

Abstract
Following activation by unique cytokines, CD4+ naïve T cells differentiate into lineages of helper/effector (Th) and regulatory T cells (Treg) that are characterized by distinct developmental pathways and unique biological functions. The trusted binary system of Th1 and Th2 has been expanded to include the IL-17 producing Th17 cell lineage, which plays a role in immune responses to infectious agents and maintenance of autoimmune diseases. Acting as counterbalance, Tregs maintain peripheral tolerance and protect the host from autoaggressive lymphocytes. Th1 cells produce IFN-γ and are involved in cell-mediated immunity; Th2 cells produce IL-4 and contribute to humoral immunity; Th17 cells generate IL-17 and play an important role in immune responses to fungi and extracellular pathogens; FOXP3+ Tregs secrete TGF-β and IL-10 and downregulate effector T cells. Autosomal dominant hyper IgE syndrome, a rare Primary Immunodeficiency Disorder, is caused by hypomorphic heterozygous mutations of STAT3, preventing Th17 lineage differentiation, and increasing susceptibility to staphylococcus and Candida infections. Mutations in FOXP3 interfere with Treg development and cause Immune dysregulation, Polyendocrinopathy, Enteropathy, X-Linked (IPEX). Other single gene defects resulting in reduced Treg function include CD25, STAT5b, AIRE, and WASP. These observations emphasize the importance of functionally distinct T cell lineages in maintaining a balanced innate and cognate immune system.
Keywords: Regulation of T effector cell lineage differentiation, Th17 cells, Regulatory T cells, Immune Dysregulation, Polyendocrinopathy, Enteropathy, X-Linked (IPEX), FOXP3, Autosomal-dominant Hyper IgE syndrome, STAT3, IL-17, TGF-β, IL-10
INTRODUCTION
Based on their pioneering work, Mosmann and Coffman proposed some 20 years ago that T helper cells could be divided into two distinct subsets, T helper type 1 (Th1) and Th2, characterized by distinct cytokine profiles and effector functions (1). Th1 cells produce large quantities of interferon (IFN)-γ, elicit delayed type hypersensitivity (DTH) responses, activate macrophages and are highly effective in clearing intracellular pathogens. Th2 cells, on the other hand, produce interleukin 4 (IL-4), IL-5, IL-13 and IL-25, and are important for IgE production, eosinophilic inflammation and the clearance of helminthic parasite infections (2). In light of recent data, the Th1/Th2 dichotomy is now being revisited. The discovery of the IL-17 family of cytokines and the analysis of IL-23-mediated effector functions on T cells have suggested the existence of an additional subset of CD4+ T cells that produce IL-17 and for this reason were designated Th17 cells (3–6). The independence of the Th17 subset with regard to Th1 and Th2 cells was firmly established with the identification of specific cytokines and transcription factors required for lineage differentiation, i.e. the combination of IL-6 and Transforming Growth Factor-β (TGF-β) (7–9), and the transcription factors, RORγt (10) and STAT3 (11, 12). Th17 effector functions are distinct from Th1 and Th2-mediated immunity. Th17 cells appear to be critical to enhance host protection against extracellular bacteria and fungi, which are not efficiently cleared by Th1 and Th2 responses. In addition, Th17 cells have emerged as potent mediators of autoimmune disease.
Including the regulatory T cell (Treg) subset (13), there are now four functionally unique populations of CD4+ T cells that are directly involved in the regulation of immune responses to pathogens, to allergens and to self-antigens. Any molecular defect involving either the entire CD4+ T cell population, e.g. Severe Combined Immune Deficiency (SCID), or individual subsets, e.g. lack of Treg cells (14) or IL-17 cells (15), may result in human disease (Table 1). In this review, we explore the biology of Th17 and Treg cells and their roles in human primary immune deficiency diseases.
Table 1.
Characteristic Features and Disease Association of CD4+ T Cell Subsets
| CD4+ T cell subsets | ||||
|---|---|---|---|---|
| Characteristic properties | Th1 | Th2 | Th17 | iTreg |
| Signature cytokines | IFN-γ | IL-4 | IL-17, IL-17F | TGF-β |
| additional cytokines produced | lymphotoxin α, IL-2 | IL-5, IL-13, IL-25 | IL-21, IL-22 | IL-10, IL-35 |
| autocrine cytokines | IFN-γ | IL-4 | IL-21 | TGF-β |
| STAT regulators | STAT1, STAT4 | STAT6, STAT5 | STAT3 | STAT5 |
| lineage specific transcription factors | T-bet | GATA-3 | RORγt | FOXP3 |
| cytokine/chemokine receptors | IL-12R, IL-18Rα, CXCR3 | IL-4Rα, IL-33Rα, CCR3, CCR4, CCR8 | IL-23R, CCR6, CCR4 | CD25 (IL-2Rα) |
| Associated Primary Immunodeficiency Diseases | IFN-γR1/2 def IL-12/23Rβ1 def IL-12p40 def STAT1 def |
Omenn Syndrome Allergic diathesis Overexpression of Th2 cells |
AD-HIES (STAT3 def) IL-12/23Rβ1def IL-12p40 def |
IPEX (FOXP3 def) IPEX-like/SCID (CD25/IL-2Rα def) Laron dwarfism (STAT5β def) WAS (WASP) APECED (AIRE) |
| T- SCID due to mutations of γc, JAK3, IL-7Rα, CD45, CD3δ/CD3ε/CD3ζ, RAG1/2, Artemis, ADA | ||||
IPEX: Immunodeficiency, Polyendocrinopathy, and Enteropathy, X-Linked Syndrome
WAS: Wiskott-Aldrich Syndrome; WASP: WAS Protein
APECED: Autoimmune Polyendocrinopathy, Candidiasis and Ectodermal Dystrophy
DIFFERENTIATION AND FUNCTION OF TH17 CELLS
Since their discovery, Th17 cells have been recognized as a unique effector T cell subset capable of producing IL-17, a cytokine originally cloned in 1995 (16). IL-17 induces stromal cells to produce pro-inflammatory and hematopoietic cytokines (17) and initiates the recruitment of neutrophils, linking adaptive and innate immunity (18).
Initial studies of Th17 cell biology, performed in mice, focused on identifying key factors required for the differentiation and function of Th17 cells. Early investigations of Th17 cell development in humans suggested that it may differ from that observed in the mouse (19–21), more recent reports however, suggest that major events controlling Th17 cell development are similar in both species (22–24).
To become Th17 cells, naïve murine CD4+ T cells have to be activated via the T cell receptor in the presence of TGF-β and IL-6, which leads to the expression of the transcription factor retinoic acid-related orphan receptor γt (RORγt) (10). Just as IFN-γ, IL-12 and T-bet control Th1 development, and IL-4 and GATA3 control Th2 development, TGF-β, IL-6 and RORγt drive naïve CD4+ T cells towards the Th17 lineage, at least in part, by directly inducing the expression of IL-17(25). The effects of IL-6 on Th17 cell differentiation are mediated by the transcription factor STAT3, which is required for RORγt expression (11, 12, 26) (Table 1). In patients with autosomal dominant Hyper IgE Syndrome (AD-HIES) due to heterozygous STAT3 mutations that cause the generation of non-functional STAT3, RORγt expression and Th17 cell development is severely impaired (15, 27).
In human effector T cell differentiation, TGF-β and IL-6 are important in the generation of Th17 cells but IL-1β also appears to play a prominent role in the induction of RORγt. This is further enhanced by IL-23 (19, 20). In mice, IL-23 seems to play a role only in activated T cells that express the IL-23R and therefore may induce Th17 differentiation only in memory but not in naïve T cells (28), suggesting that IL-23 upregulates IL-17 production and promotes survival and expansion of activated memory Th17 cells. If this assumption is correct, IL-23 must be crucial for the maintenance of autoimmune inflammation (4, 29). A recent in depth analysis has concluded that TGF-β, IL-23 and the pro-inflammatory cytokines IL-1β and IL-6 are in fact, essential mediators of human Th17 cell differentiation and are required for the expression of IL-17, IL-23R and RORγt (23). These observations were confirmed by the Littman lab, which reported that human cord blood CD4+ T cells, naïve by definition, differentiate into Th17 cells only if TGF-β, IL-1β, IL-6 and IL-23 or IL-21 are present, and that this process requires the expression of RORγt, but not T-bet or GATA3 (22). These studies demonstrate that TGF-β and IL-6 are important for Th17 development in both humans and mice while IL-1β and IL-23 play a more important role in men than mice.
CYTOKINE PRODUCTION BY TH17 CELLS
The Th17 signature cytokines, IL-17 (IL-17A) and IL-17F, are closely related and form biologic active homo- or heterodimers. By interacting with its receptor, IL-17 initiates NF-κB activation, which leads to the transcription of multiple target genes involved in innate immunity. These include chemokines such as CXCL8 (IL-8) and CCL20, the cytokines IL-6, TNF-α, G-CSF and GM-CSF, acute phase proteins such as C-reactive protein, and anti-microbial peptides and mucins (30). Thus, IL-17 plays an important role in anti-microbial defenses by recruiting and expanding the neutrophil lineage and producing anti-microbial factors. In addition, antibody responses to T-dependent antigens are defective in IL-17 deficient mice (31) and in patients with AD-HIES that lack Th17 cells (32).
In addition to IL-17, activated murine Th17 cells produce IL-21, which appears to play an important autocrine role in maintaining Th17 cell differentiation, similar to the autocrine function of IFN-γ in the generation of Th1 cells and IL-4 in promoting Th2 cells (Table 1 and Figure 1). In mice, IL-21 expression is under the control of STAT3, which binds to the IL-21 promoter in Th17 cells. While IL-21 production is RORγt independent, IL-21 itself helps to sustain expression of RORγt and IL-17 and induces IL-23 receptor expression (33–35). A role for IL-21 in the differentiation of human Th17 cells is less clear, although is likely, based on the observation that IL-21 upregulates IL-17 production and downregulates Treg function (36, 37).
Fig. 1.
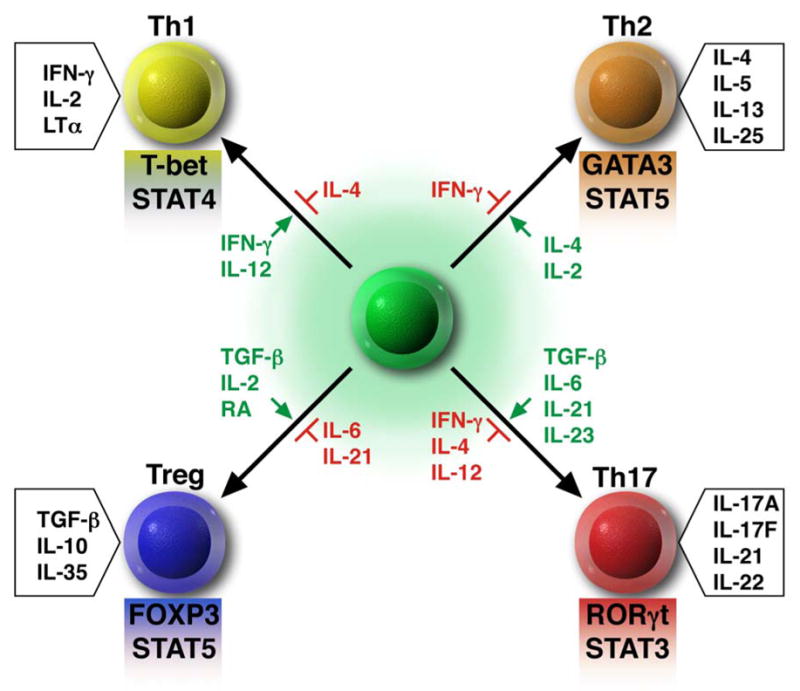
Model of CD4+ T cell differentiation through the use of cytokine and transcription factor driven pathways. Each of the four CD4+ T cell subsets generates a unique set of cytokines that control their biologic functions. Green = cytokines that promote development of each particular subset. Red = cytokines that prevent the development of each particular subset. Shaded boxes below each subset = key transcription factors involved in the development and maintenance of each subset. RA = retinoic acid. LTα = Lymphotoxin α.
Other pro-inflammatory cytokines produced by human and murine Th17 cells include TNF-α, IL-22 and IL-26, which are involved in innate immunity, and IL-6 which directs CD4+ T cell differentiation towards the Th17 lineage as discussed above(38). IL-22 has been associated with the generation of defensins, acute phase proteins and inflammatory cytokines (30, 39). Some subsets of Th17 cells may co-express IL-17 and IFN-γ (19) or IL-17 and IL-10 (40) respectively; however, the function of these “double positive” T cells remains to be determined. It has been suggested, based on mouse models, that Th17 cells are not as stable as Th1 or Th2 cells, and that in the presence of IL-12, Th17 cells may revert to Th1 like cells (41).
TRAFFICKING OF TH17 CELLS
To be effective, the adaptive immune system has the fundamental task of facilitating the encounter of antigen specific T and B lymphocytes with exogenous antigens and with one another. Exogenous antigens are picked up, processed and presented by antigen presenting cells in specialized microenvironments within secondary lymphoid organs, the skin and mucous membranes. CCR7, expressed by mature dendritic cells (DC) and T cells, including Tregs, and its ligands CCL19 and CCL21, play a major role in the homing of DCs and naïve T cells to secondary lymphoid organs where antigen presentation and effector T cell differentiation takes place (42).
Both human and mouse Th17 cells express CCR6 (43, 44) although not all CCR6+ cells are Th17 cells. Co-expression of CCR4 with CCR6 appears to correlate with a classic Th17 cell phenotype including RORγt expression and IL-17 production (43). Interestingly, CCR6 induces homing of cells to skin and mucosal sites and plays an etiologic role in many inflammatory diseases considered to be caused or maintained by IL-17, including psoriasis, ulcerative colitis, asthma and rheumatoid arthritis. The CCR6 ligand, CCL20, is expressed by Th17 cells and is upregulated in stromal cells by IL-17, allowing the attraction of additional Th17 cells into inflamed tissue (44).
FOXP3+ REGULATORY T CELLS
Whereas naïve murine CD4+ T cells differentiate into Th17 cells if co-cultured with TGF-β and IL-6 as described above, exposure to TGF-β and IL-2 causes differentiation into a regulatory T cell (Treg) phenotype including Foxp3 expression and suppressive function (8). Furthermore, addition of retinoic acid to the culture enforces the generation of Tregs and inhibits the differentiation of Th17 cells (45).
Tregs play a critical role in the maintenance of self-tolerance by suppressing, in a dominant manner, immune activation of self-aggressive T effector cells (13). Upregulation of Treg function or increasing their numbers may be beneficial for treating autoimmune diseases and allergies and for preventing allograft rejection. Conversely, inhibiting Treg function or decreasing their number may boost immunity against tumors and microorganisms.
Treg cells are characterized by the expression of the transcription factor FOXP3 (46–48), which is induced by TGF-β (49) (Table 1 and Figure 1). Absence of FOXP3 in patients with Immune dysregulation, Polyendocrinopathy, Enteropathy, X-linked (IPEX) (14) or scurfy mice (50) results in lack of functional Treg cells. Overexpression of FOXP3 in conventional T cells directs them to a Treg phenotype with suppressive activity leading to a state of immune deficiency (46, 51). The majority of Treg cells express high levels of CD25 (IL-2Rα) (13), suggesting a major influence of IL-2 for the long-term maintenance and competitiveness of these cells. STAT5, activated by IL-2, has been shown to be required for the maintenance of FOXP3 expression through binding to its promoter (52, 53). Treg cells also express TGF-β, IL-10, CTLA-4 and GITR which may play a role in their function.
Treg cells may either differentiate in the thymus and emigrate into the periphery as fully functional “natural” suppressor cells (nTreg), or they may be “induced” in the periphery from naïve T cell precursors (iTreg). The differentiation of Tregs from naïve CD4 T cells occurs when exposed to TGF-β and IL-2 (8). It has recently been demonstrated in mice that Treg cell derived TGF-β can generate de novo CD4+ Foxp3+ T cells in vitro from naïve precursor T cells (54).
Homing of Tregs to sites of inflammation is required for their suppressive function. Schneider and coworkers recently demonstrated that in CCR7 knockout mice, CD4+CD25+Foxp3+ Tregs were unable to home to lymph nodes and were unable to suppress antigen induced T cell responses. When compared with wild type Treg cells, the CCR7 deficient Tregs were less effective in preventing the development of inflammatory bowel disease when transferred into a SCID-mouse model (55). The importance of cutaneous Treg cells for the maintenance of immune homeostasis in the skin has been elegantly demonstrated in transfer experiments using FOXP3 deficient scurfy mice. Neonatal scurfy mice were injected with functional Tregs who were manipulated to no longer be able to migrate to the skin by inducing a targeted mutation of α-1, 3-fucosyltransferase VII (FuT7). This enzyme is required for the generation of the carbohydrate determinants of the E- and P- selectin ligands, which are required for optimal migration of T cells to the skin. FuT7-deficient Tregs restored the Treg cell compartment except for the skin. Loss of FuT7 selectively reduced Treg cell accumulation in the skin and resulted in severe cutaneous inflammation without developing other scurfy-associated symptoms (56).
TH17 CELLS IN PRIMARY IMMUNODEFICIENCY DISEASES
As described above, differentiation of murine Th17 cells from naïve CD4+ T cells depends on IL-6 and TGF-β signaling and the activation of STAT3. This was confirmed by the observation that CD4+ T cells conditionally deficient in STAT3 demonstrated impaired differentiation into Th17 cells and showed reduced production of IL-17 (11, 26). The recent identification of heterozygous STAT3 defects as the molecular etiology of Autosomal Dominant Hyper-IgE Syndrome (AD-HIES) raised the interesting possibility that patients with this disorder may have defective Th17 cell development and/or function. In addition, the observations that patients with AD-HIES/Job Syndrome are uniquely susceptible to Candida infections (57, 58), that Candida specific human memory T cells are predominantly present in the Th17 cell subset (43), and that IL-17 and IL-17R deficient mice had substantially reduced survival compared with control mice, when challenged with Candida albicans (59), led to a systematic assessment of Th17 cells in patients with AD-HIES (15, 27, 60, 61). AD-HIES is an autosomal dominant primary immune deficiency disorder characterized by eczema, staphylococcus aureus skin abscesses, pneumonia with pneumatocele formation, Candida infections, and skeletal and connective tissue abnormalities (58). Immunologic defects reported include markedly elevated serum IgE, eosinophilia, a neutrophil chemotactic defect (62), abnormal cytokine production (63), and abnormal antibody responses to Bacteriophage ΦX174 (32). As a result, AD-HIES patients have abnormal susceptibility to a narrow spectrum of infections including S. aureus and Candida albicans.
Flow cytometric analysis of peripheral blood lymphocytes and CD4+ T cells from patients with AD-HIES and normal control individuals showed a comparable distribution of naïve and memory T cells. However, the proportion of circulating Th17 cells was noted to be markedly diminished (15, 27, 60, 61). Circulating CD4+ T cells from normal controls, if activated with anti-CD3/anti-CD28 mAb, secreted abundant amounts of IL-17 and IL-22; in contrast, cells from AD-HIES patients failed to secrete either of these lymphokines, demonstrating that in humans, production of both IL-17 and IL-22 by activated T cells is dependent on functional STAT3 (27). Furthermore, purified naïve T cells from AD-HIES patients were unable to differentiate in vitro to Th17 cells when submitted to T cell receptor activation (anti-CD3/anti-CD28) in the presence of a cocktail of cytokines (IL-1β + IL-6 + IL-23) (15, 27). Interestingly, the expression of RORγt mRNA was also markedly impaired in AD-HIES cells cultured under these conditions (15, 27), which is in agreement with the hypothesis that impaired STAT3 function interferes with the expression of RORγt, required for Th17 cell differentiation.
To test the effect of other cytokines that govern the differentiation of Th17 cells in humans, Casanova’s group studied Th17 cell development in patients with mutations in IRAK4 or MYD88, whose cells do not respond to IL-1β, and in patients with mutations in IL12B (IL-12 p40 subunit) or IL12RB1, whose cells do not express or do not respond to IL-12 or IL-23. Results of these in vitro studies demonstrated that mutations in IRAK4/MYD88 had no detectable impact on Th17 cell generation, but that mutations in IL12B and IL12RB1 led to impaired generation of IL-17 producing cells, although less pronounced than was seen in patients with heterozygous STAT3 mutations (60). These observations suggest that IL-12 and IL-23 are important for Th17 cell differentiation in humans, but that T cells which are hyporesponsive to IL-1β can still be driven into the Th17 lineage (Table 1).
FOXP3+ REGULATORY T CELLS IN PRIMARY IMMUNODEFICIENCY DISEASES
Mutations in the FOXP3 transcription factor result in IPEX Syndrome (Table 1). The majority of affected individuals lack circulating and tissue associated FOXP3+ Treg cells and develop multiple autoimmune disorders affecting the gut, skin, endocrine organs, blood cells and joints. Death typically occurs in early childhood unless treated with aggressive immunosuppression and/or hematopoietic stem cell transplantation (14). The scurfy mouse, characterized by lymphocytic infiltrates in multiple organs and early death as a result of Treg deficiency (50), has a naturally occurring mutation of Foxp3; a two base pair insertion upstream of the forkhead domain, resulting in a frame shift and loss of the DNA binding domain (64).
A clinical syndrome resembling IPEX is associated with mutations in the α-chain of the IL-2 receptor (IL-2Rα, CD25). Patients lacking CD25 present with an IPEX-like phenotype; in addition they develop infectious complications resembling those observed in patients with T cell deficiency, such as recurrent CMV pneumonitis, Candida infections and chronic gastrointestinal disease (65, 66). CD25 deficient mice have a phenotype similar to that of scurfy mice (67). Although Treg development in the thymus and in vitro suppressive function of CD4+ Foxp3+ T cells are normal, these mice have a defect in survival, maintenance and competitive fitness of mature Treg cells (68, 69).
Mutations of STAT5b, a key mediator of IL-2 induced gene transcription, cause a rare recessive disorder characterized by dwarfism (Laron dwarfs) and low serum concentrations of insulin-like growth factor-1 (but normal serum growth hormone level) (70–72). Other physical features include a prominent forehead, a saddle nose and high pitched voice. Most patients have a marked immune deficiency characterized by recurrent varicella and herpes virus infections and pneumocystis jiroveci pneumonia, suggesting defective T cell and NK cell function (71, 72), In addition, most patients with STAT5b mutations present with diarrhea, eczema and lymphocytic interstitial pneumonitis, suggesting immune dysregulation (70–72). Patients studied for Treg pathology showed fewer CD4+CD24high cells with decreased FOXP3+ expression (70, 72) and the one patient evaluated for Treg function demonstrated reduced suppressive activity against either autologous or allogeneic effector cells (70). A likely explanation for this observation was the markedly decreased CD25 expression (20% of normal) by this patient’s T cells in response to activation, due to STAT5b deficiency as well as defective signaling from the IL-2 receptor complex. This reduced expression of CD25/IL-2Rα and defective STAT5b-mediated gene transcription interferes with IL-2 signals required for the maintenance of FOXP3 expression and Treg function (70).
Autoimmune Poly Endocrinopathy, Candidiasis and Ectodermal Dystrophy (APECED) syndrome is an autosomal recessive disorder, characterized primarily by hypoparathyroidism, adrenal insufficiency and chronic mucocutaneous Candidiasis. Type 1 diabetes, gonadal failure and pernicious anemia also occur but are less frequent. APECED is caused by mutations in the autoimmune regulator (AIRE) gene, a transcription factor responsible for the ectopic expression of tissue-specific antigens on thymic medullary epithelial cells. As a consequence, AIRE is essential for the negative selection of auto-reactive T cell clones (73, 74). To address the possibility of AIRE playing a role in the generation of functional Tregs, APECED patients’ lymphocytes were evaluated by flow cytometry and quantitative real-time PCR. Consistently, the percentages of CD4+CD24high cells were decreased, the amount of FOXP3 expressed per cell diminished, and the ability to suppress effector T cells reduced (75).
The Wiskott-Aldrich Syndrome (WAS) is a rare X-linked disorder caused by mutations in the WAS protein (WASP) gene (76). In addition to thrombocytopenia, small platelets, eczema, recurrent infections due to cellular immunodeficiency and malignancies, approximately 40–70% of the patients with classic WAS develop autoimmune diseases (77, 78). In mice, Wasp does not play a role in thymic Treg production, but is required for peripheral Treg expansion and survival, and for effective suppressive function (79–81). While WASP deficiency in humans is not associated with a decrease in the percentage of Tregs in the peripheral blood, Treg function was found to be consistently reduced, as demonstrated by the inability to suppress proliferation of effector T cells (81).
CONCLUSION
Recent advances made in our understanding of the role of T cell subsets in the regulation of immune responses against infectious agents and self-antigens have provided new insight into human immunopathology. The discovery that specific cytokines and unique transcriptional regulators control the differentiation of distinct T cell subsets have expanded and refined the aging Th1/Th2 paradigm. New lineage specific cytokines have been discovered and their role in the activation of transcription factors such as T-bet, GATA3, RORγt and FOXP3 explored.
Together with members of the STAT family of transcriptional regulators, these DNA-binding proteins direct the differentiation of naïve CD4+ T cells into Th1, Th2, Th17 and Treg cells and induce the expression of “signature” cytokines unique for each T cell lineage. This impressive progress was made possible by the development of novel biotechnologies that facilitate the study of gene regulation, protein expression and cell differentiation and by the creation of unique genetically engineered mouse models. These innovative experimental strategies were complemented by careful observation of patients with unique congenital syndromes characterized by immune dysfunction, severe infections, autoimmune diseases and allergic complications. The beneficiary of these efforts (as of February 2009, there are 60,000 hits on PubMed when searching for Th1, Th2, Th17 and Treg cells) are the clinical immunologists who have at their disposal new diagnostic techniques, more therapeutic options based on scientific medicine, and the hard facts required for authoritative genetic counseling of these complex patients. It is likely that these advances will lead to entirely new treatment strategies for severe infections, allograft rejection, cancer, and autoimmune and allergic diseases.
1) What do we know?
N aïve CD4+ T cells undergo differentiation into functionally unique lineages using distinct developmental pathways.
The signature cytokines for Th17 cells are IL-117A and IL-17F and the lineage specific STAT regulator and transcription factor for TH17 cells are STAT3 and RORγt.
The signature cytokine for Treg cells is TGF-β and the lineage specific STAT regulator/transcription factor for Treg cells are STAT5 and FOXP3.
Heterozygous “hypomorphic” STAT3 mutations result in AD-HIES/Job Syndrome, lack of TH17 cells and susceptibility to Staph aureous and Candida infections. Mutations in the X chromosome associated FOXP3 gene cause the syndrome of Immune dysregulation, Polyendocrinopathy, Enteropathy, X-Linked (IPEX).
2) What is still unknown?
It is possible that there are other CD4+ cell lineages with unique functions.
Can one functionally unique CD4+ lineage change into another unique lineage?
The mechanism by which IL17 interacts with the immune system to keep Staph aureous and Candida infections under control are unknown.
The mechanisms by which antigen-specific Treg cells achieve suppression of effector T cells are not understood.
How can one manipulate the function of Th17 and Treg cells to cure autoimmunity, immune deficiencies, overwhelming infections, graft rejection, graft vs. host disease and the development of malignancies?
ABBREVIATIONS
- AD-HIES
autosomal dominant Hyper IgE Syndrome
- AIRE
Autoimmune Regulator
- APECED
Autoimmune Polyendocrinopathy, Candidiasis, Ectodermal Dystrophy
- CMV
Cytomegalovirus
- DC
Dendritic Cells
- FOXP3
Forkhead Box Protein 3
- IL12B
IL-12p40
- IL12RB1
IL-12 Receptor β 1
- IPEX
Immune Dysregulation, Polyendocrinopathy, Enteropathy, X-Linked
- IRAK4
IL-1 Receptor-Associated Kinase 4
- MYD88
Myeloid Differentiation Primary Response Gene 88
- RORγt
Retinoic Acid-Related Orphan Receptor γ T
- SCID
Severe Combined Immune Deficiency
- STAT
Signal Transducer and Activator of Transcription
- TGF-β
Transforming Growth Factor-β
- Th
T helper cells
- TNF-α
Tumor Necrosis Factor α
- Treg
regulatory T cells
- WAS
Wiskott-Aldrich Syndrome
- WASP
WAS Protein
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Hans D. Ochs, University of Washington School of Medicine, Seattle Children’s Hospital.
Mohamed Oukka, Harvard Medical School.
Troy R. Torgerson, University of Washington School of Medicine, Seattle Children’s Hospital.
References
- 1.Mosmann TR, Coffman RL. TH1 and TH2 cells: different patterns of lymphokine secretion lead to different functional properties. Annu Rev Immunol. 1989;7:145–173. doi: 10.1146/annurev.iy.07.040189.001045. [DOI] [PubMed] [Google Scholar]
- 2.Abbas AK, Murphy KM, Sher A. Functional diversity of helper T lymphocytes. Nature. 1996;383(6603):787–793. doi: 10.1038/383787a0. [DOI] [PubMed] [Google Scholar]
- 3.Cua DJ, Sherlock J, Chen Y, Murphy CA, Joyce B, Seymour B, Lucian L, To W, Kwan S, Churakova T, Zurawski S, Wiekowski M, Lira SA, Gorman D, Kastelein RA, Sedgwick JD. Interleukin-23 rather than interleukin-12 is the critical cytokine for autoimmune inflammation of the brain. Nature. 2003;421(6924):744–748. doi: 10.1038/nature01355. [DOI] [PubMed] [Google Scholar]
- 4.Langrish CL, Chen Y, Blumenschein WM, Mattson J, Basham B, Sedgwick JD, McClanahan T, Kastelein RA, Cua DJ. IL-23 drives a pathogenic T cell population that induces autoimmune inflammation. J Exp Med. 2005;201(2):233–240. doi: 10.1084/jem.20041257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Harrington LE, Hatton RD, Mangan PR, Turner H, Murphy TL, Murphy KM, Weaver CT. Interleukin 17-producing CD4+ effector T cells develop via a lineage distinct from the T helper type 1 and 2 lineages. Nat Immunol. 2005;6(11):1123–1132. doi: 10.1038/ni1254. [DOI] [PubMed] [Google Scholar]
- 6.Park H, Li Z, Yang XO, Chang SH, Nurieva R, Wang YH, Wang Y, Hood L, Zhu Z, Tian Q, Dong C. A distinct lineage of CD4 T cells regulates tissue inflammation by producing interleukin 17. Nat Immunol. 2005;6(11):1133–1141. doi: 10.1038/ni1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Veldhoen M, Hocking RJ, Atkins CJ, Locksley RM, Stockinger B. TGFbeta in the context of an inflammatory cytokine milieu supports de novo differentiation of IL-17-producing T cells. Immunity. 2006;24(2):179–189. doi: 10.1016/j.immuni.2006.01.001. [DOI] [PubMed] [Google Scholar]
- 8.Bettelli E, Carrier Y, Gao W, Korn T, Strom TB, Oukka M, Weiner HL, Kuchroo VK. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature. 2006;441(7090):235–238. doi: 10.1038/nature04753. [DOI] [PubMed] [Google Scholar]
- 9.Mangan PR, Harrington LE, O’Quinn DB, Helms WS, Bullard DC, Elson CO, Hatton RD, Wahl SM, Schoeb TR, Weaver CT. Transforming growth factor-beta induces development of the T(H)17 lineage. Nature. 2006;441(7090):231–234. doi: 10.1038/nature04754. [DOI] [PubMed] [Google Scholar]
- 10.Ivanov II, McKenzie BS, Zhou L, Tadokoro CE, Lepelley A, Lafaille JJ, Cua DJ, Littman DR. The orphan nuclear receptor RORgammat directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell. 2006;126(6):1121–1133. doi: 10.1016/j.cell.2006.07.035. [DOI] [PubMed] [Google Scholar]
- 11.Yang XO, Panopoulos AD, Nurieva R, Chang SH, Wang D, Watowich SS, Dong C. STAT3 regulates cytokine-mediated generation of inflammatory helper T cells. J Biol Chem. 2007;282(13):9358–9363. doi: 10.1074/jbc.C600321200. [DOI] [PubMed] [Google Scholar]
- 12.Laurence A, O’Shea JJ. T(H)-17 differentiation: of mice and men. Nat Immunol. 2007;8(9):903–905. doi: 10.1038/ni0907-903. [DOI] [PubMed] [Google Scholar]
- 13.Sakaguchi S, Setoguchi R, Yagi H, Nomura T. Naturally arising Foxp3-expressing CD25+CD4+ regulatory T cells in self-tolerance and autoimmune disease. Curr Top Microbiol Immunol. 2006;305:51–66. doi: 10.1007/3-540-29714-6_3. [DOI] [PubMed] [Google Scholar]
- 14.Torgerson TR, Ochs HD. Regulatory T cells in primary immunodeficiency diseases. Curr Opin Allergy Clin Immunol. 2007;7(6):515–521. doi: 10.1097/ACI.0b013e3282f1a27a. [DOI] [PubMed] [Google Scholar]
- 15.Milner JD, Brenchley JM, Laurence A, Freeman AF, Hill BJ, Elias KM, Kanno Y, Spalding C, Elloumi HZ, Paulson ML, Davis J, Hsu A, Asher AI, O’Shea J, Holland SM, Paul WE, Douek DC. Impaired T(H)17 cell differentiation in subjects with autosomal dominant hyper-IgE syndrome. Nature. 2008;452(7188):773–776. doi: 10.1038/nature06764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yao Z, Fanslow WC, Seldin MF, Rousseau AM, Painter SL, Comeau MR, Cohen JI, Spriggs MK. Herpesvirus Saimiri encodes a new cytokine, IL-17, which binds to a novel cytokine receptor. Immunity. 1995;3(6):811–821. doi: 10.1016/1074-7613(95)90070-5. [DOI] [PubMed] [Google Scholar]
- 17.Fossiez F, Djossou O, Chomarat P, Flores-Romo L, Ait-Yahia S, Maat C, Pin JJ, Garrone P, Garcia E, Saeland S, Blanchard D, Gaillard C, Das Mahapatra B, Rouvier E, Golstein P, Banchereau J, Lebecque S. T cell interleukin-17 induces stromal cells to produce proinflammatory and hematopoietic cytokines. J Exp Med. 1996;183(6):2593–2603. doi: 10.1084/jem.183.6.2593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Matsuzaki G, Umemura M. Interleukin-17 as an effector molecule of innate and acquired immunity against infections. Microbiol Immunol. 2007;51(12):1139–1147. doi: 10.1111/j.1348-0421.2007.tb04008.x. [DOI] [PubMed] [Google Scholar]
- 19.Acosta-Rodriguez EV, Napolitani G, Lanzavecchia A, Sallusto F. Interleukins 1beta and 6 but not transforming growth factor-beta are essential for the differentiation of interleukin 17-producing human T helper cells. Nat Immunol. 2007;8(9):942–949. doi: 10.1038/ni1496. [DOI] [PubMed] [Google Scholar]
- 20.Wilson NJ, Boniface K, Chan JR, McKenzie BS, Blumenschein WM, Mattson JD, Basham B, Smith K, Chen T, Morel F, Lecron JC, Kastelein RA, Cua DJ, McClanahan TK, Bowman EP, de Waal Malefyt R. Development, cytokine profile and function of human interleukin 17-producing helper T cells. Nat Immunol. 2007;8(9):950–957. doi: 10.1038/ni1497. [DOI] [PubMed] [Google Scholar]
- 21.Chen Z, Tato CM, Muul L, Laurence A, O’Shea JJ. Distinct regulation of interleukin-17 in human T helper lymphocytes. Arthritis Rheum. 2007;56(9):2936–2946. doi: 10.1002/art.22866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Manel N, Unutmaz D, Littman DR. The differentiation of human T(H)-17 cells requires transforming growth factor-beta and induction of the nuclear receptor RORgammat. Nat Immunol. 2008;9(6):641–649. doi: 10.1038/ni.1610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Volpe E, Servant N, Zollinger R, Bogiatzi SI, Hupe P, Barillot E, Soumelis V. A critical function for transforming growth factor-beta, interleukin 23 and proinflammatory cytokines in driving and modulating human T(H)-17 responses. Nat Immunol. 2008;9(6):650–657. doi: 10.1038/ni.1613. [DOI] [PubMed] [Google Scholar]
- 24.Yang L, Anderson DE, Baecher-Allan C, Hastings WD, Bettelli E, Oukka M, Kuchroo VK, Hafler DA. IL-21 and TGF-beta are required for differentiation of human T(H)17 cells. Nature. 2008;454(7202):350–352. doi: 10.1038/nature07021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ivanov II, Zhou L, Littman DR. Transcriptional regulation of Th17 cell differentiation. Semin Immunol. 2007;19(6):409–417. doi: 10.1016/j.smim.2007.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Harris TJ, Grosso JF, Yen HR, Xin H, Kortylewski M, Albesiano E, Hipkiss EL, Getnet D, Goldberg MV, Maris CH, Housseau F, Yu H, Pardoll DM, Drake CG. Cutting edge: An in vivo requirement for STAT3 signaling in TH17 development and TH17-dependent autoimmunity. J Immunol. 2007;179(7):4313–4317. doi: 10.4049/jimmunol.179.7.4313. [DOI] [PubMed] [Google Scholar]
- 27.Ma CS, Chew GY, Simpson N, Priyadarshi A, Wong M, Grimbacher B, Fulcher DA, Tangye SG, Cook MC. Deficiency of Th17 cells in hyper IgE syndrome due to mutations in STAT3. J Exp Med. 2008;205(7):1551–1557. doi: 10.1084/jem.20080218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Aggarwal S, Ghilardi N, Xie MH, de Sauvage FJ, Gurney AL. Interleukin-23 promotes a distinct CD4 T cell activation state characterized by the production of interleukin-17. J Biol Chem. 2003;278(3):1910–1914. doi: 10.1074/jbc.M207577200. [DOI] [PubMed] [Google Scholar]
- 29.Yen D, Cheung J, Scheerens H, Poulet F, McClanahan T, McKenzie B, Kleinschek MA, Owyang A, Mattson J, Blumenschein W, Murphy E, Sathe M, Cua DJ, Kastelein RA, Rennick D. IL-23 is essential for T cell-mediated colitis and promotes inflammation via IL-17 and IL-6. J Clin Invest. 2006;116(5):1310–1316. doi: 10.1172/JCI21404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gaffen SL. An overview of IL-17 function and signaling. Cytokine. 2008;43(3):402–407. doi: 10.1016/j.cyto.2008.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nakae S, Komiyama Y, Nambu A, Sudo K, Iwase M, Homma I, Sekikawa K, Asano M, Iwakura Y. Antigen-specific T cell sensitization is impaired in IL-17-deficient mice, causing suppression of allergic cellular and humoral responses. Immunity. 2002;17(3):375–387. doi: 10.1016/s1074-7613(02)00391-6. [DOI] [PubMed] [Google Scholar]
- 32.Sheerin KA, Buckley RH. Antibody responses to protein, polysaccharide, and phi X174 antigens in the hyperimmunoglobulinemia E (hyper-IgE) syndrome. J Allergy Clin Immunol. 1991;87(4):803–811. doi: 10.1016/0091-6749(91)90126-9. [DOI] [PubMed] [Google Scholar]
- 33.Wei L, Laurence A, Elias KM, O’Shea JJ. IL-21 is produced by Th17 cells and drives IL-17 production in a STAT3-dependent manner. J Biol Chem. 2007;282(48):34605–34610. doi: 10.1074/jbc.M705100200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nurieva R, Yang XO, Martinez G, Zhang Y, Panopoulos AD, Ma L, Schluns K, Tian Q, Watowich SS, Jetten AM, Dong C. Essential autocrine regulation by IL-21 in the generation of inflammatory T cells. Nature. 2007;448(7152):480–483. doi: 10.1038/nature05969. [DOI] [PubMed] [Google Scholar]
- 35.Korn T, Bettelli E, Gao W, Awasthi A, Jager A, Strom TB, Oukka M, Kuchroo VK. IL-21 initiates an alternative pathway to induce proinflammatory T(H)17 cells. Nature. 2007;448(7152):484–487. doi: 10.1038/nature05970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Onoda T, Rahman M, Nara H, Araki A, Makabe K, Tsumoto K, Kumagai I, Kudo T, Ishii N, Tanaka N, Sugamura K, Hayasaka K, Asao H. Human CD4+ central and effector memory T cells produce IL-21: effect on cytokine-driven proliferation of CD4+ T cell subsets. Int Immunol. 2007;19(10):1191–1199. doi: 10.1093/intimm/dxm090. [DOI] [PubMed] [Google Scholar]
- 37.Peluso I, Fantini MC, Fina D, Caruso R, Boirivant M, MacDonald TT, Pallone F, Monteleone G. IL-21 counteracts the regulatory T cell-mediated suppression of human CD4+ T lymphocytes. J Immunol. 2007;178(2):732–739. doi: 10.4049/jimmunol.178.2.732. [DOI] [PubMed] [Google Scholar]
- 38.Ouyang W, Kolls JK, Zheng Y. The biological functions of T helper 17 cell effector cytokines in inflammation. Immunity. 2008;28(4):454–467. doi: 10.1016/j.immuni.2008.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wolk K, Witte E, Wallace E, Docke WD, Kunz S, Asadullah K, Volk HD, Sterry W, Sabat R. IL-22 regulates the expression of genes responsible for antimicrobial defense, cellular differentiation, and mobility in keratinocytes: a potential role in psoriasis. Eur J Immunol. 2006;36(5):1309–1323. [Google Scholar]
- 40.McGeachy MJ, Cua DJ. The link between IL-23 and Th17 cell-mediated immune pathologies. Semin Immunol. 2007;19(6):372–376. doi: 10.1016/j.smim.2007.10.012. [DOI] [PubMed] [Google Scholar]
- 41.Lee YK, Turner H, Maynard CL, Oliver JR, Chen D, Elson CO, Weaver CT. Late developmental plasticity in the T helper 17 lineage. Immunity. 2009;30(1):92–107. doi: 10.1016/j.immuni.2008.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sanchez-Sanchez N, Riol-Blanco L, Rodriguez-Fernandez JL. The multiple personalities of the chemokine receptor CCR7 in dendritic cells. J Immunol. 2006;176(9):5153–5159. doi: 10.4049/jimmunol.176.9.5153. [DOI] [PubMed] [Google Scholar]
- 43.Acosta-Rodriguez EV, Rivino L, Geginat J, Jarrossay D, Gattorno M, Lanzavecchia A, Sallusto F, Napolitani G. Surface phenotype and antigenic specificity of human interleukin 17-producing T helper memory cells. Nat Immunol. 2007;8(6):639–646. doi: 10.1038/ni1467. [DOI] [PubMed] [Google Scholar]
- 44.Hirota K, Yoshitomi H, Hashimoto M, Maeda S, Teradaira S, Sugimoto N, Yamaguchi T, Nomura T, Ito H, Nakamura T, Sakaguchi N, Sakaguchi S. Preferential recruitment of CCR6-expressing Th17 cells to inflamed joints via CCL20 in rheumatoid arthritis and its animal model. J Exp Med. 2007;204(12):2803–2812. doi: 10.1084/jem.20071397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Xiao S, Jin H, Korn T, Liu SM, Oukka M, Lim B, Kuchroo VK. Retinoic acid increases Foxp3+ regulatory T cells and inhibits development of Th17 cells by enhancing TGF-beta-driven Smad3 signaling and inhibiting IL-6 and IL-23 receptor expression. J Immunol. 2008;181(4):2277–2284. doi: 10.4049/jimmunol.181.4.2277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fontenot JD, Gavin MA, Rudensky AY. Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nat Immunol. 2003;4(4):330–336. doi: 10.1038/ni904. [DOI] [PubMed] [Google Scholar]
- 47.Hori S, Nomura T, Sakaguchi S. Control of regulatory T cell development by the transcription factor Foxp3. Science. 2003;299(5609):1057–1061. doi: 10.1126/science.1079490. [DOI] [PubMed] [Google Scholar]
- 48.Khattri R, Cox T, Yasayko SA, Ramsdell F. An essential role for Scurfin in CD4+CD25+ T regulatory cells. Nat Immunol. 2003;4(4):337–342. doi: 10.1038/ni909. [DOI] [PubMed] [Google Scholar]
- 49.Chen W, Jin W, Hardegen N, Lei KJ, Li L, Marinos N, McGrady G, Wahl SM. Conversion of peripheral CD4+CD25- naive T cells to CD4+CD25+ regulatory T cells by TGF-beta induction of transcription factor Foxp3. J Exp Med. 2003;198(12):1875–1886. doi: 10.1084/jem.20030152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Clark LB, Appleby MW, Brunkow ME, Wilkinson JE, Ziegler SF, Ramsdell F. Cellular and molecular characterization of the scurfy mouse mutant. J Immunol. 1999;162(5):2546–2554. [PubMed] [Google Scholar]
- 51.Khattri R, Kasprowicz D, Cox T, Mortrud M, Appleby MW, Brunkow ME, Ziegler SF, Ramsdell F. The amount of scurfin protein determines peripheral T cell number and responsiveness. J Immunol. 2001;167(11):6312–6320. doi: 10.4049/jimmunol.167.11.6312. [DOI] [PubMed] [Google Scholar]
- 52.Yao Z, Kanno Y, Kerenyi M, Stephens G, Durant L, Watford WT, Laurence A, Robinson GW, Shevach EM, Moriggl R, Hennighausen L, Wu C, O’Shea JJ. Nonredundant roles for Stat5a/b in directly regulating Foxp3. Blood. 2007;109(10):4368–4375. doi: 10.1182/blood-2006-11-055756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Burchill MA, Yang J, Vogtenhuber C, Blazar BR, Farrar MA. IL-2 receptor beta-dependent STAT5 activation is required for the development of Foxp3+ regulatory T cells. J Immunol. 2007;178(1):280–290. doi: 10.4049/jimmunol.178.1.280. [DOI] [PubMed] [Google Scholar]
- 54.Andersson J, Tran DQ, Pesu M, Davidson TS, Ramsey H, O’Shea JJ, Shevach EM. CD4+ FoxP3+ regulatory T cells confer infectious tolerance in a TGF-beta-dependent manner. J Exp Med. 2008;205(9):1975–1981. doi: 10.1084/jem.20080308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Schneider MA, Meingassner JG, Lipp M, Moore HD, Rot A. CCR7 is required for the in vivo function of CD4+ CD25+ regulatory T cells. J Exp Med. 2007;204(4):735–745. doi: 10.1084/jem.20061405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Dudda JC, Perdue N, Bachtanian E, Campbell DJ. Foxp3+ regulatory T cells maintain immune homeostasis in the skin. J Exp Med. 2008;205(7):1559–1565. doi: 10.1084/jem.20072594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Buckley RH. The hyper-IgE syndrome. Clin Rev Allergy Immunol. 2001;20(1):139–154. doi: 10.1385/CRIAI:20:1:139. [DOI] [PubMed] [Google Scholar]
- 58.Grimbacher B, Holland SM, Gallin JI, Greenberg F, Hill SC, Malech HL, Miller JA, O’Connell AC, Puck JM. Hyper-IgE syndrome with recurrent infections--an autosomal dominant multisystem disorder. N Engl J Med. 1999;340(9):692–702. doi: 10.1056/NEJM199903043400904. [DOI] [PubMed] [Google Scholar]
- 59.Huang W, Na L, Fidel PL, Schwarzenberger P. Requirement of interleukin-17A for systemic anti-Candida albicans host defense in mice. J Infect Dis. 2004;190(3):624–631. doi: 10.1086/422329. [DOI] [PubMed] [Google Scholar]
- 60.de Beaucoudrey L, Puel A, Filipe-Santos O, Cobat A, Ghandil P, Chrabieh M, Feinberg J, von Bernuth H, Samarina A, Janniere L, Fieschi C, Stephan JL, Boileau C, Lyonnet S, Jondeau G, Cormier-Daire V, Le Merrer M, Hoarau C, Lebranchu Y, Lortholary O, Chandesris MO, Tron F, Gambineri E, Bianchi L, Rodriguez-Gallego C, Zitnik SE, Vasconcelos J, Guedes M, Vitor AB, Marodi L, Chapel H, Reid B, Roifman C, Nadal D, Reichenbach J, Caragol I, Garty BZ, Dogu F, Camcioglu Y, Gulle S, Sanal O, Fischer A, Abel L, Stockinger B, Picard C, Casanova JL. Mutations in STAT3 and IL12RB1 impair the development of human IL-17-producing T cells. J Exp Med. 2008;205(7):1543–1550. doi: 10.1084/jem.20080321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Renner ED, Rylaarsdam S, Anover-Sombke S, Rack AL, Reichenbach J, Carey JC, Zhu Q, Jansson AF, Barboza J, Schimke LF, Leppert MF, Getz MM, Seger RA, Hill HR, Belohradsky BH, Torgerson TR, Ochs HD. Novel signal transducer and activator of transcription 3 (STAT3) mutations, reduced T(H)17 cell numbers, and variably defective STAT3 phosphorylation in hyper-IgE syndrome. J Allergy Clin Immunol. 2008;122(1):181–187. doi: 10.1016/j.jaci.2008.04.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hill HR, Ochs HD, Quie PG, Clark RA, Pabst HF, Klebanoff SJ, Wedgwood RJ. Defect in neutrophil granulocyte chemotaxis in Job’s syndrome of recurrent “cold” staphylococcal abscesses. Lancet. 1974;2(7881):617–619. doi: 10.1016/s0140-6736(74)91942-4. [DOI] [PubMed] [Google Scholar]
- 63.Holland SM, DeLeo FR, Elloumi HZ, Hsu AP, Uzel G, Brodsky N, Freeman AF, Demidowich A, Davis J, Turner ML, Anderson VL, Darnell DN, Welch PA, Kuhns DB, Frucht DM, Malech HL, Gallin JI, Kobayashi SD, Whitney AR, Voyich JM, Musser JM, Woellner C, Schaffer AA, Puck JM, Grimbacher B. STAT3 mutations in the hyper-IgE syndrome. N Engl J Med. 2007;357(16):1608–1619. doi: 10.1056/NEJMoa073687. [DOI] [PubMed] [Google Scholar]
- 64.Brunkow ME, Jeffery EW, Hjerrild KA, Paeper B, Clark LB, Yasayko SA, Wilkinson JE, Galas D, Ziegler SF, Ramsdell F. Disruption of a new forkhead/winged-helix protein, scurfin, results in the fatal lymphoproliferative disorder of the scurfy mouse. Nat Genet. 2001;27(1):68–73. doi: 10.1038/83784. [DOI] [PubMed] [Google Scholar]
- 65.Roifman CM. Human IL-2 receptor alpha chain deficiency. Pediatr Res. 2000;48(1):6–11. doi: 10.1203/00006450-200007000-00004. [DOI] [PubMed] [Google Scholar]
- 66.Caudy AA, Reddy ST, Chatila T, Atkinson JP, Verbsky JW. CD25 deficiency causes an immune dysregulation, polyendocrinopathy, enteropathy, X-linked-like syndrome, and defective IL-10 expression from CD4 lymphocytes. J Allergy Clin Immunol. 2007;119(2):482–487. doi: 10.1016/j.jaci.2006.10.007. [DOI] [PubMed] [Google Scholar]
- 67.Willerford DM, Chen J, Ferry JA, Davidson L, Ma A, Alt FW. Interleukin-2 receptor alpha chain regulates the size and content of the peripheral lymphoid compartment. Immunity. 1995;3(4):521–530. doi: 10.1016/1074-7613(95)90180-9. [DOI] [PubMed] [Google Scholar]
- 68.D’Cruz LM, Klein L. Development and function of agonist-induced CD25+Foxp3+ regulatory T cells in the absence of interleukin 2 signaling. Nat Immunol. 2005;6(11):1152–1159. doi: 10.1038/ni1264. [DOI] [PubMed] [Google Scholar]
- 69.Fontenot JD, Rasmussen JP, Gavin MA, Rudensky AY. A function for interleukin 2 in Foxp3-expressing regulatory T cells. Nat Immunol. 2005;6(11):1142–1151. doi: 10.1038/ni1263. [DOI] [PubMed] [Google Scholar]
- 70.Cohen AC, Nadeau KC, Tu W, Hwa V, Dionis K, Bezrodnik L, Teper A, Gaillard M, Heinrich J, Krensky AM, Rosenfeld RG, Lewis DB. Cutting edge: Decreased accumulation and regulatory function of CD4+ CD25(high) T cells in human STAT5b deficiency. J Immunol. 2006;177(5):2770–2774. doi: 10.4049/jimmunol.177.5.2770. [DOI] [PubMed] [Google Scholar]
- 71.Hwa V, Little B, Adiyaman P, Kofoed EM, Pratt KL, Ocal G, Berberoglu M, Rosenfeld RG. Severe growth hormone insensitivity resulting from total absence of signal transducer and activator of transcription 5b. J Clin Endocrinol Metab. 2005;90(7):4260–4266. doi: 10.1210/jc.2005-0515. [DOI] [PubMed] [Google Scholar]
- 72.Bernasconi A, Marino R, Ribas A, Rossi J, Ciaccio M, Oleastro M, Ornani A, Paz R, Rivarola MA, Zelazko M, Belgorosky A. Characterization of immunodeficiency in a patient with growth hormone insensitivity secondary to a novel STAT5b gene mutation. Pediatrics. 2006;118(5):e1584–1592. doi: 10.1542/peds.2005-2882. [DOI] [PubMed] [Google Scholar]
- 73.Liston A, Lesage S, Wilson J, Peltonen L, Goodnow CC. Aire regulates negative selection of organ-specific T cells. Nat Immunol. 2003;4(4):350–354. doi: 10.1038/ni906. [DOI] [PubMed] [Google Scholar]
- 74.Anderson MS, Venanzi ES, Klein L, Chen Z, Berzins SP, Turley SJ, von Boehmer H, Bronson R, Dierich A, Benoist C, Mathis D. Projection of an immunological self shadow within the thymus by the aire protein. Science. 2002;298(5597):1395–1401. doi: 10.1126/science.1075958. [DOI] [PubMed] [Google Scholar]
- 75.Kekalainen E, Tuovinen H, Joensuu J, Gylling M, Franssila R, Pontynen N, Talvensaari K, Perheentupa J, Miettinen A, Arstila TP. A defect of regulatory T cells in patients with autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy. J Immunol. 2007;178(2):1208–1215. doi: 10.4049/jimmunol.178.2.1208. [DOI] [PubMed] [Google Scholar]
- 76.Ochs HD, Thrasher AJ. The Wiskott-Aldrich syndrome. J Allergy Clin Immunol. 2006;117(4):725–738. doi: 10.1016/j.jaci.2006.02.005. quiz 739. [DOI] [PubMed] [Google Scholar]
- 77.Sullivan KE, Mullen CA, Blaese RM, Winkelstein JA. A multiinstitutional survey of the Wiskott-Aldrich syndrome. J Pediatr. 1994;125(6 Pt 1):876–885. doi: 10.1016/s0022-3476(05)82002-5. [DOI] [PubMed] [Google Scholar]
- 78.Dupuis-Girod S, Medioni J, Haddad E, Quartier P, Cavazzana-Calvo M, Le Deist F, de Saint Basile G, Delaunay J, Schwarz K, Casanova JL, Blanche S, Fischer A. Autoimmunity in Wiskott-Aldrich syndrome: risk factors, clinical features, and outcome in a single-center cohort of 55 patients. Pediatrics. 2003;111(5 Pt 1):e622–627. doi: 10.1542/peds.111.5.e622. [DOI] [PubMed] [Google Scholar]
- 79.Maillard MH, Cotta-de-Almeida V, Takeshima F, Nguyen DD, Michetti P, Nagler C, Bhan AK, Snapper SB. The Wiskott-Aldrich syndrome protein is required for the function of CD4(+)CD25(+)Foxp3(+) regulatory T cells. J Exp Med. 2007;204(2):381–391. doi: 10.1084/jem.20061338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Humblet-Baron S, Sather B, Anover S, Becker-Herman S, Kasprowicz DJ, Khim S, Nguyen T, Hudkins-Loya K, Alpers CE, Ziegler SF, Ochs H, Torgerson T, Campbell DJ, Rawlings DJ. Wiskott-Aldrich syndrome protein is required for regulatory T cell homeostasis. J Clin Invest. 2007;117(2):407–418. doi: 10.1172/JCI29539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Marangoni F, Trifari S, Scaramuzza S, Panaroni C, Martino S, Notarangelo LD, Baz Z, Metin A, Cattaneo F, Villa A, Aiuti A, Battaglia M, Roncarolo MG, Dupre L. WASP regulates suppressor activity of human and murine CD4(+)CD25(+)FOXP3(+) natural regulatory T cells. J Exp Med. 2007;204(2):369–380. doi: 10.1084/jem.20061334. [DOI] [PMC free article] [PubMed] [Google Scholar]