

Abstract
We analyzed the defensive role of the cytosolic innate recognition receptor nucleotide oligomerization domain 1 (NOD1) during infection with Listeria monocytogenes. Mice lacking NOD1 showed increased susceptibility to systemic intraperitoneal and intravenous infection with high or low doses of L. monocytogenes, as measured by the bacterial load and survival. NOD1 also controlled dissemination of L. monocytogenes into the brain. The increased susceptibility to reinfection of NOD1−/− mice was not associated with impaired triggering of listeria-specific T cells, and similar levels of costimulatory molecules or activation of dendritic cells was observed. Higher numbers of F480+ Gr1+ inflammatory monocytes and lower numbers of F480− Gr1+ neutrophils were recruited into the peritoneum of infected WT mice than into the peritoneum of infected NOD1−/− mice. We determined that nonhematopoietic cells accounted for NOD1-mediated resistance to L. monocytogenes in bone marrow radiation chimeras. The levels of NOD1 mRNA in fibroblasts and bone marrow-derived macrophages (BMM) were upregulated after infection with L. monocytogenes or stimulation with different Toll-like receptor ligands. NOD1−/− BMM, astrocytes, and fibroblasts all showed enhanced intracellular growth of L monocytogenes compared to WT controls. Gamma interferon-mediated nitric oxide production and inhibition of L. monocytogenes growth were hampered in NOD1−/− BMM. Thus, NOD1 confers nonhematopoietic cell-mediated resistance to infection with L. monocytogenes and controls intracellular bacterial growth in different cell populations in vitro.
Innate immunity initiates defense mechanisms on the basis of nonclonal recognition of microbial components. Microbial identification involves host receptors that recognize conserved molecular motifs present on a wide range of different microbes. Toll-like receptors (TLR) and nucleotide oligomerization domain (NOD)-like receptors (NLR) form two molecular host protein families involved in innate immune detection. The TLR are a family of membrane-bound receptors, whereas NLR molecules occur in the cytoplasm and detect microbial motifs that gain entry into the host cell (22).
NOD1 and NOD2 are two NLR proteins that sense bacterial molecules produced during the synthesis and/or degradation of peptidoglycan (PGN) (2, 20). NOD1 is expressed ubiquitously in adult tissues and recognizes a meso-diaminopimelate (DAP)-containing structure that is found mainly in the PGN of gram-negative bacteria but also is found in the gram-positive bacterium Listeria monocytogenes (3, 8, 13-15). Both NOD1 and NOD2 signal via the adaptor protein RIP2 (also designated RICK or CARDIAK) (55). Like TLR signaling, NOD signaling can activate transcription factors, such as NF-κB and various mitogen-activated protein kinases. The signals induce a variety of inflammatory cytokines, including tumor necrosis factor alpha (TNF-α), interleukin-1 (IL-1), IL-6, and IL-12, and expression of costimulatory molecules, such as CD40, CD80, and CD86, on the surface of antigen-presenting cells (42).
While the NODs formally constitute a recognition system for innate defense, few studies have shown an actual role for NOD proteins in the control of infectious diseases. Several reports have shown that NOD1 and NOD2 contribute to the innate immune responses induced by various pathogenic bacteria in vitro (9, 23, 34, 35, 50). The evidence that NOD1 or NOD2 is important in host defense in vivo is restricted mainly to evidence obtained with experimental models of gastrointestinal infection, where the pathogen interacts with cells largely devoid of TLR signaling (26, 54). Thus, the importance of NOD1 and NOD2 in regulation of systemic bacterial infections is still unknown.
We have analyzed the role of NOD1 in resistance to L. monocytogenes infection. L. monocytogenes is a facultative intracellular pathogen that escapes from phagosomes and replicates in the cytosolic compartment of hematopoietic and nonhematopoietic cells. L. monocytogenes may cause severe infections in the nervous system and also causes systemic disease, showing a preference for macrophage-rich organs, such as the liver and spleen.
Here we demonstrate that NOD1 has a protective role during infection of mice with L. monocytogenes and can control intracellular bacterial growth in different populations of cells in vitro.
MATERIALS AND METHODS
Mice.
A mutant mouse strain without NOD1 (4) was generated by homologous recombination in embryonic stem cells. Animals were bred and kept under specific-pathogen-free conditions. NOD1−/+ mice used in this study were backcrossed with C57BL/6 mice for six generations as heterozygotes. Then heterozygous mice were bred, and NOD1−/− and NOD1+/+ littermates were genotyped by using PCR and selected. The following primers were used for genotyping: Sense NOD1 WT (5′ GCT TGG CTC CTT TGT CAT TG 3′), Antisense NOD1 WT (5′ ACT GCT GCT TGG CTT TAT TCT C 3′), Sense mutant (5′ TTG GTG GTC GAA TGG GCA GGT A 3′), and Antisense mutant (5′ CGC GCT GTT CTC CTC TTC CTC A 3′).
All experiments were conducted by using protocols that received institutional approval and were authorized by the local animal ethics committee (Stockholms Norra Djurförsöksetiska Nämnd).
Generation of mouse BMM.
Mouse bone marrow-derived macrophages (BMM) were obtained from 6- to 10-week-old mice as described previously (40). Mice were euthanized, and the femurs and tibias of the hind legs were dissected. Bone marrow cavities were flushed with 5 ml cold, sterile phosphate-buffered saline (PBS). The bone marrow cells were washed and resuspended in Dulbecco's modified Eagle's medium (DMEM) containing glucose and supplemented with 2 mM l-glutamine, 10% fetal bovine serum (FBS), 10 mM HEPES, 100 μg/ml streptomycin, and 100 U/ml penicillin (all obtained from Sigma, St. Louis, MO), as well as 20 to 30% L929 cell-conditioned medium (as a source of macrophage colony-stimulating factor). Bone marrow cells were passed through a 100-μm cell strainer, plated in 24-well plates (5 × 104 to 8 × 104 cells in 2 ml), and incubated for 7 days at 37°C in the presence of 5% CO2. Before use, BMM cultures were washed vigorously to remove nonadherent cells. Cells were also harvested from several wells and counted by using trypan blue exclusion. The yields of BMM differentiated in 24-well plates for L. monocytogenes infection were 2 × 104 to 4 × 105 BMM per well. We have previously shown by using immunofluorescence staining that these BMM are F4/80+ and Mac-3+ (40).
Embryonic astrocytes.
Astrocytes were cultured from brains of day 12 to 14 embryos as previously described (18).
Embryonic fibroblasts.
Murine embryonic fibroblasts were isolated from embryos by trypsin-EDTA treatment and cultured as described previously (16).
Differentiation of mouse bone marrow-derived DC.
Mouse bone marrow-derived dendritic cells (DC) were differentiated as previously described (31). Briefly, bone marrow was extracted from tibias and femurs, and cell suspensions were cultured in IMDM (Cambrex, East Rutherford, NJ) containing 10% FCS, 100 U/ml penicillin, 100 μg/ml streptomycin, and 10 ng/ml granulocyte-macrophage colony-stimulating factor (Peprotech, London, United Kingdom). Fresh medium and cytokine were added every 2 to 3 days. On days 3 to 7, loosely adherent cells were harvested and infected with L. monocytogenes as described above for BMM.
Infection and infectivity assay.
L. monocytogenes strain EGD (BUG600; serotype 1/2a; kindly provided by Chakraborti, Gießen, Germany) was grown in brain heart infusion (BHI) broth and on BHI agar (Difco, Becton Dickinson, Maryland) at 37°C. Before infection, bacteria were grown at 37°C in BHI broth to late exponential phase (optical density at 600 nm, 0.8). The bacteria were washed once with PBS and resuspended in broth with 15% glycerol, and bacterial stocks were stored at −70°C until they were used. Frozen bacterial stocks were diluted in PBS, plated on BHI agar plates, and quantified after 48 h of incubation at 37°C.
BMM, embryonic fibroblasts, and embryonic astrocytes cultured in 24-well plates were cocultured with L. monocytogenes in DMEM containing 5% FBS for 1 h at 37°C in the presence of 5% CO2. The cells were then extensively washed with PBS to remove the extracellular bacteria. To prevent extracellular bacterial growth, the cells were then cultivated in DMEM containing 5% FBS and 5 μg gentamicin per ml. At different time points after infection, cells were washed with PBS and lysed with 0.1% Triton X-100 in PBS, and aliquots of the lysates were plated on BHI agar plates. The plates were incubated for 48 h at 37°C, and the numbers of CFU were determined.
Organs were obtained at different time points after infection with L. monocytogenes and minced in PBS containing 0.1% Triton X-100, and aliquots of lysates were plated on BHI agar plates. The plates were incubated for 48 h at 37°C, and the numbers of CFU were determined.
Real-time RT-PCR.
Cytokine and NOD1 transcripts in cells and spleen lysates before or after infection with L. monocytogenes or after stimulation with lipopolysaccharide (LPS) (Sigma), CpG (Cybergene, Huddinge, Sweden), poly(I-C) (Sigma), Pam3CysLys4 (Pam3) (Alexis Biochemicals, Nottingham, United Kingdom), or MALP-2 (Alexis) were quantified by real-time reverse transcription (RT)-PCR. Freshly extracted RNA was reverse transcribed to obtain cDNA as described previously (41). The real-time RT-PCR was performed using duplicate 25-μl reaction mixtures containing Platinum SYBR Green quantitative PCR Supermix-UDG (Applied Biosystems, Foster City, CA), forward and reverse primers, and 0.5 μl of cDNA with an ABI Prism 7500 sequence detection system (Applied Biosystems). The following primers were used at concentrations between 50 and 250 nM: Sense IFN-β (5′ CTG GAG CAG CTG AAT GGA AAG 3′), Antisense IFN-β (5′ TCC GTC ATC TCC ATA GGG ATCT 3′), Sense IL-1β (5′ TGG TGT GTG ACG TTC CCA TT 3′), Antisense IL-1β (5′ CAG CAC GAG GCT TTT TTG TTG 3′), Sense IL-6 (5′ ACA AGT CGG AGG CTT AAT TAC ACA T 3′), Antisense IL-6 (5′ TTG CCA TTG CAC AAC TCT TTT C 3′), Sense CXCL1 (5′ GGC GCC TAT CGC CAA TG 3′), Antisense CXCL1 (5′ CTG GAT GTT CTT GAG GTG AAT CC 3′), Sense NOD1 (5′ TGA CGT TCC TGG GTT TAT ACA ACA 3′), Antisense NOD1 (5′ CCA GGA TTT GGG CCA CAT AC 3′), Sense HPRT (5′ CCC AGC GTC GTG ATT AGC 3′), and Antisense HPRT (5′ GGA ATA AAC ACT TTT TCC AAA TCC 3′).
Serial dilutions of a cDNA sample were amplified to control the amplification efficiency for each primer pair. After this the Ct values for all cDNA samples were determined. The hypoxanthine-guanine phosphoribosyltransferase (HPRT) gene was used as a control gene to calculate the ΔCt values for individual samples. The relative amounts of cytokine and HPRT transcripts were calculated using the 2−ΔΔCTmethod as described previously (28). The resulting values were then used to calculate the relative expression of cytokine or NOD1 mRNA in uninfected and infected BMM or in ligand-stimulated BMM.
Harvest of peritoneal exudates.
Immediately after mice were killed, they were peritoneally lavaged with 5 ml of ice-cold PBS. For recovery of cellular contents, recovered lavage fluid was centrifuged at 200 × g for 10 min at 4°C. Fluorescence-activated cell sorting analyses using anti-Gr1-phycoerythrin (Biolegend, San Diego, CA) and anti-F4/80-fluorescein isothiocyanate (Serotec, Oxford, United Kingdom) were performed to differentiate between neutrophils (Gr1+ F480−), inflammatory monocytes (Gr1+ F480+), and resident monocytes and macrophages (Gr1− F480+) in the lavage fluid.
ELISPOT assay.
Briefly, mice were infected with a low dose of L. monocytogenes (2 × 103 CFU) and 2 months later were reinfected with a high dose of L. monocytogenes (105 CFU). Twelve days after reinfection spleens were collected. The concentration of splenocytes was adjusted to 2 × 105 cells per well, and splenocytes were added to precoated (anti-gamma interferon [IFN-γ]; MabTech, Stockholm, Sweden) and blocked enzyme-linked immunospot assay (ELISPOT assay) plates (Millipore, Billerica, MA). Cells were stimulated with 2 μg concanavalin A (Sigma) per ml, 2 μM listeriolysin 296-304 (LLO 296-304) VAYGRQVYL H-2b restricted CD8 T-cell epitope, 2 μM LLO 190-201 NEKYAQAYPNVS H-2b restricted CD4 T-cell epitope (11), or different concentrations of heat-killed L. monocytogenes (HKL) or were not stimulated (medium only). After 20 h of incubation in a 5% CO2 incubator at 37°C, a biotinylated anti-IFN-γ antibody (MabTech) was added, and the plates were incubated for 1 h at room temperature. After washing and incubation with an avidin-peroxidase complex (ABC kit; Vector Laboratories, Burlingame, CA) for another hour, the spots were developed with aminoethyl carbazole substrate, and the enzymatic reaction was stopped after 4 min by washing the plates in water. Spots were counted using an ELISPOT reader (Axioplan 2 Imaging; Zeiss, Oberkochen, Germany), and the results were expressed as the number of spot-forming cells (SFC) per 106 cells.
DC enrichment and flow cytometry analysis.
Spleens from NOD1−/− and wild-type (WT) mice sacrificed 4 days after infection with L. monocytogenes were digested with a mixture of 20 μg/ml DNase I and 25 μg/ml Liberase (Roche Diagnostics Scandinavia AB, Bromma, Sweden) in RPMI 1640 medium (Sigma) supplemented with 10% FBS before they were passed through a 70-μm nylon strainer. Red blood cells were lysed in NH4Cl, and DC were recovered by centrifugation using 11.5% OptiPrep (Axis-Shield PoC, Oslo, Norway) as recommended by the manufacturer. The DC were stained using anti-CD11c antibodies labeled with phycoerythrin, and the surface expression of CD80, CD86, major histocompatibility complex (MHC) class II, or CD40 was detected with fluorescein isothiocyanate-labeled monoclonal antibodies (BD Biosciences PharMingen, San Jose, CA) and analyzed using flow cytometry (FACScan; BD).
Nitrite assay.
Nitrite concentrations in BMM culture supernatants were measured using the Griess reagent and a previously described colorimetric assay. Aliquots (100 μl) of culture medium were mixed in 96-well plates with an equal volume of 0.5% sulfanilamide dihydrochloride and 0.05% naphthylethylenediamide dihydrochloride in 2.5% phosphoric acid, and the absorbance at 540 nm was determined. Sodium nitrite, dissolved in DMEM, was used to generate a standard concentration curve. The lower limit of detection of the assay was 0.5 μM NO2.
Generation of bone marrow radiation chimeras.
Bone marrow cells were harvested from uninfected NOD1−/− and WT mice by flushing bone marrow cavities with cold PBS, and red blood cells were lysed. To create bone marrow chimeras, WT and NOD1−/− mice were irradiated with 900 cGy and 4 h later inoculated in the tail vein with 2 ×107 bone marrow cells from WT or NOD1−/− mice. Six weeks after reconstitution, mice were infected intraperitoneally (i.p.) with 105 CFU of L. monocytogenes. Mice were sacrificed 5 days after infection, and the numbers of CFU in their spleens and livers were determined. Spleen cells were obtained from chimeric and control mice, and the presence of NOD1 or the neomycin gene was determined by PCR using genomic material.
RESULTS
NOD1 protects mice against infection with L. monocytogenes.
The role of NOD1 in the control of L. monocytogenes infection in vivo was studied first. Livers and spleens from NOD1−/− mice infected i.p. with L. monocytogenes contained higher bacterial levels than livers and spleens from infected WT controls (Fig. 1A and B). While 85% of the NOD1−/− mice infected i.p. with 105 CFU L. monocytogenes died between 6 and 15 days after infection, all WT mice survived (Fig. 1C). NOD1−/− mice infected with a 50-fold-lower dose of L. monocytogenes (2 × 103 CFU) had increased levels of bacteria in their spleens and livers compared to controls (Fig. 1D), and 40% of these mice died before 15 days after infection. Also, spleens and livers from NOD1−/− mice infected intravenously (i.v.) with 2 × 104 L. monocytogenes CFU contained higher levels of bacteria than spleens and livers from WT controls (Fig. 1E, F) and died before 13 days after infection (data not shown).
FIG. 1.

NOD1 is required for resistance to infection with L. monocytogenes in vivo. The course of infection with L. monocytogenes in NOD1−/− and WT mice was analyzed. Mice were infected i.p. (A to D) or i.v. (E and F) with 105 (A to C), 2 × 104 (E and F), or 2 × 103 (D) L. monocytogenes CFU and sacrificed at indicated time points (A, B, E, and F) or 4 days after infection (D). The mean L. monocytogenes CFU titers in livers and spleens of six mice per group are shown. The error bars indicate standard errors of the means (SEM). *, P < 0.05 for a comparison with WT mice (Student's t test). (C) Cumulative mortality of NOD1−/− and WT mice (eight mice per group) after i.p. infection with 105 L. monocytogenes CFU. (G) Numbers of L. monocytogenes CFU in the snout, trigeminal nerve (TG), and brain of six NOD1−/− mice and six WT mice 4 days after infection via the snout. The bacterial loads are expressed in CFU per g of tissue (mean ± standard error of the mean). The differences between the NOD1−/− and WT groups are significant for all tissues except the snout (P < 0.05, Student's t test; indicated by an asterisk).
L. monocytogenes can spread to the brainstem along the trigeminal nerve and invade the central nervous system from the bloodstream (6, 21). Whether NOD1 controls listerial dissemination into the brain after infection via the snout was studied next. NOD1−/− mice infected via the snout had higher listerial levels in their trigeminal ganglia and brains, but not in their snouts, than WT mice (Fig. 1G). The brains of NOD1−/− mice also contained higher bacterial levels than the brains of WT mice after i.p. infection with L. monocytogenes (data not shown).
The levels of IFN-γ, IL-1β, and IL-6 mRNA in spleens from infected WT and NOD1−/− mice were then measured. We found that the levels of IFN-γ, IL-1β, and IL-6 all increased in the spleens of L. monocytogenes-infected mice 5 days after infection. However, the level of none of these cytokines was reduced in infected NOD1−/− mice compared to the level in infected WT mice (data not shown).
Next, the involvement of NOD1 in the generation of protective memory responses was investigated. For this purpose, mutant and WT mice were infected i.p. with 2 × 103 L. monocytogenes CFU. Twenty days after infection, when bacteria were not detected in WT or NOD1−/− animals, mice were reinfected with 105 L. monocytogenes CFU. Reinfected NOD1−/− mice had higher L. monocytogenes loads in their spleens than WT controls (Fig. 2A) but showed no mortality after reinfection.
FIG. 2.

NOD1 is redundant for induction of adaptive immune responses. Mice infected with 2 × 103 L. monocytogenes CFU were reinfected i.p. with a 50-fold-larger bacterial inoculum 20 days after the primary infection. (A) Mean numbers of L. monocytogenes CFU in livers and spleens from mice sacrificed 4 days after reinfection. The error bars indicate standard errors of the means (SEM). *, P < 0.05 for a comparison with WT mice (Student's t test), indicating a significant difference. (B) Spleen cells from uninfected WT or NOD1−/− mice or from mice 12 days after reinfection with 105 L. monocytogenes CFU (six mice per group) were cultured overnight with or without 2 μM LLO 296-304, 2 μM LLO 190-201, or 0.2 (low) or 0.8 (high) HKL per cell. The data are the numbers of IFN-γ-secreting cells as determined by an ELISPOT assay. All mice showed concanavalin A responses well above 100 SFC per 105 cells, confirming the viability of the splenocyte cultures (data not shown). Uninfected mice showed no SFC following addition of L. monocytogenes whole-cell lysates or following peptide-specific stimulation (data not shown). (C) Spleen CD11c+ DC from NOD1−/− and WT mice at 0 or 4 days after infection with L. monocytogenes were examined by flow cytometry for expression of CD40, CD80, and CD86.
To investigate if impaired adaptive immune responses account for the increased susceptibility of NOD1−/− mice to reinfection with L. monocytogenes, the frequency of specific IFN-γ-producing T cells was determined. Splenocytes from mice that had been reinfected with L. monocytogenes were incubated with HKL, an MHC class I-restricted listerial peptide (LLO 296-304), or an MHC class II-restricted listerial peptide (LLO 190-201). Similar frequencies of HKL-specific or MHC class I- or MHC class II-restricted peptide-specific IFN-γ-secreting cells were detected in spleens from reinfected NOD1−/− and WT mice (Fig. 2B). Analysis of splenic CD11c+ DC obtained from WT and NOD1−/− mice at 4 days postinfection showed an increase in expression of CD40, CD80, and CD86 similar to that shown by DC from uninfected animals (Fig. 2C and data not shown). On the other hand, NOD1−/− bone marrow-derived DC showed decreased levels of IL-6 mRNA compared to infected WT controls (data not shown).
Whether the NOD1-mediated control of infection was associated with recruitment of inflammatory cells to the site of infection was studied next. Prior to infection peritoneal exudates of WT mice contained higher levels of Gr1− F4/80+ resident macrophages than peritoneal exudates of NOD1−/− mice. Neutrophils (Gr1+ F4/80−) and Gr1+ F4/80+ inflammatory monocytes were not present in the peritoneal exudates of uninfected mice (Fig. 3A and B). After i.p. infection with 105 L. monocytogenes CFU, neutrophils and inflammatory monocytes accumulated in the peritoneum (Fig. 3A and B). In infected NOD1−/− mice the frequencies of inflammatory and resident monocytes were decreased, whereas the percentage of neutrophils in the peritoneal cavity was increased compared with WT mice (Fig. 3A and B).
FIG. 3.

NOD1 affects L. monocytogenes-induced recruitment of granulocytes and resident and inflammatory monocytes to the inflammatory site. NOD1−/− and WT peritoneal exudate cells were harvested from mice before (five WT mice and four NOD1−/− mice) or 2 days after (6 WT mice and six NOD1−/− mice) i.p. infection with 105 L. monocytogenes CFU. The mean percentages of cells that were stained with anti-Gr1 and/or anti-F4/80 to differentiate between polymorphonuclear leukocytes and inflammatory monocytes and resting macrophages and monocytes in the peritoneal exudates from individual mice are shown. Differences between the total numbers of harvested peritoneal cells from L. monocytogenes-infected and uninfected NOD1−/− and WT animals are not significant. Differences between the percentages of F4/80+ Gr1− cells in infected and uninfected peritoneal exudates from WT or NOD1−/− mice are significant (P < 0.001, Student's t test). Differences between the percentages of F4/80+ Gr1− cells in peritoneal exudates from infected or uninfected WT and NOD1−/− mice are significant (P < 0.001, Student's t test). Differences between the percentages of Gr1+ F4/80− cells from infected WT and NOD1−/− mice are significant (P < 0.001, Student's t test). Differences between the percentages of Gr1+ F4/80+ peritoneal exudate cells from WT and NOD1−/− mice are significant (P < 0.02, Student's t test). The results of one of two independent experiments are shown. (B) Levels of expression of F4/80+ and Gr1+ cells in the peritoneal exudates of representative mice before or after infection with L. monocytogenes. FITC, fluorescein isothiocyanate; PE, phycoerythrin.
Whether in vivo NOD1-dependent protection is mediated by hematopoietic cells and/or by nonhematopoietic cells was investigated. Reciprocal bone marrow radiation chimeras of WT and NOD1−/− mice were generated by inoculation of bone marrow cells into irradiated recipients. The NOD1 WT allele was detected in spleens from WT (donor)→NOD1−/− (recipient) mice 8 weeks after transfer of bone marrow cells, confirming that there was successful repopulation by inoculated stem cells (data not shown). Nonhematopoietic cells were responsible for the NOD1-dependent resistance, since NOD1−/−→WT chimeras and WT→WT controls had similar low levels of L. monocytogenes in their spleens and livers, whereas high levels of L. monocytogenes were detected for the WT→NOD1−/− and NOD1−/−→NOD1−/− combinations (Fig. 4).
FIG. 4.

NOD1-mediated resistance to infection with L. monocytogenes is due to the activity of nonhematopoietic cells. NOD1−/− or WT bone marrow cells were inoculated i.v. into irradiated NOD1−/− or WT mice. Six weeks after inoculation of the cells, mice were infected i.p. with 105 L. monocytogenes CFU. Mice were sacrificed 5 days after infection. The numbers of CFU in lungs from mice treated as described above were determined. The data are the means ± standard errors of the means (SEM) and are the numbers of CFU per liver (A) or spleen (B) for six mice per group. *, P < 0.05 for a comparison of chimeric mice and WT→WT sham chimera mice (Student's t test), indicating a significant difference.
NOD1 controls the intracellular growth of L. monocytogenes in vitro.
The role of NOD1 in macrophage control of L. monocytogenes infection was tested next. We found that NOD1−/− BMM contained higher levels of intracellular bacteria than WT control BMM after infection with L. monocytogenes (Fig. 5A and B), whereas the initial numbers of L. monocytogenes bacteria taken up by NOD1−/− and WT BMM at different multiplicities of infection were similar (Fig. 5C).
FIG. 5.

NOD1 controls the growth of L. monocytogenes in BMM. (A and B) NOD1−/− and WT BMM in triplicate wells were incubated with L. monocytogenes at a multiplicity of infection (MOI) of 0.2 (A) or 2 (B) and lysed at the indicated time points after infection, and the numbers of L. monocytogenes CFU in the BMM lysates were determined. Two independent experiments were performed, and the results of one of them are shown. (C) NOD1−/− and WT BMM were infected with L. monocytogenes at the indicated multiplicities of infection. After 1 h, cells were extensively washed and lysed, and the numbers of CFU were determined. (D) NOD1−/− and WT BMM were incubated with 100 U recombinant IFN-γ (BD, Pharmingen) 18 h before infection with L. monocytogenes at a multiplicity of infection of 0.2. One hour after infection cells were extensively washed, and IFN-γ was replenished. Cells were lysed at the indicated time points after infection, and the numbers of L. monocytogenes CFU in the BMM lysates were determined. *, P < 0.05 for a comparison with WT mice (Student's t test), indicating a significant difference. (E and F) NO2− levels in culture supernatants of BMM incubated with 100 U IFN-γ 18 h before incubation with HKL (multiplicity of infection, 10:1), of BMM incubated with IFN-γ, or of BMM incubated with HKL alone 24 h (E) and 48 h (F) after addition of the bacterial lysate.
IFN-γ-activated macrophages restrict the intracellular growth of intracellular bacterial and protozoan pathogens, including L. monocytogenes (38). While coincubation with IFN-γ dramatically decreased the levels of bacteria in WT BMM, NOD1−/− BMM treated with IFN-γ showed only a small reduction in the bacterial load, demonstrating that NOD1 is required for efficient IFN-γ-mediated bacterial control in BMM (Fig. 5D). IFN-γ-mediated killing of L. monocytogenes is dependent on nitric oxide (NO) release (1). The amount of NO released by macrophages cultured in the presence of listerial antigens was greatly increased in the presence of IFN-γ, while similarly treated NOD1−/− BMM showed lower NO2 levels (Fig. 5E and F).
L. monocytogenes-infected WT BMM contained increased levels of IL-1β, IL-6, IFN-β, and CXCL1 (formerly KC) mRNA (Fig. 6A to D). Infected NOD1−/− BMM contained lower levels of IL-1β and IL-6 mRNA (Fig. 6A and B) than infected WT BMM, while the levels of IFN-β and CXCL1 mRNA in mutant and WT BMM were similar (Fig. 6 C, D).
FIG. 6.

NOD1 modulates IL-6 and IL-1β responses after incubation with L. monocytogenes but not after incubation with heat-killed bacterial lysates or with bacteria unable to escape from the phagosome into the cytosol. Total RNA from BMM was extracted before and 6 h after infection with L. monocytogenes at a multiplicity of infection of 0.2 (A to D) or with Δhly L. monocytogenes at a multiplicity of infection of 10 (F) or after incubation with HKL (E). The titers of IL-1β, IL-6, IFN-β, CXCL1 (A to F) and HPRT mRNA were determined by real-time RT-PCR. The mean increases in cytokine or HPRT levels for triplicate (A to D) or duplicate (E and F) wells are shown. Two independent experiments were performed, and the results of one of them are shown.
LLO is a pore-forming toxin that is largely responsible for rupture of the phagosomal membrane and is required for bacterial access to the cytosol (30). We next studied if NOD1-mediated cytokine responses depend on bacterial cytosolic localization. For this, NOD1−/− and WT BMM were incubated with LLO-defective (Δhly) L. monocytogenes or HKL. Similar levels of IL-6 mRNA were observed when NOD1−/− and WT BMM were incubated with Δhly L. monocytogenes or HKL (Fig. 6E and F).
Whether NOD1 also controls intracellular growth of L. monocytogenes in other cellular populations was then studied. NOD1−/− embryonic astrocytes and embryonic fibroblasts both contained higher levels of bacteria than NOD1+/+ controls (Fig. 7A to C). At some time points, the levels of bacteria in NOD1−/− fibroblasts were more than 2 logs higher than those in WT cells.
FIG. 7.

NOD1 controls growth of L. monocytogenes in murine embryonic fibroblasts and astrocytes. NOD1−/− and WT fibroblasts (A and B) and astrocytes (C) in triplicate wells were incubated with L. monocytogenes at multiplicities of infection (MOI) of 30 (A), 0.3 (B), and 10 (C) and lysed at the indicated time points after infection. The numbers of L. monocytogenes CFU in the cell lysates were determined. *, P < 0.05 for a comparison with WT mice (Student's t test), indicating a significant difference. SEM, standard error of the mean.
We next analyzed if infection of BMM or fibroblasts with L. monocytogenes increased the expression of NOD1, since this could enhance the sensitivity of the cells to NOD1 ligands. BMM and fibroblasts both contained increased levels of NOD1 mRNA after infection with L. monocytogenes (Fig. 8A and B). Whether stimulation of a TLR in the absence of NOD1 ligands can induce NOD1 expression was then tested. Indeed, increased NOD1 mRNA levels were found in CpG-, LPS-, and poly(I-C)-stimulated BMM and, to a lesser extent, in Pam3- and MALP-2-stimulated BMM compared to untreated controls (Fig. 8C and D). A high level of NOD1 expression is known to activate NF-κB in a dose-dependent manner in the absence of the DAP-containing ligands (19). NOD1, however, does not seem to regulate TLR responses in the absence of NOD1 ligands since similar levels of IL-1β, IL-6, and IFN-β mRNA were detected in CpG-, poly(I-C)-, and Pam3-stimulated WT and NOD1−/− BMM (data not shown).
FIG. 8.

NOD1 mRNA levels are increased in fibroblasts and macrophages after stimulation with L. monocytogenes or incubation with TLR ligands. The levels of NOD1 mRNA transcripts in WT BMM (A, C, and D) or fibroblasts (B) infected with L. monocytogenes (A and B) or stimulated with CpG (C and D), poly(I-C) (C), LPS (C), Pam3 (D), or MALP-2 (D) were determined by real-time RT-PCR. The mean increases in the level of NOD1 mRNA for duplicate wells for each condition compared to the level for unstimulated wells are shown. SEM, standard error of the mean.
DISCUSSION
We show here that NOD1 signaling protects against subcutaneous, i.v., and i.p. infection with L. monocytogenes and that NOD1-dependent protection in vivo is mediated by nonhematopoietic cells. Mice lacking the NOD1 adaptor protein RIP2 were previously shown to be highly susceptible to infection with L. monocytogenes (5). NOD2−/− mice were shown to be more susceptible to infection than WT mice when they were challenged with L. monocytogenes given intragastrically, but not when they were challenged by i.v. infection (26). The restricted expression of NOD2 in the intestinal crypts, monocytes, and DC (26), in contrast to the ubiquitous expression of NOD1, might explain the difference between the susceptibility of NOD1−/− mice and the resistance of NOD2−/− mice to systemic infection with L. monocytogenes, as well as the selective vulnerability of NOD2−/− mice infected intragastrically with L. monocytogenes (26). Using radiation chimeric mice, we demonstrated that nonhematopoietic cells account for the NOD1-mediated protection against L. monocytogenes infection in vivo, supporting the hypothesis that NOD1 and NOD2 have nonredundant roles in bacterial control. In agreement with our data, it has recently been shown that nonhematopoietic cells mediate T-cell responses to ovalbumin when animals are inoculated with a NOD1 ligand (10).
In experiments performed in the 1970s to study the active moiety of PGN for adjuvant activity, DAP-containing muropeptides prepared from Escherichia coli together with a test antigen in a water-in-oil emulsion were reported to induce specific delayed-type hypersensitivity in guinea pigs (27). Consistent with this, RIP2-mediated responses have been shown to regulate adaptive immune responses (5, 25, 26). NOD1 has been shown to be required for optimal generation of immune responses to soluble antigens with Freund's adjuvant (10). Although NOD1−/− mice contained higher levels of bacteria in their spleens after reinfection with 105 L. monocytogenes CFU than WT mice, similar frequencies of IFN-γ-secreting spleen T cells specific to MHC class I- and class II-restricted listerial epitopes were found for reinfected NOD1−/− and WT mice. Thus, NOD1 was not required for generation of normal adaptive cellular immune responses as measured by IFN-γ secretion. Our data, however, do not rule out the possibility that NOD1 has a regulatory role in other immune parameters. NOD1 and NOD2 signaling has recently been shown to participate in the generation of IL-6- and transforming growth factor β-dependent Th17 inflammatory cells (10, 52).
Gr1 is expressed by neutrophils and inflammatory monocytes. Gr1+ F4/80+ monocytes are proinflammatory and, after they emerge from the bone marrow, home to sites of inflammation (12), as also shown during infection with L. monocytogenes (45).
We found that Gr1− F4/80+ tissue resident macrophages were underrrepresented in the peritoneal exudates of NOD1−/− mice, suggesting that NOD1 has a role in the homeostasis of this leukocyte population.
The resistance to i.p. infection with L. monocytogenes conferred by NOD1 was also associated with increased recruitment of inflammatory monocytes and decreased recruitment of neutrophils into the peritoneal cavity. Recruitment of inflammatory cells could be directly mediated by NOD1 in a process in which resident monocytes might be involved. To further explore this possibility, we studied recruitment of inflammatory cells using an air pouch model, in which resident macrophages are not present before infection. Similar recruitment of inflammatory monocytes was observed in the air pouches of NOD1−/− and WT mice 24 h after inoculation of L. monocytogenes, supporting the hypothesis that resident macrophages have a role in the extravasation of inflammatory monocytes to the infected peritoneum. It is interesting that a failure to recruit Gr1+ monocytes resulted in greatly increased mortality with Toxoplasma gondii despite the induction of normal Th1 cell responses leading to high levels of IL-12, TNF-α, and IFN-γ (39). Gr1+ F4/80+ inflammatory monocytes contribute to host defense by secretion of TNF-α and NO and exerting bacterial phagocytosis and killing (7). Hence, reduced recruitment of these cells may be one explanation for the reduced survival of NOD1−/− mice infected with L. monocytogenes.
CCR2, CCR5, and CX3CR1 are crucial for extravasation of inflammatory monocytes (46). Although CCL2, CX3CL1, and CCL5 have been reported to be under NOD1 control (37, 56), the levels of expression of these chemokines in infected NOD1−/− fibroblasts and in WT fibroblasts were similar (data not shown).
Next, we studied the effect of NOD1 at the cellular level and showed that NOD1 is required for efficient control of listerial intracellular growth in hematopoietic and nonhematopoietic cells in vitro. NOD1 signaling does not modulate L. monocytogenes uptake into macrophages. Listeria activates at least three signaling pathways in macrophages: one that requires TLR/MyD88 signaling, particularly through TLR2 (29, 43), one that requires RIP2 (5, 25), and one that is independent of both TLR and RIP2 and involves recognition of cytosolic DNA in which the newly described double-stranded DNA receptor DAI might be involved (33, 47-49).
NOD1−/− BMM and DC exhibited reduced titers of IL-6 mRNA in response to Listeria infection in vitro. Invasive L. monocytogenes and noninvasive L. monocytogenes activate distinct signaling pathways, and cytosolic bacteria trigger a unique cytokine response that includes production of IFN-α/β and CCL2 (36, 44). Penetration of Listeria into the cytosol was required for the NOD1-mediated increase in the levels of IL-6 mRNA in BMM (Fig. 6). On the other hand, in agreement with previous data, Listeria induced IFN-α/β expression in macrophages independent of NOD1 (33, 48, 56). It is important that diminished levels of other proinflammatory molecules were not detected in NOD1−/− BMM, and NOD1 deficiency was not related to diminished IL-6 expression in fibroblasts. The levels of IL-6 and IFN-γ were not decreased in spleens from L. monocytogenes-infected NOD1−/− mice compared to spleens from L. monocytogenes-infected WT mice. Moreover, the levels of mRNA of a number of inflammatory cytokines, chemokine and cytokine receptors, transcription factors, and matrix metalloproteases were increased in infected NOD1−/− fibroblasts compared to WT cells (data not shown), probably reflecting a stronger host cell NOD1-independent response resulting from higher levels of intracellular L. monocytogenes.
It has recently been shown that NOD2 signaling could be triggered by ligands generated by bacterial degradation in the phagosome of IFN-γ-activated macrophages (17). While WT BMM treated with IFN-γ showed a robust reduction in the bacterial load and increased NO release, only a moderate decrease in the bacterial load and lower levels of NO release were observed in NOD1−/− BMM treated with IFN-γ compared to untreated NOD1−/− BMM controls. This suggests that NOD1 signaling is indeed involved in bacterial clearance in IFN-γ-treated cells.
Primary macrophages have been shown to have a very weak response to the synthetic NOD1 ligand iE-DAP (D-γ-Glu-DAP) compared to TLR activation (51, 53). However, NOD1 activation by iE-DAP has been shown to positively regulate TLR signaling (32, 51). The cytokine responses induced by L. monocytogenes were abolished in MyD88-deficient macrophages (43). The latter results appear to be at odds with a model in which RIP2 and MyD88 function in parallel signaling pathways, but they support a model in which NOD1 synergizes with TLR signaling for optimal innate defense. We show that NOD1 mRNA levels are increased in BMM stimulated with TLR2, -3, -4, and -9 ligands. An increase in NOD1 expression in BMM and fibroblasts also occurred after infection with L. monocytogenes. In accordance with our results, NOD1 mRNA expression increases after pneumococcal infection in mouse lung tissue, as well as in the bronchial epithelial cell line BEAS-2B (35). However, in line with previous results (37), the presence of NOD1 did not increase BMM responses to pure TLR agonists.
Mice lacking NOD1 and NOD2 exhibit impaired bacterial clearance and survival after systemic administration of L. monocytogenes when LPS- or E. coli-treated mice are used (24). In contrast, a deficiency of NOD1 and NOD2 had only a modest effect on bacterial clearance when the mice were infected i.p. with 104 L. monocytogenes CFU in the absence of TLR ligand administration (24). On the other hand, RIP2−/− mice showed increased cumulative mortality and increased bacterial loads after i.v. inoculation of 2 × 103 L. monocytogenes CFU (5). In the present study, NOD1−/− mice were found to be very susceptible to i.v., subcutaneous, or i.p. infection with L. monocytogenes even without exposure to other pathogens or TLR.
In summary, NOD1 confers nonhematopoietic cell-mediated resistance to infection with L. monocytogenes. It plays a role in the control of L. monocytogenes in macrophages, astrocytes, and especially fibroblasts and is required for IFN-γ-mediated control of L. monocytogenes growth in macrophages. NOD1-mediated control was associated with increased levels of resident macrophages/monocytes, recruitment of inflammatory monocytes, and decreased levels of granulocytes at the site of infection. However, NOD1 is redundant for triggering adaptive immunity.
Acknowledgments
NOD1−/− mice were kindly provided by W. C. Yeh, Department of Medical Biophysics, University of Toronto, Toronto, Ontario, Canada. We thank Laura Plant, Karolinska Institute, for her comments concerning the manuscript.
This work was supported by European Community grant QLK2-CT-2002-00846, by the Karolinska Institute, by the Swedish health insurance company AFA, by the Swedish Research Council, and by NRJ. O.S. was a recipient of postdoctoral grant SO 876/1-1 from the Deutsche Forschungsgemeinschaft. This work was also partially supported by International Research Training Group 1273, funded by the German Research Foundation and the Swedish Research Council.
Editor: J. L. Flynn
Footnotes
Published ahead of print on 27 April 2009.
REFERENCES
- 1.Beckerman, K. P., H. W. Rogers, J. A. Corbett, R. D. Schreiber, M. L. McDaniel, and E. R. Unanue. 1993. Release of nitric oxide during the T cell-independent pathway of macrophage activation. Its role in resistance to Listeria monocytogenes. J. Immunol. 150888-895. [PubMed] [Google Scholar]
- 2.Carneiro, L. A., L. H. Travassos, and D. J. Philpott. 2004. Innate immune recognition of microbes through Nod1 and Nod2: implications for disease. Microbes Infect. 6609-616. [DOI] [PubMed] [Google Scholar]
- 3.Chamaillard, M., S. E. Girardin, J. Viala, and D. J. Philpott. 2003. Nods, Nalps and Naip: intracellular regulators of bacterial-induced inflammation. Cell. Microbiol. 5581-592. [DOI] [PubMed] [Google Scholar]
- 4.Chamaillard, M., M. Hashimoto, Y. Horie, J. Masumoto, S. Qiu, L. Saab, Y. Ogura, A. Kawasaki, K. Fukase, S. Kusumoto, M. A. Valvano, S. J. Foster, T. W. Mak, G. Nunez, and N. Inohara. 2003. An essential role for NOD1 in host recognition of bacterial peptidoglycan containing diaminopimelic acid. Nat. Immunol. 4702-707. [DOI] [PubMed] [Google Scholar]
- 5.Chin, A. I., P. W. Dempsey, K. Bruhn, J. F. Miller, Y. Xu, and G. Cheng. 2002. Involvement of receptor-interacting protein 2 in innate and adaptive immune responses. Nature 416190-194. [DOI] [PubMed] [Google Scholar]
- 6.Dons, L., Y. Jin, K. Kristensson, and M. E. Rottenberg. 2007. Axonal transport of Listeria monocytogenes and nerve-cell-induced bacterial killing. J. Neurosci. Res. 852529-2537. [DOI] [PubMed] [Google Scholar]
- 7.Dunay, I. R., R. A. Damatta, B. Fux, R. Presti, S. Greco, M. Colonna, and L. D. Sibley. 2008. Gr1(+) inflammatory monocytes are required for mucosal resistance to the pathogen Toxoplasma gondii. Immunity 29306-317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dziarski, R. 2003. Recognition of bacterial peptidoglycan by the innate immune system. Cell. Mol. Life Sci. 601793-1804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ferwerda, G., S. E. Girardin, B.-J. Kullberg, L. Le Bourhis, D. J. de Jong, D. M. L. Langenberg, R. van Crevel, G. J. Adema, T. H. M. Ottenhoff, J. W. M. Van der Meer, and M. G. Netea. 2005. NOD2 and Toll-like receptors are nonredundant recognition systems of Mycobacterium tuberculosis. PLoS Pathog. 1e34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fritz, J. H., L. Le Bourhis, G. Sellge, J. G. Magalhaes, H. Fsihi, T. A. Kufer, C. Collins, J. Viala, R. L. Ferrero, S. E. Girardin, and D. J. Philpott. 2007. Nod1-mediated innate immune recognition of peptidoglycan contributes to the onset of adaptive immunity. Immunity 26445-459. [DOI] [PubMed] [Google Scholar]
- 11.Geginat, G., S. Schenk, M. Skoberne, W. Goebel, and H. Hof. 2001. A novel approach of direct ex vivo epitope mapping identifies dominant and subdominant CD4 and CD8 T cell epitopes from Listeria monocytogenes. J. Immunol. 1661877-1884. [DOI] [PubMed] [Google Scholar]
- 12.Geissmann, F., S. Jung, and D. R. Littman. 2003. Blood monocytes consist of two principal subsets with distinct migratory properties. Immunity 1971-82. [DOI] [PubMed] [Google Scholar]
- 13.Girardin, S. E., I. G. Boneca, L. A. Carneiro, A. Antignac, M. Jehanno, J. Viala, K. Tedin, M. K. Taha, A. Labigne, U. Zathringer, A. J. Coyle, P. S. DiStefano, J. Bertin, P. J. Sansonetti, and D. J. Philpott. 2003. Nod1 detects a unique muropeptide from gram-negative bacterial peptidoglycan. Science 3001584-1587. [DOI] [PubMed] [Google Scholar]
- 14.Girardin, S. E., and D. J. Philpott. 2004. Mini-review: the role of peptidoglycan recognition in innate immunity. Eur. J. Immunol. 341777-1782. [DOI] [PubMed] [Google Scholar]
- 15.Girardin, S. E., L. H. Travassos, M. Herve, D. Blanot, I. G. Boneca, D. J. Philpott, P. J. Sansonetti, and D. Mengin-Lecreulx. 2003. Peptidoglycan molecular requirements allowing detection by Nod1 and Nod2. J. Biol. Chem. 27841702-41708. [DOI] [PubMed] [Google Scholar]
- 16.Harada, H., M. Matsumoto, M. Sato, Y. Kashiwazaki, T. Kimura, M. Kitagawa, T. Yokochi, R. S. Tan, T. Takasugi, Y. Kadokawa, C. Schindler, R. D. Schreiber, S. Noguchi, and T. Taniguchi. 1996. Regulation of IFN-alpha/beta genes: evidence for a dual function of the transcription factor complex ISGF3 in the production and action of IFN-alpha/beta. Genes Cells 1995-1005. [DOI] [PubMed] [Google Scholar]
- 17.Herskovits, A. A., V. Auerbuch, and D. A. Portnoy. 2007. Bacterial ligands generated in a phagosome are targets of the cytosolic innate immune system. PLoS Pathog. 3e51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hertz, L., L. Peng, and J. C. Lai. 1998. Functional studies in cultured astrocytes. Methods 16293-310. [DOI] [PubMed] [Google Scholar]
- 19.Inohara, N., T. Koseki, L. del Peso, Y. Hu, C. Yee, S. Chen, R. Carrio, J. Merino, D. Liu, J. Ni, and G. Nunez. 1999. Nod1, an Apaf-1-like activator of caspase-9 and nuclear factor-kappaB. J. Biol. Chem. 27414560-14567. [DOI] [PubMed] [Google Scholar]
- 20.Inohara, N., and G. Nunez. 2003. NODs: intracellular proteins involved in inflammation and apoptosis. Nat. Rev. Immunol. 3371-382. [DOI] [PubMed] [Google Scholar]
- 21.Jin, Y., L. Dons, K. Kristensson, and M. E. Rottenberg. 2001. Neural route of cerebral Listeria monocytogenes murine infection: role of immune response mechanisms in controlling bacterial neuroinvasion. Infect. Immun. 691093-1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kanneganti, T.-D., M. Lamkanfi, and G. Nunez. 2007. Intracellular NOD-like receptors in host defense and disease. Immunity 27549-559. [DOI] [PubMed] [Google Scholar]
- 23.Kim, J. G., S. J. Lee, and M. F. Kagnoff. 2004. Nod1 is an essential signal transducer in intestinal epithelial cells infected with bacteria that avoid recognition by Toll-like receptors. Infect. Immun. 721487-1495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kim, Y.-G., J.-H. Park, M. H. Shaw, L. Franchi, N. Inohara, and G. N′Òez. 2008. The cytosolic sensors Nod1 and Nod2 are critical for bacterial recognition and host defense after exposure to Toll-like receptor ligands. Immunity 28246-257. [DOI] [PubMed] [Google Scholar]
- 25.Kobayashi, K., N. Inohara, L. D. Hernandez, J. E. Galan, G. Nunez, C. A. Janeway, R. Medzhitov, and R. A. Flavell. 2002. RICK/Rip2/CARDIAK mediates signalling for receptors of the innate and adaptive immune systems. Nature 416194-199. [DOI] [PubMed] [Google Scholar]
- 26.Kobayashi, K. S., M. Chamaillard, Y. Ogura, O. Henegariu, N. Inohara, G. Nunez, and R. A. Flavell. 2005. Nod2-dependent regulation of innate and adaptive immunity in the intestinal tract. Science 307731-734. [DOI] [PubMed] [Google Scholar]
- 27.Kotani, S., Y. Watanabe, T. Shimono, T. Narita, and K. Kato. 1975. Immunoadjuvant activities of cell walls, their water-soluble fractions and peptidoglycan subunits, prepared from various gram-positive bacteria, and of synthetic N-acetylmuramyl peptides. Z. Immunitaetsforsch. Exp. Klin. Immunol. 149302-319. [PubMed] [Google Scholar]
-
28.Livak, K. J., and T. D. Schmittgen. 2001. Analysis of relative gene expression data using real-time quantitative PCR and the
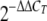 method. Methods 25402-408. [DOI] [PubMed] [Google Scholar]
method. Methods 25402-408. [DOI] [PubMed] [Google Scholar] - 29.McCaffrey, R. L., P. Fawcett, M. O'Riordan, K. D. Lee, E. A. Havell, P. O. Brown, and D. A. Portnoy. 2004. A specific gene expression program triggered by Gram-positive bacteria in the cytosol. Proc. Natl. Acad. Sci. USA 10111386-11391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mengaud, J., J. Chenevert, C. Geoffroy, J. L. Gaillard, and P. Cossart. 1987. Identification of the structural gene encoding the SH-activated hemolysin of Listeria monocytogenes: listeriolysin O is homologous to streptolysin O and pneumolysin. Infect. Immun. 553225-3227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Montoya, M., G. Schiavoni, F. Mattei, I. Gresser, F. Belardelli, P. Borrow, and D. F. Tough. 2002. Type I interferons produced by dendritic cells promote their phenotypic and functional activation. Blood 993263-3271. [DOI] [PubMed] [Google Scholar]
- 32.Netea, M. G., G. Ferwerda, D. J. de Jong, T. Jansen, L. Jacobs, M. Kramer, T. H. Naber, J. P. Drenth, S. E. Girardin, B. J. Kullberg, G. J. Adema, and J. W. Van der Meer. 2005. Nucleotide-binding oligomerization domain-2 modulates specific TLR pathways for the induction of cytokine release. J. Immunol. 1746518-6523. [DOI] [PubMed] [Google Scholar]
- 33.O'Connell, R. M., S. A. Vaidya, A. K. Perry, S. K. Saha, P. W. Dempsey, and G. Cheng. 2005. Immune activation of type I IFNs by Listeria monocytogenes occurs independently of TLR4, TLR2, and receptor interacting protein 2 but involves TNFR-associated NF kappa B kinase-binding kinase 1. J. Immunol. 1741602-1607. [DOI] [PubMed] [Google Scholar]
- 34.Opitz, B., S. Forster, A. C. Hocke, M. Maass, B. Schmeck, S. Hippenstiel, N. Suttorp, and M. Krull. 2005. Nod1-mediated endothelial cell activation by Chlamydophila pneumoniae. Circ. Res. 96319-326. [DOI] [PubMed] [Google Scholar]
- 35.Opitz, B., A. Puschel, B. Schmeck, A. C. Hocke, S. Rosseau, S. Hammerschmidt, R. R. Schumann, N. Suttorp, and S. Hippenstiel. 2004. Nucleotide-binding oligomerization domain proteins are innate immune receptors for internalized Streptococcus pneumoniae. J. Biol. Chem. 27936426-36432. [DOI] [PubMed] [Google Scholar]
- 36.O'Riordan, M., C. H. Yi, R. Gonzales, K. D. Lee, and D. A. Portnoy. 2002. Innate recognition of bacteria by a macrophage cytosolic surveillance pathway. Proc. Natl. Acad. Sci. USA 9913861-13866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Park, J. H., Y. G. Kim, C. McDonald, T. D. Kanneganti, M. Hasegawa, M. Body-Malapel, N. Inohara, and G. Nunez. 2007. RICK/RIP2 mediates innate immune responses induced through Nod1 and Nod2 but not TLRs. J. Immunol. 1782380-2386. [DOI] [PubMed] [Google Scholar]
- 38.Portnoy, D. A., R. D. Schreiber, P. Connelly, and L. G. Tilney. 1989. Gamma interferon limits access of Listeria monocytogenes to the macrophage cytoplasm. J. Exp. Med. 1702141-2146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Robben, P. M., M. LaRegina, W. A. Kuziel, and L. D. Sibley. 2005. Recruitment of Gr-1+ monocytes is essential for control of acute toxoplasmosis. J. Exp. Med. 2011761-1769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rothfuchs, A. G., D. Gigliotti, K. Palmblad, U. Andersson, H. Wigzell, and M. E. Rottenberg. 2001. IFN-αβ-dependent, IFN-γ secretion by bone marrow-derived macrophages controls an intracellular bacterial infection. J. Immunol. 1676453-6461. [DOI] [PubMed] [Google Scholar]
- 41.Rottenberg, M. E., A. C. Gigliotti Rothfuchs, D. Gigliotti, C. Svanholm, L. Bandholtz, and H. Wigzell. 1999. Role of innate and adaptive immunity in the outcome of primary infection with Chlamydia pneumoniae, as analyzed in genetically modified mice. J. Immunol. 1622829-2836. [PubMed] [Google Scholar]
- 42.Schnare, M., G. M. Barton, A. C. Holt, K. Takeda, S. Akira, and R. Medzhitov. 2001. Toll-like receptors control activation of adaptive immune responses. Nat. Immunol. 2947-950. [DOI] [PubMed] [Google Scholar]
- 43.Seki, E., H. Tsutsui, N. M. Tsuji, N. Hayashi, K. Adachi, H. Nakano, S. Futatsugi-Yumikura, O. Takeuchi, K. Hoshino, S. Akira, J. Fujimoto, and K. Nakanishi. 2002. Critical roles of myeloid differentiation factor 88-dependent proinflammatory cytokine release in early phase clearance of Listeria monocytogenes in mice. J. Immunol. 1693863-3868. [DOI] [PubMed] [Google Scholar]
- 44.Serbina, N. V., W. Kuziel, R. Flavell, S. Akira, B. Rollins, and E. G. Pamer. 2003. Sequential MyD88-independent and -dependent activation of innate immune responses to intracellular bacterial infection. Immunity 19891-901. [DOI] [PubMed] [Google Scholar]
- 45.Serbina, N. V., and E. G. Pamer. 2006. Monocyte emigration from bone marrow during bacterial infection requires signals mediated by chemokine receptor CCR2. Nat. Immunol. 7311-317. [DOI] [PubMed] [Google Scholar]
- 46.Soehnlein, O., and C. Weber. 24 February 2009. Myeloid cells in atherosclerosis: initiators and decision shapers. Semin. Immunopathol. [Epub ahead of print.] doi: 10.1007/s00281-009-0141-z. [DOI] [PubMed]
- 47.Stetson, D. B., and R. Medzhitov. 2006. Recognition of cytosolic DNA activates an IRF3-dependent innate immune response. Immunity 2493-103. [DOI] [PubMed] [Google Scholar]
- 48.Stockinger, S., B. Reutterer, B. Schaljo, C. Schellack, S. Brunner, T. Materna, M. Yamamoto, S. Akira, T. Taniguchi, P. J. Murray, M. Muller, and T. Decker. 2004. IFN regulatory factor 3-dependent induction of type I IFNs by intracellular bacteria is mediated by a TLR- and Nod2-independent mechanism. J. Immunol. 1737416-7425. [DOI] [PubMed] [Google Scholar]
- 49.Takaoka, A., Z. Wang, M. K. Choi, H. Yanai, H. Negishi, T. Ban, Y. Lu, M. Miyagishi, T. Kodama, K. Honda, Y. Ohba, and T. Taniguchi. 2007. DAI (DLM-1/ZBP1) is a cytosolic DNA sensor and an activator of innate immune response. Nature 448501-505. [DOI] [PubMed] [Google Scholar]
- 50.Travassos, L. H., L. A. M. Carneiro, S. E. Girardin, I. G. Boneca, R. Lemos, M. T. Bozza, R. C. P. Domingues, A. J. Coyle, J. Bertin, D. J. Philpott, and M. C. Plotkowski. 2005. Nod1 participates in the innate immune response to Pseudomonas aeruginosa. J. Biol. Chem. 28036714-36718. [DOI] [PubMed] [Google Scholar]
- 51.Uehara, A., S. Yang, Y. Fujimoto, K. Fukase, S. Kusumoto, K. Shibata, S. Sugawara, and H. Takada. 2005. Muramyldipeptide and diaminopimelic acid-containing desmuramylpeptides in combination with chemically synthesized Toll-like receptor agonists synergistically induced production of interleukin-8 in a NOD2- and NOD1-dependent manner, respectively, in human monocytic cells in culture. Cell. Microbiol. 753-61. [DOI] [PubMed] [Google Scholar]
- 52.van Beelen, A. J., Z. Zelinkova, E. W. Taanman-Kueter, F. J. Muller, D. W. Hommes, S. A. J. Zaat, M. L. Kapsenberg, and E. C. de Jong. 2007. Stimulation of the intracellular bacterial sensor NOD2 programs dendritic cells to promote interleukin-17 production in human memory T cells. Immunity 27660-669. [DOI] [PubMed] [Google Scholar]
- 53.van Heel, D. A., S. Ghosh, M. Butler, K. Hunt, B. M. Foxwell, D. Mengin-Lecreulx, and R. J. Playford. 2005. Synergistic enhancement of Toll-like receptor responses by NOD1 activation. Eur. J. Immunol. 352471-2476. [DOI] [PubMed] [Google Scholar]
- 54.Viala, J., C. Chaput, I. G. Boneca, A. Cardona, S. E. Girardin, A. P. Moran, R. Athman, S. Memet, M. R. Huerre, A. J. Coyle, P. S. DiStefano, P. J. Sansonetti, A. Labigne, J. Bertin, D. J. Philpott, and R. L. Ferrero. 2004. Nod1 responds to peptidoglycan delivered by the Helicobacter pylori cag pathogenicity island. Nat. Immunol. 51166-1174. [DOI] [PubMed] [Google Scholar]
- 55.Viala, J., P. Sansonetti, and D. J. Philpott. 2004. Nods and ‘intracellular’ innate immunity. C. R. Biol. 327551-555. [DOI] [PubMed] [Google Scholar]
- 56.Werts, C., L. le Bourhis, J. Liu, J. G. Magalhaes, L. A. Carneiro, J. H. Fritz, S. Stockinger, V. Balloy, M. Chignard, T. Decker, D. J. Philpott, X. Ma, and S. E. Girardin. 2007. Nod1 and Nod2 induce CCL5/RANTES through the NF-kappaB pathway. Eur. J. Immunol. 372499-2508. [DOI] [PubMed] [Google Scholar]