

Abstract
Borrelia burgdorferi has developed efficient mechanisms for evading the innate immune response during mammalian infection and has been shown to be resistant to the complement-mediated bactericidal activity of human serum. It is well recognized that B. burgdorferi expresses multiple lipoproteins on its surface that bind the human complement inhibitors factor H and factor H-like protein 1 (FH/FHL-1). The binding of FH/FHL-1 on the surface of B. burgdorferi is thought to enhance its ability to evade serum-mediated killing during the acute phase of infection. One of the key B. burgdorferi FH/FHL-1 binding proteins identified thus far was designated CspA. While it is known that CspA binds FH/FHL-1, it is unclear how the interaction between CspA and FH/FHL-1 specifically enhances serum resistance. To better understand how CspA mediates serum resistance in B. burgdorferi, we inactivated cspA in a virulent strain of B. burgdorferi. An affinity ligand blot immunoassay and indirect immunofluorescence revealed that the CspA mutant does not efficiently bind human FH to its surface. Consistent with the lack of FH binding, the CspA mutant was also highly sensitive to killing by human serum. Additionally, the deposition of complement components C3, C6, and C5b-9 was enhanced on the surface of the CspA mutant compared to that of the wild-type strain. The combined data lead us to conclude that the CspA-mediated binding of human FH confers serum resistance by directly inhibiting complement deposition on the surface of B. burgdorferi.
Lyme disease, or Lyme borreliosis, is an arthropod-borne infection caused by pathogenic spirochetes of the Borrelia burgdorferi sensu lato complex (62). The spirochete is transmitted to humans by the bite of infected Ixodes scapularis ticks, and the earliest disease manifestations include a skin rash, termed erythema migrans, along with concomitant flu-like symptoms (61). Infected individuals that do not receive antibiotic therapy are at risk for developing chronic forms of the disease, which can result in various disorders of the heart, nervous system, and joints (50, 60). During infection, B. burgdorferi is capable of disseminating and persisting in a variety of host tissues, suggesting that B. burgdorferi has developed efficient mechanisms for evading the host innate immune response. In fact, with regard to the major borrelial genospecies that cause Lyme disease, B. burgdorferi and Borrelia afzelii are resistant to the complement-mediated bactericidal activity of serum, while most strains of Borrelia garinii are killed by human serum (4, 7, 10, 48).
Many human pathogens, including serum-resistant B. burgdorferi, evade complement destruction by binding complement factor H (FH) and FH-like protein 1 (FHL-1) (4, 29, 32, 40, 55). Both FH and FHL-1 (which is a truncated version of FH that results from an alternative splice site in the FH transcript [71]) are serum proteins that negatively regulate the alternative pathway of complement. It has been suggested by several labs, including our own, that B. burgdorferi binds FH/FHL-1 on its surface to inhibit the activation of the alternative pathway of complement and prevent its destruction during the earliest stages of mammalian infection (4, 10, 40, 48, 71). The binding of FH/FHL-1 on the cell surface can also inhibit alternative pathway activation by promoting factor I-mediated cleavage of surface-bound C3b to iC3b (51, 71). Ultimately, it is thought that the inactivation of C3b to iC3b inhibits the deposition of terminal complement components, which then prevents the formation of the bactericidal membrane attack complex. Consistent with this notion, the terminal complement components (C5b, C6, C7, C8, and C9) are deposited more efficiently and abundantly on the surface of serum-sensitive strains of Borrelia spp. than on serum-resistant strains (6, 7, 30).
Since Meri and coworkers first described FH binding surface proteins in B. burgdorferi (29), several proteins have been identified that bind human FH and/or FHL-1. Collectively, these proteins are referred to as complement regulator-acquiring surface proteins (CRASPs) (39). In the B. burgdorferi type strain B31-MI, the FH/FHL-1 binding proteins identified to date are all lipoproteins and include CspA (36), three OspE-related proteins (29), and CspZ (24). CspA is a 27-kDa protein encoded by open reading frame (ORF) BBA68 located on linear plasmid 54 (23). The OspE-related lipoproteins are encoded by ORFs BBL39, BBP38, and BBN38 on different 32-kb circular plasmids (13, 23). The most recent FH binding protein to be identified was termed CspZ (24, 56, 59), which is encoded by ORF BBH06 (23). CspZ was shown to enhance serum resistance in vitro when expressed from a shuttle vector in the serum-sensitive strain B. burgdorferi B313 as well as in the serum-sensitive strain B. garinii G1 (24). However, more recently it was reported that CspZ is not required for the experimental infection of mice (14). The OspE-related proteins and CspA, on the other hand, have been shown by multiple laboratories to play an important role in serum resistance in vitro (2, 10, 24, 31, 49). However, even with the combined studies of the past several years from many different groups, it is still not clear what role CspA and/or the OspE-related proteins play with regard to serum resistance throughout the complex borrelial enzootic life cycle.
To extend these prior studies and help elucidate the role of CspA in FH binding and serum resistance, we inactivated cspA in a virulent strain of B. burgdorferi. We examined the CspA mutant for both its ability to acquire FH from human serum and its ability to resist the surface deposition of various terminal complement components. The CspA mutant generated was highly sensitive to serum and was observed to bind less FH on its surface compared to the parental, wild-type B. burgdorferi strain. Additionally, in contrast to the wild-type strain, complement components C3, C6, and C5b-9 were abundantly deposited on the cell surface of the CspA mutant. The combined data lead us to conclude that the CspA-mediated binding of human FH confers serum resistance by directly inhibiting complement deposition on the surface of B. burgdorferi.
MATERIALS AND METHODS
Bacterial strains and growth conditions.
B. burgdorferi strains 5A4NP1 (34) and 5A15 (53) were kindly provided by Steven Norris (University of Texas Medical School at Houston, Houston, TX). B. burgdorferi strain 5A15 organisms were cultivated at 34°C in BSK-H medium containing 6% heat-inactivated rabbit serum (BSK-H complete medium) (Sigma, St. Louis, MO) (5). The 5A4NP1 strain, which has the kanamycin resistance cassette inserted into ORF BBE02 to enhance transformation with exogenous DNA (34), was cultivated in BSK-H complete medium with the addition of 200 μg/ml kanamycin. All cloning experiments performed with Escherichia coli used strain DH5α grown using tryptone-yeast broth or agar supplemented with the appropriate antibiotic.
cspA mutagenesis and complementation in B. burgdorferi 5A4NP1.
The cspA construct used for gene disruption was previously described (10). The inactivation construct, however, was modified because strain 5A4NP1 is kanamycin resistant as indicated above (34). Therefore, the kanamycin resistance cassette was replaced with the streptomycin resistance cassette (22). The streptomycin resistance cassette was amplified from the borrelial shuttle vector pKFSS-1 (22) using primers FlgB 5′ X and Strep 3′ (Table 1). The resulting amplicon was digested with XbaI and cloned into the original cspA inactivation construct (10) that was digested with XbaI to remove the kanamycin resistance cassette. The construct was electroporated into E. coli DH5α, and a clone was identified that contained the streptomycin resistance cassette in the opposite orientation of the cspA gene. The resulting clone was subsequently used as a template for the PCR amplification of the complete cspA inactivation construct using primers CspA-US 5′ and CspA-DS 3′ (Table 1). Fifteen micrograms of the cspA inactivation construct was electroporated into B. burgdorferi 5A4NP1 as described previously (58). Following electroporation, spirochetes were allowed to recover in BSK-H complete medium for 16 h at 34°C. After they recovered, streptomycin (100 μg/ml) was added and spirochetes were aliquoted onto microtiter plates and screened as previously described (70). One clonal isolate was identified that contained the streptomycin-resistant cassette in the cspA gene, which was confirmed by PCR using primers CspA-US 5′ and CspA-DS 3′ (Table 1). Subsequently, a complete plasmid analysis of the cspA mutant was performed as previously described (10, 21), which revealed that lp21 was lost during mutagenesis. Since it was previously shown that lp21 is not related to B. burgdorferi virulence or Lyme disease pathogenesis (53), the cspA mutant was used for all further experiments. B. burgdorferi strain 5A15, which has the same plasmid content as the cspA mutant (53), was therefore utilized as the wild-type control for all experiments.
TABLE 1.
Oligonucleotide primers used in this study
| Primer | Sequence (5′-3′)a | Description |
|---|---|---|
| FlgB5′X | GCGTCTAGATACCCGAGCTTCAAGGAAGAT | Nucleotides 1-21 of the streptomycin resistance cassette plus the XbaI site |
| Strep 3′ | GCGTCTAGATTATTTGCCGACTACCTTGGTGAT | Complementary to nucleotides 1176-1199 of the streptomycin resistance cassette plus the XbaI site |
| CspA-US 5′ | TTACAGCTACAAGAAAAGTTTAAA | Nucleotides 377-400 upstream of B. burgdorferi cspA |
| CspA-DS 3′ | ATTTGCATTAGCAATGATTTAGAT | Complementary to nucleotides 576-599 downstream of B. burgdorferi cspA |
| BbB19 5′ | GCGCCGCGGGATTTTAAAATCAAATTAAGACAATA | Nucleotides 16751-16776 of B. burgdorferi cp26 plus the SacII site |
| BbB20 3′ | GCGTCTAGAAATTTTGTTTATATTTAAATGCTTAAA | Complementary to nucleotides 17725-17751 of B. burgdorferi cp26 plus the XbaI site |
| BbB21 5′ | GCGCTCGAGGGGTGCTAGAAATTTGATTTTAA | Nucleotides 17752-17774 of B. burgdorferi cp26 plus the XhoI site |
| BbB22 3′ | GCGGGTACCCAATTAAATATAAGGGGAAGTATA | Complementary to nucleotides 18728-18751 of B. burgdorferi cp26 plus the KpnI site |
| FlaB 5′ | GCGTCTAGATGTCTGTCGCCTCTTGTGG | Nucleotides 1-19 of the gfp expression construct plus the XbaI site |
| GFP 3′ | GCGGGATCCCTATTTGTATAGTTCATCCATGCC | Complementary to nucleotides 1048-1071 of the gfp expression construct plus the BamHI site |
| FIgB5′B | GCGGGATCCTACCCGAGCTTCAAGGAAGAT | Nucleotides 1-21 of the gentamicin resistance cassette plus the BamHI site |
| Gent 3′ | GCGGGATCCTTAGGTGGCGGTACTTGGGTCGA | Complementary to nucleotides 919-941 of the gentamicin resistance cassette plus the BamHI site |
| CspA-comp 5′ | GCGAAGCTTTTACAGCTACAAGAAAAGTTTAAA | Nucleotides 377-400 upstream of B. burgdorferi cspA plus the HindIII site |
| CspA-comp 3′ | GCGAAGCTTAGAAGAATTAACTTCTCTTTTTAM | Complementary to nucleotides 792-816 downstream of B. burgdorferi cspA plus the HindIII site |
Nucleotides in bold indicate restriction sites used for cloning.
To complement the mutant cspA strain, the native cspA gene was inserted and expressed from the B. burgdorferi circular plasmid 26 (cp26). The insertion was accomplished using a crossover vector, designated pSPCG-cspA, which was constructed in pBluescript-II SK+ (Stratagene, La Jolla, CA). To generate pSPCG-cspA, a 1,000-bp fragment corresponding to regions of BBB19 and BBB20 of cp26 was amplified using primers BbB19 5′ and BbB20 3′, respectively (Table 1). The amplicon was subsequently purified, digested, and cloned into the SacII and XbaI sites of the pBluescript-II SK+ vector. Next, the gene encoding green fluorescent protein (GFP) fused to the borrelial flaB promoter was amplified from vector pMC1919 using primers FlaB 5′ and GFP 3′ (19) and cloned into the XbaI and BamHI sites of the pBluescript-II SK+ vector. The gentamicin resistance cassette was then amplified from the borrelial shuttle vector pBSV2G (20) using primers FlgB 5′ B and Gent 3′ (Table 1) and inserted into the BamHI restriction site of pBluescript-II SK+. Subsequently, a 1,000-bp region of cp26 containing regions of BBB21 and BBB22 was amplified using primers BbB21 5′ and BbB22 3′ (Table 1) and cloned into the XhoI and KpnI sites of pBluescript-II SK+. Finally, the complete cspA gene and a 400-bp upstream region was amplified with primers CspA-comp 5′ and CspA-comp 3′ (Table 1) and cloned into the HindIII restriction site. The final construct was designated pSPCG-cspA. The cspA gene and the 400-bp upstream region of the final construct were subjected to nucleotide sequence analysis to confirm that no mutations had been incorporated during the PCR. To generate a complemented strain, pSPCG-cspA was electroporated into the cspA mutant as described above, and several gentamicin-resistant clones were selected. The presence of the cspA gene in the mutant strain was confirmed by PCR using primers FlaB 5′ and BbB22 3′ (Table 1). Subsequently, a complete plasmid analysis of the various clones indicated that all but one cspA complemented strain examined was lacking multiple plasmids, including lp25, which is needed for mammalian infection (43, 47, 53). The single isolate that contained all of the plasmids needed for full virulence was lacking cp32-6 but otherwise had the same plasmid profile as the cspA mutant. Given that no FH binding proteins are located on cp32-6, combined with the observation that cp32-6 is not needed for mammalian infection (65), we chose to use this complemented strain for further analyses.
Human sera and antibodies.
Normal human serum (NHS) was purchased from PAA Laboratories (New Bradford, MA) and used for serum sensitivity assays, serum adsorption assays, and the detection of FH bound to the surfaces of B. burgdorferi isolates. For the complement deposition assays, sera from 20 healthy human blood donors that were negative for anti-Borrelia antibodies were combined and used for detecting complement components C3, C6, and C5b-9.
Rat anti-CspA and rabbit anti-FlaB antibodies were generated as previously described (10, 54). The goat anti-human FH, goat anti-human C3, and goat anti-human C6 antibodies were purchased from Calbiochem (Gibbstown, NJ). The monoclonal anti-human C5b-9 antibody was purchased from Quidel (San Diego, CA).
Immunoblotting.
Whole-cell lysates from the wild-type, CspA mutant, and complemented strains (1 × 107) were boiled for 10 min in final sample buffer (62 mM Tris-HCl [pH 6.8], 10% [vol/vol] glycerol, 100 mM dithiothreitol, 2% sodium dodecyl sulfate, 0.001% bromophenol blue) and subjected to sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) through a 2.4% stacking and 12.5% separating gel before being transferred onto a nitrocellulose membrane. To confirm that CspA was not expressed in the mutant strain, the membrane was blocked for 1 h in 5% nonfat dry milk dissolved in phosphate-buffered saline (PBS; pH 7.4) containing 0.2% Tween 20 (blocking buffer) before being incubated with a 1:5,000 dilution of rat anti-CspA antibodies for 1 h. The membrane was subsequently washed three times in blocking buffer and incubated with a 1:20,000 dilution of horseradish peroxidase (HRP)-conjugated goat anti-rat secondary antibodies (Invitrogen, Carlsbad, CA). Following incubation with the secondary antibodies, the membrane was washed three times with blocking buffer and three times with PBS and then developed by enhanced chemiluminescence (ECL) according to the manufacturer's instructions (G-Biosciences, Maryland Heights, MO). To ensure that samples were loaded equivalently, the same membrane was also subjected to immunoblot analysis for the constitutively expressed FlaB protein using rabbit anti-FlaB antibodies.
Surface localization assays.
B. burgdorferi cells were enumerated and diluted to a final concentration of 5 × 106 cells per ml in BSK-H complete medium. Wild-type and CspA mutant cell suspensions were coincubated with rat anti-CspA antibodies (dilution of 1:200) and rabbit anti-FlaB antibodies (dilution of 1:200) for 1 h. The cells were subsequently harvested at 4,000 × g for 4 min and gently washed three times using PBS. The final pellet was resuspended in 100 μl of PBS, and 10-μl aliquots were spotted onto glass slides and allowed to air dry. Samples were fixed for 10 min with acetone and blocked for 30 min with PBS containing 0.5% bovine serum albumin. After blocking was complete, the samples were incubated for 45 min with a 1:750 dilution of Alexa Fluor 488-conjugated goat anti-rat antibodies and a 1:750 dilution of Alexa Fluor 568-conjugated goat anti-rabbit antibodies (Invitrogen). After incubation, all samples were washed three times with blocking buffer and mounted in buffered glycerol containing the DNA binding dye 4′,6-diamidino-2-phenylindole dihydrochloride (DAPI; Vector Laboratories, Burlingame, CA). Because the CspA-complemented strain expresses GFP, Alexa Fluor 488-conjugated antibodies could not be used for the localization assays as outlined above for the wild-type and CspA mutant strains. To overcome this caveat, the CspA-complemented strain was first incubated separately with a 1:200 dilution of rat anti-CspA antibodies or rabbit anti-FlaB antibodies for 1 h. The samples were then spotted on glass slides, fixed with acetone, and incubated with blocking buffer for 30 min. The samples were incubated for 45 min with a 1:750 dilution of Alexa Fluor 568-conjugated goat anti-rat antibodies (for detection of CspA) or Alexa Fluor 568-conjugated goat anti-rabbit antibodies (for detection of FlaB). After the samples were incubated with the appropriate secondary antibodies, the slides were washed, mounted, and sealed as described above. For all localization experiments, spirochetes were visualized at ×1,000 magnification using an Olympus BX60 fluorescent microscope (Olympus America, Inc., Center Valley, PA).
ALBI.
An FH affinity ligand blot immunoassay (ALBI) was performed as described previously (10, 48). Briefly, membranes were incubated for 1 h with 10 μg of human FH (hFH; Calbiochem) and washed three times with blocking buffer. FH binding proteins were then identified in the wild-type, CspA mutant, and complemented strains by incubating the membrane for 1 h with a 1:3,000 dilution of goat anti-hFH antibodies before being washed three times. This was followed by a 45-min incubation in a 1:10,000 dilution of HRP-conjugated rabbit anti-goat antibodies before washing three times in blocking buffer and three times in PBS. The final immunoblot was developed using ECL according to the manufacturer's instructions (G-Biosciences). To ensure that samples were loaded equivalently, the same membrane was subjected to immunoblot analysis for the constitutively expressed FlaB protein using rabbit anti-FlaB antibodies.
Serum sensitivity assays.
Serum sensitivity assays were performed as previously described (10). Briefly, cell suspensions were harvested at 4,000 × g for 5 min and washed three times in BSK-H medium before organisms (5 × 106) were incubated at 34°C with either 50% NHS or 50% heat-inactivated NHS. All assays were performed in triplicate. The samples were examined 1, 4, and 16 h after the addition of serum, and the number of motile spirochetes was enumerated by dark field microscopy. Significant differences between samples incubated with NHS or the corresponding heat-inactivated NHS control were determined using an unpaired, two-tailed Student's t test (17).
Complement deposition assays.
Spirochetes were grown to mid-exponential phase (∼5 × 107 per ml), harvested by centrifugation at 5,000 × g for 30 min, and washed three times before being resuspended in 300 μl PBS. After enumerating the spirochetes, 6 × 106 cells were incubated with 25% NHS for 30 min at 37°C with gentle agitation, washed three times with PBS containing 1% bovine serum albumin, and resuspended in 100 μl of the same buffer. Aliquots of 10 μl were then spotted on microscope slides and allowed to air dry overnight. After fixation for 10 min with methanol, the slides were dried and incubated for 1 h with antibodies against complement components C3 (dilution of 1:1,000), C6 (dilution of 1:200), or C5b-9 (dilution of 1:50). Following four washes with PBS, the slides were incubated for 1 h at room temperature with 1:2,000 dilutions of the appropriate Alexa Fluor 488-conjugated secondary antibodies (Molecular Probes, Leiden, The Netherlands). The slides were then washed four times with PBS and mounted in ProLong Gold antifade reagent containing DAPI (Molecular Probes) before being sealed with coverslips. The slides were visualized using an Olympus CX40 fluorescence microscope mounted with a DS-5Mc charge-coupled device camera (Nikon Precision Europe GmbH, Germany). For the detection of hFH on the cell surface, a similar assay was completed. However, after the samples were fixed to the slides, the samples were incubated with goat anti-hFH antibodies (dilution of 1:200), washed, and incubated with Alexa Fluor 488-conjugated rabbit anti-goat antibodies (Invitrogen) at a dilution of 1:250. The slides were mounted in buffered glycerol containing DAPI (Vector Laboratories) and sealed with a coverslip, and images were visualized and captured with an Olympus BX-60 fluorescence microscope (Olympus America, Inc.).
Serum adsorption assays.
Serum adsorption assays were performed as described previously with minor modifications (30). Briefly, spirochetes were grown to mid-log phase and harvested at 5,000 × g for 20 min. The cell pellet was gently resuspended in PBS, bacteria were enumerated, and 2 × 109 organisms were pelleted by centrifugation at 5,000 × g. The organisms were then resuspended in 850 μl NHS containing 34 mM EDTA and incubated for 1 h at room temperature. After incubation, the cells were harvested by centrifugation and washed three times in PBS containing 0.02% sodium azide and 0.05% Tween 20. The final pellet was then resuspended and incubated in the presence of 0.1 M glycine-HCl (pH 2.0) for 15 min. After incubation, the cells were centrifuged at 15,000 × g for 10 min, the supernatant was removed, and proteins were precipitated using 100% ethanol. Equivalent amounts of precipitated proteins from each sample were then subjected to SDS-PAGE through a 10.5% separating gel. The proteins were transferred to a nitrocellulose membrane and incubated with goat anti-hFH antibodies at a 1:5,000 dilution for 1 h before being washed three times and incubated for 45 min with HRP-conjugated rabbit anti-goat antibodies at a 1:10,000 dilution. After three washes in blocking buffer and three washes in PBS, the membrane was developed by ECL. The same membrane was also immunoblotted with rabbit anti-FlaB antibodies to ensure equal loading between all samples.
RESULTS
Inactivation and complementation of cspA.
CspA has been intensively studied in recent years and several groups have shown that CspA is a major FH/FHL-1 binding protein in B. burgdorferi (15, 16, 25, 35, 38, 41, 67, 67, 67, 71). Consistent with these prior observations, we previously generated a CspA mutant in an avirulent strain of B. burgdorferi and reported that CspA confers resistance to human serum in vitro (10). To extend these prior studies, we inactivated cspA in the virulent B. burgdorferi strain 5A4NP1, which is a highly transformable strain that only lacks plasmid cp9 (34). The virulent cspA mutant strain was generated by electroporating a cspA inactivation construct into B. burgdorferi 5A4NP1. Several streptomycin-resistant clones were identified, and to ensure that the plasmids relevant to mammalian infection (i.e., lp25, lp28-1) were not lost during electroporation (43, 47, 53), we next performed a complete plasmid analysis on all the isolates identified. One cspA mutant was identified that only lost plasmid lp21, and it was chosen for all further analyses. As a wild-type control for the studies outlined below, we utilized B. burgdorferi strain 5A15 because it not only has the same plasmid content as the cspA mutant, but it is also fully virulent (53).
To confirm that the streptomycin resistance cassette was inserted into the cspA gene, a PCR analysis of the mutant strain was performed using primers CspA-US5′ and CspA-DS3′ (Table 1). As shown in Fig. 1A, an amplicon of approximately 1 kb was observed for the wild-type strain, as expected (Fig. 1A, lane 2), while the mutant strain produced an amplicon of approximately 2.2 kb, which is consistent with the insertion of the streptomycin resistance cassette into the cspA gene (Fig. 1A, lane 3).
FIG. 1.
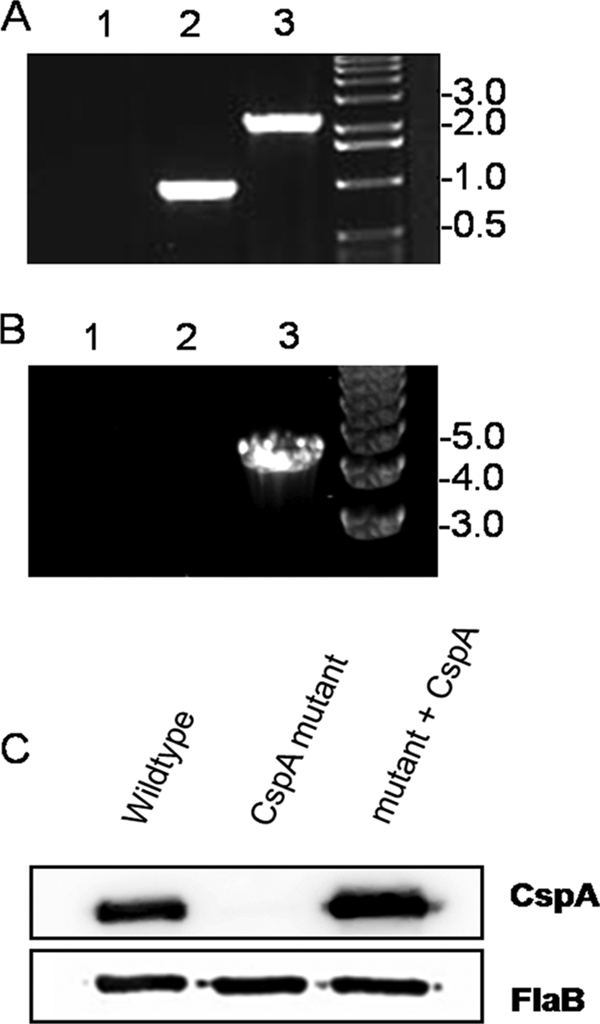
Generation of a CspA mutant in virulent B. burgdorferi and complementation of the CspA mutant. (A) Insertion of the streptomycin resistance cassette into cspA was verified by the PCR amplification of the wild-type or cspA mutant strains using primers CspA-US 5′ and CspA-DS 3′. The wild-type strain produced the expected 1-kb amplicon (lane 2), while the cspA mutant produced a 2.2-kb amplicon (lane 3), which is consistent with the insertion of the streptomycin resistance cassette. As a negative control, a PCR lacking a DNA template was included (lane 1). Molecular weight standards, in kb, are indicated at the right. (B) Insertion of the cspA complementation construct into cp26 was verified by PCR using primers FlaB 5′ and BbB22 3′. No amplicon was detected when the wild-type (lane 1) or cspA mutant (lane 2) strain was subjected to PCR. The cspA complemented strain, however, produced an amplicon of approximately 4.2 kb (lane 3), which is consistent with the insertion of the cspA complementation construct into cp26. Molecular weight standards, in kb, are indicated at the right. (C) B. burgdorferi whole-cell lysates (1 × 107) from the wild-type, CspA mutant, and complemented strains were immunoblotted with rat anti-CspA antibodies (top panel). As expected, CspA was expressed by the wild-type and complemented strains but not by the CspA mutant. The same membrane was also immunoblotted with rabbit anti-FlaB antibodies to verify that equivalent amounts were loaded in each lane (bottom panel).
The cspA mutant strain was complemented with a native copy of the cspA gene by inserting it between ORFs BBB20 and BBB21 in the essential cp26 (12, 33). To confirm that the cspA gene had recombined correctly into the cp26 locus, the cspA-complemented strain was subjected to a PCR with primers FlaB 5′ and BbB22 3′ (Table 1). As shown in Fig. 1B, no amplicon was produced from the wild-type or mutant strains (Fig. 1B, lanes 1 and 2), while the expected 4.2-kb product was detected in the cspA-complemented strain (Fig. 1B, lane 3). Additionally, an immunoblot analysis using rat anti-CspA antibodies confirmed that CspA was expressed by the CspA-complemented strain and not by the CspA mutant (Fig. 1C, upper panel). As a control, the same membrane was subjected to an immunoblot analysis for the constitutively expressed flagellin protein (FlaB) to confirm that equivalent amounts of whole-cell lysates were loaded in all lanes (Fig. 1C, lower panel).
CspA expressed from cp26 is localized to the B. burgdorferi surface.
To confirm that the complemented strain expressed and translocated CspA to the surface of B. burgdorferi, we next performed surface localization assays using indirect immunofluorescence on the wild-type, CspA mutant, and complemented strains. As expected, CspA localized to the outer surface of the wild-type organisms but was not detected in the CspA mutant strain (Fig. 2A). The complemented strain also displayed CspA on its surface (Fig. 2B), indicating that the altered genomic location of this gene did not affect the expression or translocation of the lipoprotein. To ensure that the fragile outer membrane of B. burgdorferi was not disrupted, antibodies specific for the periplasmic protein FlaB were also included in the surface localization experiments. When rabbit anti-FlaB antibodies were coincubated with the rat anti-CspA antibodies, no FlaB was identified, indicating that the outer membranes were intact (Fig. 2A). Because the CspA-complemented strain also expresses GFP from the cp26 locus, Alexa Fluor 488-conjugated secondary antibodies could not be used with this strain. Therefore, aliquots of the complemented strain were incubated separately with either anti-FlaB or anti-CspA antibodies followed by incubation with Alexa Fluor 568 secondary antibodies as the conjugate for both. As shown in Fig. 2B, FlaB was not detected, indicating that the outer membranes remained intact during the surface localization assay. The DNA-binding dye DAPI was also included in the mounting media for the immunofluorescent experiments so that all spirochetes could be visualized in a given microscopic field.
FIG. 2.

CspA is surface localized in wild-type and CspA-complemented B. burgdorferi. The wild-type (A, top panel) and CspA mutant (A, bottom panel) were coincubated with rat anti-CspA antibodies and rabbit anti-FlaB antibodies. The CspA-complemented strain (B) was incubated with either rat anti-CspA antibodies or rabbit anti-FlaB antibodies. After incubation in primary antibodies, organisms were fixed to the slide with acetone and samples were probed with the appropriate Alexa Fluor-conjugated secondary antibodies. Antibodies to the periplasmic FlaB protein were included to ensure that spirochetes were negative for FlaB, which confirmed the outer-membrane integrity of the spirochetes during the surface localization experiments. Spirochetes were counterstained with DAPI to visualize cells in a given field of view, and images were visualized by fluorescence microscopy at ×1,000 magnification.
hFH ALBI.
To examine the ability of the wild-type, mutant, and complemented strains to bind hFH, whole-cell lysates were subjected to ALBI analysis as previously described (10, 48). As shown in Fig. 3 (top panel), when lysates were incubated with hFH, a 27-kDa protein corresponding to CspA was identified in the wild-type and CspA-complemented strains but not in the mutant strain. Interestingly, all strains examined also expressed two additional FH binding proteins. These ∼20-kDa proteins are consistent with the known sizes of the OspE-related lipoproteins, which are also known to bind hFH (1-3,29). Detection of the OspE-related proteins is expected given that low levels of these proteins are typically expressed during the in vitro cultivation of B. burgdorferi (28). To confirm that whole-cell lysates were loaded equivalently, the same membrane was also subjected to immunoblotting with anti-FlaB antibodies (Fig. 3, bottom panel).
FIG. 3.

FH ALBI. Whole-cell lysates from 1 × 107 wild-type, CspA mutant, and CspA-complemented strains were subjected to FH ALBI. The wild-type and CspA-complemented strains expressed a 27-kDa hFH binding protein consistent with the size of CspA. The CspA mutant strain did not express an FH binding protein corresponding to CspA. The membrane was also immunoblotted with rabbit anti-FlaB antibodies to ensure equal loading of the whole-cell lysates.
CspA confers resistance to human serum.
Serum sensitivity assays (4, 10) were next used to examine serum resistance in the wild-type, CspA mutant, and complemented strains. Each of the strains were cultivated in triplicate in 50% NHS or 50% heat-inactivated NHS. Since spirochetes that are sensitive to human serum are immobilized and degraded, serum sensitivity is easily measured by examining the number of intact and motile spirochetes that remain after incubation in serum. For the serum sensitivity assays, aliquots were removed after 1, 4, and 16 h and enumerated for surviving spirochetes. While the wild-type strain was resistant to killing by human serum, as expected (Fig. 4A), the CspA mutant strain was highly sensitive to serum-mediated killing (Fig. 4B). In fact, when the CspA mutant was cultivated in normal serum versus heat-inactivated serum, there was a 90% reduction in survival within 16 h. Importantly, when the mutant strain was complemented with the cspA gene, serum resistance was restored (Fig. 4C). As a control for the killing activity of the serum utilized for these assays, we also included a serum-sensitive, avirulent B. burgdorferi strain for all experiments (10). As expected, the serum-sensitive organisms were all killed within 1 h (Fig. 4D). The combined data indicate that CspA greatly enhances serum resistance in B. burgdorferi.
FIG. 4.

CspA is required for resistance to human serum. Wild-type (A), CspA mutant (B), CspA-complemented (C), and serum-sensitive (D) strains of B. burgdorferi were subjected to serum sensitivity assays. A total of 5 × 106 spirochetes were incubated with either 50% normal serum (•) or 50% heat-inactivated serum (▪) in triplicate. Samples were analyzed for the presence of motile spirochetes by dark field microscopy after 1, 4, and 16 h. Asterisks in panels B and D denote significant differences (P ≤ 0.05) in viability between similar samples incubated with NHS or heat-inactivated NHS.
FH is bound to the B. burgdorferi surface by CspA.
The observation that the CspA mutant was highly sensitive to killing by human serum led us to examine the mechanism by which CspA might mediate serum resistance. Therefore, we examined the ability of the wild-type and mutant strains to bind and adsorb hFH to their surface when incubated in human serum. As shown in Fig. 5A (left panels), when indirect surface immunofluorescence assays were performed, the wild-type and CspA mutant strains displayed a dramatic difference in the amount of hFH bound to their surfaces. While the wild-type strain acquired abundant amounts of hFH on its surface, hFH could barely be detected on the surface of the mutant strain. The small amount of hFH that was detected on the surface of the CspA mutant strain is most likely due to the low-level expression observed for the OspE-related proteins (Fig. 3). The samples examined by immunofluorescence for surface hFH binding were also counterstained with the DNA-binding dye DAPI so that all spirochetes could be visualized in each field of view (Fig. 5A, right panels).
FIG. 5.

CspA is required for the efficient adsorption of FH to the surface of B. burgdorferi. (A) Human FH adsorbed to the surface of wild-type and CspA mutant spirochetes was detected by immunofluorescence. Spirochetes were incubated with 25% NHS, and FH on the surface of B. burgdorferi was detected with anti-hFH antibodies. The DNA-binding dye DAPI was used to visualize all spirochetes in each field. Images were visualized by fluorescence microscopy at ×1,000 magnification. (B) The amount of FH bound to wild-type and CspA mutant organisms was examined by an immunoblot assay. Whole-cell lysates (2 × 109) from wild-type and CspA mutant strains were resuspended in NHS containing EDTA before FH was eluted. The final supernatant was separated by SDS-PAGE, and FH was detected using anti-hFH antibodies (top panel). To verify that equivalent amounts of protein were loaded in each lane, the membrane was also immunoblotted with anti-FlaB antibodies (bottom panel).
Consistent with the immunofluorescence assays, when hFH serum adsorption experiments were performed with the wild-type and CspA mutant organisms (30), an abundant amount of hFH could be eluted from the wild-type strain, while almost no hFH was eluted from the CspA mutant strain (Fig. 5B, top panel). To confirm that equivalent amounts of the wild-type and mutant strains were analyzed in the serum adsorption experiments, the same membrane was subjected to immunoblotting with anti-FlaB antibodies (Fig. 5B, bottom panel). Collectively, these results indicate that CspA is required for the efficient adsorption of hFH to the B. burgdorferi surface.
CspA inhibits the deposition of complement on the surface of B. burgdorferi.
The combined studies thus far indicated that the CspA mutant is highly sensitive to serum-mediated killing and is severely impaired in its ability to acquire FH from human serum. To determine if the decreased binding of FH on the surface of the CspA mutant correlates with an increase in complement deposition, we next examined the surfaces of the wild-type and CspA mutant organisms for the deposition of complement components C3, C6, and membrane attack complex C5b-9. As shown in Fig. 6, no fluorescent staining was observed for the majority of the wild-type cells and those that were positive were only weakly fluorescent. In contrast, all CspA mutant cells stained strongly positive for C3, C6, and C5b-9. In addition, most spirochetes that were covered with complement component proteins stained negative with DAPI and likely represent DNA-free cell ghosts. We also observed strong fluorescent staining of blebs with DAPI in the CspA mutant strain (indicated by arrowheads in the middle panels), suggesting that there is a high concentration of DNA in these particles. These data are consistent with a previous report showing that borrelial cells displaying a serum-resistant phenotype show the low-level deposition of complement activation products, while serum-sensitive strains show an abundance of C3, C6, and C5b-9 deposition (30).
FIG. 6.

Identification of activated complement components on the surface of B. burgdorferi. The deposition of complement components C3, C6, and C5b-9 on the surfaces of the wild-type and CspA mutant strains was detected by immunofluorescence. Spirochetes were incubated in the presence of 25% NHS, and complement proteins bound to the cell surface were detected with antibodies directed against C3, C6, or C5b-9. DAPI stain was used to detect all spirochetes within a given field, and the bottom panels display merged images of the fluorescent and DAPI images. Arrowheads in the C6 and C5b-9 DAPI panels for the CspA mutant indicate blebs tightly packed with DNA. Spirochetes were visualized by fluorescence microscopy at ×1,000 magnification.
DISCUSSION
The B. burgdorferi type strain B31-MI expresses five lipoproteins that bind FH/FHL-1, including CspA, CspZ, and three OspE-related proteins (13, 23, 24, 59). While several studies have reported on the FH binding activity of these lipoproteins in vitro (1-3, 9, 24, 29, 56, 59, 63), only CspZ has been mutated in a virulent strain and assessed for its role in vivo using an animal model (14). Therefore, we generated a CspA mutant in a virulent B. burgdorferi strain that expresses all of the other identified FH/FHL-1 binding proteins. The CspA mutant characterized was highly serum sensitive, impaired in its ability to adsorb FH onto the borrelial surface, and hypersensitive to the deposition of downstream complement components, including the bactericidal membrane attack complex.
B. burgdorferi has evolved complex parasitic strategies for evading the innate immune response of mammals since small rodents, especially mice, are the typical reservoir for this organism (44). In this regard, several surface proteins have been implicated in helping B. burgdorferi evade the innate immune response during the acute phase of mammalian infection. These include outer surface lipoprotein C (OspC) (66) and the five FH/FHL-1 binding proteins (13, 15, 23, 24, 37). While it is unclear how OspC enhances evasion of the innate immune response (66, 69), it has been suggested by several laboratories that the FH/FHL-1 binding proteins of B. burgdorferi inhibit activation of the alternative pathway of complement by binding FH/FHL-1 to its surface (3, 4, 10, 24, 29, 31, 37, 41, 42, 48, 56, 71). Consistent with this speculation, not only was the CspA mutant strain dramatically impaired in its ability to adsorb FH to the borrelial surface, but it was also hypersensitive to the deposition of C3, the downstream complement component C6, and the membrane attack complex (i.e., complement components C5b-9). These observations strongly suggest that CspA greatly enhances the acquisition and accumulation of FH onto the surface of B. burgdorferi, which provides protection against the bactericidal activity of the membrane attack complex. A question that remains, however, is whether FH bound to CspA on the surface of B. burgdorferi inhibits activation of the alternative pathway by (i) enhancing factor I-mediated degradation of C3 to iC3b, (ii) preventing factor B binding to C3b, or (iii) displacing factor B from C3bBb (71). Future studies are needed to determine whether only one or a combination of these FH regulatory activities is utilized by B. burgdorferi to evade complement-mediated destruction.
The virulent CspA mutant displayed a serum-sensitive phenotype compared to that of wild-type B. burgdorferi, which is similar to what we previously reported when we inactivated CspA in an avirulent strain (10). Interestingly, while both the virulent and avirulent CspA mutants were serum sensitive, the avirulent CspA mutant strain was completely destroyed by human serum, while ∼10% of the virulent CspA mutant spirochetes survived when incubated with serum. Furthermore, it took more than 10-fold longer (1 h versus 16 h) for maximal serum-killing activity when CspA was mutated in the virulent strain than in the avirulent strain. While there are several possibilities that may explain this difference, it is important to note that the avirulent strain lacks cp32-9, which encodes one of the OspE-related FH binding proteins. In addition to cp32-9, however, there are numerous other plasmids that are contained in the virulent strain but are missing from the avirulent strain, including lp5, lp25, lp28-1, lp28-2, lp28-3, lp28-4, lp36, lp38, lp56, cp32-6, and cp32-7 (9, 18). Therefore, while it is tempting to speculate that the lack of one of the OspE-related proteins accounts for the differences observed in serum sensitivity, it cannot be ruled out that one of the missing plasmids encodes one or more proteins that renders the avirulent strain so sensitive to serum-mediated killing.
It is still unclear if the FH binding property of CspA and the OspE-related proteins is directly related to B. burgdorferi virulence and, ultimately, evasion of complement-mediated destruction during mammalian infection. In this regard, it has been reported by Woodman et al. (68) that FH-deficient mice are susceptible to B. burgdorferi infection. This finding led the authors to conclude that FH and the various FH-binding proteins are not relevant to B. burgdorferi infection. While this study did show that FH is not relevant in controlling or altering B. burgdorferi infection in FH-deficient mice, it must be noted that these mice have extremely low levels of the central complement component C3 (52). In fact, C3 levels in the serum of FH-deficient mice are typically well below minimal detection levels (52). The overall lack of C3 expression in FH-deficient mice not only renders them severely impaired in their abilities to activate both the classical and alternative pathways of complement, but it also makes them unable to opsonize invading pathogens, such as B. burgdorferi. These combined observations indicate that the FH-deficient mouse is a poor model for examining the role of FH binding proteins in B. burgdorferi serum resistance and underscores the need for utilizing other animal models to fully characterize the role of FH in B. burgdorferi virulence in future studies.
Prior reports by us and others have shown that CspA is expressed abundantly in the tick, but it is dramatically downregulated soon after tick feeding and during transmission to the mammal (8, 11, 45, 46, 57). Interestingly, at the same time that CspA expression is being downregulated, the OspE-related FH binding proteins are upregulated (8, 26-28, 63, 64). In fact, the OspE-related proteins become maximally expressed as the organism migrates from the tick salivary glands and is transmitted into the mammalian host (27). The combined expression data lead us to propose a model for serum resistance in B. burgdorferi in which CspA is expressed abundantly in the tick midgut and prevents serum-mediated killing during the tick blood meal. Then, as CspA is downregulated during tick feeding, the OspE-related proteins are upregulated and provide serum resistance in the mammalian host. We are currently using the virulent CspA mutant and complemented strains characterized here to examine the role of CspA during the tick blood meal. Consistent with our proposed model, preliminary studies have revealed that the CspA mutant is severely impaired in its ability to be transmitted from the tick to the mammal because mutants that do not express CspA are serum sensitive and killed in the midgut by the incoming blood meal during tick feeding (M. R. Kenedy, S. R. Vuppala, and D. R. Akins, unpublished data). To complete a full analysis of the model we have proposed, virulent borrelial strains that lack various combinations of CspA and/or the OspE-related proteins are necessary so that the role(s) of these FH binding proteins in virulence and Lyme disease pathogenesis can be examined throughout the tick and mammalian phases of the complex B. burgdorferi life cycle.
Acknowledgments
We thank Steven Norris for providing B. burgdorferi 5A4NP1 and 5A15, Scott Samuels for providing vector pKFSS-1, Justin Radolf and Melissa Caimano for providing the flaB-GFP plasmid construct pMC1919, and Philip Stewart and Patricia Rosa for providing pBSV2G.
This work was supported by grants AI059373 and AI064629 from the National Institutes of Health (NIAID) to D.R.A. and by grant Kr3383/1-1 from the Deutsche Forschungsgemeinschaft (DFG) to P.K.
Editor: J. B. Bliska
Footnotes
Published ahead of print on 18 May 2009.
REFERENCES
- 1.Alitalo, A., T. Meri, T. Chen, H. Lankinen, Z. Z. Cheng, T. S. Jokiranta, I. J. Seppala, P. Lahdenne, P. S. Hefty, D. R. Akins, and S. Meri. 2004. Lysine-dependent multipoint binding of the Borrelia burgdorferi virulence factor outer surface protein E to the C terminus of factor H. J. Immunol. 1726195-6201. [DOI] [PubMed] [Google Scholar]
- 2.Alitalo, A., T. Meri, P. Comstedt, L. Jeffery, J. Tornberg, T. Strandin, H. Lankinen, S. Bergstrom, M. Cinco, S. R. Vuppala, D. R. Akins, and S. Meri. 2005. Expression of complement factor H binding immunoevasion proteins in Borrelia garinii isolated from patients with neuroborreliosis. Eur. J. Immunol. 353043-3053. [DOI] [PubMed] [Google Scholar]
- 3.Alitalo, A., T. Meri, H. Lankinen, I. Seppala, P. Lahdenne, P. S. Hefty, D. R. Akins, and S. Meri. 2002. Complement inhibitor factor H binding to Lyme disease spirochetes is mediated by inducible expression of multiple plasmid-encoded outer surface protein E paralogs. J. Immunol. 1693847-3853. [DOI] [PubMed] [Google Scholar]
- 4.Alitalo, A., T. Meri, L. Ramo, T. S. Jokiranta, T. Heikkila, I. J. Seppala, J. Oksi, M. Viljanen, and S. Meri. 2001. Complement evasion by Borrelia burgdorferi: serum-resistant strains promote C3b inactivation. Infect. Immun. 693685-3691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Barbour, A. G. 1984. Isolation and cultivation of Lyme disease spirochetes. Yale J. Biol. Med. 57521-525. [PMC free article] [PubMed] [Google Scholar]
- 6.Brade, V., I. Kleber, and G. Acker. 1992. Differences of two Borrelia burgdorferi strains in complement activation and serum resistance. Immunobiology 185453-465. [DOI] [PubMed] [Google Scholar]
- 7.Breitner-Ruddock, S., R. Wurzner, J. Schulze, and V. Brade. 1997. Heterogeneity in the complement-dependent bacteriolysis within the species of Borrelia burgdorferi. Med. Microbiol. Immunol. 185253-260. [DOI] [PubMed] [Google Scholar]
- 8.Brooks, C. S., P. S. Hefty, S. E. Jolliff, and D. R. Akins. 2003. Global analysis of Borrelia burgdorferi genes regulated by mammalian host-specific signals. Infect. Immun. 713371-3383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brooks, C. S., S. R. Vuppala, A. M. Jett, and D. R. Akins. 2006. Identification of Borrelia burgdorferi outer surface proteins. Infect. Immun. 74296-304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brooks, C. S., S. R. Vuppala, A. M. Jett, A. Alitalo, S. Meri, and D. R. Akins. 2005. Complement regulator-acquiring surface protein 1 imparts resistance to human serum in Borrelia burgdorferi. J. Immunol. 1753299-3308. [DOI] [PubMed] [Google Scholar]
- 11.Bykowski, T., M. E. Woodman, A. E. Cooley, C. A. Brissette, V. Brade, R. Wallich, P. Kraiczy, and B. Stevenson. 2007. Coordinated expression of Borrelia burgdorferi complement regulator-acquiring surface proteins during the Lyme disease spirochete's mammal-tick infection cycle. Infect. Immun. 754227-4236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Byram, R., P. E. Stewart, and P. Rosa. 2004. The essential nature of the ubiquitous 26-kilobase circular replicon of Borrelia burgdorferi. J. Bacteriol. 1863561-3569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Casjens, S., N. Palmer, R. van Vugt, W. M. Huang, B. Stevenson, P. Rosa, R. Lathigra, G. Sutton, J. Peterson, R. J. Dodson, D. Haft, E. Hickey, M. Gwinn, O. White, and C. M. Fraser. 2000. A bacterial genome in flux: the twelve linear and nine circular extrachromosomal DNAs in an infectious isolate of the Lyme disease spirochete Borrelia burgdorferi. Mol. Microbiol. 35490-516. [DOI] [PubMed] [Google Scholar]
- 14.Coleman, A. S., X. Yang, M. Kumar, X. Zhang, K. Promnares, D. Shroder, M. R. Kenedy, J. F. Anderson, D. R. Akins, and U. Pal. 2008. Borrelia burgdorferi complement regulator-acquiring surface protein 2 does not contribute to complement resistance or host infectivity. PLoS ONE 33010e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cordes, F. S., P. Kraiczy, P. Roversi, M. M. Simon, V. Brade, O. Jahraus, R. Wallis, L. Goodstadt, C. P. Ponting, C. Skerka, P. F. Zipfel, R. Wallich, and S. M. Lea. 2006. Structure-function mapping of BbCRASP-1, the key complement factor H and FHL-1 binding protein of Borrelia burgdorferi. Int. J. Med. Microbiol. 296(Suppl. 40)177-184. [DOI] [PubMed] [Google Scholar]
- 16.Cordes, F. S., P. Roversi, P. Kraiczy, M. M. Simon, V. Brade, O. Jahraus, R. Wallis, C. Skerka, P. F. Zipfel, R. Wallich, and S. M. Lea. 2005. A novel fold for the factor H-binding protein BbCRASP-1 of Borrelia burgdorferi. Nat. Struct. Mol. Biol. 12276-277. [DOI] [PubMed] [Google Scholar]
- 17.Daniel, W. W. 1999. Biostatistics: a foundation for analysis in the health sciences. John Wiley and Sons, Inc., New York, NY.
- 18.Eggers, C. H., M. J. Caimano, M. L. Clawson, W. G. Miller, D. S. Samuels, and J. D. Radolf. 2002. Identification of loci critical for replication and compatibility of a Borrelia burgdorferi cp32 plasmid and use of a cp32-based shuttle vector for the expression of fluorescent reporters in the Lyme disease spirochaete. Mol. Microbiol. 43281-295. [DOI] [PubMed] [Google Scholar]
- 19.Eggers, C. H., M. J. Caimano, and J. D. Radolf. 2006. Sigma factor selectivity in Borrelia burgdorferi: RpoS recognition of the ospE/ospF/elp promoters is dependent on the sequence of the −10 region. Mol. Microbiol. 591859-1875. [DOI] [PubMed] [Google Scholar]
- 20.Elias, A. F., J. L. Bono, J. J. Kupko III, P. E. Stewart, J. G. Krum, and P. A. Rosa. 2003. New antibiotic resistance cassettes suitable for genetic studies in Borrelia burgdorferi. J. Mol. Microbiol. Biotechnol. 629-40. [DOI] [PubMed] [Google Scholar]
- 21.Elias, A. F., P. E. Stewart, D. Grimm, M. J. Caimano, C. H. Eggers, K. Tilly, J. L. Bono, D. R. Akins, J. D. Radolf, T. G. Schwan, and P. Rosa. 2002. Clonal polymorphism of Borrelia burgdorferi strain B31 MI: implications for mutagenesis in an infectious strain background. Infect. Immun. 702139-2150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Frank, K. L., S. F. Bundle, M. E. Kresge, C. H. Eggers, and D. S. Samuels. 2003. aadA confers streptomycin resistance in Borrelia burgdorferi. J. Bacteriol. 1856723-6727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fraser, C. M., S. Casjens, W. M. Huang, G. G. Sutton, R. Clayton, R. Lathigra, O. White, K. A. Ketchum, R. Dodson, E. K. Hickey, M. Gwinn, B. Dougherty, J.-F. Tomb, R. D. Fleischmann, D. Richardson, J. Peterson, A. R. Kerlavage, J. Quackenbush, S. Salzberg, M. Hanson, R. van Vugt, N. Palmer, M. D. Adams, J. Gocayne, J. Weidman, T. Utterback, L. Watthey, L. McDonald, P. Artiach, C. Bowman, S. Garland, C. Fujii, M. D. Cotton, K. Horst, K. Roberts, B. Hatch, H. O. Smith, and J. C. Venter. 1997. Genomic sequence of a Lyme disease spirochaete, Borrelia burgdorferi. Nature 390580-586. [DOI] [PubMed] [Google Scholar]
- 24.Hartmann, K., C. Corvey, C. Skerka, M. Kirschfink, M. Karas, V. Brade, J. C. Miller, B. Stevenson, R. Wallich, P. F. Zipfel, and P. Kraiczy. 2006. Functional characterization of BbCRASP-2, a distinct outer membrane protein of Borrelia burgdorferi that binds host complement regulators factor H and FHL-1. Mol. Microbiol. 611220-1236. [DOI] [PubMed] [Google Scholar]
- 25.Haupt, K., P. Kraiczy, R. Wallich, V. Brade, C. Skerka, and P. F. Zipfel. 2007. Binding of human factor H-related protein 1 to serum-resistant Borrelia burgdorferi is mediated by borrelial complement regulator-acquiring surface proteins. J. Infect. Dis. 196124-133. [DOI] [PubMed] [Google Scholar]
- 26.Hefty, P. S., C. S. Brooks, A. M. Jett, G. L. White, S. K. Wikel, R. C. Kennedy, and D. R. Akins. 2002. OspE-related, OspF-related, and Elp lipoproteins are immunogenic in baboons experimentally infected with Borrelia burgdorferi and in human Lyme disease patients. J. Clin. Microbiol. 404256-4265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hefty, P. S., S. E. Jolliff, M. J. Caimano, S. K. Wikel, and D. R. Akins. 2002. Changes in temporal and spatial patterns of outer surface lipoprotein expression generate population heterogeneity and antigenic diversity in the Lyme disease spirochete, Borrelia burgdorferi. Infect. Immun. 703468-3478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hefty, P. S., S. E. Jolliff, M. J. Caimano, S. K. Wikel, J. D. Radolf, and D. R. Akins. 2001. Regulation of OspE-related, OspF-related, and Elp lipoproteins of Borrelia burgdorferi strain 297 by mammalian host-specific signals. Infect. Immun. 693618-3627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hellwage, J., T. Meri, T. Heikkila, A. Alitalo, J. Panelius, P. Lahdenne, I. J. Seppala, and S. Meri. 2001. The complement regulator factor H binds to the surface protein OspE of Borrelia burgdorferi. J. Biol. Chem. 2768427-8435. [DOI] [PubMed] [Google Scholar]
- 30.Herzberger, P., C. Siegel, C. Skerka, V. Fingerle, U. Schulte-Spechtel, A. van Dam, B. Wilske, V. Brade, P. F. Zipfel, R. Wallich, and P. Kraiczy. 2007. Human pathogenic Borrelia spielmanii sp. nov. resists complement-mediated killing by direct binding of immune regulators factor H and factor H-like protein 1. Infect. Immun. 754817-4825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hovis, K. M., E. Tran, C. M. Sundy, E. Buckles, J. V. McDowell, and R. T. Marconi. 2006. Selective binding of Borrelia burgdorferi OspE paralogs to factor H and serum proteins from diverse animals: possible expansion of the role of OspE in Lyme disease pathogenesis. Infect. Immun. 741967-1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jarva, H., T. S. Jokiranta, R. Wurzner, and S. Meri. 2003. Complement resistance mechanisms of streptococci. Mol. Immunol. 4095-107. [DOI] [PubMed] [Google Scholar]
- 33.Jewett, M. W., R. Byram, A. Bestor, K. Tilly, K. Lawrence, M. N. Burtnick, F. Gherardini, and P. A. Rosa. 2007. Genetic basis for retention of a critical virulence plasmid of Borrelia burgdorferi. Mol. Microbiol. 66975-990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kawabata, H., S. J. Norris, and H. Watanabe. 2004. BBE02 disruption mutants of Borrelia burgdorferi B31 have a highly transformable, infectious phenotype. Infect. Immun. 727147-7154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kraiczy, P., C. Hanssen-Hubner, V. Kitiratschky, C. Brenner, S. Besier, V. Brade, M. M. Simon, C. Skerka, P. Roversi, S. M. Lea, B. Stevenson, R. Wallich, and P. F. Zipfel. 2009. Mutational analyses of the BbCRASP-1 protein of Borrelia burgdorferi identify residues relevant for the architecture and binding of host complement regulators FHL-1 and factor H. Int. J. Med. Microbiol. 299255-268. [DOI] [PubMed] [Google Scholar]
- 36.Kraiczy, P., J. Hellwage, C. Skerka, H. Becker, M. Kirschfink, M. M. Simon, V. Brade, P. F. Zipfel, and R. Wallich. 2004. Complement resistance of Borrelia burgdorferi correlates with the expression of BbCRASP-1, a novel linear plasmid-encoded surface protein that interacts with human factor H and FHL-1 and is unrelated to Erp proteins. J. Biol. Chem. 2792421-2429. [DOI] [PubMed] [Google Scholar]
- 37.Kraiczy, P., J. Hellwage, C. Skerka, M. Kirschfink, V. Brade, P. F. Zipfel, and R. Wallich. 2003. Immune evasion of Borrelia burgdorferi: mapping of a complement-inhibitor factor H-binding site of BbCRASP-3, a novel member of the Erp protein family. Eur. J. Immunol. 33697-707. [DOI] [PubMed] [Google Scholar]
- 38.Kraiczy, P., E. Rossmann, V. Brade, M. M. Simon, C. Skerka, P. F. Zipfel, and R. Wallich. 2006. Binding of human complement regulators FHL-1 and factor H to CRASP-1 orthologs of Borrelia burgdorferi. Wien. Klin. Wochenschr. 118669-676. [DOI] [PubMed] [Google Scholar]
- 39.Kraiczy, P., C. Skerka, V. Brade, and P. F. Zipfel. 2001. Further characterization of complement regulator-acquiring surface proteins of Borrelia burgdorferi. Infect. Immun. 697800-7809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kraiczy, P., C. Skerka, M. Kirschfink, V. Brade, and P. F. Zipfel. 2001. Immune evasion of Borrelia burgdorferi by acquisition of human complement regulators FHL-1/reconectin and factor H. Eur. J. Immunol. 311674-1684. [DOI] [PubMed] [Google Scholar]
- 41.Kraiczy, P., C. Skerka, M. Kirschfink, P. F. Zipfel, and V. Brade. 2001. Mechanism of complement resistance of pathogenic Borrelia burgdorferi isolates. Int. Immunopharmacol. 1393-401. [DOI] [PubMed] [Google Scholar]
- 42.Kurtenbach, K., S. De Michelis, S. Etti, S. M. Schafer, H. S. Sewell, V. Brade, and P. Kraiczy. 2002. Host association of Borrelia burgdorferi sensu lato—the key role of host complement. Trends Microbiol. 1074-79. [DOI] [PubMed] [Google Scholar]
- 43.Labandeira-Rey, M., and J. T. Skare. 2001. Decreased infectivity in Borrelia burgdorferi strain B31 is associated with loss of linear plasmid 25 or 28-1. Infect. Immun. 69446-455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lane, R. S., J. Piesman, and W. Burgdorfer. 1991. Lyme borreliosis: relation of its causative agent to its vectors and hosts in North America and Europe. Annu. Rev. Entomol. 36587-609. [DOI] [PubMed] [Google Scholar]
- 45.Lederer, S., C. Brenner, T. Stehle, L. Gern, R. Wallich, and M. M. Simon. 2005. Quantitative analysis of Borrelia burgdorferi gene expression in naturally (tick) infected mouse strains. Med. Microbiol. Immunol. 19481-90. [DOI] [PubMed] [Google Scholar]
- 46.McDowell, J. V., K. M. Hovis, H. Zhang, E. Tran, J. Lankford, and R. T. Marconi. 2006. Evidence that the BBA68 protein (BbCRASP-1) of the Lyme disease spirochetes does not contribute to factor H-mediated immune evasion in humans and other animals. Infect. Immun. 743030-3034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.McDowell, J. V., S. Y. Sung, M. Labandeira-Rey, J. T. Skare, and R. T. Marconi. 2001. Analysis of mechanisms associated with loss of infectivity of clonal populations of Borrelia burgdorferi B31MI. Infect. Immun. 693670-3677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.McDowell, J. V., J. Wolfgang, E. Tran, M. S. Metts, D. Hamilton, and R. T. Marconi. 2003. Comprehensive analysis of the factor H binding capabilities of Borrelia species associated with Lyme disease: delineation of two distinct classes of factor H binding proteins. Infect. Immun. 713597-3602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Metts, M. S., J. V. McDowell, M. Theisen, P. R. Hansen, and R. T. Marconi. 2003. Analysis of the OspE determinants involved in binding of factor H and OspE-targeting antibodies elicited during Borrelia burgdorferi infection in mice. Infect. Immun. 713587-3596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Nocton, J. J., and A. C. Steere. 1995. Lyme disease. Adv. Intern. Med. 4069-117. [PubMed] [Google Scholar]
- 51.Pangburn, M. K., R. D. Schreiber, and H. J. Muller-Eberhard. 1977. Human complement C3b inactivator: isolation, characterization, and demonstration of an absolute requirement for the serum protein beta1H for cleavage of C3b and C4b in solution. J. Exp. Med. 146257-270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Pickering, M. C., H. T. Cook, J. Warren, A. E. Bygrave, J. Moss, M. J. Walport, and M. Botto. 2002. Uncontrolled C3 activation causes membranoproliferative glomerulonephritis in mice deficient in complement factor H. Nat. Genet. 31424-428. [DOI] [PubMed] [Google Scholar]
- 53.Purser, J. E., and S. J. Norris. 2000. Correlation between plasmid content and infectivity in Borrelia burgdorferi. Proc. Natl. Acad. Sci. USA 9713865-13870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Radolf, J. D., K. W. Bourell, D. R. Akins, J. S. Brusca, and M. V. Norgard. 1994. Analysis of Borrelia burgdorferi membrane architecture by freeze-fracture electron microscopy. J. Bacteriol. 17621-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ram, S., A. K. Sharma, S. D. Simpson, S. Gulati, D. P. McQuillen, M. K. Pangburn, and P. A. Rice. 1998. A novel sialic acid binding site on factor H mediates serum resistance of sialylated Neisseria gonorrhoeae. J. Exp. Med. 187743-752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Rogers, E. A., and R. T. Marconi. 2007. Delineation of species-specific binding properties of the CspZ protein (BBH06) of Lyme disease spirochetes: evidence for new contributions to the pathogenesis of Borrelia spp. Infect. Immun. 755272-5281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Rossmann, E., V. Kitiratschky, H. Hofmann, P. Kraiczy, M. M. Simon, and R. Wallich. 2006. Borrelia burgdorferi complement regulator-acquiring surface protein 1 of the Lyme disease spirochetes is expressed in humans and induces antibody responses restricted to nondenatured structural determinants. Infect. Immun. 747024-7028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Samuels, D. S. 1995. Electrotransformation of the spirochete Borrelia burgdorferi. Methods Mol. Biol. 47253-259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Siegel, C., J. Schreiber, K. Haupt, C. Skerka, V. Brade, M. M. Simon, B. Stevenson, R. Wallich, P. F. Zipfel, and P. Kraiczy. 2008. Deciphering the ligand-binding sites in the Borrelia burgdorferi complement regulator-acquiring surface protein 2 required for interactions with the human immune regulators factor H and factor H-like protein 1. J. Biol. Chem. 28334855-34863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Steere, A. C. 1989. Lyme disease. N. Engl. J. Med. 321586-597. [DOI] [PubMed] [Google Scholar]
- 61.Steere, A. C. 2005. Borrelia burgdorferi (Lyme disease, Lyme borreliosis), p. 2798-2809. In G. L. Mandell, J. E. Bennett, and R. Dolin (ed.), Principles and practice of infectious diseases. Churchill Livingstone, Philadelphia, PA.
- 62.Steere, A. C., J. Coburn, and L. Glickstein. 2004. The emergence of Lyme disease. J. Clin. Investig. 1131093-1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Stevenson, B., N. El Hage, M. A. Hines, J. C. Miller, and K. Babb. 2002. Differential binding of host complement inhibitor factor H by Borrelia burgdorferi Erp surface proteins: a possible mechanism underlying the expansive host range of Lyme disease spirochetes. Infect. Immun. 70491-497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Stevenson, B., T. G. Schwan, and P. A. Rosa. 1995. Temperature-related differential expression of antigens in the Lyme disease spirochete, Borrelia burgdorferi. Infect. Immun. 634535-4539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Terekhova, D., R. Iyer, G. P. Wormser, and I. Schwartz. 2006. Comparative genome hybridization reveals substantial variation among clinical isolates of Borrelia burgdorferi sensu stricto with different pathogenic properties. J. Bacteriol. 1886124-6134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Tilly, K., A. Bestor, M. W. Jewett, and P. Rosa. 2007. Rapid clearance of Lyme disease spirochetes lacking OspC from skin. Infect. Immun. 751517-1519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wallich, R., J. Pattathu, V. Kitiratschky, C. Brenner, P. F. Zipfel, V. Brade, M. M. Simon, and P. Kraiczy. 2005. Identification and functional characterization of complement regulator-acquiring surface protein 1 of the Lyme disease spirochetes Borrelia afzelii and Borrelia garinii. Infect. Immun. 732351-2359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Woodman, M. E., A. E. Cooley, J. C. Miller, J. J. Lazarus, K. Tucker, T. Bykowski, M. Botto, J. Hellwage, R. M. Wooten, and B. Stevenson. 2007. Borrelia burgdorferi binding of host complement regulator factor H is not required for efficient mammalian infection. Infect. Immun. 753131-3139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Xu, Q., K. McShan, and F. T. Liang. 2008. Essential protective role attributed to the surface lipoproteins of Borrelia burgdorferi against innate defences. Mol. Microbiol. 6915-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Yang, X. F., U. Pal, S. M. Alani, E. Fikrig, and M. V. Norgard. 2004. Essential role for OspA/B in the life cycle of the Lyme disease spirochete. J. Exp. Med. 199641-648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Zipfel, P. F., C. Skerka, J. Hellwage, S. T. Jokiranta, S. Meri, V. Brade, P. Kraiczy, M. Noris, and G. Remuzzi. 2002. Factor H family proteins: on complement, microbes and human diseases. Biochem. Soc. Trans. 30971-978. [DOI] [PubMed] [Google Scholar]