates in virions, suggesting unexpected MBD effects on the regulation of later Pol protein cleavage steps. Our results indicate that while the membrane-targeting and assembly functions of HIV-1 MA can be replaced, alternative MBDs compromise virus infectivity by perturbation of virus core structure and function.
Results
Particle release directed by alternative membrane binding domains
To analyze whether the AKTPH, PLCPH, or PKCCBD membrane-binding domains could replace HIV-1 MA in directing the assembly and budding of HIV virus-like particles, we initially examined VLPs released from cells transfected with the matrix substitution constructs GFP-AKTPH-Gag, GFP-PLCPH-Gag, and PKCCBD-GFP-Gag. As illustrated in Figure 1, these plasmids express PrGag proteins from PR- constructs in which the HIV-1 wild type (wt) Gag domains from CA through p6 were linked to N-terminal modules composed of green fluorescence protein (GFP) and MBD units. For controls in these studies, we employed a wt HIV-1 Gag expression construct, HIVgpt (McDermott et al., 1996; Page et al., 1990; Wang and Barklis, 1993; Wang et al., 1993, 1994; Zhang and Barklis, 1997), as well as HIVgpt-MAGFP (Figure 1), in which the GFP coding region has been inserted near the C-terminus of MA, in a fashion similar to the GFP-Gag construct described by Muller et al (2004).
Figure 1. Recombinant DNA constructs.

All recombinant constructs express variants of the HIV-1 HXB2 gag gene that ordinarily is translated into the precursor Gag (PrGag) protein. Wild type (wt) versions of the HIV-1gag gene, expressed in the presence of pol gene products, were produced from the previously described HIVgpt construct (Scholz et al., 2005; Wang and Barklis, 1993; Wang et al., 1993). In control experiments, HIV-1 protease-minus (PR-) proteins were expressed from the previously described HIVgpt2498T plasmid (Wang and Barklis, 1993; Zhang and Barklis, 1997). GFP-AKTPH-Gag, GFP-PLCPH-Gag, and PKCCBD-GFP-Gag generate PR- Gag fusion proteins in which the HIV MA domain has been replaced with the green fluorescence protein (GFP) plus the AKT protein kinase pleckstrin homology (PH) domain (Hermida-Matsumato and Resh, 1999; Marsharll et al., 2001), the phospholipase Cδ1 (PLC) PH domain (Stauffer et al., 1998), or the cysteine-rich membrane-binding domain (CBD) from phosphokinase Cγ (PKC; Oancea and Meyer, 1998; Oancea et al., 1998). The depicted HIVgpt constructs are variants of the wt HIVgpt plasmid (Page et al., 1990; Wang and Berwin, 1993; Wang et al., 1993), and express proteins in the presence of HIV-1 pol gene products. HIVgpt-MAGFP carries the GFP coding region in-frame as an insert near the C-terminal end of MA. HIVgpt-AKTPH, HIVgpt-PLCPH, and HIVgpt-PKCCBD derive from a myristoylation-minus HIVgpt construct (Wang and Barklis, 1993), and replace matrix residues downstream from codon 16 and upstream from the MA/CA juncture region (codon 120) with the AKTPH, PLCPH, or PKCCBD membrane-binding domains. Juncture sequences and construct details are provided in the Materials and Methods.
Consistent with previous results (McDermott et al., 1996; Wang and Barklis, 1993; Wang et al., 1993; Zhang and Barklis, 1997), in transfected cells, the wt HIVgpt construct expressed Gag proteins and efficiently assembled and released virus particles. When cells transfected with the wt construct were treated with the phorbol esters PMA or PDB, we did not observe any noticeable changes on wt virus release levels (Figure 2). Relative to the wt construct, our HIVgpt-MAGFP construct appeared to release slightly reduced Gag protein levels, while results with our matrix replacement constructs were varied (Figure 2). With the PH substitutions GFP-AKTPH-Gag and GFP-PLCPH-Gag VLPs were assembled and released efficiently (Figure 2). In contrast, PKCCBD-GFP-Gag proteins failed to release VLPs to media samples. However, because PKCCBD binds DAG and is activated by phorbol esters (Colon-Gonzalez and Kazanietz, 2006; Edwards and Newton, 1997; Hurley and Meyer, 2001; Oancea and Meyer, 1998; Oancea et al., 1998), we also tested whether phorbol myristate acetate (PMA) or phorbol dibutyrate (PDB) could mobilize the release of PKCCBD-GFP-Gag VLPs. Interestingly, while the phorbol esters had no apparent effect on the release of wt HIV particles, they clearly increased particle release directed by the PKCCBD (Figure 2).
Figure 2. Release of virus-like particles.
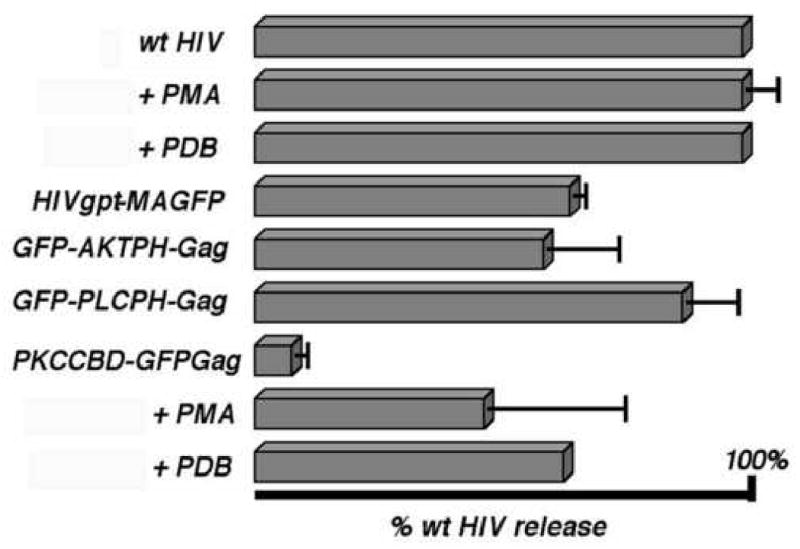
Cells were transfected with wt HIVgpt (wt HIV) or the indicated constructs and either untreated or treated with 1 μM PMA or PDB. At 72 h post-transfection, cell and VLP samples were collected, subjected to SDS-PAGE and anti-HIVCA immunoblotting. Cellular PrGag and VLP PrGag plus CA levels were quantitated densitometrically, raw release levels ([VLP PrGag + CA]/[Cell PrGag]) were calculated and are shown, normalized to untreated wt HIVgpt release levels. Results derive from one (wt HIV + PDB; PKCCBD-GFP-Gag + PDB), two (wt HIV + PMA; HIVgpt-MAGFP), three (PCKCBD-GFP-Gag + PMA), or four (GFP-AKTPH-Gag; GFP-PLCPH-Gag; PKCCBD-GFPGag) separate experiments. Standard deviations are as shown.
To verify that GFP modules were retained on the full-length PrGag proteins expressed by the matrix replacement constructs, VLP samples were electrophoresced in parallel and then immunoblotted for detection of either HIV-1 CA or GFP. Not surprisingly, none of the wt HIV-1 bands detected with our CA antibody were observed when the anti-GFP antibody was employed whereas with matrix replacement VLPs, each of the lowest mobility PrGag proteins visualized with anti-CA, also were seen with anti-GFP, supporting the conclusion that these represent the full-length PrGag proteins (data not shown). The incorporation of GFP into the VLPs of GFP-AKTPH-Gag, GFP-PLCPH-Gag, and PKCCBD-GFP-Gag permitted us monitor VLP formation via fluorescence microscopy, and as a control, we took advantage of the availability of HIVgpt-MAGFP particles. When subjected to fluorescence microscopy, HIVgpt-MAGFP VLPs were observed as bright green spots (data not shown), similar to the appearance of vpr-GFP-labeled HIV-1 particles (Muthumani et al., 2000). Particles assembled and released by the PH replacement Gag proteins also were imaged as green spots, while for PKCCBD-GFP-Gag, fluorescent VLP release required phorbol ester induction. These results lend qualitative support to the results presented in Figure 2.
Cellular localization of alternative Gag proteins
Where do matrix replacement Gag proteins accumulate within cells? We addressed this question via microscopic tracking of fluorescent proteins in transfected cells. With HIVgpt-MAGFP, the fluorescent proteins were observed at cell surfaces and perinuclear regions, as well as in a heterogeneously staining pattern throughout cells (Figure 3A). This staining profile was similar to that observed previously for wt HIVgpt CA (McDermott et al., 1996; Wang and Barklis, 1993; Wang et al., 1993; Zhang and Barklis, 1997), and for the MAGFP Gag protein of Muller et al. (2004), and was not altered appreciably by the addition of phorbol esters (Figure 3B). Interestingly, AKT PH, which binds to 3′ PIs, and PLC PH, which binds PI(4,5)P2 both targeted their respective Gag proteins to the PM, as indicated by the strong cell surface staining of the GFP-AKTPH-Gag (Figure 3C) and GFP-PLCPH-Gag proteins (Figure 3D). For PKCCBD-GFP-Gag, proteins accumulated in large aggregates, often adjacent to cell nuclei (Figure 3E), but this pattern was shifted somewhat to smaller aggregates and more surface staining on PMA treatment (Figure 3F).
Figure 3. Fluorescence localization of proteins in transfected cells.
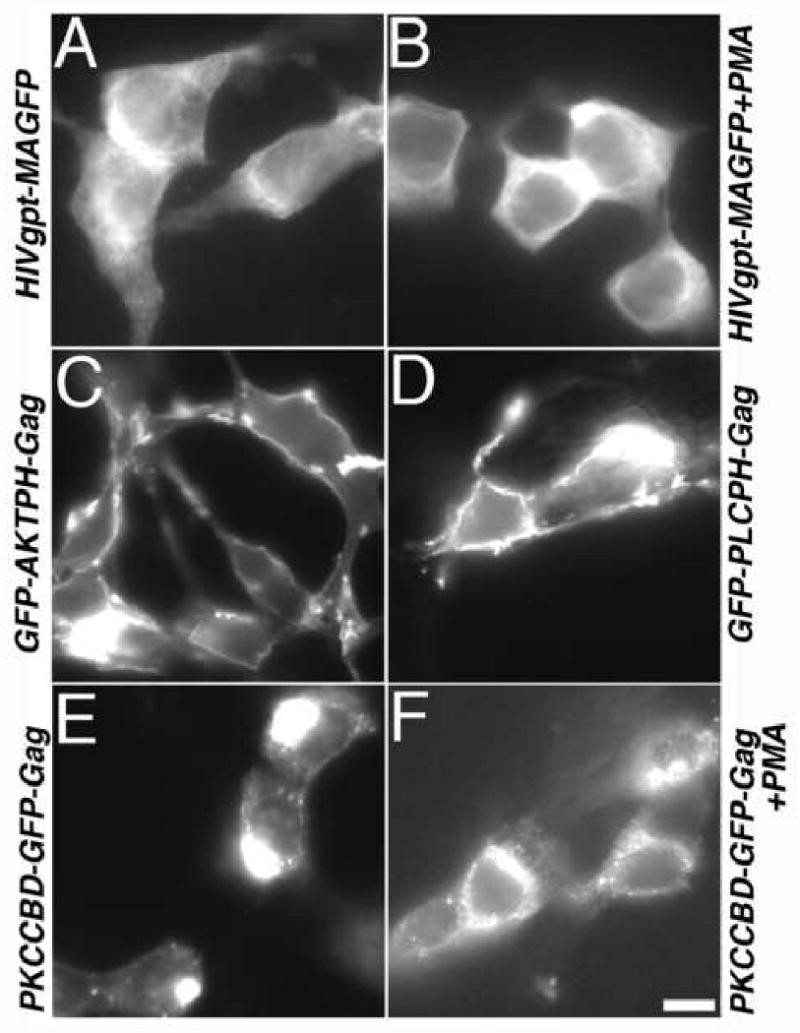
Cells on coverslips that were transfected with the indicated constructs were either untreated (Panels A, C, D, E) or treated with 1 uM PMA (Panels B, F). Subsequently, cells were processed for microscopic analysis of GFP proteins as described in the Materials and Methods section, imaged on a Zeiss Axioplan fluorescence microscope, and photographed using Improvision OpenLab software. A 20 micron size bar for all panels is provided at the bottom of the panel.
In an effort to quantitate our fluorescence staining observations, we calculated cell surface staining levels. To do so, fluorescence levels corresponding to the outermost 20% of cell radii were expressed as percentages of total cell fluorescence signals (see Materials and Methods) to give percentages of cell surface staining. For HIVgpt-MAGFP, we calculated the cell surface staining percentage to be 29 ± 4 %, presumably reflecting the fact that wt PrGag proteins localize both to the PM and to intracellular membrane and vesicle compartments (Dong et al., 2005; Jouvenet et al., 2006; Lindwasser and Resh, 2004; Nydegger et al., 2003, Ono and Freed, 2004). PMA treatment did not alter this surface staining percentage (28 ± 5 %). Relative to these levels, the PH replacement proteins demonstrated higher surface staining, with percentages of 36 ± 7 % for GFP-AKTPH-Gag and 39 ± 6 % for GFP-PLCPH-Gag. Not surprisingly, surface staining levels were low (15 ± 5 %) for PKCCBD-GFP-Gag, but increased slightly to 23 ± 4 % on PMA treatment.
As another approach to the localization of Gag proteins within cells, we resorted to electron microscopy (EM), relying on the tendency of Gag protein aggregates to stain darkly in negatively stained thin sections (Arvidson et al., 2003; Facke et al., 1993; Hockley et al., 1998). For a control in these studies, we used PR- but otherwise wt HIV-1 Gag expression construct (HIVgpt PR-; McDermott et al., 1996; Wang and Barklis, 1993; Wang et al., 1994; Zhang and Barklis, 1997). In agreement with previous work (Arvidson et al., 2003), wt PR- HIV-1 particles were observed budding and at cell surfaces (Figure 4A-C). In contrast, the GFP-AKTPH-Gag proteins showed a variety of structures (Figure 4D-F). Occasionally, large vesicles containing black rimmed smaller vesicles and darkly staining virus-sized spherical structures were observed (Figure 4D). However, virus-like budding structures, or VLPs within budding structures (Figures 4E, F) also were seen. Relative to the structures assembled in GFP-AKTPH-Gag-transfected cells, those formed by GFP-PLCPH-Gag (Figure 4G-I) and PKCCBD-GFP-Gag (Figure 4J-L) were more consistent. In particular, for GFP-PLCPH-Gag, particle assembly occurred at the PM. This was indicated by the appearance of electron dense nascent buds at the PM (Figure 4G), as well as small (Figure 4H) and large (Figure 4I) budding structures. For PKCCBD-GFP-Gag, the picture was quite different. Rather than assembly at the PM, these proteins associated in large (Figures 4J,K) or small clusters or aggresomes (Johnston et al., 1998). We did not observe any obvious reduction in the numbers of aggresomes in PMA treated PKCCBD-GFP-Gag-transfected cells (data not shown), suggesting that the quantitative effects of phorbol esters on the release (Figure 2) and localization (Figure 3) were not dramatic enough to be scored by EM.
Figure 4. Electron microscopy of transfected cells.
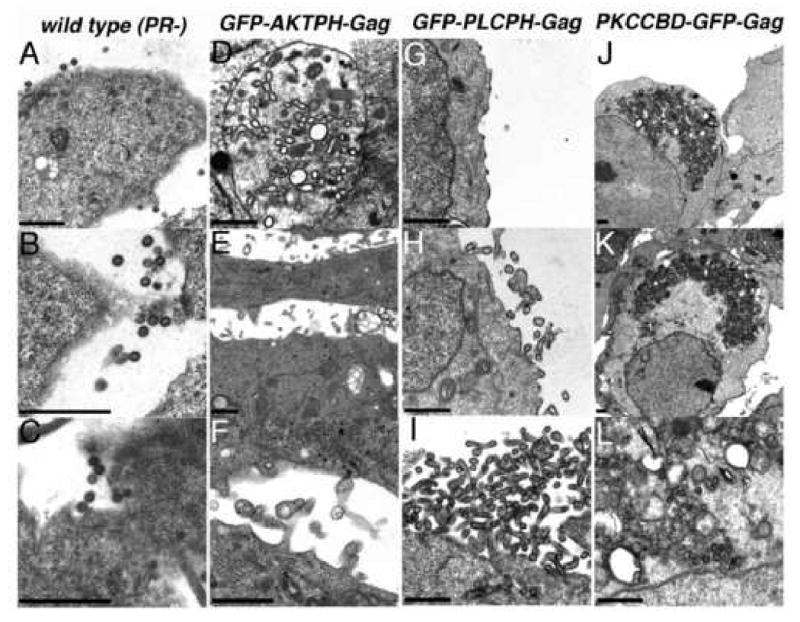
Cells were transfected with constructs expressing protease-minus but otherwise wild type HIV Gag (wild type Pr-; panels A-C), GFP-AKTPH-Gag (panels D-F), GFP-PLCPH-Gag (panels G-I), or PKCCBD-GFP-Gag (panels J-L). At 3 days post-transfection, cells were fixed, post-fixed, dehydrated, embedded, sectioned, and stained for electron microscopy as described in the Materials and Methods. As illustrated, relatively homogeneous wild type PR- particles were observed at the surfaces of cells (A-C), while structures assembled by GFP-AKTPH-Gag proteins were pleiomorphic and appeared as VLPs or darkly rimmed vesicles within intracellular vacuoles (D), particles released at the cell surface (E), or preformed VLPs near the cell surface or within attached or sheared filopodia (F). In contrast, assembly of spherical or tubular GFP-PLCPH-Gag VLPs occurred specifically at cell surfaces (G-I), and PKCCBD-GFP-Gag proteins assembled into large intracellular aggresomes (J-L).
Despite the localization differences between the wt and matrix replacement Gag proteins, it was of interest to assess whether the proteins might cross paths within cells. We examined this by coexpression of wt HIV-1 Gag-β-galactosidase (Gag-β-gal) fusion proteins along with wt or MA substitution Gag proteins in cotransfected HEK 293 cells. As previously observed (Wang et al., 1994), expression of HIV-1 Gag-β-gal in the absence of any helper proteins resulted in very low levels of Gag-β-gal release from cells, and treatment with PMA or coexpression with murine leukemia virus (MuLV) Gag proteins failed to increase levels of release. However, cellular coexpression of HIV-1 Gag-β-gal and wt HIV-1 Gag proteins caused the efficient assembly and release of β-gal-positive (β-gal+) VLPs (data not shown), as described previously (Wang et al., 1994). The Gag proteins with PH domains also efficiently facilitated the efficient release of β-gal+ VLPs, while PKCCBD-GFP-Gag proteins induced β-gal release to a lower extent, and in a phorbol ester-dependent fashion (data not shown). Altogether, these results demonstrate that despite their different membrane-targeting signals, wt and MA substitution proteins are able to associate with each other within cells.
Replication defects of chimeric Gag proteins
Since alternative MBDs were able to direct the release of VLPs, albeit to different degrees, and with potentially different morphologies, we next tested whether they were compatible with virus replication. For these studies, we replaced the MA domain in HIVgpt (Page et al., 1990; Wang and Barklis, 1993) with MBDs, excluding GFP domains. The resulting constructs, HIVgpt-AKTPH, HIVgpt-PLCPH, and HIVgpt-PKCCBD, retained the wt HIV-1 MA-CA cleavage region and the MA N-terminus, but bore the glycine-to-alanine mutation at gag codon 2, to prevent Gag protein myristoylation (Bryant and Ratner, 1990; Wang and Barklis, 1993; Wang et al., 1994). We scored for replication in single cycle infections via cotransfection of HIVgpt-derived constructs with a Vesicular Stomatitis virus glycoprotein (VSV-G) expression construct (pVSV-G) into HEK 293 cells, collection of released virions for infection of CD4+ Hela (HiJ; Scholz et al., 2005; Wang and Barklis, 1993; Wang et al., 1993) cells, and selection for gpt gene expression. As illustrated in Figure 5, while mock-infected HiJ cells were killed in gpt selective media, numerous darkly staining colonies derived from wt HIVgpt infected cells survived selection. In our hands, with Gag-normalized input virus, our GFP insertion construct HIVgpt-MAGFP (Figure 1) proved to be about 10% as infectious as wt (Figure 5). This level of infection was about 10-fold higher than that observed by Muller et al. (2004), but those investigators used a slightly different MAGFP construct, employed the wt HIV-1 Env protein, and scored infection in an alternative fashion. In contrast to HIVgpt and HIVgpt-MAGFP, our matrix replacement HIVgpt constructs failed to demonstrate any infectivity (Figure 5), and identical results were observed when we pseudotyped the viruses with either HIV-1 Env, or an amphotropic MuLV Env (data not shown). We then verified that vRNAs transcribed by MA substitution constructs could be encapsidated, reverse transcribed, integrated and expressed in target cells. To do so, constructs were cotransfected with pVSV-G plus a wt gpt-minus HIV-1 Gag expression construct (HIV-Luc; Scholz et al., 2005), and infectivities of released viruses were scored by selection for gpt. In these studies, wt Gag proteins efficiently fostered the transduction of HIVgpt-ADKPH, HIVgpt-PLCPH, and HIVGPT-PKCCBD genomes to target cells (data not shown), implying that the matrix replacement vRNAs did not carry a cis-active defect for replication.
Figure 5. Virus infectivity.

Viruses were produced by mock transfection (mock) or cotransfection of 293T cells with a VSV G expression vector plus HIVgpt (WT), HIVgpt-MAGFP (MAGFP), HIVgpt-AKTPH (AKTPH), HIVgpt-PLCPH (PLCPH) or HIVgpt-PKCCBD (PKCCBD). For production of PKCCBD viruses, transfected cells were treated with 1 μM PMA as described in the Materials and Methods to improve particle release. At 3 days post-transfection, virus-containing media supernatants were collected, filtered through 0.45 micron sterile filters and used to infect HiJ cells. Three days post-infection, HiJ cells were split 1:10 into gpt selective media, and colonies expressing proviral gpt genes were grown for 7-10 d prior to staining.
The lack of infectivity for HIVgpt-AKTPH, HIVgpt-PLCPH, and HIVgpt-PKCCBD viruses did not appear to be a consequence of impaired particle assembly and release, since the constructs yielded detectable levels of PrGag and CA proteins in pelleted media samples (Figure 6, top panel; lanes E-H). Compared with cellular Gag protein levels (Figure 6, top panel, lanes A-D), HIVgpt wt and HIVgpt-PLCPH showed similar VLP release levels (Figure 6, middle panel), HIVgpt-AKTPH showed about a two-fold reduction in VLP release (Figure 6, middle panel), and HIVgpt-PKCCBD showed at least a ten-fold release reduction (Figure 6, middle panel) even in the presence of PMA. However, these release reductions were comparatively small, relative to the observed infectivity block of over a thousand-fold (Figure 5).
Figure 6. Virus-like particle release and processing.
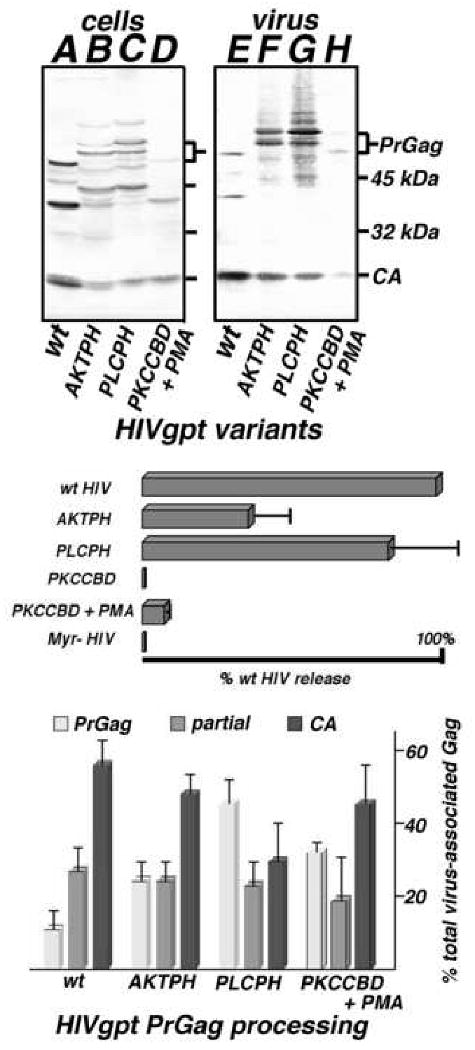
Top panel. Cell lysate (A-D) and VLP (E-H) samples were collected from cells transfected with wt HIVgpt (wt), HIVgpt-AKTPH (AKTPH), HIVgpt-PLCPH (PLCPH) or HIVgpt-PKCCBD (PKCCBD), and either untreated or treated with 1 μM PMA (+PMA). Gag proteins in samples were fractionated by SDS-PAGE, and detected by immunoblotting using an anti-HIVCA primary antibody. Size marker mobilities, as well as PrGag and CA bands, are indicated. Middle panel. Virus-like particle assembly and release levels, normalized to that of wt HIVgpt, were determined as described in Figure 2, including a myristoylation-minus HIVgpt construct (Myr- HIV) as a negative control. Values derive from two (PKCCBD, Myr- HIV), three (PKCCBD), or four (AKTPH, PKCCBD + PMA) independent experiments, with standard deviations as shown. Bottom panel. PrGag processing levels for VLPs produced from cells expressing wt HIVgpt (wt), AKTPH, PLCPH, and PKCCBD (in the presence of PMA) were determined by densitometric quantitation of PrGag (light gray), processing intermediates (medium gray), and CA bands (dark gray) from immunoblots. Values represent percentages of the total virus-associated Gag levels (plus standard deviations) and derive from three (PKCCBD +PMA), four (AKTPH, PLCPH), or seven (wt) independent VLP preparations.
Despite the appearance of PrGag and CA bands for the matrix substitution HIVgpt variants, several anomalies were evident. In particular, HIVgpt-AKTPH and HIVgpt-PLCPH VLP preparations both showed immunoreactive bands of slightly slower mobility than their respective predicted 57-58 kDa PrGag proteins (Figure 6, top panel, lanes F, G). Because the intensities of these low mobility species varied from preparation to preparation, we hypothesized that they might represent variable post-translational modification forms; but they were resistant to endoglycosidase F, and were unreactive to anti-phosphotyrosine, anti-phosphothreonine, and anti-phosphoserine antibodies, suggesting that they did not result from N-glycosidation or phosphorylation (data not shown). In addition to these low mobility species, some Gag processing differences were noted. Although all of the MA replacement particle preparations looked similar to wt with regard to their levels of partial processing intermediates, PrGag percentages were increased, at the expense of mature CA (Figure 6, bottom panel). These processing differences appeared relatively minor for HIVgpt-AKTPH, but were clearly skewed for HIVgpt-PLCPH, where only a third of the particle-associated Gag protein was in the mature form (Figure 6, bottom panel).
Because MA substitution viruses were functionally impaired, we decided to examine VLP morphologies of HIVgpt wt, and the two well-released MBD variants (AKTPH and PLCPH) by EM. To do so, pelleted particles from transfected cell media samples were applied to EM grids, dried, stained and imaged, as we have done previously (Scholz et al., 2005). Examination of 100 particles of each virus type showed that they had comparable diameters (Figure 7, lower right). We also scored for the percentages of particles with roughly conical or cylindrical cores, and found that wt and HIVgpt-AKTPH results were similar (65%), while the percentage for HIVgpt-PLCPH was slightly reduced (50%; Figure 7, upper right). However, we noted some differences that were difficult to quantitate. In particular, HIVgpt-AKTPH cores occasionally appeared to have abnormalities (Figure 7, AKTPH, leftmost image), and frequently stained poorly at the narrow cone ends (right two images). HIVgpt-PLCPH cores seemed even more aberrant, and were often short (PLCPH, leftmost two images) or poorly staining (rightmost image) at narrow ends.
Figure 7. Electron microscopy of virus-like particles.
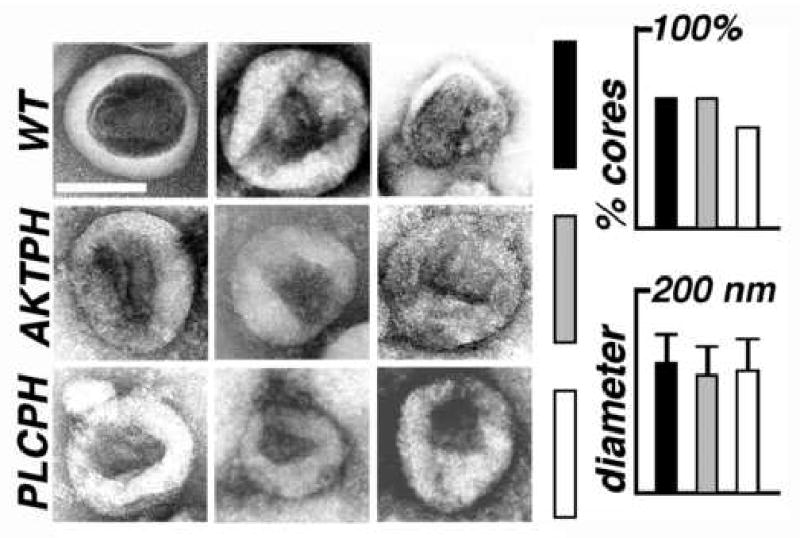
Wild type (wt; black bars) HIVgpt, HIVgpt-AKTPH (AKTPH; gray bars), and HIVgpt-PLCPH (PLCPH; white bars) virus-like particles (VLPs) were lifted onto EM grids, stained, dried, and imaged by electron microscopy (EM). Micrographs show VLPs with central, roughly conical cores, and the white size bar in the upper right panel corresponds to 100 nm for all pictures. The graphs on the right shows the average VLP diameters, and the percentages of VLPs with discernable, roughly conical or cylindrical cores: each value was derived from 100 separate VLP images.
The above observations suggest that alternative MBDs interfere with the formation of mature infectious virus cores. To examine the replication defects of HIVgpt-AKTPH and HIVgpt-PLCPH particles in more detail, viruses produced by these constructs were subjected to additional assays: we excluded HIVgpt-PKCCBD from these analyses, due to difficulties in obtaining sufficient material. For quantitation of vRNA encapsidation, virus particle vRNA levels were determined by RNAse protection, as we have done previously (Scholz et al., 2005 Wang et al., 1993; Zhang and Barklis, 1997). However, since we have demonstrated that HIV-1 MA is not needed for efficient vRNA encapsidation (Wang et al., 1993), it was not surprising that our PH HIVgpt variants incorporated 83% (AKTPH) to 104% (PLCPH) wt amounts of vRNA into viruses. Another parameter that conceivably could have affected infectivity was the efficiency of VSV-G pseudotyping. We assayed this via parallel immunoblotting of virus samples for detection of Gag and VSV-G, but found that VSV-G-to-Gag ratios either were not reduced (AKTPH) or only slightly reduced (PLCPH) relative to wt ratios. These observations were consistent with our entry assay results. Here, we followed previously reported methodology (Munk et al., 2002; Scholz et al., 2005; see Materials and Methods) to score for virus-mediated β-lactamase Vpr (BlaM-Vpr) fusion protein delivery to HiJ target cells. Although entry levels ranged between two-thirds (PLCPH) and 170% (AKTPH) that of wt levels, these differences were small compared to the infectivity block (Figure 5). Thus, the dominant defect of our MA substitution variants does not appear to be virus core delivery to new cells.
While delivery of HIVgpt-AKTPH and HIVgpt-PLCPH VLP cores to HiJ cells was not impaired appreciably, the replicative capacities of the cores were compromised. One indication of this defect was a reduction in the RT levels associated with matrix replacement VLPs. Indeed, Gag-normalized RT levels for HIVgpt-AKTPH particles were reduced two-fold from wt, while activities for HIVgpt-PLCPH were only 15% of wt levels. These RT defects were more evident when reverse transcription products were monitored in infected cells. For these assays, one long terminal repeat (1-LTR) RT byproducts (Butler et al., 2001) in infected cells were measured after PCR amplification as described in the Materials and Methods. Importantly, 1-LTR products were detected readily for HIVgpt wt, but were undetectable with the MBD variants, indicating that their virus cores failed to complete RT steps in target cells. We further examined this RT defect by immunoblot analysis of RT proteins in HIVgpt wt, HIVgpt-AKTPH, HIVgpt-PLCPH, and HIVgpt-PKCCBD VLPs. Surprisingly, whereas wt VLPs showed the expected 66 kDa RT-RNAse H (RT-RH) and 51 kDa RT species, our three MBD variant VLPs each revealed a single immunoreactive band at a mobility of about 75 kDa (Figure 8). Because species with this mobility also were detected with an anti-PR antibody (data not shown), our data indicate that these bands represent PR-RT-RH processing intermediates (Pettit et al., 2004, 2005). These results suggest that MA replacements exert an influence on PrGagPol cleavages that occur after Gag and Pol are separated, and are consistent with reduced RT and PrGag processing levels in our chimeric viruses.
Figure 8. Analysis of viral reverse transcriptase proteins.

VLP samples were collected from cells transfected with wt HIVgpt (A), HIVgpt-AKTPH (B), HIVgpt-PLCPH (C), or HIVgpt-PKCCBD (D), and either untreated or treated with 1 μM PMA (+PMA). Proteins in Gag-normalized samples were fractionated by SDS-PAGE, and detected by immunoblotting using an anti-HIVRT primary antibody. Size marker mobilities, as well as 66 kDa RT-RH and 51 kDa RT bands, are indicated. Note that no other bands were detected with the anit-RT antibody, but that similarly sized bands for the MBD variants (B-D) were detected in parallel immunoblots with an anti-HIVPR antibody.
Discussion
The HIV-1 matrix domain targets PrGag proteins to assembly sites on cellular membranes, and is required for incorporation of full-length HIV-1 Env proteins into virus particles (Bryant and Ratner, 1990; Davis et al., 2006; Dorfman et al., 1994; Freed and Martin, 1995, 1996; Mammano et al., 1995; Ono and Freed, 1999, 2004; Ono et al., 1997, 2000a, 2000b; Pal et al., 1990; Wang and Barklis, 1993; Wang et al., 1993; Wyma et al., 2000; Yu et al., 1992; Zhou et al., 1994). However, PrGag proteins with MA deletions that retain myristoylation signals and MA/CA cleavage sites are able to direct the assembly of infectious virions, provided that the particles are pseudotyped with heterologous Env proteins or C-terminally truncated HIV-1 Env proteins (Reil et al., 1998; Wang et al., 1993). These results may suggest that both full-length and MA-deleted, myristoylated HIV-1 PrGag proteins are targeted to membrane sites that are permissive for the assembly of infectious virions. Alternatively, virus assembly simply may require the delivery of the C-terminal Gag domains to a membrane surface, with few additional requirements.
Our results suggest that the HIV-1 assembly machinery is relatively tolerant of the MBD used to foster membrane association. Specifically, the AKT PH, which binds 3′ PI lipids (Haugh et al., 2000; Hurley and Meyer, 2001; Lemon, 2003; Marshall et al., 2001), and the PLC PH, which binds PI(4,5)P2 (Hurley and Meyer, 2001; Lemmon, 2003; Stauffer et al., 1998), both mediated the assembly and release of VLPs (Figures 2, 6). Efficient VLP release occurred in the absence of PI 3-kinase and PI(4)P 5-kinase activation, implying that basal levels of PM PI(3,4,5)P3 and PI(4,5)P2 were sufficient for cell surface localization. In contrast to the two PH domains, the cysteine-rich membrane-binding domain of PKC (PKCCBD; Colon-Gonzalez and Kazanietz, 2006; Edwards and Newton, 1997; Hurley and Meyer, 2001; Oancea and Meyer, 1998; Oancea et al., 1998) directed Gag protein accumulation into large aggresomes (Johnston et al., 1998; Figures 3, 4). However, consistent with the ability of the PKCCPD to bind DAG and phorbol esters (Colon-Gonzalez and Kazanietz, 2006; Edwards and Newton, 1997; Hurley and Meyer, 2001; Oancea and Meyer, 1998; Oancea et al., 1998), cell treatment with PMA or PDB boosted VLP release from cells (Figures 2, 6). Despite their different cellular itineraries and membrane-binding mechanisms, all of our MA substitution Gag proteins were able to facilitate the release of wt HIV-1 Gag-β-gal proteins from cotransfected cells, which demonstrates that there is some overlap in the pathway taken by Gag-β-gal proteins with wt MA domains, and the assembly and release routes taken by the matrix replacement proteins.
Although our alternative MBDs sufficed for VLP assembly and release, they failed to support virus replication (Figure 5). For the PH variants, vRNA encapsidation, VSV-G pseudotyping, and entry all occurred at wt or slightly diminished levels. However, reverse transcription in infected cells was greatly impaired, and viruses showed reduced RT activities, moderate to severe PrGag processing defects (Figure 6), and the appearance of altered mature cores (Figure 7). Notably, VLPs assembled by all three MA replacements carried PR-RT-RH proteins, and did not show the normal 66 kDa RT-RH and 51 kDa RT products (Figure 8). This finding is consistent with the observed RT and PrGag processing detects of MBD variant viruses. However, our results imply that MBDs influence PrGagPol processing steps that occur after Pol domains are freed from their co-translated partners. Formally, MBDs could interfere with later PrGagPol cleavages via a direct interaction with PR domains. Alternatively, they may exert their effects as a consequence of their specific membrane assembly sites, or through flexibility constraints imposed on the viral proteins.
Previous reports have demonstrated that while the globular head of HIV-1 MA can exert a negative effect on virus assembly in some cellular contexts (Hatziioannou et al., 2005; Hubner and Chen, 2006; Perez-Caballero et al., 2004), large sequence insertions into MA can be accommodated for the purposes of VLP assembly and release (Liao et al., 2004; Muller et al., 2004; Wang et al., 2000). Moreover, matrix domain swapping studies have revealed that some chimeric retrovirus PrGag proteins efficiently assemble virions that can be infectious (Chen et al., 2001; Reed et al., 2002). We have extended these studies by showing that the HIV-1 assembly machinery is flexible with regard to its means of membrane association, but that alternative MBDs directly or indirectly may interfere with the elaboration of infectious virus cores.
Materials and Methods
Recombinant DNA constructs
Wild type (wt) versions of the HIV-1 gag gene, expressed in the presence of pol gene products were produced from the previously described HIVgpt construct (Page et al., 1990; Wang and Barklis, 1993; Wang et al., 1993). The myristoylation-minus (Myr- HIV) and protease-minus (PR-; HIVgpt2498T) variants of HIVgpt also have been described before (McDermott et al., 1996; Wang and Barklis, 1993; Wang et al., 1994; Zhang and Barklis, 1997). HIVgpt-MAGFP, encodes a Gag-GFP fusion protein similar to that reported by Muller et al. (2004), in which the green fluorescent protein (GFP) open reading frame (orf) has been inserted near the C-terminus of the gag matrix domain. In HIVgpt-MAGFP, the backbone construct is HIVgpt (Page et al., 1990; Wang and Barklis, 1993; Wang et al., 1993), and the source of the GFP orf was pEGFP-c1 (Clontech). The 5′ juncture sequence is GCT GCG CTA GCG CTA CCG GTC GCC ACC ATG, the first codon corresponds to HIV HXB2 gag codon 120, while the last codon is the first GFP codon. The 3′ juncture sequence is AAG TCC GGA CTC AGA TCC GCT GAC, where the first codon corresponds to the final coding codon of wt GFP, and the last codon is gag codon 121.
Alternative membrane-binding domain (MBD) constructs derived from pEGFPc1-AKTPH, pEGFPc1-PLCδPH, and GFP-pcDNA3-PKCγCys1ACys1B, which all were kindly provided by Tobias Meyer (Haugh et al., 2000; Hurley and Meyer, 2001; Marshall et al., 2001; Oancea and Meyer, 1998; Oancea et al., 1998; Stauffer et al., 1998). For GFP-AKTPH-Gag and GFP-PLCPH-Gag, the MA-truncated HXB2 gag orf was inserted C-terminal to the different pleckstrin homology (PH) domains, yielding constructs in which the pEGFPc1 cytomegalovirus (CMV) promoter drives expression of GFP-PH-Gag fusion proteins. The PH-gag juncture sequences for GFP-AKTPH-Gag and GFP-PLCPH-Gag are respectively CTG GGC CCA ACG AAT TCA TCT AGA GCT, and AAG GAG CTC GGG ATA TCT AGA GCT, where the final codon in each sequence corresponds to gag codon 120. For PKCCBD-GFP-Gag, the pcDNA3 (Invitrogen) CMV promoter drives expression of a fusion composed of PKC cysteine-rich membrane-binding domain (CBD), GFP, and the MA-truncated HIV gag orf: the GFP-gag juncture sequence is CTG TAC ACC GCG GGA TCC TCT AGA GCT, where the final codon is gag codon 120. Note that the HIV sequences in these constructs extend to the BclI site at HXBC2 nt 2432, but that PR activity is killed via fusion to a translation termination codon in an XbaI site, with juncture sequences of TAT GAT CTC TAG A.
To create the MA replacement constructs HIVgpt-AKTPH, HIVgpt-PLCPH, and HIVgpt-PKCCBD in the HIVgpt (Page et al., 1990; Wang and Barklis, 1993; Wang et al., 1993) backbone, the myristoylation-minus, gag second codon glycine-to-alanine HIVgpt mutant plasmid (Wang and Barklis, 1993; Wang et al., 1994) was used as a parent. These constructs harbor alternative MBDs between gag codons 15 and 120. The N-terminal and C-terminal juncture sequences, with codons 15 and 120 italicized are as follows: AKTPH and PLCPH N-junctures, CGA TGG GGA TCC; AKTPH and PLCPH C-junctures, TCT AGA GCT; PKCCBD N-juncture, CGA TAC ATG GGG GCC CGG TAC CTT; PKCCBD C-juncture, CGG TAC CAT GCC GCG GCC GGA TCC TCT AGA GCT. Other constructs used in these studies have been reported previously. They include the following: Blam-vpr (Munk et al., 2002; Scholz et al., 2005); the MuLV gag expression construct, pXMGPE (Hansen and Barklis, 1995; McDermott et al., 2000); the HIV Gag-β-galactosidase expression construct, HIVGBG (Wang et al., 1994); pBluescribeHIV831-680 (Wang et al., 1993; Zhang and Barklis, 1997), for RNAse protection probe production; and the glycoprotein expression constructs pVSV-G (Scholz et al., 2005), SV-A-MLVenv (Wang and Barklis, 1993; Wang et al., 1993), and HIV-env (Wang and Barklis, 1993; Wang et al., 1993).
Cells and viruses
Human embryonic kidney (HEK) 293T (Scholz et al., 2005) and HiJ (Scholz et al., 2005; Wang and Barklis, 1993; Wang et al., 1993) HeLa cells expressing human CD4 were grown at 37°C in 5% CO2 in culture medium composed of Dulbecco's modified Eagle medium (DMEM) supplemented with 10% fetal calf serum, penicillin, streptomycin, and 10 mM HEPES (pH 7.4). For transfections, 10 cm dishes of 293T cells were transfected by the calcium phosphate method (Scholz et al., 2005; Wang and Barklis, 1993; Wang et al., 1993). For Gag analysis, 24 μg of plasmid DNAs (Figure 1) were used; for infection assays, 16 μg HIV-derived DNAs were cotransfected with 8 μg envelope protein expression plasmids; for entry assays, transfections employed 12 μg of HIV-derived DNAs, plus 6 μg of pVSV-G expression plasmid (Scholz et al., 2005), plus 6 μg of BlaM-Vpr DNA (Munk et al., 2002; Scholz et al., 2005); while for Gag-β-galactosidase (Gag-β-gal) experiments, 16 μg of Gag expression plasmids were cotransfected with 8 μg of HIVGBG (Wang et al., 1994). Routinely, cell and virus samples were collected 3 d post-transfection. In some cases, the effects of phorbol esters were tested by treatment with a 1:1000 dilution of 1 mM phorbol myristate acetate (PMA; Sigma) or phorbol dibutyrate (PDB) at 24 h and again at 6 h prior to sample collection. Cell pellets were collected and washed in phosphate-buffered saline (PBS; 9.5 mM sodium potassium phosphate [pH 7.4], 137 mM NaCl, 2.7 mM KCl). Virus-containing media were filtered through 0.45 μm-pore filters (Gelman) and concentrated by centrifugation through 20% sucrose in PBS cushions (2 h at 82,500 × g [25,000 rpm in a Beckman SW28 rotor, 4 ml cushions] or 45 min at 197,000 × g [40,000 rpm, SW41 rotor, 2 ml cushions]). Virus pellets were resuspended in 0.1 ml PBS per cell plate and stored in aliquots at -80°C.
To test virus infectivities, confluent 10 cm dishes of HiJ cells were split 1:40 onto 35 mm dishes the day before infections. For infections, cells were incubated at 37°C for 72 h in the presence of filtered virus in a total volume of 2 ml culture media. Subsequently, cells were transferred to 10 cm dishes and fed with selection media containing 9.35 ml of culture media, 0.65 ml of gpt supplement solution (3.85 mg/ml xanthine, 0.046 mg/ml hypoxanthine, 0.062 mg/ml thymidine, 0.154 mg/ml glycine, 2.308 mg/ml glutamine), and 15 μl of mycophenolic acid (Gibco) per plate. Cells were selected for 10-14 d, with selection media changed at 3-4 d intervals. After selection, gpt-positive colonies were stained with 0.5 % methylene blue in 50 % methanol, gently rinsed in water and dried prior to colony counting. Infectivity was calculated as follows: Virus titer = (colonies)(virus dilution factor)(cell split ratio)/(cell proliferation between infection and selection). Results were normalized to input virus Gag protein levels.
Protein analysis
For protein analysis, cell samples (20% of pellets from each 10 cm plate) were suspended in IPB (20 mM Tris-HCl [pH 7.5], 150 mM NaCl, 1 mM ethylenediamine tetraacetic acid [EDTA], 0.1% sodium dodecyl sulfate [SDS], 0.5% sodium deoxylate, 1.0% Triton X-100, 0.02% sodium azide), incubated on ice for 5 min, vortexed, and subjected to centrifugation 13,000 × g for 15 min at 4°C. Soluble material was collected, mixed with 1 volume of 2 × sample buffer (12.5 mM Tris-HCl [pH 6.8], 2% SDS, 20% glycerol, 0.25% bromophenol blue) and 0.1 volume of β-mercaptoethanol (β-Me). For virus samples, 40 μl aliquots of resuspended virus pellets were mixed with one volume of 2 × sample buffer and 0.1 volume of β-Me. Cell and virus samples were heated to 95°C for 3 to 5 min, and subjected to SDS-polyacrylamide electrophoresis (SDS-PAGE). After SDS-PAGE fractionation, proteins were electroblotted, and immunoblotted following previously described methods (Hansen and Barklis, 1995; Jones et al., 1990; McDermott et al., 1996, 2000; Scholz et al., 2005; Wang and Barklis, 1993; Wang et al., 1993, 1994; Zhang and Barklis, 1997). The antibody used for detection of HIV-1 CA was Hy183 (kindly provided by Bruce Chesebro), used at 1:15 dilution from the hybridoma culture medium. Antibodies obtained through the NIH AIDS Research and Reference Reagent Program, Division of AIDS, NIAID, NIH were the HIV-1 RT monoclonal antibody (MAb21) from Dr. Stephen Hughes, used at 1:300; and HIV-1 protease antisera from HIVSF2 from BioMolecular Technology, used at 1:1000. Other primary antibodies were anti-VSV-G (Roche), used at 1:400; and anti-GFP (Invitrogen #A11121), used at 1:1000. In all cases, the secondary antibody was an anti-mouse IgG alkaline phosphatase-conjugated secondary antibody (Promega) was used at 1:15,000. Color reactions for visualization of antibody-bound bands employed nitroblue tetrazolium plus 5-bromo-4-chloro-3-indolyl phosphate in AP buffer (100 mM Tris-hydrochloride [pH 9.5], 100 mM NaCl, 5 mM MgCl2). For quantitation, immunoblots were air-dried and scanned using an Epson G810A scanner. Band intensities of scanned TIFF images were quantitated using NIH Image 1.61 software.
Nucleic acid analysis
For RNA analysis, viral RNA samples were obtained from virus pellets that were resuspended in 200 μl PBS, and supplemented with 2 μl of 10% SDS plus1 μl of 10 mg/ml carrier E. coli tRNA (Roche). Samples were extracted twice with 200 μl of 1:1 phenol:chloroform, extracted twice with 200 μl chloroform, ethanol-precipitated, dried, and resuspended in 50 μl of TE buffer (10 mM Tris [pH 7.8], 0.1 mM EDTA). To isolate cellular RNA samples, 10 cm dishes of cell monolayers were washed, suspended in 1 ml of GTC mix (4 M guanidium thiocyanate, 25 mM sodium citrate [pH 7.0], 0.5% sarkosyl, 0.1 M β-Me), layered onto CsCl/EDTA (6.2 M CsCl, 0.1 M EDTA [pH 7.0]) cushions, and centrifuged at 115,000 × g (35,000 rpm in a Beckman SW50.1 rotor) for 18 h at 15°C. After centrifugation, supernatants were removed, and pellets were resuspended in 400 μl TE, phenol-choloroform-extracted twice, chloroform-extracted once, ethanol-precipitated, dried, and resuspended in 100 μl TE. Samples were quantitated by measuring UV absorbance at 260 nm in a Beckman DU-64 spectrophotometer, assuming an extinction coefficient of 1 optical density (O.D.) unit per 40 ug/ml RNA at a pathlength of 1 cm.
Probes for RNase detection were made by incubation of 1 μg of EcoRI-linearized template plasmid (pBluescribeHIV831-680; Wang et al., 1993; Zhang and Barklis, 1997) with transcription buffer (40 mM Tris [pH 7.4], 10 mM DTT, 6 mM MgCl2, 0.8 mM spermidine), 100 μCi of [α-32P]rGTP, 0.5 mM each of rATP, rCTP, and rUTP, 1 μl of RNasin (Promega), 1 mM dithiothreotol [DTT], and 20 U of T3 polymerase (Promega) at 37°C for 1 h. Probes then were ethanol precipitated, dried, resuspended, separated on 5% sequencing gels (Wang et al., 1993; Zhang and Barklis, 1997), eluted, and reprecipitated prior to use (Wang et al., 1993; Zhang and Barklis, 1997). For RNAse protections, RNA-normalized cell samples or Gag-normalized viral samples were precipitated with 10 μg of tRNA. Pellets were resuspended in 30 μl of 80% formamide, 400 mM NaCl, 40 mM piperazine-N,N-bis(2-ethanesulfonic acid) (PIPES, pH 6.4), mixed with probe, incubated at 75-85°C for 5 min, and then incubated at 30°C overnight. Overnight incubation samples were supplemented with 350 μl RNase treatment buffer (300 mM NaCl, 10 mM Tris [pH 7.5], 5 mM EDTA, 40 μg/ml RNAse A [Rochel, 2 μg/ml RNAse T1 [Roche]) and incubated at 30°C for 30 min, followed by the addition of 2.5 μl of 20 mg/ml proteinase K (Boehringer) plus 20 μl of 10% SDS and further incubation at 37°C for 15 min. Samples subsequently were phenol-chloroform extracted, chloroform extracted, ethanol precipitated, dried, fractionated on 6% acrylamide sequencing gels, and autoradiographed. Viral genomic RNA bands on autoradiographs were scanned using an Epson G810A scanner, and quantitated using NIH Image 1.61 software.
For detection of one long terminal repeat (1-LTR) circles in infected cells, HiJ cells were split 1:40 onto 35-mm plates one day prior to infection. Cells on plates were infected 72 h in a total volume of 2 ml virus plus media, and then washed, collected in 500 μl PBS, pelleted, suspended in 200 μl PCR lysis buffer (10 mM Tris [pH 8.3], 50 mM KCl, 1.5 mM MgCl2, 0.01% gelatin, 0.45% NP-40, 0.45% Tween-20, 100 μg/ml proteinase K [Sigma]), incubated at 55°C 2-20 h, heated to 100°C for 10 min, and then stored at -80°C. One and two LTR circles in 0-4 μl lysate samples were amplified by polymerase chain reaction (PCR). Reactions employed Taq DNA polymerase (New England Biolabs; NEB) plus primers corresponding to HIV-1 HXB2 nucleotides 9038-9058 and 1221-1202, and were performed for 35-45 cycles with cycle times of 95°C 1 min, 55 °C 1 min, 72 °C 2.5 min. Aliquots of PCR reactions were fractionated electrophoretically on horizontal 0.9% agarose-TBE (89 mM Tris, 89 mM boric acid, 2 mM EDTA, pH 8.2) gels in parallel with Lambda HindIII and PhiX HaeIII DNA size standards, and stained with ethidium bromide. Stained DNA bands were imaged under UV light using a Kodak EDAS 290 Gel Photo system, and 1-LTR circle products were quantitated as a marker for reverse transcription in infected cells (Butler et al., 2001) using NIH Image 1.61 software.
Enzyme assays
Exogenous reverse transcriptase (RT) assays were performed with poly(A) and oligo(dT) templates and primers and detergent-disrupted virions (Scholz et al., 2005; Wang and Barklis, 1993). Gag-normalized virus samples in PBS were incubated at 37°C for 2 h in RT assay cocktail (50 mM Tris [pH 8.3], 20 mM dithiothreitol [DTT], 0.6 mM MnCl2, 60 mM NaCl, 0.05% NP-40, 2.5 μg/ml of oligo[dT] [Pharmacia], 10 μg/ml of poly[A][Pharmacia], 10 μM dTTP [1 Ci/mM {α-32P}dTTP]). Dilutions of avian myeloblastosis virus (AMV) RT (Roche) were run in parallel as controls. Following incubations, samples were precipitated by addition of 0.1 volume of 100% trichloroacetic acid (TCA) and incubated overnight at 4°C. TCA precipitates were pelleted by centrifugation for 10 min at 13,600 × g at 4°C, washed five times with 10% TCA, and quantitated in a scintillation counter (Beckman).
For β-galactosidase (β-gal) assays, cells and viruses were collected from 10 cm plates and processed as described above. Sample aliquots (half of each virus sample, or 10% of each cell sample) in 150 μl PBS were mixed with 1.5 μl of 10% SDS and 600 μl of PM2 buffer (33 mM NaH2PO4, 66 mM Na2HPO4, 0.1 mM MnCl2, 2 mM MgSO4, 40 mM β-Me), and vortexed. Reactions were initiated by the addition of 150 μl of 4 mg/ml o-nitrophenyl β-D-galactopyranoside (ONPG; Sigma) in PM2; incubations proceeded at 37°C until color changes were observed; reactions were quenched with 375 μl of 1 M Na2CO3; and β-gal activities were calculated from 420 nm absorbance readings as described previously (Jones et al., 1990; Wang et al.,1994).
Enzymatic entry assays followed previously reported protocols and scored for β-lactamase activity delivered to target cells by BlaM-Vpr fusion proteins (Munk et al., 1002; Scholz et al., 2005). Confluent 10 cm dishes of HiJ cells were split 1:40 onto 35-mm dishes the day before infections. Cells were infected overnight at 37°C in a total volume of 2 ml cell culture media. After infections, supernatants were removed, cells were washed with Hank's balanced salt solution (HBSS) without calcium or magnesium, and then incubated in 1 ml CCF2/AM loading solution (2 μM CCF2/AM; Invitrogen) at 26°C and in 5% CO2 for 4 h. Loading solution then was removed, cells were washed in HBSS, trypsinized, pelleted, fixed in 1% paraformaldehyde in PBS for 20 min, pelleted, washed in HBSS, pelleted, and resuspended in 500 μl HBSS. For analysis, fluorescence profiles of infected cells were monitored on a Becton-Dickinson Turbo Vantage flow cytometer to quantitate cleaved product as blue fluorescence and uncleaved substrate as green fluorescence signals. Cells were gated on the infected, unstained control samples, and the percentages of cells positive for product were normalized to Gag protein levels in input virus samples.
Microscopy
To examine GFP-positive virus particles, 20 μl aliquots of suspended virus particle preparations were deposited on microscope slides, covered with 22 mm × 22 mm coverslips (Fisher), viewed on a Zeiss Axioplan fluorescence microscope, and imaged using Improvision OpenLab software. For fluorescence microscopy of transfected cells, 22 mm × 22 mm coverslips in 6 well dishes were pretreated with 0.7% porcine gelatin (Sigma) at 4°C for 30 minutes, after which gelatin was replaced with media. Transfected cells were split 1:40 or 1:80 from 10 cm plates onto coverslips one day post-transfection, incubated for another two days, and then either mounted for GFP fluorescence or processed for immunofluorescence and then mounted. For GFP fluorescence, cells were washed once in PBS, fixed in 4% paraformaldehyde (Sigma) in PBS at room temperature for 1 h, washed in PBS, and mounted on slides (Fisher) in Fluoro-G mounting medium (Southern Biotech). For immunofluorescence, cells were fixed and washed as above, and then permeabilized in 0.2% Triton X-100 in PBS at room temperature for 10 min, washed once, and incubated in culture medium for 10 min. Subsequently, anti-HIV-1 CA primary antibody (Hy183, from Bruce Chesebro) in culture medium or medium alone for negative controls was added, and cells were incubated at 37°C for 1 h, rocking every 15 min. After primary antibody incubations, coverslips were washed three times in culture medium, then secondary antibody (anti-mouse AlexaFluor 594; Invitrogen) diluted 1:500 in media was added, and the samples were incubated at 37°C for 30 min, rocking once at 15 min. Following incubations, the cells were washed twice in culture medium and three times in PBS, followed by mounting onto microscope slides in Fluoro-G mounting medium. Samples were viewed on a Zeiss Axioplan fluorescence microscope, and photographed using Improvision OpenLab software. For quantitation of cell surface staining, rectangular sections from cell centers to cell edges taken from 10 cells per sample were cut and analyzed. To do so, brightness intensity signals ([number of pixels] × [pixel brightness – backround brightness]) from the outermost 20% of sampled areas were divided by brightness intensity signals from the entire sample areas. These ratios, multiplied by 100 were designated as percentages of cell surface staining.
Electron microscopy (EM) of negatively stained virus particles was performed as described by Scholz et al. (2005). Concentrated virus particle samples were lifted for 2 min onto carbon-coated UV-treated EM grids, rinsed for 15 s in water, stained for 1 min in filtered 1.3% uranyl acetate, wicked, and dried. EM images were collected on a Philips CM120/Biotwin TEM equipped with a Gatan 794 multiscan charge-coupled device (CCD) camera. Virus particle diameters were determined with the aid of Gatan digital micrograph software, and particles were scored as having conical or cylindrical cores if projection images showed negatively stained triangular, rectangular, or trapezoidal structures. For EM of cells, transfected cells were fixed, postfixed, prestained, dehydrated, infiltrated, embedded, sectioned and stained as described by Arvidson et al. (2003). EM was performed on a Philips CM120/Biotwin, and 1024 pixel × 1024 pixel images were collected electronically on a Gatan 794 multiscan CCD camera.
Acknowledgments
We are grateful to Tobias Meyer (Stanford) for the AKTPH, PLCPH and PKCCBD plasmids, and advice concerning their use. We also appreciate the assistance and advice from lab members Ayna Alfadhli, Ben Kukull, and Carolyn McQuaw. This work was supported by National Institutes of Health Grants GM060170 and AI071798 to EB.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Arvidson B, Seeds J, Webb M, Finlay L, Barklis E. Analysis of the retrovirus capsid interdomain linker region. Virology. 2003;308:166–77. doi: 10.1016/s0042-6822(02)00142-3. [DOI] [PubMed] [Google Scholar]
- Batonick M, Favre M, Boge M, Spearman P, Honing S, Thali M. Interaction of HIV-1 Gag with the clathrin-associated adaptor AP-2. Virology. 2005;342:190–200. doi: 10.1016/j.virol.2005.08.001. [DOI] [PubMed] [Google Scholar]
- Booth AM, Fang Y, Fallon JK, Yang JM, Hildreth JE, Gould SJ. Exosomes and HIV Gag bud from endosome-like domains of the T cell plasma membrane. J Cell Biol. 2006;172:923–35. doi: 10.1083/jcb.200508014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brugger B, Glass B, Haberkant P, Leibrecht I, Wieland F, Krausslich HG. The HIV lipidome: a raft with an unusual composition. Proc Natl Acad Sci USA. 2006;103:2641–2646. doi: 10.1073/pnas.0511136103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryant M, Ratner L. Myristoylation-dependent replication and assembly of human immunodeficiency virus 1. Proc Natl Acad Sci U S A. 1990;87:523–7. doi: 10.1073/pnas.87.2.523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler SL, Hansen MS, Bushman FD. A quantitative assay for HIV DNA integration in vivo. Nat Med. 2001;7:631–4. doi: 10.1038/87979. [DOI] [PubMed] [Google Scholar]
- Chen B, Rousso I, Shim S, Kim P. Efficient assembly of an HIV-1/MLV Gag-chimeric virus in murine cells. Proc Natl Acad Sci USA. 2001;98:15239–15244. doi: 10.1073/pnas.261563198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colon-Gonzalez F, Kazanietz MG. C1 domains exposed: from diacylglycerol binding to protein-protein interactions. Biochim Biophys Acta. 2006;1761:827–37. doi: 10.1016/j.bbalip.2006.05.001. [DOI] [PubMed] [Google Scholar]
- Davis MR, Jiang J, Zhou J, Freed EO, Aiken C. A mutation in the human immunodeficiency virus type 1 Gag protein destabilizes the interaction of the envelope protein subunits gp120 and gp41. J Virol. 2006;80:2405–17. doi: 10.1128/JVI.80.5.2405-2417.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong X, Li H, Derdowski A, Ding L, Burnett A, Chen X, Peters T, Dermody T, Woodruff E, Wang J, Spearman P. AP-3 directs the intracellular trafficking of HIV-1 Gag and plays a key role in particle assembly. Cell. 2005;120:663–674. doi: 10.1016/j.cell.2004.12.023. [DOI] [PubMed] [Google Scholar]
- Dorfman T, Mammano F, Haseltine WA, Gottlinger HG. Role of the matrix protein in the virion association of the human immunodeficiency virus type 1 envelope glycoprotein. J Virol. 1994;68:1689–96. doi: 10.1128/jvi.68.3.1689-1696.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards AS, Newton AC. Regulation of protein kinase C betaII by its C2 domain. Biochemistry. 1997;36:15615–23. doi: 10.1021/bi9718752. [DOI] [PubMed] [Google Scholar]
- Facke M, Janetzko A, Shoeman RL, Krausslich HG. A large deletion in the matrix domain of the human immunodeficiency virus gag gene redirects virus particle assembly from the plasma membrane to the endoplasmic reticulum. J Virol. 1993;67:4972–80. doi: 10.1128/jvi.67.8.4972-4980.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freed EO, Martin MA. Virion incorporation of envelope glycoproteins with long but not short cytoplasmic tails is blocked by specific, single amino acid substitutions in the human immunodeficiency virus type 1 matrix. J Virol. 1995;69:1984–9. doi: 10.1128/jvi.69.3.1984-1989.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freed EO, Martin MA. Domains of the human immunodeficiency virus type 1 matrix and gp41 cytoplasmic tail required for envelope incorporation into virions. J Virol. 1996;70:341–51. doi: 10.1128/jvi.70.1.341-351.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen MS, Barklis E. Structural interactions between retroviral Gag proteins examined by cysteine cross-linking. J Virol. 1995;69:1150–9. doi: 10.1128/jvi.69.2.1150-1159.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatziioannou T, Martin-Serrano J, Zang T, Bieniasz PD. Matrix-induced inhibition of membrane binding contributes to human immunodeficiency virus type 1 particle assembly defects in murine cells. J Virol. 2005;79:15586–9. doi: 10.1128/JVI.79.24.15586-15589.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haugh J, Codazzi F, Teruel M, Meyer T. Spatial sensing in fibroblasts mediated by 3′ phosphoinositides. J Cell Biol. 2000;151:1269–1279. doi: 10.1083/jcb.151.6.1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hermida-Matsumato L, Resh M. Human immunodeficiency virus type 1 protease triggers a myristoyl switch that modulates membrane binding of Pr55Gag and p17MA. J Virol. 1999;73:1902–1908. doi: 10.1128/jvi.73.3.1902-1908.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hockley DJ, Wood RD, Jacobs JP, Garrett AJ. Electron microscopy of human immunodeficiency virus. J Gen Virol. 1988;69(Pt 10):2455–69. doi: 10.1099/0022-1317-69-10-2455. [DOI] [PubMed] [Google Scholar]
- Holm K, Weclewicz K, Hewson R, Suomalainen R. HIV-1 assemblyand lipid rafts: Pr55Gag complexes associate with membrane-domains that are largely resistant to Brij 98, but sensitive to Triton X-100. J Virol. 2003;77:4805–4817. doi: 10.1128/JVI.77.8.4805-4817.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubner W, Chen BK. Inhibition of viral assembly in murine cells by HIV-1 matrix. Virology. 2006;352:27–38. doi: 10.1016/j.virol.2006.04.024. [DOI] [PubMed] [Google Scholar]
- Hurley JH, Meyer T. Subcellular targeting by membrane lipids. Current Opinion in Cell Biology. 2001;13:146–152. doi: 10.1016/s0955-0674(00)00191-5. [DOI] [PubMed] [Google Scholar]
- Johnston JA, Ward CL, Kopito RR. Aggresomes: a cellular response to misfolded proteins. J Cell Biol. 1998;143:1883–98. doi: 10.1083/jcb.143.7.1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones T, Blaug G, Hansen M, Barklis E. Assembly of gag-B galactosidase proteins into retrovirus particles. J Virol. 1990;64:2265–2279. doi: 10.1128/jvi.64.5.2265-2279.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jouvenet N, Neil SJ, Bess C, Johnson MC, Virgen CA, Simon SM, Bieniasz PD. Plasma membrane is the site of productive HIV-1 particle assembly. PLoS Biol. 2006;4:e435. doi: 10.1371/journal.pbio.0040435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemmon MA. Phosphoinositide recognition domains. Traffic. 2003;4:201–13. doi: 10.1034/j.1600-0854.2004.00071.x. [DOI] [PubMed] [Google Scholar]
- Liao WH, Chiu HC, Wang CT. Effects of mutations in an HIV-1 gag gene containing a 107-codon tandem repeat in the matrix region on assembly and processing of the protein product. J Med Virol. 2004;74:528–35. doi: 10.1002/jmv.20209. [DOI] [PubMed] [Google Scholar]
- Lindwasser O, Resh M. Multimerization of human immunodeficiency virus type 1 Gag promotes its localization to barges, raft-like membrane microdomains. J Virol. 2001;75:7913–7924. doi: 10.1128/JVI.75.17.7913-7924.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindwasser OW, Resh MD. Myristoylation as a target for inhibiting HIV assembly: unsaturated fatty acids block viral budding. Proc Natl Acad Sci U S A. 2002;99:13037–42. doi: 10.1073/pnas.212409999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindwasser O, Resh M. Human immunodeficiency virus type 1 Gag contains a dileucine-like mitif that regulates association with multivesicular bodies. J Virol. 2004;78:6013–6023. doi: 10.1128/JVI.78.11.6013-6023.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Verges S, Camus G, Blot G, Beauvoir R, Benarous R, Berlioz-Torrent C. Tail-interacting protein TIP47 is a connector between Gag and Env and is required for Env incorporation into HIV-1 virions. Proc Natl Acad Sci USA. 2006;103:14947–14952. doi: 10.1073/pnas.0602941103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mammano F, Kondo E, Sodroski J, Bukovsky A, Gottlinger HG. Rescue of human immunodeficiency virus type 1 matrix protein mutants by envelope glycoproteins with short cytoplasmic domains. J Virol. 1995;69:3824–30. doi: 10.1128/jvi.69.6.3824-3830.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall J, Booth J, Stambolic V, Mak T, Balla T, Schreiber A, Meyer T, Grinstein S. Restricted accumulation of phosphatidylinositol 3-kinase products in a plasmalemmal subdomain during Fcγ receptor-mediated phagocytosis. J Cell Biol. 2001;153:1369–1380. doi: 10.1083/jcb.153.7.1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDermott J, Farrell L, Ross R, Barklis E. Structural analysis of human immunodeficiency virus type 1 Gag protein interactions, using cysteine-specific reagents. J Virol. 1996;70:5106–14. doi: 10.1128/jvi.70.8.5106-5114.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDermott J, Karanjia S, Love Z, Barklis E. Crosslink analysis of N-terminal, C-terminal, and N/B determining regions of the Moloney murine leukemia virus capsid protein. Virology. 2000;269:190–200. doi: 10.1006/viro.2000.0212. [DOI] [PubMed] [Google Scholar]
- Muller B, Daecke J, Fackler OT, Dittmar MT, Zentgraf H, Krausslich HG. Construction and characterization of a fluorescently labeled infectious human immunodeficiency virus type 1 derivative. J Virol. 2004;78:10803–13. doi: 10.1128/JVI.78.19.10803-10813.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munk C, Brandt SM, Lucero G, Landau NR. A dominant block to HIV-1 replication at reverse transcription in simian cells. Proc Natl Acad Sci U S A. 2002;99:13843–8. doi: 10.1073/pnas.212400099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray P, Li L, Wang J, Tang C, Honig B, Murray D. Retroviral matrix domains share electrostatic homology: models for membrane binding function throughout the viral life cycle. Structure. 2005;13:1521–1531. doi: 10.1016/j.str.2005.07.010. [DOI] [PubMed] [Google Scholar]
- Muthumani K, Montaner LJ, Ayyavoo V, Weiner DB. Vpr-GFP virion particle identifies HIV-infected targets and preserves HIV-1Vpr function in macrophages and T-cells. DNA Cell Biol. 2000;19:179–88. doi: 10.1089/104454900314564. [DOI] [PubMed] [Google Scholar]
- Nguyen D, Hildreth J. Evidence for budding of human immunodeficiency virus type 1 selectively from glycolipid-enriched membrane lipid rafts. J Virol. 2000;74:3264–3272. doi: 10.1128/jvi.74.7.3264-3272.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nydegger S, Foti M, Derdowski A, Spearman P, Thali M. HIV-1 egress is gated through late endosomal membranes. Traffic. 2003;4:902–910. doi: 10.1046/j.1600-0854.2003.00145.x. [DOI] [PubMed] [Google Scholar]
- Oancea E, Meyer T. Protein kinase C as a molecular machine for decoding calcium and diacylglycerol signals. Cell. 1998;95:307–18. doi: 10.1016/s0092-8674(00)81763-8. [DOI] [PubMed] [Google Scholar]
- Oancea E, Teruel MN, Quest AF, Meyer T. Green fluorescent protein (GFP)-tagged cysteine-rich domains from protein kinase C as fluorescent indicators for diacylglycerol signaling in living cells. J Cell Biol. 1998;140:485–98. doi: 10.1083/jcb.140.3.485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ono A, Ablan S, Lockett S, Nagashima K, Freed E. Phosphatidylinositol (4,5) bisphosphate regulates HIV-1 Gag targeting to the plasma membrane. Proc Natl Acad Sci USA. 2004;101:14889–14894. doi: 10.1073/pnas.0405596101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ono A, Demirov D, Freed E. Relationship between human immunodeficiency virus type 1 Gag multimerization and membrane binding. J Virol. 2000;74:5142–5150. doi: 10.1128/jvi.74.11.5142-5150.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ono A, Freed E. Binding of human immunodeficiency virus type 1 Gag to membrane: role of the matrix amino terminus. J Virol. 1999;73:4136–4144. doi: 10.1128/jvi.73.5.4136-4144.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ono A, Freed E. Plasma membrane rafts play a critical role in HIV-1 assembly and release. Proc Natl Acad Sci USA. 2001;98:13925–13930. doi: 10.1073/pnas.241320298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ono A, Freed EO. Cell-type-dependent targeting of human immunodeficiency virus type 1 assembly to the plasma membrane and the multivesicular body. J Virol. 2004;78:1552–63. doi: 10.1128/JVI.78.3.1552-1563.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ono A, Huang M, Freed E. Characterization of human immunodeficiency virus type 1 matrix revertants: effects on virus assembly, Gag processing, and Env incorporation into virions. J Virol. 1997;71:4409–4418. doi: 10.1128/jvi.71.6.4409-4418.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ono A, Orenstein JM, Freed EO. Role of the Gag matrix domain in targeting human immunodeficiency virus type 1 assembly. J Virol. 2000;74:2855–66. doi: 10.1128/jvi.74.6.2855-2866.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Page KA, Landau NR, Littman DR. Construction and use of a human immunodeficiency virus vector for analysis of virus infectivity. J Virol. 1990;64:5270–6. doi: 10.1128/jvi.64.11.5270-5276.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paillart JC, Gottlinger HG. Opposing effects of human immunodeficiency virus type 1 matrix mutations support a myristyl switch model of gag membrane targeting. J Virol. 1999;73:2604–12. doi: 10.1128/jvi.73.4.2604-2612.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pal R, Reitz MS, Jr, Tschachler E, Gallo RC, Sarngadharan MG, Veronese FD. Myristoylation of gag proteins of HIV-1 plays an important role in virus assembly. AIDS Res Hum Retroviruses. 1990;6:721–30. doi: 10.1089/aid.1990.6.721. [DOI] [PubMed] [Google Scholar]
- Pettit S, Everitt L, Choudhury S, Dunn B, Kaplan A. Initial cleavage of the human immunodeficiency virus type 1 GagPol precursor by its activated protease occurs by an intramolecular mechanism. J Virol. 2004;78:8477–8485. doi: 10.1128/JVI.78.16.8477-8485.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettit S, Clemente J, Jeung J, Dunn B, Kaplan A. Ordered processing of the human immunodeficiency virus type 1 GagPol precursor is influenced by the context of the embedded viral protease. J Virol. 2005;79:10601–10607. doi: 10.1128/JVI.79.16.10601-10607.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Caballero D, Hatziioannou T, Martin-Serrano J, Bieniasz PD. Human immunodeficiency virus type 1 matrix inhibits and confers cooperativity on gag precursor-membrane interactions. J Virol. 2004;78:9560–3. doi: 10.1128/JVI.78.17.9560-9563.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reed M, Mariani R, Sheppard L, Pekrun K, Landau NR, Soong NW. Chimeric human immunodeficiency virus type 1 containing murine leukemia virus matrix assembles in murine cells. J Virol. 2002;76:436–43. doi: 10.1128/JVI.76.1.436-443.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reil H, Bukovsky AA, Gelderblom HR, Gottlinger HG. Efficient HIV-1 replication can occur in the absence of the viral matrix protein. EMBO J. 1998;17:2699–708. doi: 10.1093/emboj/17.9.2699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saad JS, Miller J, Tai J, Kim A, Ghanam RH, Summers MF. Structural basis for targeting HIV-1 Gag proteins to the plasma membrane for virus assembly. Proc Natl Acad Sci U S A. 2006;103:11364–9. doi: 10.1073/pnas.0602818103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scholz I, Arvidson B, Huseby D, Barklis E. Virus particle core defects caused by mutations in the human immunodeficiency virus capsid N-terminal domain. J Virol. 2005;79:1470–9. doi: 10.1128/JVI.79.3.1470-1479.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spearman P, Horton R, Ratner L, Kuli-Zade I. Membrane binding of human immunodeficiency virus type 1 matrix protein in vivo supports a conformational myristyl switch mechanism. J Virol. 1997;71:6582–6592. doi: 10.1128/jvi.71.9.6582-6592.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stauffer T, Ahn S, Meyer T. Receptor-induced transient reduction in plasma membrane PtdIns(4,5)P2 concentration monitored in living cells. Curr Biology. 1998;8:343–346. doi: 10.1016/s0960-9822(98)70135-6. [DOI] [PubMed] [Google Scholar]
- Tang C, Loeliger E, Luncsford P, Kinde I, Beckett D, Summers MF. Entropic switch regulates myristate exposure in the HIV-1 matrix protein. Proc Natl Acad Sci U S A. 2004;101:517–22. doi: 10.1073/pnas.0305665101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang Y, Winkler U, Freed E, Torrey T, Kim W, Li H, Goff S, Morse H. Cellular motor protein KIF-4 associates with retroviral Gag. J Virol. 1999;73:10508–10513. doi: 10.1128/jvi.73.12.10508-10513.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang CT, Barklis E. Assembly, processing, and infectivity of human immunodeficiency virus type 1 gag mutants. J Virol. 1993;67:4264–73. doi: 10.1128/jvi.67.7.4264-4273.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang CT, Chen S, Chiang C. Assembly and release of human immunodeficiency virus type 1 Gag proteins containing tandem repeats of the matrix protein coding sequences in the matrix domain. Virology. 2000;278:289–98. doi: 10.1006/viro.2000.0655. [DOI] [PubMed] [Google Scholar]
- Wang CT, Stegeman-Olsen J, Zhang Y, Barklis E. Assembly of HIV GAG-B-galactosidase fusion proteins into virus particles. Virology. 1994;200:524–34. doi: 10.1006/viro.1994.1215. [DOI] [PubMed] [Google Scholar]
- Wang CT, Zhang Y, McDermott J, Barklis E. Conditional infectivity of a human immunodeficiency virus matrix domain deletion mutant. J Virol. 1993;67:7067–76. doi: 10.1128/jvi.67.12.7067-7076.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyma D, Kotov A, Aiken C. Evidence for a stable interaction of gp41 with Pr55(Gag) in immature human immunodeficiency virus type 1 particles. J Virol. 2000;74:9381–7. doi: 10.1128/jvi.74.20.9381-9387.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu X, Yuan X, Matsuda Z, Lee TH, Essex M. The matrix protein of human immunodeficiency virus type 1 is required for incorporation of viral envelope protein into mature virions. J Virol. 1992;66:4966–4971. doi: 10.1128/jvi.66.8.4966-4971.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Barklis E. Effects of nucleocapsid mutations on human immunodeficiency virus assembly and RNA encapsidation. J Virol. 1997;71:6765–76. doi: 10.1128/jvi.71.9.6765-6776.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou W, Parent LJ, Wills JW, Resh MD. Identification of a membrane-binding domain within the amino-terminal region of human immunodeficiency virus type 1 Gag protein which interacts with acidic phospholipids. J Virol. 1994;68:2556–69. doi: 10.1128/jvi.68.4.2556-2569.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]