

Abstract
Terminal restriction fragment length polymorphism (TRFLP) profiling of the internally transcribed spacer (ITS) ribosomal DNA of unknown fungal communities is currently unsupported by a broad-range enzyme-choosing rationale. An in silico study of terminal fragment size distribution was therefore performed following virtual digestion (by use of a set of commercially available 135 type IIP restriction endonucleases) of all published fungal ITS sequences putatively annealing to primers ITS1 and ITS4. Different diversity measurements were used to rank primer-enzyme pairs according to the richness and evenness that they showed. Top-performing pairs were hierarchically clustered to test for data dependency. The enzyme set composed of MaeII, BfaI, and BstNI returned much better results than randomly chosen enzyme sets in computer simulations and is therefore recommended for in vitro TRFLP profiling of fungal ITSs.
Terminal restriction fragment length polymorphism (TRFLP) profiling was originally developed as a means of genotyping mixed DNA samples (30) and is currently being employed in fungal community ecology studies (3, 5, 6, 7, 10, 13, 19, 22, 26, 27, 29, 33, 38), despite a number of technical and conceptual difficulties (11). Briefly, TRFLP profiling involves amplifying the DNA in pools of mixed genetic material with fluorescently labeled primers, digesting the products with restriction endonucleases, and sizing the labeled terminal fragments in a sequencer. The difference in the positions at which the different restriction enzymes cleave DNA is thought to provide enough variability for such DNA mixtures to be characterized and the contributing organisms to be identified.
However, the technique is not without its problems. DNA extraction and PCR amplification biases burden most modern molecular techniques, including TRFLPs (18, 25). Additionally, concerns exist regarding the ability of the differences between primer-enzyme pairs (PEPs) to generate sufficiently different fragment sizes (2), the success of enzymatic cleavage (2), the dependency on the detection threshold of the sequencer (4), and the accuracy of DNA sizing (1). The choice of the primer pairs and restriction enzymes to be used has also been a matter of concern since the appearance of TRFLP profiling. Liu et al. (30) performed virtual digestion of all the bacterial RNA sequences in the Ribosomal Database Project database (release V) with 10 different enzymes and four primer pairs. This pioneering work showed the importance of avoiding enzymes with highly conserved target motifs, something that later became recognized as a major source of TRFLP bias (2, 14, 16, 32). Similar studies have been performed by Osborn et al. (36), Dunbar et al. (12), Engebretson and Moyer (15), and Cardinale et al. (8).
The first virtual TRFLP analysis involving a database of fungal DNA sequences was performed by Edwards and Turco (14). This consisted of virtual digestion, by use of six restriction endonucleases, of 316 internally transcribed spacer (ITS) sequences belonging to a number of ectomycorrhizal genera. Avis et al. (2) found only small differences in the diversity of the TRFLPs produced in silico by three PEPs when using their own fungal ITS database, although these differences increased with sample number in iterative analysis. Recent advances using automated resources, such as REPK software (9), have allowed optimal enzyme selection for TRFLP profiling of previously defined communities of organisms. This software selects up to four restriction endonucleases capable of discriminating a desired number of sequence groups. However, this system relies on a priori information, which in real biological communities may not available.
The aim of the present work was to improve selection of restriction enzymes for use in the TRFLP profiling of the ITS sequences of unknown fungal communities.
MATERIALS AND METHODS
Sequence acquisition and processing.
The International Nucleotide Sequence Database (INSD) was searched via the NCBI web service (http://www.ncbi.nlm.nih.gov/) in November 2007 for ITS sequences of dicaryal fungi thought to anneal with the ITS1-ITS4 primer pair. While these are not fungal DNA-specific primers, many more fungal ITS sequences contain their complementary sequences than those of the truly fungal specific primer pair ITS1F-ITS4. The diversity of the fragment sizes obtained with these primer pairs should be the same as that obtained with ITS1F-ITS4 since the length of the region between ITS1F and ITS1 is conserved (in fact, it contains the ITS5 universal primer locus). The search strings included the following terms and their etymological variations: (“ascomycete” [organism] AND “internal” [all fields]) and (“basidiomycete” [organism] AND “internal” [all fields]). These were additively employed. The FASTA format files produced were exported to a Microsoft Excel spreadsheet and filtered for the presence of the ITS1 (5′-TCCGTAGGTGAACCTGCGG-3′) and ITS4 (5′-TCCTCCGCTTATTGATATGC-3′) sequences.
Extra bases beyond the primer complementary sequence positions were deleted. ITS sequences only identified above the genus level were discarded, while sequences annotated as cf. or aff. were assumed to be properly identified. The entire database was virtually digested individually with 135 commercially available type IIP restriction endonucleases (see Table S1 in the supplemental material), using the string-finding functions in Excel. If no target was found in an amplicon, its length was assumed to be the size that would be recorded by the sequence analyzer in in vitro analysis. Additionally, sequences were discarded if they contained ambiguous nucleotides (12). Sequences identified at the genus level were verified not to contain redundant information, i.e., no fully identified sequences of the same genus presented the same TRFLP size (see the supplemental material). Finally, each set of data obtained from virtual digestion of the entire database with each PEP was independently filtered so that, for size diversity analysis purposes, a single record was obtained for each size type in each taxon.
The influence of the filtered PEP data set size on the diversity of fragment sizes was corrected by means of random selection of data from each PEP data set to provide final data sets of equal size (i.e., containing the same number of data as the smallest of all PEP data sets). This rarefaction was performed in quintuplet for each PEP data set. The relative abundance (pi) of each size type in each rarefacted PEP data set was obtained using equation 1, where n represents the number of different taxa sharing that TRFL, Sn is the number of different TRFLs in each of these taxa, and m is the number of different TRFLs in the PEP-TRFL data set. A final filtering step was performed to avoid analyzing fragments outside the size range of the internal standard employed (50 to 1,200 bp) (24). Outsiders were pooled in a single “zero” category.
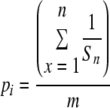 |
(1) |
Diversity measurements.
Hill's effective numbers of classes (20), which are calculated from classic ecological indices (21, 23), were used to measure the diversity of the fragments' sizes. Hill's indices were calculated for diversity orders representing α values of 0, 1, 2, and ∞ (Table 1) by using the fragment size frequency data set obtained from each of the five rarefacted PEP data sets; the median value was calculated for each index. The PEPs were then ordered according to each of these four values, and the 20 top-scoring PEPs within each diversity rank were pooled together in a top-50 group for further analysis. The square root of Jensen-Shannon's divergence (28) between the averages for the five frequency data sets for each PEP pair in this pool was calculated using equation 2, where JSπ represents Jensen-Shannon's divergence, p1 and p2 are two probability distributions, H is Shannon's entropy [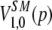 ], and π is the weighting of each probability distribution (here, 0.5) (28).
], and π is the weighting of each probability distribution (here, 0.5) (28).
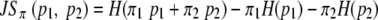 |
(2) |
The resulting divergence matrix was subjected to multidimensional scaling in two dimensions by using the ALSCAL and PROXSCAL routines in the SPSS 11.5 software package (with 50,000 random starting points). Tentative sorting was performed by calculating the product of the arctangent-transformed multidimensional scaling coordinates of each PEP. This transformation was intended to standardize both coordinates in order to invest them with equal weight.
TABLE 1.
Hill's effective number of classes and related classical indices
| Diversity order | Hill's number of classesa | Ecological index |
|---|---|---|
| α | 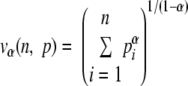 |
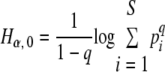 |
| 0 | 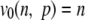 |
Richness |
| 1 | 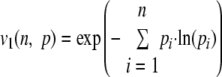 |
Shannon |
| 2 | 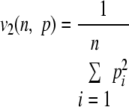 |
Simpson |
| ∞ | 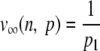 |
Berger-Parker |
n is the number of classes of a given distribution, pi is the frequency of the class, and i, vα (n, p) is the efficient number of classes of order α diversity for the given distribution.
Enzyme set selection.
PEPs were tested for data independence by hierarchical clustering of the original (unfiltered) TRFLP data since some enzymes have been reported to provide redundant data (14). Free hierarchical clustering was performed using interval distance measurements, and employing the unweighted-pair group method using average linkages for clustering. One enzyme in each of the main clusters was selected, and the optimal number of these in a putative optimal set was analyzed by comparing the ribotype profiles resulting from fingerprinting differently sized databases. The ribotype richness (the number of different profiles), the percentage of unique ribotypes, and the ratio between congeneric and noncongeneric shared ribotypes were calculated using optimal enzyme sets composed of one to six enzymes. The ITS1- and ITS4-primed TRFLP data for each enzyme were employed in this analysis.
TRAMPR simulations.
The fragments produced by the selected set of enzymes and five randomly generated sets of enzymes were compared by means of TRAMPR software simulations (11, 17). This software allows for computer-assisted matching of TRFLP profiles, relating them to a given molecular database organized by taxonomic and ribotypic sequence similarity. Matching stringency and algorithmic distance measurements can be set by the user. “Knowns” files were generated from the databases mentioned above and used as input for the TRAMPR program. “Sample” files with 30 replicate random communities of 10, 20, and 50 entries each were also generated and loaded. Matching was performed with an error of 0.5 bp and by using the “maximum” distance computing method. Results were exported to Excel and original taxon frequencies calculated using equation 1, where n is the number of original taxa in each TRAMPR grouping, Sn the number of taxa in each TRAMPR grouping, and m the number of TRAMPR groupings in each virtual community. Finally, the resolving power of each set was measured as the square root of Jensen-Shannon's divergence between the original frequency distribution (homogeneous) and the TRAMPR-biased frequency distribution of the original taxa.
RESULTS
The INSD search of fungal ITS sequences retrieved a total of 61,752 entries. Filtering for the presence of sequences complementary to primers ITS1 and ITS4 in two subsets of ITS sequences (totaling 11,298 and 12,716 entries, respectively) was performed. Sufficiently identified sequences with both complementary sequences (4,618 distinct entries) (see Table S2 in the supplemental material) were virtually digested with 135 different endonucleases (see Table S1 in the supplemental material), redundancy filtered, and randomly rarefacted to a common size of 1,659 entries (presented by ITS4-ITS1 MseI) in quintuplet. The top performers from the four different median diversity measurements (Table 1) were pooled together, giving 48 distinct PEPs (Table 2).
TABLE 2.
Pooled diversity for top-scoring PEPs, sorted by effective number of classes of Renyi's order 1 diversitya
| Name | Target | H0 | H1 | H2 | H∞ |
|---|---|---|---|---|---|
| ITS1 MaeIII | /GTNAC | 562 | 384.4 | 265.9 | 36.55 |
| ITS1 MaeII | A/CGT | 544 | 370.9 | 253.8 | 37.74 |
| ITS1 ApyI | /CCWGG | 552 | 364.2 | 237.9 | 33.61 |
| ITS1 BstNI | CC/WGG | 543 | 363.1 | 240.0 | 35.22 |
| ITS4 BstNI | CC/WGG | 533 | 361.3 | 213.8 | 24.28 |
| ITS4 ScrFI | CC/NGG | 554 | 360.6 | 186.7 | 20.75 |
| ITS4 ApyI | /CCWGG | 538 | 359.5 | 202.6 | 21.96 |
| ITS4 BfaI | C/TAG | 532 | 354.8 | 263.8 | 68.59 |
| ITS4 MaeII | A/CGT | 524 | 349.1 | 236.2 | 48.82 |
| ITS4 StyI | C/CWWGG | 517 | 348.3 | 263.0 | 74.09 |
| ITS4 RsaI | GT/AC | 505 | 347.4 | 266.4 | 81.04 |
| ITS4 DraII | RG/GNCCY | 484 | 340.1 | 264.3 | 63.40 |
| ITS1 StyI | C/CWWGG | 504 | 338.3 | 261.8 | 73.77 |
| ITS4 BmyI | GDGCH/C | 496 | 337.4 | 243.0 | 41.39 |
| ITS4 DdeI | C/TNAG | 553 | 336.5 | 181.9 | 27.14 |
| ITS4 AflI | G/GWCC | 495 | 333.6 | 217.8 | 31.25 |
| ITS4 BanII | GRGCY/C | 488 | 331.7 | 243.3 | 49.20 |
| ITS1 AcyI | GR/CGYC | 511 | 331.6 | 224.4 | 33.15 |
| ITS4 BstUI | CG/CG | 515 | 329.2 | 230.1 | 40.56 |
| ITS1 DdeI | C/TNAG | 578 | 328.9 | 111.5 | 12.84 |
| ITS1 BsiEI | CGRY/CG | 481 | 325.9 | 253.5 | 93.78 |
| ITS1 NspBII | CMG/CKG | 477 | 325.2 | 252.0 | 80.70 |
| ITS1 NgoMI | G/CCGGC | 483 | 323.8 | 255.3 | 93.85 |
| ITS4 AcyI | GR/CGYC | 476 | 323.2 | 248.7 | 70.00 |
| ITS1 BanI | G/GYRCC | 485 | 323.2 | 248.8 | 62.65 |
| ITS1 CfrI | Y/GGCCR | 493 | 321.7 | 238.7 | 59.20 |
| ITS1 HaeII | RGCGC/Y | 479 | 318.4 | 241.7 | 66.89 |
| ITS1 RsaI | GT/AC | 526 | 317.6 | 169.1 | 21.06 |
| ITS1 BstUI | CG/CG | 552 | 315.8 | 140.1 | 16.47 |
| ITS1 AflI | G/GWCC | 515 | 314.9 | 127.9 | 14.29 |
| ITS4 BsrFI | R/CCGGY | 461 | 313.2 | 239.6 | 75.93 |
| ITS4 DsaI | C/CRYGG | 477 | 313.1 | 234.5 | 75.69 |
| ITS1 ScrFI | CC/NGG | 546 | 308.7 | 140.9 | 19.66 |
| ITS1 AhaIII | TTT/AAA | 461 | 305.9 | 232.9 | 80.02 |
| ITS1 SfcI | C/TRYAG | 461 | 298.6 | 230.7 | 88.51 |
| ITS1 EagI | C/GGCCG | 438 | 291.7 | 232.8 | 88.62 |
| ITS4 MaeIII | /GTNAC | 527 | 286.8 | 83.6 | 10.91 |
| ITS1 BfaI | C/TAG | 566 | 282.5 | 92.6 | 12.89 |
| ITS4 KasI | G/GCGCC | 415 | 275.7 | 214.3 | 77.64 |
| ITS4 AatI | AGG/CCT | 403 | 272.9 | 212.8 | 74.55 |
| ITS1 PaeR7I | C/TCGAG | 407 | 272.7 | 217.2 | 85.55 |
| ITS1 SnaBI | TAC/GTA | 405 | 268.7 | 213.6 | 82.19 |
| ITS1 KspI | CCGC/GG | 403 | 260.0 | 202.5 | 75.60 |
| ITS4 BsePI | G/CGCGC | 393 | 254.6 | 200.1 | 76.12 |
| ITS1 AatII | GACGT/C | 387 | 249.7 | 196.3 | 76.36 |
| ITS1 NlaIV | GGN/NCC | 515 | 245.6 | 60.6 | 8.680 |
| ITS1 SspI | AAT/ATT | 385 | 243.2 | 190.2 | 75.55 |
| ITS4 PvuII | CAG/CTG | 371 | 242.5 | 195.6 | 81.03 |
| ITS1 BamHI | G/GATCC | 374 | 239.7 | 189.6 | 75.45 |
| ITS4 Bsp1407I | T/GTACA | 366 | 235.2 | 186.7 | 76.55 |
H1, exponential Shannon index; H0, richness; H2, reciprocal Simpson index; H∞, reciprocal Berger-Parker index; /, enzyme's cleavage point.
As an indirect estimate of diversity, two-dimensional scaling of Jensen-Shannon's divergences (28) between the averaged fragment size frequency distributions was performed, resulting in a consistent pattern (Fig. 1) in which dimension 1 relates to TRFLP richness (order 0 diversity) and dimension 2 represents TRFL evenness (in the sense of the Hill series). Tentative final scores were calculated from the rescaled coordinates (Table 3). A putatively optimal group of enzymes formed by MaeIII, MaeII, BfaI, BstNI, StyI, and DdeI was selected from among the independently clustered candidates (see Fig. S1 in the supplemental material), although MaeIII was replaced by RsaI on the basis of cost. The group formed by the ITS1- and ITS4-primed MaeII, BfaI, and BstNI data sets showed the greatest increases in both the number of ribotypes and the number of unique ribotypes (Fig. 2), indicating that the inclusion of any more enzymes would be ineffective. Similarly, the trend of the noncongeneric/congeneric shared ribotype ratio (Fig. 2) suggests that TRFLP profiles are increasingly shared by phylogenetically related sequences when up to three enzymes are used; no major changes are seen if more are included.
FIG. 1.

Common space plot of PROXSCAL (distances as interval; 50,000 random starting points; SPSS 11.5) multidimensional scaling of the Jensen-Shannon square root divergence matrix computed from the average frequency distributions for the top 50 PEPs. Each point represents a different PEP.
TABLE 3.
Single PEP tentative final sortinga
| PEP | Target | MDSX | MDSY | Score |
|---|---|---|---|---|
| ITS1-ITS4 MaeIII | /GTNAC | −0.7423 | −0.2583 | 0.1614 |
| ITS1-ITS4 MaeII | A/CGT | −0.6506 | −0.2471 | 0.1397 |
| ITS4-ITS1 MaeII | A/CGT | −0.4964 | −0.2542 | 0.1147 |
| ITS4-ITS1 BfaI | C/TAG | −0.3415 | −0.3564 | 0.1126 |
| ITS1-ITS4 BstNI | CC/WGG | −0.6566 | −0.1642 | 0.0946 |
| ITS4-ITS1 RsaI | GT/AC | −0.1920 | −0.3770 | 0.0684 |
| ITS4-ITS1 StyI | C/CWWGG | −0.2076 | −0.3394 | 0.0669 |
| ITS1-ITS4 ApyI | /CCWGG | −0.6226 | −0.1208 | 0.0669 |
| ITS4-ITS1 BmyI | GDGCH/C | −0.2478 | −0.1201 | 0.0290 |
| ITS4-ITS1 DraII | RG/GNCCY | −0.0714 | −0.3508 | 0.0240 |
| ITS4-ITS1 BstNI | CC/WGG | −0.6872 | −0.0322 | 0.0194 |
| ITS4-ITS1 BanII | GRGCY/C | −0.1163 | −0.1462 | 0.0168 |
| ITS1-ITS4 StyI | C/CWWGG | −0.0448 | −0.3259 | 0.0141 |
| ITS4-ITS1 BstUI | CG/CG | −0.1410 | −0.0513 | 0.0071 |
| ITS1-ITS4 AcyI | GR/CGYC | −0.2136 | −0.0309 | 0.0065 |
| ITS4-ITS1 ApyI | /CCWGG | −0.7172 | −0.0081 | 0.0050 |
MDS-X and MDS-Y are the abscissa and ordinate coordinates, respectively, obtained by multidimensional scaling. /, enzyme's cleavage point.
FIG. 2.

Comparisons of differently sized enzyme sets. Databases of 4,618 entries (squares), 2,500 entries (triangles), and 1,000 entries (diamonds) were constructed.
The enzyme set composed of MaeII, BfaI, and BstNI (using both ITS1 and ITS4 TRFLP data) outperformed other randomly selected sets at identifying the members of model communities via their TRAMPR profiles, irrespective of the number of community members. This was true despite the fact that random enzyme sets were expected to be more data independent, since they were formed using either ITS1 or ITS4 TRFLP data (but not both) of six different enzymes. The enzyme set composed of MaeII, BfaI, and BstNI was, in turn, slightly less accurate than a single-primed six-enzyme optimal set (Table 4), in accordance with the simulations shown in Fig. 2.
TABLE 4.
Square roots of Jensen-Shannon's divergence between original sample communities and TRAMPR-biased communities
| Set | PEPa | Mean square root (SD) for indicated virtual community size
|
|||||||
|---|---|---|---|---|---|---|---|---|---|
| 10 sequences | 20 sequences | 50 sequences | |||||||
| 3E | MaeII-14 | MaeII-41 | BfaI-14 | BfaI-41 | BstNI-14 | BstNI-41 | 0.3084 (0.0622) | 0.2900 (0.0448) | 0.2803 (0.0411) |
| 6E | MaeII-14 | BfaI-41 | BstNI-14 | RsaI-41 | StyI-41 | DdeI-14 | 0.2520 (0.0546) | 0.2471 (0.0420) | 0.2501 (0.0283) |
| Rand1 | BssGI-41 | CfoI-41 | EcoNI-41 | PacI-41 | SwaI-41 | TaqI14 | 0.3072 (0.0532) | 0.3129 (0.0380) | 0.3253 (0.0199) |
| Rand2 | ApyI-41 | BanII-41 | BsiWI-14 | MfeI-41 | NdeI-41 | NruI-14 | 0.4136 (0.0601) | 0.4114 (0.0419) | 0.4145 (0.0249) |
| Rand3 | AlwNI-14 | AspI-41 | Fnu4HI-14 | HpaI-41 | NarI-14 | SrfI-14 | 0.4013 (0.0480) | 0.4022 (0.0322) | 0.4287 (0.1024) |
| Rand4 | AspI-41 | AspHI-14 | HaeII-14 | Psp1406I-41 | ScaI-14 | SfcI-14 | 0.4199 (0.0604) | 0.4292 (0.0379) | 0.4268 (0.0212) |
| Rand5 | BsaI-41 | BspLU11I-41 | CfrI-14 | HpaI-41 | NciI-41 | NlaIII-14 | 0.3772 (0.0477) | 0.3629 (0.0346) | 0.3776 (0.0201) |
3E, three-enzyme, double-sided PEP set; 6E, six-enzyme, single-sided PEP set; Rand1 to Rand5, randomly chosen PEP sets. “14” represents the ITS1-ITS4 primer pair, and “41” represents the ITS4-ITS1 primer pair.
DISCUSSION
An optimal, maximally cost-effective set of enzymes for TRFLP analysis of fungal ITS, formed by MaeII, BfaI, and BstNI, is here proposed. The results of in silico TRFLP diversity measurements, hierarchical clustering, and TRAMPR simulations all support the choice of this set of enzymes. Some of the individually top-performing enzymes were the same as those reported in other simulations involving fungi and even bacteria. Moyer et al. (34) reported HhaI, RsaI, and BstUI to be the best enzymes for RFLP profiling when performed virtually using a local bacterial sequence database. MspI, HhaI, RsaI, and BstUI were reported as top performers for TRFLP of bacterial samples by Liu et al. (30), and later, BstUI, DdeI, Sau96I, and MspI were identified as such by Engebretson and Moyer (15). Edwards and Turco (14), whose work involved profiling fungal ITS sequences, identified HaeIII as the top-performing enzyme. The same was reported by Avis et al. (2) and Dickie and FitzJohn (11), together with HpyCH4IV. However, the most-used enzymes in real fungal TRFLP profiling have been HinfI (3, 10, 13, 19, 26, 27, 29, 38), HaeIII (3, 5, 6, 7, 10, 13, 27), AluI (6, 7, 29, 38), TaqI (13, 19, 26, 29, 33), CfoI (29), HhaI and MspI (31), BsuRI (29), and Hsp92II (22), among others. While the outstanding performance of HpyCH4IV (an isoschizomer of MaeII) is in accordance with the present results, a comparison of Tables 2 and 5 shows that most of the other most-used enzymes cannot be considered optimal. Interestingly, the present results for RsaI contrast with those obtained by Edwards and Turco (14), who declared this enzyme unsuitable for TRFLP profiling of fungal ITS sequences. This discrepancy might be due to differences in fragment size detection range; detection thresholds can impose critical limitations on measurement of diversity (4, 15).
TABLE 5.
Diversity values for the most-frequently used TRFLP enzymes reported to be employed with fungi
| Name | Hill's effective no. of classes fora:
|
|||
|---|---|---|---|---|
| D0 | D1 | D2 | D∞ | |
| ITS1-ITS4 HinfI | 350 | 210.1 | 119.6 | 17.37 |
| ITS4-ITS1 HinfI | 324 | 201.3 | 132.1 | 23.86 |
| ITS4-ITS1 HaeIII | 491 | 251.9 | 114.6 | 17.18 |
| ITS1-ITS4 HaeIII | 492 | 206.2 | 47.6 | 7.68 |
| ITS1-ITS4 AluI | 487 | 312.7 | 184.2 | 23.16 |
| ITS4-ITS1 AluI | 429 | 261.4 | 162.3 | 30.29 |
| ITS4-ITS1 TaqI | 332 | 194.0 | 119.5 | 22.03 |
| ITS1-ITS4 TaqI | 301 | 164.8 | 69.2 | 10.29 |
| ITS4-ITS1 CfoI | 376 | 240.1 | 173.5 | 41.59 |
| ITS1-ITS4 CfoI | 412 | 24.3 | 102.0 | 13.75 |
D0, order 0 diversity; D1, order 1 diversity; D2, order 2 diversity; D∞, order ∞ diversity.
As Marsh (32) and, later, Engebretson and Moyer (15) indicated for bacterial in silico TRFLP simulations, databases afford biased views of true diversity; not all organisms' DNA have received the same sequencing interest (37). Moreover, Nilsson et al. (35) reported a worrying percentage of misidentified fungal sequences in public databases, some 10 to 21% of all those deposited. Database bias may affect the present results in a PEP evenness-dependent manner, since overestimation of diversity more probably occurs in more-diverse PEP environments.
A more realistic community would show size frequencies dependent on abundance of local taxa, number of ITS copies per taxon, and success of DNA extraction and amplification (4, 6, 18, 36). Assuming the frequencies derived from redundancy filtering, a conservative estimate of the resolving power of the TRFLPs was made.
Real biological communities are available as population sets (PopSets) at the NCBI website, although few of them meet the requirements for use in the current simulation. Only 10 POP sets (search performed in December 2008) corresponding to dicaryal fungi have a minimum of 60% of sequences (a total of 77 sequences) simultaneously showing the complementary sequences to the ITS1 and ITS4 primers, lack ambiguous nucleotides, and have been sufficiently well identified. If sufficient POP sets could be obtained, it would be interesting to select enzyme sets for use in identifying the organisms present in broad ecological systems, e.g., European temperate forests or decaying meat.
It would certainly be possible to select a different optimal enzyme set. MaeIII was rejected on the basis of cost, but it was in fact the best enzyme tested in the current simulation. It has been shown that a set of enzymes selected using six one-sided PEPs can outperform the proposed double-sided three-enzyme set, but this would make the method too expensive, and the gain in accuracy would only be very small. Other factors, such as the enzyme's optimal buffer and working conditions, could be interesting too. If an alternative enzyme set based on richness was constructed, the size distribution of the fragments produced might be uneven and data dependent, while a set based on diversity alone might suffer the same problem. In either case, lower diversity values would be returned and the results would show greater variability. Some of the results of the present work bear this out (see Tables S3 and S4 in the supplemental material).
The proposed enzyme set is nonoptimal in two ways. First, the entire diversity of the INSD ITS database is not fully reproduced by the TRFLPs, and second, it is insufficiently large to include all real diversity. As shown above, two or three enzymes can reflect most of the variation between sequences in this database but still cannot reflect it all, probably due to similarities between close-relative and improperly identified data in the INSD. Greater efficiency might be achieved by using more enzymes (15), but broader in silico simulations searching for more-diverse and data-independent PEPs may lead to requirement of fewer enzymes for achievement of the same resolving power. The number of enzymes to be used is therefore open to discussion, but certainly there must come a point at which too many could be used if databases are only small (14), or too few could be used in an attempt to profile the huge diversity of the world's fungi (37). Thus, the number of enzymes required in TRFLP profiling depends on the combined efficiency of those selected.
Supplementary Material
Acknowledgments
This work was supported by project PAI08-0240-5097 of Junta de Comunidades de Castilla-La Mancha, the Empleaverde Truficulture project from the Fundación Diversidad (cofinanced by the European Social Fund and the FGUA Cátedra de Medio Ambiente), and FPU grant AP2006-00890 from the Ministerio de Educación y Ciencia of Spain.
We express our gratitude to D. Abarca, M. C. Alonso, E. Ferrer, and M. Martín of the University of Alcalá for their comments on the manuscript.
Footnotes
Published ahead of print on 22 May 2009.
Supplemental material for this article may be found at http://aem.asm.org/.
REFERENCES
- 1.Abdo, Z., U. M. E. Schüette, S. J. Bent, C. J. Williams, L. J. Forney, and P. Joyce. 2006. Statistical methods for characterizing diversity of microbial communities by analysis of terminal restriction fragment length polymorphisms of 16S rRNA genes. Environ. Microbiol. 8:929-938. [DOI] [PubMed] [Google Scholar]
- 2.Avis, P. G., I. A. Dickie, and G. M. Mueller. 2006. A ‘dirty’ business: testing the limitations of terminal restriction length polymorphism (TRFLP) analysis of soil fungi. Mol. Ecol. 15:873-882. [DOI] [PubMed] [Google Scholar]
- 3.Avis, P. G., G. M. Mueller, and J. Lussenhop. 2008. Ectomycorrhizal fungal communities in two North American oak forests respond to nitrogen addition. New Phytol. 179:472-483. [DOI] [PubMed] [Google Scholar]
- 4.Blackwood, C. B., D. Hudleston, D. R. Zak, and J. S. Buyer. 2007. Interpreting ecological diversity indices applied to terminal restriction fragment length polymorphism data: insights from simulated microbial communities. Appl. Environ. Microbiol. 73:5276-5283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Buchan, A., S. Y. Newell, J. I. L. Moreta, and M. A. Moran. 2002. Analysis of internal transcribed spacer (ITS) regions of rRNA genes in fungal communities in a Southeastern U.S. salt marsh. Microb. Ecol. 43:329-340. [DOI] [PubMed] [Google Scholar]
- 6.Burke, D. J., K. J. Martin, P. T. Rygiewicz, and M. A. Topa. 2005. Ectomycorrhizal fungi identification in single and pooled root samples: terminal restriction fragment length polymorphism (TRFLP) and morphotyping compared. Soil Biol. Biochem. 37:1683-1694. [Google Scholar]
- 7.Burke, D. J., K. J. Martin, P. T. Rygiewicz, and M. A. Topa. 2006. Relative abundance of ectomycorrhizas in a managed loblolly pine (Pinus taeda) genetics plantation as determined through terminal restriction fragment length polymorphism profiles. Can. J. Bot. 84:924-932. [Google Scholar]
- 8.Cardinale, M., L. Brusetti, P. Quatrini, S. Borin, A. M. Puglia, A. Rizzi, E. Zanardini, C. Sorlini, C. Corselli, and D. Daffonchio. 2004. Comparison of different primer sets for use in automated ribosomal intergenic spacer analysis of complex bacterial communities. Appl. Environ. Microbiol. 70:6147-6156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Collins, R. E., and G. Rocap. 2007. REPK: an analytical web server to select restriction endonucleases for terminal restriction fragment length polymorphism analysis. Nucleic Acids Res. 35:W58-W62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dickie, I. A., W. Xu, and R. T. Koide. 2002. Vertical niche differentiation of ectomycorrhizal hyphae in soils as shown by T-RFLP analysis. New Phytol. 156:527-535. [DOI] [PubMed] [Google Scholar]
- 11.Dickie, I. A., and R. G. FitzJohn. 2007. Using terminal restriction fragment length polymorphism (T-RFLP) to identify mycorrhizal fungi: a methods review. Mycorrhiza 17:259-270. [DOI] [PubMed] [Google Scholar]
- 12.Dunbar, J., L. O. Ticknor, and C. R. Kuske. 2001. Phylogenetic specificity and reproducibility and new method for analysis of terminal restriction fragment profiles of 16S rRNA genes from bacterial communities. Appl. Environ. Microbiol. 67:190-197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Edwards, I. P., J. L. Cripliver, A. R. Gillespie, K. H. Johnsen, M. Scholler, and R. F. Turco. 2004. Nitrogen availability alters macrofungal basidiomycete community structure in optimally fertilized loblolly pine forests. New Phytol. 162:755-770. [DOI] [PubMed] [Google Scholar]
- 14.Edwards, I. P., and R. F. Turco. 2005. Inter- and intraspecific resolution of nrDNA TRFLP assessed by computer-simulated restriction analysis of a diverse collection of ectomycorrhizal fungi. Mycol. Res. 109:212-226. [DOI] [PubMed] [Google Scholar]
- 15.Engebretson, J. J., and C. L. Moyer. 2003. Fidelity of select restriction endonucleases in determining microbial diversity by terminal-restriction fragment length polymorphism. Appl. Environ. Microbiol. 69:4823-4829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Farmer, D. J., and D. M. Sylvia. 1998. Variation in the ribosomal DNA internal transcribed spacer of a diverse collection of ectomycorrhizal fungi. Mycol. Res. 102:859-862. [Google Scholar]
- 17.FitzJohn, R. G., and I. A. Dickie. 2007. TRAMPR: an R package for analysis and matching of terminal-restriction fragment length polymorphism (TRFLP) profiles. Mol. Ecol. Notes. doi: 10.1111/j.1471-8286.2007.01744.x. [DOI]
- 18.Frey, J. C., E. R. Angert, and A. N. Pell. 2006. Assessment of biases associated with profiling simple, model communities using terminal-restriction fragment length polymorphism-based analyses. J. Microbiol. Methods 67:9-19. [DOI] [PubMed] [Google Scholar]
- 19.Genney, D. R., I. C. Anderson, and I. J. Alexander. 2006. Fine-scale distribution of pine ectomycorrhizas and their extramatrical mycelium. New Phytol. 170:381-390. [DOI] [PubMed] [Google Scholar]
- 20.Hill, M. 1973. Diversity and evenness: a unifying notation and its consequences. Ecology 54:427-431. [Google Scholar]
- 21.Hoffmann, S., and A. Hoffman. 2008. Is there a “true” diversity? Ecol. Econ. 65:213-215. [Google Scholar]
- 22.Johnson, D., P. J. Vandenkoornhuyse, J. R. Leake, L. Gilbert, R. E. Booth, J. P. Grime, J. P. W. Young, and D. J. Read. 2003. Plant communities affect arbuscular mycorrhizal fungal diversity and community composition in grassland microcosms. New Phytol. 161:503-515. [DOI] [PubMed] [Google Scholar]
- 23.Jost, L. 2006. Entropy and diversity. Oikos 113:363-375. [Google Scholar]
- 24.Karudapuram, S., R. Padilla, S. Chen, S. Koepf, J. Hauser, Y. Wang, K. Jacobson, M. White, R. Bordoni, M. Wenz, A. Shah, and L. Joe. 2007. A new high-density size standard for sizing large fragments across multiple fragment analysis capillary electrophoresis applications, poster abstr. P48-T. J. Biomol. Tech. 18: 17. [Google Scholar]
- 25.Kirk, J. L., L. A. Beaudette, M. Hart, P. Moutoglis, J. N. Klironomos, H. Lee, and J. T. Trevors. 2004. Methods of studying soil microbial diversity. J. Microbiol. Methods 58:169-188. [DOI] [PubMed] [Google Scholar]
- 26.Klamer, M., and K. Hedlund. 2004. Fungal diversity in set-aside agricultural soil investigated using terminal-restriction fragment length polymorphism. Soil Biol. Biochem. 36:983-988. [Google Scholar]
- 27.Koide, R. T., B. Xu, and J. Sharda. 2005. Contrasting below-ground views of and ectomycorrhizal fungal community. New Phytol. 166:251-262. [DOI] [PubMed] [Google Scholar]
- 28.Lin, J. 1991. Divergence measures based on the Shannon entropy. IEEE Trans. Inf. Theory 37:145-151. [Google Scholar]
- 29.Lindahl, B. D., K. Ihrmark, J. Boberg, S. E. Trumbore, P. Högberg, J. Stenlid, and R. D. Finlay. 2007. Spatial separation of litter decomposition and mycorrhizal nitrogen uptake in a boreal forest. New Phytol. 173:611-620. [DOI] [PubMed] [Google Scholar]
- 30.Liu, W., T. L. Marsh, H. Cheng, and L. J. Forney. 1997. Characterization of microbial diversity by determining terminal restriction fragment length polymorphisms of genes encoding 16S rRNA. Appl. Environ. Microbiol. 63:4516-4522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.MacDonald, L. M., B. K. Singh, N. Thomas, M. J. Brewer, C. D. Campbell, and L. A. Dawson. 2008. Microbial DNA profiling by multiplex terminal restriction fragment length polymorphism for forensic comparison of soil and the influence of sample condition. J. Appl. Microbiol. 105:813-821. [DOI] [PubMed] [Google Scholar]
- 32.Marsh, T. L. 1999. Terminal restriction fragment length polymorphism (T-RFLP): an emerging method for characterizing diversity among homologous populations of amplification products. Curr. Opin. Microbiol. 2:323-327. [DOI] [PubMed] [Google Scholar]
- 33.Midgley, D. J., J. A. Saleeba, M. I. Stewart, A. E. Simpson, and P. A. McGee. 2007. Molecular diversity of soil basidiomycete communities in northern-central New South Wales, Australia. Mycol. Res. 111:370-378. [DOI] [PubMed] [Google Scholar]
- 34.Moyer, C. L., J. M. Tiedje, F. C. Dobbs, and D. M. Karl. 1996. A computer-simulated restriction fragment length polymorphism analysis of bacterial small-subunit rRNA genes: efficacy of selected tetrameric restriction enzymes for studies of microbial diversity in nature. Appl. Environ. Microbiol. 62:2501-2507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nilsson, R. H., M. Ryberg, E. Kristiansson, K. Abarenkov, K. H. Larsson, and U. Köljalg. 2006. Taxonomic reliability of DNA sequences in public sequence databases: a fungal perspective. PLoS ONE 1:e59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Osborn, A. M., E. R. B. Moore, and K. N. Timmis. 2000. An evaluation of terminal-restriction fragment length polymorphism (T-RFLP) analysis for the study of microbial community structure and dynamics. Environ. Microbiol. 2:39-50. [DOI] [PubMed] [Google Scholar]
- 37.Schmit, J. P., and G. M. Mueller. 2007. An estimate of the lower limit of global fungal diversity. Biodivers. Conserv. 16:99-111. [Google Scholar]
- 38.Zhou, Z., and T. Hogetsu. 2002. Subterranean community structure of ectomycorrhizal fungi under Suillus grevillei sporocarps in a Larix kaempferi forest. New Phytol. 154:529-539. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.