

Abstract
Listeria monocytogenes is an intracellular bacterial pathogen whose virulence depends on the regulated expression of numerous secreted bacterial factors. As for other gram-positive bacteria, many proteins secreted by L. monocytogenes are translocated across the bacterial membrane in an unfolded state to the compartment existing between the membrane and the cell wall. This compartment presents a challenging environment for protein folding due to its high density of negative charge, high concentrations of cations, and low pH. We recently identified PrsA2 as a gene product required for L. monocytogenes virulence. PrsA2 was identified based on its increased secretion by strains containing a mutationally activated form of prfA, the key regulator of L. monocytogenes virulence gene expression. The prsA2 gene product is one of at least two predicted peptidyl-prolyl cis/trans-isomerases encoded by L. monocytogenes; these proteins function as posttranslocation protein chaperones and/or foldases. In this study, we demonstrate that PrsA2 plays a unique and important role in L. monocytogenes pathogenesis by promoting the activity and stability of at least two critical secreted virulence factors: listeriolysin O (LLO) and a broad-specificity phospholipase. Loss of PrsA2 activity severely attenuated virulence in mice and impaired bacterial cell-to-cell spread in host cells. In contrast, mutants lacking prsA1 resembled wild-type bacteria with respect to intracellular growth and cell-to-cell spread as well as virulence in mice. PrsA2 is thus distinct from PrsA1 in its unique requirement for the stability and full activity of L. monocytogenes-secreted factors that contribute to host infection.
Listeria monocytogenes is a gram-positive, food-borne facultative intracellular bacterial pathogen that is capable of crossing the intestinal epithelial barrier of susceptible humans to cause serious and life-threatening infections (65). While healthy individuals primarily exhibit mild forms of disease such as gastroenteritis, immunocompromised and elderly individuals often suffer more severe forms of illness that include meningoencephalitis and septicemia (42, 71). Central to its ability to cause serious infections is the bacterium's capacity to gain access to its protected replication niche within the cytosol of infected host cells (12, 71). Bacterial mutants that fail to establish and exploit this replication niche are severely attenuated in host infection models (10, 19, 48, 50, 57, 58, 80).
A number of secreted bacterial factors that contribute to L. monocytogenes cell invasion, entry into the cytosol, intracellular bacterial replication, and cell-to-cell spread have been described (71). These factors include the internalins InlA and InlB, which mediate entry into nonprofessional phagocytic cells, and listeriolysin O (LLO; encoded by hly), which is required for bacterial escape from host cell vacuoles into the cytosol (17, 18, 20, 33, 37, 48). Two phospholipases, a phosphatidylinositol-specific phospholipase (PI-PLC; encoded by plcA) and a broad-specificity phospholipase (PC-PLC; encoded by plcB) also contribute to vacuole lysis (21, 41, 61, 70). A bacterial surface protein known as ActA is required for bacterial actin-based motility and L. monocytogenes spread to adjacent cells (11, 29, 30, 68). The expression of these secreted gene products is regulated by a transcriptional activator known as PrfA (16, 35, 43). PrfA exists in a low activity state in bacteria grown outside of host cells; upon bacterial entry into the cytosol of infected cells, PrfA becomes activated and induces the expression of the gene products required for intracellular bacterial replication and cell-to-cell spread (54). PrfA thus serves a critical role in regulating the expression of L. monocytogenes virulence gene products within the appropriate cellular location.
We recently identified a secreted protein, PrsA2, based on its abundant expression by bacteria containing prfA mutations that result in the constitutive expression of PrfA-dependent gene products (prfA* mutations) (47). Insertional disruption of lmo2219, which encodes PrsA2, was found to significantly reduce L. monocytogenes virulence in mice (47). PrsA2 had previously been identified by Chatterjee et al. (7) and was found to contribute to L. monocytogenes intracellular growth and/or cell-to-cell spread in tissue culture cells. PrsA2 is one of two predicted peptidyl-prolyl cis/trans-isomerases of the Parvulin family encoded within the L. monocytogenes genome (the other is encoded by prsA1, or lmo1444); both share significant homology with the single PrsA protein produced by Bacillus subtilis (Fig. 1A). In B. subtilis, prsA is an essential gene that assists in the secretion, folding, and sequestration of a number of secreted proteins (69, 73-75).
FIG. 1.

Amino acid sequence alignment and construction of prsA1 and prsA2 loss-of-function mutants. (A) Amino acid sequence alignment of B. subtilis PrsA and the two PrsA homologues in L. monocytogenes (PrsA1 and PrsA2) using the ClustalW program (http://www.ebi.ac.uk/Tools/clustalw/index.html). Amino acid residues that are identical between at least two of the three PrsA proteins are shaded in black; asterisks indicate amino acid residues that are identical in all three proteins. Residues that are similar between protein sequences are shaded in gray. (B) Schematic of the ΔprsA1 deletion mutant, the prsA2::T targetron insertion mutant, and the ΔprsA2::erm mutant. The gray and white center arrows denote the open reading frames of prsA1 and prsA2, respectively, while the white and black flanking arrows denote flanking gene sequences. Predicted transcriptional terminators are depicted by stem-loops. The dashed bracket indicates the deletion of the prsA1 open reading frame, the vertical line through the lmo2219 open reading frame and T denote the intron insertion at nucleotide 457 in prsA2, and the solid broken line designates the ΔprsA2::erm deletion.
In this work, we demonstrate that PrsA2 plays a unique and distinguishable role from PrsA1 by promoting the activity and stability of L. monocytogenes-secreted factors critical for bacterial virulence.
MATERIALS AND METHODS
Bacterial strains, plasmids, and media.
All bacterial strains used in this study are listed in Table 1. L. monocytogenes 10403S containing an actA-gus transcriptional fusion (NF-L476) was used as the parent strain for genetic manipulations unless otherwise specified (60). Escherichia coli alpha select (BioLine, Boston, MA), One Shot TOP10 (Invitrogen, Carlsbad, CA), and SM10 were used as host strains for recombinant plasmids. All strains were grown in Luria broth (LB) (Invitrogen Corp., Carlsbad, CA) or brain heart infusion (BHI) medium (Difco Laboratories, Detroit, MI) and supplemented with the appropriate antibiotic. Antibiotics were used at the following concentrations unless otherwise noted: ampicillin, 100 μg/ml; carbenicillin, 50 μg/ml; chloramphenicol, 10 and 7.5 μg/ml; erythromycin, 5 μg/ml; and streptomycin, 200 μg/ml. All L. monocytogenes strains were grown overnight at 37°C without agitation prior to in vitro and in vivo assays. The temperature-sensitive shuttle plasmid pKSV7 (62) was used for generation of L. monocytogenes mutants via allelic exchange, and the integration plasmid pPL2 (34) was used for genetic complementation.
TABLE 1.
Bacterial strains and plasmids used in this work
| Strain or plasmid | Genotype | Designation | Source or reference |
|---|---|---|---|
| Strains | |||
| TOP10 | E. coli host strain used for recombinant pPL2 plasmids | ||
| DH5α | E. coli host strain for recombinant pKSV7 plasmids | ||
| SM10 | E. coli host strain for conjugation of pPL2 plasmids | ||
| NF-E1254 | E. coli BL21 Star (DE3) with pNF-E1251 for PrsA2 protein expression and purification | This work | |
| NF-L100 | L. monocytogenes 10403S parent strain | 2 | |
| NF-L476 | L. monocytogenes 10403S actA-gus-plcB | 59 | |
| NF-L1167 | 10403S actA-gus-neo prfA(L140F) | L140F | 45 |
| DP-L2161 | 10403S containing in-frame deletion of hly gene | Δhly | 27 |
| NF-L1456 | NF-L476 with group II intron (targetron) insertion into lmo2219 (prsA2) at position 457 | prsA2::T | This work |
| NF-L1475 | NF-L1456 with integrated pPL2-prsA2. | prsA2::T + prsA2 | This work |
| NF-L1587 | NF-L1456 with prsA2::T replaced with a ΔprsA2::erm mutation | This work | |
| NF-L1635 | DP-L2161 (Δhly) with ΔprsA2::erm | ΔprsA2::erm Δhly | This work |
| NF-L1637 | NF-L1167 [prfA(L140F) with ΔprsA2::erm] | prfA(L140F) ΔprsA2::erm | This work |
| NF-L1659 | NF-L1637 [prfA(L140F) ΔprsA2::erm] with integrated pPL2-prsA2 (pNF1255) | prfA(L140F) ΔprsA2::erm + pPL2-prsA2 | This work |
| NF-L1651 | 10403S with ΔprsA2::erm | ΔprsA2::erm | This work |
| NF-L1656 | NF-L1651 (ΔprsA2::erm) with integrated pPL2-prsA2 (pNF1255) | ΔprsA2::erm + pPL2-prsA2 | This work |
| HEL 402 | 10403S with prsA1 in-frame deletion | ΔprsA1 | This work |
| Plasmids | |||
| pNF1256 | pKSV7 with ΔprsA2 SOE fragment with internal SalI restriction site | This work | |
| pNF1264 | pNF1256 with erm resistance cassette inserted at SalI restriction site | This work | |
| pHY304 | Plasmid pHY304 containing constitutive erm cassette | 26 | |
| pNF1251 | pET-TOPO N-His with prsA2 open reading frame | This work | |
| pNF1255 | pPL2-prsA2 containing prsA2 with 636 bp upstream of start ATG and 68 bp downstream of stop TAA | This work | |
| pNF1520 | pNL9146 with targeted targetron group II intron for gene disruption of prsA2 at nucleotide position 457 | This work |
Construction of the ΔprsA1 in-frame deletion mutant.
A 789-bp internal in-frame deletion was generated in lmo1444 as follows: initially two DNA fragments were generated by PCR with primer pairs Marq 161/162 and Marq 163/164 using L. monocytogenes 10403S chromosomal DNA as the template. (All oligonucleotides are listed in Table S1 in the supplemental material.) The two fragments were purified and used in a splicing-by-overlap extension (SOE) PCR with primer pair Marq 161/164, generating a 1,250-bp fragment encompassing the 5′ end of lmo1444 with upstream sequence and the 3′ end of the same gene with downstream sequence. The 1,250-bp fragment was cloned into the shuttle vector pKSV7 using KpnI and PstI restriction sites, generating pMC10. L. monocytogenes strain 10403S was electroporated with pMC10 and the ΔprsA1 mutation was introduced into the bacterial chromosome in single copy by allelic exchange as previously described (6) to generate strain HEL-402. Chromosomal deletion of a 789-bp fragment within lmo1444 (prsA1) was confirmed by PCR. This deletion encompasses 90% of the structural gene.
Construction of the prsA2 targetron and ΔprsA2::erm mutants.
Repeated attempts to generate a ΔprsA2 in-frame deletion mutant using standard approaches based on homologous recombination (as described above for ΔprsA1) were unsuccessful. As an alternative approach, a targeted insertional disruption of prsA2 (lmo2219) was generated using the targetron gene knockout system (Sigma, St. Louis, MO) following the recommendations made by the supplier, with minor modifications (8, 56, 79, 83). Briefly, a retargeted stable group II intron derived from Lactococcus lactis was generated by PCR using oligonucleotide primers (prsA2-457IBS, prsA2-457EBS1D, and prsA2-457EBS2) in the temperature-sensitive shuttle vector pNL9146 (Sigma, St. Louis, MO). The plasmid containing the retargeted intron with its expression under the control of a cadmium-inducible promoter (pNF1520) was introduced into L. monocytogenes via electroporation. Overnight cultures of L. monocytogenes transformants containing the pNF1520 shuttle vector were diluted 1:20 in BHI broth and grown at 32°C to an approximate optical density at 600 nm (OD600) of 0.5, at which point 10 μM cadmium chloride (Sigma, St. Louis, MO) was added to the growth medium to induce targeted insertion of the intron into prsA2 between nucleotides 457 and 458. Approximately 20 bacterial colonies were screened for the presence of the intron using primers 457checkF and 457checkR. All strains positive for the insertional disruption were cured of the plasmid by shifting growth conditions to the nonpermissive temperature 41°C as follows: intron-containing colonies were inoculated into 2 ml of BHI broth and grown to stationary phase at 41°C, and then 2-μl aliquots were removed and diluted into 2 ml of fresh medium and once again allowed to reach stationary phase. This procedure was repeated five times. Individual colonies were then screened for erythromycin susceptibility signifying loss of plasmid-encoded antibiotic resistance. The presence of the stable insertional disruption was then reconfirmed by DNA sequencing of PCR products derived from the target region using primers 457checkF and 457checkR, and the prsA2 targetron mutant (prsA2::T) was designated NF-L1456.
Based on the successful construction of the prsA2 targetron mutation, we sought to generate additional prsA2 mutations that lacked the majority of prsA2 coding sequence and that could be marked with antibiotic resistance genes to facilitate transduction into new genetic backgrounds. Using the NF-L1456 prsA2::T mutant as the parent strain, a gene replacement mutant was constructed wherein the majority of the prsA2 coding sequence (nucleotides 31 to 849 of 879 total, representing 93% of the coding region) was replaced with a constitutive erm cassette. Five-hundred-base-pair fragments upstream and downstream of prsA2, including the first and last 30 nucleotides of the coding sequence as well as PstI and XbaI restriction sites, respectively, were PCR amplified from L. monocytogenes strain 10403S genomic DNA using primer pairs 2219SOEaPst1/2219SOEbSalint and 2219SOEcSalint/2219SOEdXbaI. The two fragments were purified and used in a SOE PCR with primer pair 2219SOEaPstI/2219SOEdXbaI, generating a 1,031-bp fragment encompassing the 5′ end of lmo2219 with upstream sequence and the 3′ end of the same gene with downstream sequence. The 1,031-bp fragment was cloned into the shuttle vector pKSV7 using PstI and XbaI restriction sites, generating pNF1256. (An internal SalI restriction site was included in the overlap region of the SOE reaction such that a SalI site would be present in between the upstream and downstream fragments.) A constitutively expressed erm resistance gene was then amplified from plasmid pHY304 (26) (a kind gift of Amanda Jones, Seattle Children's Research Hospital, Seattle, WA) using primers ermSalI5 and ermSalI3, each containing a SalI restriction site. The fragment was purified and cloned into pNF-1256 to generate pNF-1264. pNF1264 was introduced into L. monocytogenes strain NF-L1456 prsA2::T by electroporation, and transformants were isolated on BHI agar containing chloramphenicol. The ΔprsA2::erm mutation was introduced into the L. monocytogenes chromosome in single copy to replace the targetron insertion using allelic exchange as previously described (6). The resulting in-frame prsA2 deletion mutant with the antibiotic resistance gene replacement was confirmed by PCR analysis and sequencing of genomic DNA and was designated NF-L1587.
For construction of a plasmid vector containing prsA2 for complementation of all subsequent prsA2 mutations, primers 2219pPL2SacIa and 2219pPL2SmaIb were used to amplify the entire open reading frame of prsA2 from L. monocytogenes strain 10403S genomic DNA, including the putative PrfA box located 206 nucleotides upstream of the predicted ATG translational start site. The PCR-amplified product was digested with SacI and SmaI and subcloned into appropriately digested shuttle plasmid vector pPL2 to generate pNF1255. pPL2 integrates in single copy into the L. monocytogenes phage attachment site located within the tRNAArg gene following conjugation, resulting in single-copy gene complementation within this neutral site in the L. monocytogenes chromosome (14, 34).
Construction of prfA(L140F) ΔprsA1, prfA(L140F) ΔprsA2::erm, and ΔprsA2::erm Δhly double mutants.
The ΔprsA2::erm mutation was transduced into strain 10403S, NF-L1167 [10403S actA-gus-neo, prfA(L140F)] (45), and DP-L2161 (10403S Δhly) (27) using U153 bateriophage-mediated phage transduction as previously described, with some minor modifications (78). Briefly, phage lysates (108 to 109 PFU per ml) were prepared from NF-L1587 (NF-L476 with ΔprsA2::erm) and mixed with 108 CFU of each of the indicated strains at a final ratio of 108 phage to 108 bacteria. CaCl2 and MgSO4 were then added to a final concentration of 10 mM, and the mixture was incubated at room temperature for 40 min. After 40 min, the bacterium-phage mixture was plated onto BHI plates containing 1 μg/ml erythromycin and resultant colonies were picked after 1 to 2 days and screened for the presence of the erythromycin marker by PCR. The resultant strains were designated NF-L1651 (10403S ΔprsA2::erm), NF-L1637 [prfA(L140F) ΔprsA2::erm], and NF-L1635 (ΔprsA2::erm Δhly). Similarly, phage lysates were prepared from NF-L1167 [10403S actA-gus-neo, prfA(L140F)] in order to transduce the prfA(L140F) allele into HEL-402 (ΔprsA1). The transduction was carried out in a similar fashion with minor modifications. After incubation of the bacteria and phage for 40 min at room temperature, the mixture was plated onto BHI plates containing 5 μg/ml neomycin and 50 μg/ml 5-bromo-4-chloro-3-indolyl-β-d-glucuronide (Inalco, Milano Italy). Positive transductants that were both neomycin resistant and blue in color (indicative of constitutively active PrfA activity) were isolated and used for subsequent analysis.
Purification of PrsA2 and generation of rabbit polyclonal antiserum directed against PrsA2.
Oligonucleotides listed in Table S1 in the supplemental material were used to amplify prsA2 from L. monocytogenes strain 10403S genomic DNA, and the resulting PCR fragment was subcloned into pET100 using the Champion pET directional TOPO expression kit (Invitrogen, Carlsbad, CA) according to the manufacturer's protocol. Plasmids containing prsA2 coding sequences were transformed into E. coli BL21 Star (DE3) expression cells, and recombinant protein expression was induced by addition of 1 mM IPTG (isopropyl-β-d-thiogalactopyranoside) to mid-log-phase cultures for 5 h. Bacteria were recovered following centrifugation at 3,000 × g for 10 min at 4°C, the supernatant was discarded, and bacterial pellets were frozen at −80°C, thawed, resuspended in 25 ml phosphate-buffered saline (PBS), followed by bacterial cell disruption via French press. Protein extracts were passed over nickel columns, and recombinant PrsA2 was eluted off at pH 6.0 under denaturing conditions using 6 M guanidinium HCl, 8 M urea, 20 mM NaH2PO4, and 500 mM NaCl. Eluted protein fractions were concentrated using a Centricon centrifugal filter (Millipore, Billerica, MA), and proteins were analyzed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and Coomassie staining. Five hundred micrograms of recombinant PrsA2 protein was used to generate rabbit polyclonal antiserum through a commercial supplier (Cocalico Biologicals, Reamstown, PA).
Mouse intravenous infections.
All animal procedures were IACUC approved and performed in the Biological Resources Laboratory at the University of Illinois at Chicago. Overnight cultures of bacteria grown in BHI broth at 37°C were diluted 1:20 into fresh medium and grown to an OD600 of approximately 0.6. One milliliter of culture corresponding to 6 × 108 CFU/ml was washed twice in PBS (0.144 g/ml KH2PO4, 9.00 g/ml NaCl, 0.795 g/ml Na2HPO4 [anhydrous]), diluted, and resuspended in PBS to a final concentration of 1 × 105 CFU/ml. Eight- to 10-week-old female ND4 Swiss Webster mice (Harlan, Madison, WI) were injected with 200 μl PBS containing 2 × 104 CFU of L. monocytogenes via the tail vein. Seventy-two hours postinfection, mice were sacrificed and livers and spleens were dissected. Organs were homogenized with a Tissue Master 125 homogenizer (Omni, Marietta, GA) and 10-fold serial dilutions were plated onto BHI plates containing 200 μg/ml streptomycin (all 10403S derivatives are streptomycin resistant).
Assessment of bacterial intracellular growth in tissue culture cells.
The J774 and Henle 407 cell lines were maintained as previously described (4, 67). Monolayers of cells were grown on acid-washed coverslips and infected with L. monocytogenes strains at a multiplicity of infection (MOI) of 0.1 bacterium per 10 macrophage for J774s and 30 bacteria per epithelial cell in Henle 407s for wild-type (WT) and ΔprsA2::erm strains or 90 bacteria per epithelial cell for all strains containing an hly deletion (to compensate for the loss of LLO-mediated enhancement of bacterial invasion) (46). Infection was carried out for 30 min followed by washing three times with warm PBS and addition of fresh medium containing 50 μg/ml gentamicin to kill extracellular bacteria at 1 h. To measure bacterial intracellular growth, coverslips were removed at the time points indicated and host cells were lysed in 5 ml of sterile H2O by vigorous vortexing. Dilutions of these lysates were then plated on LB agar plates for enumeration of bacterial counts per coverslip. Results were obtained from at least three independent experiments. Coverslips of infected Henle 407 cells (10.5 h postinfection) were also processed for immunofluorescence microscopy as previously described (46). Processed coverslips were then viewed on a Leica DM4000B wide-field epifluorescence microscope (Leica Microsystems, Wetzlar, Germany). Representative images of all fields viewed were captured using SlideBook image acquisition software (Intelligent Imaging Innovations, Inc., Denver, CO).
Plaque assays.
Plaque assays were conducted as previously described (64). Monolayers of L2 fibroblast cells were grown on six-well tissue culture plates and infected for 1 h with L. monocytogenes at a multiplicity of 30 bacteria to 1 fibroblast. Gentamicin was added to single wells after infection (20 μg/ml) to kill extracellular bacteria. Plaque size was measured using a micrometer (Finescale, Orange, CA). Results were obtained from at least three independent experiments.
Hemolysin assays.
Hemolytic activity was measured as previously described (5, 28). Briefly, overnight L monocytogenes cultures grown in BHI broth at 37°C were diluted 1:10 into fresh BHI broth and grown at 37°C with vigorous shaking for 5 h. OD600 readings were taken, and 1 ml of bacterial culture was spun down at 16,000 × g for 5 min. Supernatants were removed from bacterial pellets, and dilutions of bacterial supernatants (standardized for OD600) were incubated with PBS-washed sheep red blood cells (RBCs; in Alsevers) for 30 min at 37°C (Cocalico Biologicals, Reamstown, PA). After incubation, RBCs were removed by centrifugation and hemolytic units were measured as the reciprocal of the dilutions that gave 50% lysis of RBCs as determined by visual inspection of RBC pellets after centrifugation. Results were obtained from at least three independent experiments.
Detection of PC-PLC activity.
plcB-dependent phospholipase production was visualized using an egg yolk overlay agar plate assay (46). Chicken egg yolk was added in a 1:1 (vol/vol) ratio to PBS and vortexed to generate a suspension. Five milliliters of egg yolk suspension was then added to 100 ml of molten LB medium (45°C to 48°C), and 3 ml of egg yolk-agar suspension was overlaid onto an LB agar plate. Egg yolk agar used for L. monocytogenes strains containing a WT prfA allele was additionally supplemented with 0.2% activated charcoal (Fisher Scientific, Pittsburgh, PA) and 25 mM glucose-6-phosphate (Sigma, St Louis, MO) to enhance prfA-dependent expression of plcb in these strains. Following solidification of the medium, bacterial strains were gently streaked onto the surface of the plate and incubated at 37°C for 48 h for strains containing a WT prfA allele and 24 h for those containing a prfA(L140F) allele. Phospholipase activity was detected as a zone of opacity surrounding bacterial streaks. Results were obtained from at least three independent experiments.
Isolation of supernatant and bacterial cell-associated proteins.
Overnight cultures of L. monocytogenes strains grown in BHI broth were diluted 1:20 into 30 ml of fresh BHI broth and were grown at 37°C with shaking for 5 h (approximate OD600 of 1.4). Twenty-five milliliters of each culture was centrifuged at 12,350 × g for 15 min at 4°C. Supernatants were recovered, and secreted proteins were obtained by precipitation using trichloroacetic acid (TCA) (Sigma Chemical Co., St. Louis, MO) at a final percentage of 10%; after TCA addition, culture supernatants were incubated on ice for 30 min. Precipitated protein pellets were recovered by centrifugation at 12,350 × g for 10 min, the supernatant was discarded, and protein pellets were allowed to air dry. Four milliliters of ice-cold acetone was used to wash pellets, which were then centrifuged again at 12,350 × g for 10 min. The TCA-precipitated protein pellet was then resuspended in 200 μl of 1× SDS boiling buffer (5% SDS, 5% β-mercaptoethanol, 10% glycerol, 60 mM Tris [pH 6.8]). For bacterial cell-associated protein fractions, bacterial pellets were resuspended in 200 μl of 2% SDS boiling buffer and boiled at 100°C for 5 min. This resulting cell-associated fraction was clarified by centrifugation at 10,000 × g for 20 min to remove the remaining insoluble material.
Western analysis.
All protein samples were run on 4 to 12% gradient SDS-polyacrylamide gels (Invitrogen, Carlsbad, CA) for 1 h at 200 V. To ensure equivalent total protein was analyzed for each strain, equal volumes of the previously normalized supernatant and surface-associated proteins (OD of ∼1.4) were loaded onto the gels. For visualization of total protein, gels were fixed in a 50% methanol and 7% acetic acid solution for 30 min, washed three times for 15 min each in deionized H2O, and stained with GelCode Blue staining reagent (Thermo Scientific, Rockford, IL) for 1 h. For Western analysis, proteins were transferred to polyvinylidene difluoride membranes at 30 V (constant) for 1 h. Membranes were blocked for 1 h in PBS containing 0.05% Tween 20 (PBST) and 5% milk. Membranes were then probed with the following antibodies and described dilutions: primary polyclonal anti-PrsA2 antibody was used at a 1:2,500 dilution, primary polyclonal anti-LLO antibody (AbCam, Cambridge, MA) was used at a 1:2,500 dilution, and primary polyclonal anti-PC-PLC antibody (a kind gift from Helene Marquis, Cornell University, NY) was used at a 1:2,500 dilution. Primary antibody was added to 20 ml of PBST and incubated at room temperature with shaking for 2 h. Membranes were then washed three times for 15 min each in PBST. Secondary antibody conjugated to alkaline phosphatase was added at a 1:2,500 dilution for 1.5 h (Southern Biotech, Birmingham, AL). Detection was carried out via colorimetric development in the presence of BCIP-NBTPlus (5-bromo-4-chloro-3-indolylphosphate-nitroblue tetrazolium) for approximately 5 min (Southern Biotech, Birmingham, AL). Images of blots were then photographed using an AlphaImager 2200 (Alpha Innotech, San Leandro, CA). Results were obtained from at least three independent experiments. Densitometry was performed on all Western blots from three independent experiments using NIH ImageJ Software (http://rsbweb.nih.gov/ij/). The area under the curve was used to calculate arbitrary units for both the WT and L140F mutant for each blot, which was then set to a value of 1.00. All other strains were then compared according to that value and are thus shown as a deviation from 1.00. The average values from three blots and standard deviations are shown.
RESULTS
Construction of prsA1 and prsA2 stable gene disruption mutants.
L. monocytogenes prsA1 and prsA2 have significant amino acid sequence similarity to B. subtilis PrsA (43% and 45% identity and 63% and 65% similarity, respectively) and even higher amino acid identity and similarity to each other (58% identity and 75% similarity) (Fig. 1A). To explore the functional contributions of the prsA1 and prsA2 gene products to L. monocytogenes pathogenesis, we attempted to introduce in-frame deletion mutations into prsA1 and prsA2 within the L. monocytogenes chromosome by allelic exchange. Whereas the construction of the L. monocytogenes ΔprsA1 strain proved straightforward and was rapidly accomplished, efforts to generate ΔprsA2 strains were unsuccessful despite the fact that prsA2 loss-of-function mutations via plasmid insertion had been readily generated (47). A similar failure to generate an L. monocytogenes ΔprsA2 mutant was also reported by Milohanic et al. (44), although Chatterjee et al. (7) reported the successful construction of a ΔprsA2 mutant in EGDe. As an alternative approach for the generation of a stable prsA2 mutant, the commercially available targetron gene knockout system was used to insert a Lactococcus lactis group II intron into prsA2 at nucleotide position 457 (prsA2::T) (Fig. 1B). The L. monocytogenes ΔprsA1 and prsA2::T mutant strains thus generated exhibited normal patterns of growth in broth culture and on solid media (see Fig. S1A in the supplemental material). Subsequent use of the prsA2::T mutant as the starting strain for allelic exchange enabled the construction of an in-frame prsA2 deletion mutant containing an erythromycin resistance cassette (NF-L1587). This strain was constructed so as to facilitate transduction of the antibiotic resistance-marked ΔprsA2 mutation into other L. monocytogenes genetic backgrounds. Although prsA2 mutants were indistinguishable from WT strains for growth in broth culture, it was observed that the mutants exhibited a subtle fitness defect in mixed cultures such that after several cycles of bacterial growth and dilution, the WT strain dominated the cultures (see Fig. S1B in the supplemental material). The fitness defect of prsA2 mutant strains in mixed cultures thus accounts for the difficulties encountered by our lab and the labs of other investigators in the isolation of in-frame deletion mutants of prsA2 when using the repeated cycles of bacterial outgrowth required for allelic exchange.
The prsA2 strain is unique from the prsA1 strain in its requirement for L. monocytogenes virulence in mice.
ΔprsA1 and ΔprsA2::erm mutants were assessed for virulence in mice following intravenous inoculation. Bacteria (2 × 104 CFU) were injected into the tail vein of Swiss mice, and after 72 h, bacterial burdens were determined in both livers and spleens. In contrast to mice infected with either the WT strain or the ΔprsA1 mutant, mice infected with the ΔprsA2::erm mutant appeared healthy at 3 days postinfection, with no evident signs of illness (data not shown). Enumeration of bacterial CFU from the livers and spleens of infected animals resulted in the recovery of approximately 5 to 6 logs fewer bacteria for the ΔprsA2::erm mutant compared to mice infected with the WT parent strain or the ΔprsA1 mutant (Fig. 2). The virulence defect of the ΔprsA2::erm mutant was fully complemented by the introduction of prsA2 in single copy on the integrative plasmid vector pPL2 (Fig. 2), thus indicating that the ΔprsA2::erm virulence defect was the result of the loss of the functional prsA2 gene product. These data indicate that prsA2, but not prsA1, is essential for L. monocytogenes pathogenesis in vivo.
FIG. 2.

ΔprsA2::erm mutants are severely attenuated for virulence in mice. Mice were intravenously infected with 2 × 104 CFU of the WT or ΔprsA2::erm, ΔprsA2::erm + pPL2-prsA2, or ΔprsA1 mutant strain, and the bacterial burden in livers and spleens was determined at 72 h postinfection. The scatter plot shows CFU obtained from the livers and spleens of five individual mice; the data are representative of three independent experiments. Solid lines and brackets represent the mean and standard error of the mean, respectively, for the data points in each group. ***, statistically significant value (P < 0.0001) for ΔprsA2::erm in both the liver and the spleen compared to WT using a one-way analysis of variance with Dunnett's posttest (GraphPad V.5.0A). Differences between WT and the ΔprsA2::erm + pPL2 prsA2 mutant or the WT and the ΔprsA1 mutant were not statistically significant (P > 0.05).
prsA2 mutants exhibit altered patterns of intracellular growth and cell-to-cell spread in tissue culture cells.
The severe virulence defect associated with the loss of prsA2 prompted examination of the mutant for intracellular growth in tissue culture cell lines. Although uptake of a ΔprsA2::erm mutant was reduced in J774 mouse macrophage-like cells, mutant growth closely resembled that of the WT and the ΔprsA1 mutant, with comparable rates of bacterial replication up to 9 h postinfection (Fig. 3A). Inspection of infected cell monolayers by microscopy indicated that the ΔprsA2::erm mutant strain was capable of intracellular replication, actin-based motility, and spread to adjacent cells (data not shown). At 9 h postinfection, the number of mutant bacteria was seen to decline; this phenotype was consistently observed in multiple independent experiments, whereas WT and ΔprsA1 numbers increased or remained constant up to the 11-h time point (Fig. 3A). Consistent with reduced bacterial growth at late time points of infection in J774 cells, the ΔprsA2::erm mutant was observed to be defective for intracellular growth and cell-to-cell spread in monolayers of infected mouse L2 fibroblasts (Fig. 3B). In this cell line, L. monocytogenes host cell invasion, intracellular growth, and cell-to-cell spread are readily visible after infection of the monolayers via the formation of small, distinct zones of cell clearing (plaques) that arise as a result of cell death following bacterial replication and spread (64). Plaques formed by the ΔprsA2::erm mutant were approximately 20% of the size of those formed by the WT L. monocytogenes strain, the ΔprsA1 mutant, or the complemented prsA2 mutant (Fig. 3B). A similar plaque formation defect was reported for the EDGe ΔprsA2 mutant generated by Chatterjee et al. (7). The ΔprsA2::erm mutant strain is therefore capable of host cell entry, intracellular growth, and cell-to-cell spread; however, it is impaired for these activities in comparison to WT L. monocytogenes.
FIG. 3.
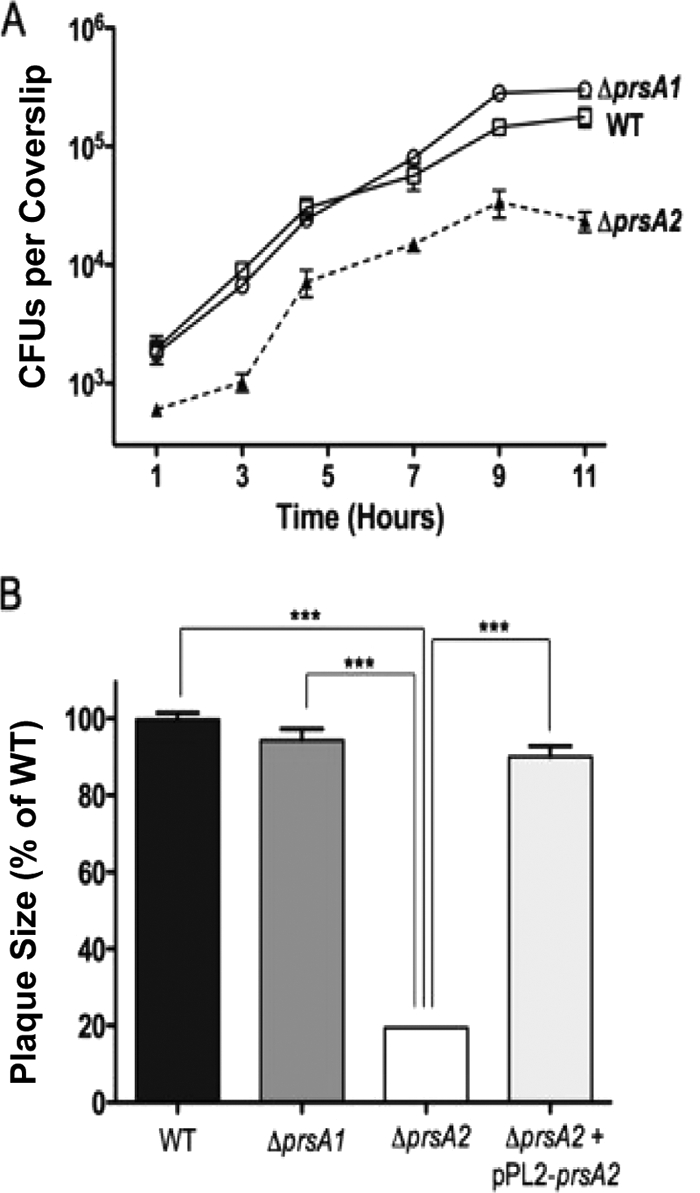
The ΔprsA2::erm mutant is capable of intracellular growth but is defective for cell-to-cell spread in tissue culture cells. (A) J774 macrophage-like cells were infected with the WT or the ΔprsA1 or ΔprsA2::erm mutant at an MOI of 0.1. After 30 min, the monolayers were washed and gentamicin (50 μg/ml) was added at 1 h postinfection. The data shown are representative of at least three independent experiments done in duplicate. □, wild type; ○, prsA1; ▴, ΔprsA2::erm. (B) Plaque formation in mouse L2 fibroblast cells. Monolayers of L2 cells were infected with the WT or ΔprsA1, ΔprsA2::erm, or ΔprsA2::erm + pPL2-prsA2 mutant for 1 h and washed with PBS, and 20 μg/ml gentamicin was added. At least 15 plaques were measured in three independent experiments for all strains; measurements represent plaque size comparisons with respect to the WT (set at 100%). ***, statistically significant value (P < 0.0001) as calculated using a one-way way analysis of variance with Tukey's multiple comparison test (GraphPad v.6.0A).
PrsA2 is required for optimal secreted LLO and PC-PLC activity.
In B. subtilis, PrsA has been shown to contribute to the proper folding of secreted proteins at the membrane-cell wall interface (24, 31, 49). To explore the potential role of L. monocytogenes PrsA2 as a posttranslocational molecular chaperone, we examined the activity of two secreted L. monocytogenes gene products critical for bacterial virulence, LLO and PC-PLC (9, 21, 48, 80). Measurement of LLO-associated hemolytic activity in supernatants derived from ΔprsA2::erm strains indicated an approximate twofold decrease in activity in comparison to WT and ΔprsA1 strains, based on the lysis of sheep RBCs (33 ± 2.5 U for the ΔprsA2::erm mutant versus 60 ± 3.7 U for the parent strain and 65 ± 5.0 U for ΔprsA1) (Fig. 4A). The introduction of pPL2-prsA2 fully restored hemolytic activity (55 ± 6.5 U). The modest reduction in secreted hemolytic activity was likewise evident in the presence of the mutationally activated prfA(L140F) allele, which confers constitutive high-level expression of PrfA-dependent gene products, including LLO and PC-PLC (45, 47, 72, 78). When the ΔprsA2::erm mutation was introduced into the prfA(L140F) strain by phage transduction, hemolytic activity in prfA(L140F) ΔprsA2::erm-derived supernatants was decreased approximately threefold in comparison to that of the prfA(L140F) strain containing WT prsA2 or the prsA1 deletion [111 ± 8.9 U for prfA(L140F) ΔprsA2::erm versus 373 ± 53.3 U for prfA(L140F) ΔprsA1 and 396 ± 29.5 U for prfA(L140F)] and again was fully restored upon complementation with pPL2-prsA2 (360 ± 40.0 U) (Fig. 4A). The presence of prfA(L140F) increased LLO-dependent hemolytic activity four- to fivefold for all strains in comparison to strains carrying the WT prfA allele.
FIG. 4.
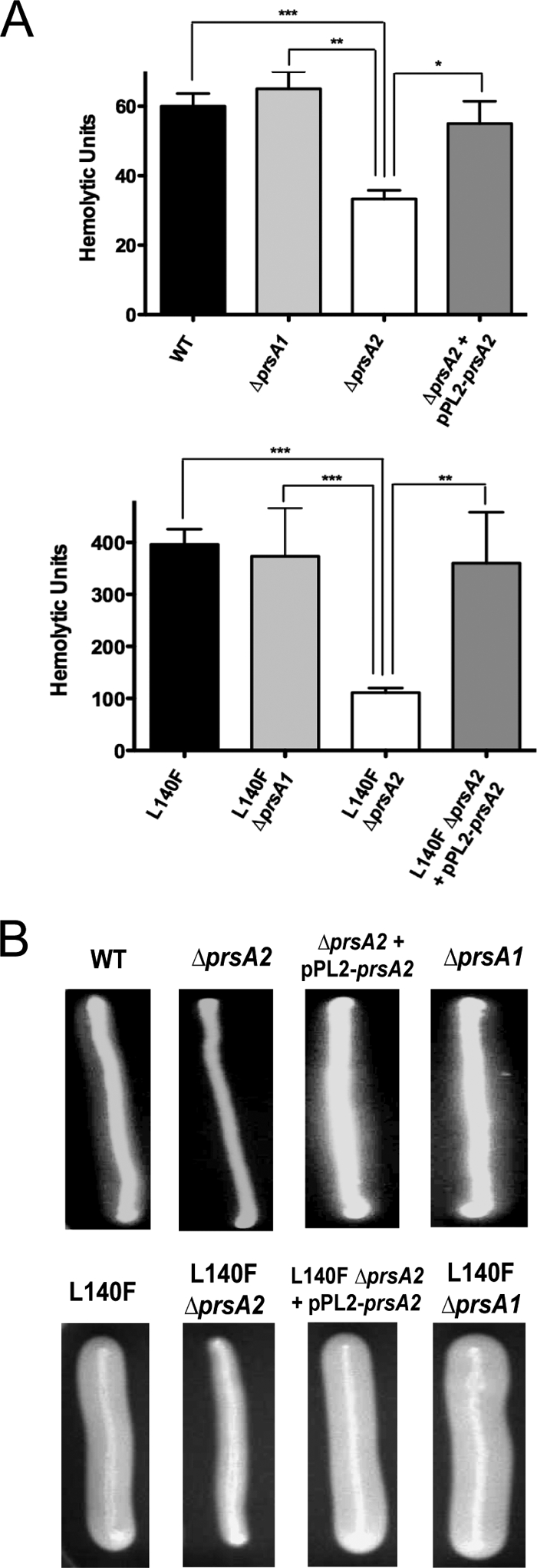
ΔprsA2::erm strains have decreased LLO and PC-PLC activity. (A) The hemolytic activity of culture supernatants derived from WT and prfA(L140F) strains in the presence and absence of prsA1 or prsA2 and its complement was determined based on 50% lysis of sheep RBCs. The average values of four independent experiments are shown. Statistically significant differences are indicated as determined by one-way analysis of variance with Tukey's multiple comparison test (*, P < 0.01; **, P < 0.001; ***, P < 0.0001) (GraphPad V.5.0). (B) Detection of PC-PLC-related phospholipase activity of the ΔprsA1 or ΔprsA2::erm mutant and its complement in the presence of the WT (top panel) and mutationally activated prfA [prfA(L140F)] (bottom panel). prfA(L140F), prfA(L140F) ΔprsA2::erm, prfA(L140F) ΔprsA2::erm + pPL2-prsA2, and prfA(L140F) ΔprsA1 strains incubated on egg yolk agar plates exhibited zones of opacity indicative of PC-PLC activity. A representative image from 1 of 10 plates is shown.
PC-PLC activity was assessed following growth of the bacteria on solid medium containing egg yolk and was based on the ability of bacterially secreted PC-PLC-associated lecithinase activity to yield a white precipitate. PC-PLC activity is difficult to detect for strains containing WT prfA as the expression of plcB (which encodes PC-PLC) is low in bacteria grown on standard agar media; however, the addition of charcoal and phosphorylated glucose has been shown to enhance PC-PLC expression (81), as does the introduction of the prfA(L140F) allele (72, 78). PC-PLC activity was detected for all strains as visible zones of precipitation (Fig. 4B). Strains lacking prsA2 exhibited significantly reduced levels of PC-PLC activity; these levels could be fully restored following the introduction of the pPL2-prsA2 plasmid (Fig. 4B). Strains lacking prsA1 exhibited levels of secreted PC-PLC activity equivalent to those of strains containing WT prsA1 in the presence of either WT prfA or the prfA(L140F) allele (Fig. 4B). PrsA2 is therefore unique from PrsA1 in its requirement for optimal activity of both secreted LLO and PC-PLC, most notably under conditions where PrfA is activated.
PrsA2 contributes to LLO stability and processing of pro-PC-PLC to its mature form.
Based on the apparent reduction in activity of LLO and PC-PLC observed for strains containing prsA2 mutations, we next examined the amount of LLO and PC-PLC protein secreted by WT and ΔprsA2::erm strains as well as protein associated with the bacterial cell surface. In strains expressing the WT prfA allele, Western blot analysis of secreted LLO revealed approximately 2.5-fold-lower levels of LLO secreted by the ΔprsA2::erm mutant in comparison to WT strains (Fig. 5). LLO secretion was restored to the mutant by the introduction of the pPL2-prsA2 plasmid. No reduction in LLO was observed for protein associated with the bacterial cell surface of prsA2 mutants. Levels of detectable PC-PLC in secreted fractions were similar for all strains, with no visible protein retained at the cell surface (Fig. 5). Under these conditions, PC-PLC was only detected as the unprocessed pro-form of the enzyme based on its molecular weight. Western blot analysis of secreted levels of LLO and PC-PLC for ΔprsA1 strains indicated no difference from WT bacteria (data not shown).
FIG. 5.

Strains lacking PrsA2 secrete reduced amounts of LLO and PC-PLC. Bacterial supernatant and surface-associated proteins from the WT or ΔprsA2::erm or ΔprsA2::erm + pPL2-prsA2 mutant were isolated by TCA precipitation (secreted proteins) or boiling in 2% SDS (surface-associated proteins). Sample volumes were adjusted to reflect equivalent bacterial densities (OD600 of ∼1.4). Western analysis of protein fractions subjected to SDS-PAGE was done using rabbit polyclonal anti-LLO, rabbit polyclonal anti-PrsA2, and rabbit polyclonal anti-PC-PLC antibody to detect LLO, PrsA2, and PC-PLC, respectively. Arrows indicate bands of interest (either LLO, PrsA2, or PC-PLC) in each panel. The amount of protein detected for each sample in comparison to the WT lane (set at 1.0) as determined by densitometry is indicated at the bottom of each panel. Lane 1, WT; lane 2, ΔprsA2::erm; lane 3, ΔprsA2::erm + pPL2-prsA2.
In the presence of the activated prfA(L140F) allele, levels of both secreted and cell-associated LLO and PC-PLC were increased in strains with WT prsA2 as well as the ΔprsA2::erm mutant (Fig. 6). However, a number of lower-molecular-weight bands that reacted with the anti-LLO antibody were evident in the secreted protein fractions, suggesting that degradation of LLO was occurring along with the increase in LLO synthesis. Interestingly, degradation products were more abundant and exhibited a different peptide banding pattern for secreted LLO isolated from the ΔprsA2::erm mutant from the parent strain (Fig. 6). LLO degradation products were also evident for cell-associated protein fractions. Comparison of relative amounts of full-length LLO between WT and ΔprsA2::erm strains with the prfA(L140F) allele indicated an approximately 2.5-fold decrease in LLO protein secreted by the ΔprsA2::erm strain, with no significant difference observed for cell-associated LLO (Fig. 6). Overall, these results indicate that (i) bacteria with WT prsA2 secrete more full-length LLO than strains lacking prsA2, (ii) induction of LLO synthesis results in increasingly greater amounts of LLO degradation for strains with and without prsA2, and (iii) induction of LLO synthesis in strains lacking prsA2 results in increased amounts of LLO degradation and different degradation peptide patterns in comparison to bacteria with WT prsA2. The altered banding patterns of the LLO degradation products in prsA2 mutant strains may be indicative of an altered LLO structure that is more sensitive to proteolytic cleavage, a result that would be consistent with a role for PrsA2 in proper LLO folding.
FIG. 6.

Strains lacking PrsA2 exhibit increased LLO degradation and reduced processing of pro-PC-PLC in the presence of activated PrfA. Bacterial supernatant and surface-associated proteins from the mutationally activated prfA(L140F), prfA(L140F) ΔprsA2::erm, or prfA(L140F) ΔprsA2::erm + pPL2-prsA2 mutant were isolated by TCA precipitation (secreted proteins) or boiling in 2% SDS (surface-associated proteins). Sample volumes were adjusted to reflect equivalent bacterial densities (OD600 of ∼1.4). Western analysis of protein fractions subjected to SDS-PAGE was done using rabbit polyclonal anti-LLO antibody, rabbit polyclonal anti-PrsA2, and rabbit polyclonal anti-PC-PLC antibody to detect LLO, PrsA2, and PC-PLC respectively. Arrows indicate bands of interest (either LLO, PrsA2, pro-PC-PLC, or PC-PLC) in each panel. Small stars indicate the altered LLO degradation pattern found in prfA(L140F) ΔprsA2::erm strains compared to that of prfA(L140F) and complement strains. The amount of protein detected for each sample in comparison to the WT lane (set at 1.0) as determined by densitometry is indicated at the bottom of each panel. The top and bottom rows of numbers located at the bottom of the PC-PLC Western blot represent the total amounts of pro-PC-PLC and PC-PLC, respectively. Lane 1, prfA(L140F); lane 2, prfA(L140F) ΔprsA2::erm; lane 3, prfA(L140F) ΔprsA2::erm + pPL2-prsA2.
Secretion of PC-PLC was increased in the presence of prfA(L140F) for both WT and prsA2 mutant strains, with the predominant form of the enzyme remaining the unprocessed pro-form (Fig. 6). Strains lacking prsA2 secreted slightly reduced amounts of pro-PC-PLC in comparison to strains with WT prsA2 and approximately three- to fourfold-less mature PC-PLC. Examination of cell-associated PC-PLC in the presence of prfA(L140F) revealed slightly reduced amounts of pro-PC-PLC associated with the cell surface of ΔprsA2::erm strains; however, strains with WT prsA2 had significantly increased levels of processed PC-PLC associated with the cell surface in comparison to ΔprsA2::erm strains (approximately three- to fourfold-more mature PC-PLC enzyme) (Fig. 6). L. monocytogenes is known to synthesize and sequester PC-PLC until the bacterium encounters the appropriate host environment for the release of active enzyme; thus, the cell-associated processed PC-PLC observed in Fig. 6 may reflect sequestered enzyme (41). These results implicate a role for PrsA2 in promoting the processing of pro-PC-PLC to its mature form.
Reduced PC-PLC activity in prsA2 strains is functionally detectable following L. monocytogenes infection of human epithelial cells.
Infection of J774 macrophages with the ΔprsA2::erm strain indicated that the mutant was fully capable of mediating bacterial escape from the primary phagosome (Fig. 3A). Although reduced numbers of bacteria were internalized, bacterial replication rates for the first 9 h of infection were similar (Fig. 3A), and examination of bacteria associated with host cell actin (a marker for cytosolic entry) indicated no difference in the rate of escape of the ΔprsA2::erm mutant from primary phagosomes in comparison to that of WT L. monocytogenes (data not shown). Consistent with these observations, efficient phagosome lysis has been previously demonstrated with L. monocytogenes mutants expressing low levels of LLO in broth culture (approximately 10-fold below WT levels) (15), and the ΔprsA2::erm mutant exhibits only an approximately two- to threefold reduction in secreted LLO activity in comparison to WT L. monocytogenes (Fig. 4A). However, the decline in bacterial numbers in J774 cells beginning approximately 9 h postinfection and the small-plaque phenotype of the mutant in fibroblast cells (Fig. 3B) are strongly suggestive of a defect in bacterial cell-to-cell spread. PC-PLC activity has been shown to be an important contributor to lysis of secondary vacuoles formed during bacterial spread to adjacent cells (40, 41, 63, 80). To more fully investigate the potential consequences on cellular infection of reduced PC-PLC activity in ΔprsA2::erm mutants, we examined intracellular growth of ΔprsA2::erm mutants in Henle human epithelial tissue culture cells in the presence and absence of hly. Unlike most tissue culture cell lines, LLO (encoded by hly) is not required for vacuole lysis in human epithelial cells; this function instead can be fully provided by active PC-PLC, thereby enabling intracellular assessment of PC-PLC activity based on intracellular bacterial growth and cell-to-cell spread (39, 46, 48). Henle human epithelial cell monolayers were infected with WT and ΔprsA2::erm mutant strains in the presence and absence of hly. Bacterial growth was monitored in the presence of 50 μg/ml gentamicin added at 1 h postinfection. As previously reported, Δhly strains lacking LLO activity efficiently escaped the vacuoles of Henle cells and replicated within the cytosol (Fig. 7A). Strains lacking prsA2 grew similarly to WT L. monocytogenes, with a slight reduction in the number of bacteria initially entering host cells observable at 1 h postinfection and a consistent leveling off of bacterial numbers by 11 h postinfection. In contrast, the Δhly ΔprsA2::erm double mutant exhibited a pronounced defect in intracellular growth, with bacterial numbers increasing modestly only after 7 h postinfection and continuing to lag behind the WT up to 11 h postinfection (Fig. 7A). The Δhly ΔprsA2::erm double mutant was, however, capable of limited intracellular bacterial replication based on its increased numbers in comparison to those of strains completely lacking LLO and PC-PLC activity (the Δhly ΔplcB double mutant), which remain trapped within host cell vacuoles (39) (Fig. 7A). Visual inspection of infected monolayers by microscopy at 10.5 h postinfection indicated cells infected with the Δhly ΔprsA2::erm double mutant primarily contained single bacteria, suggesting that the double mutants were significantly impaired for vacuolar escape and hence intracellular replication (Fig. 7B). These results indicate that the secretion of active PC-PLC is significantly reduced in ΔprsA2::erm strains during cellular infection.
FIG. 7.
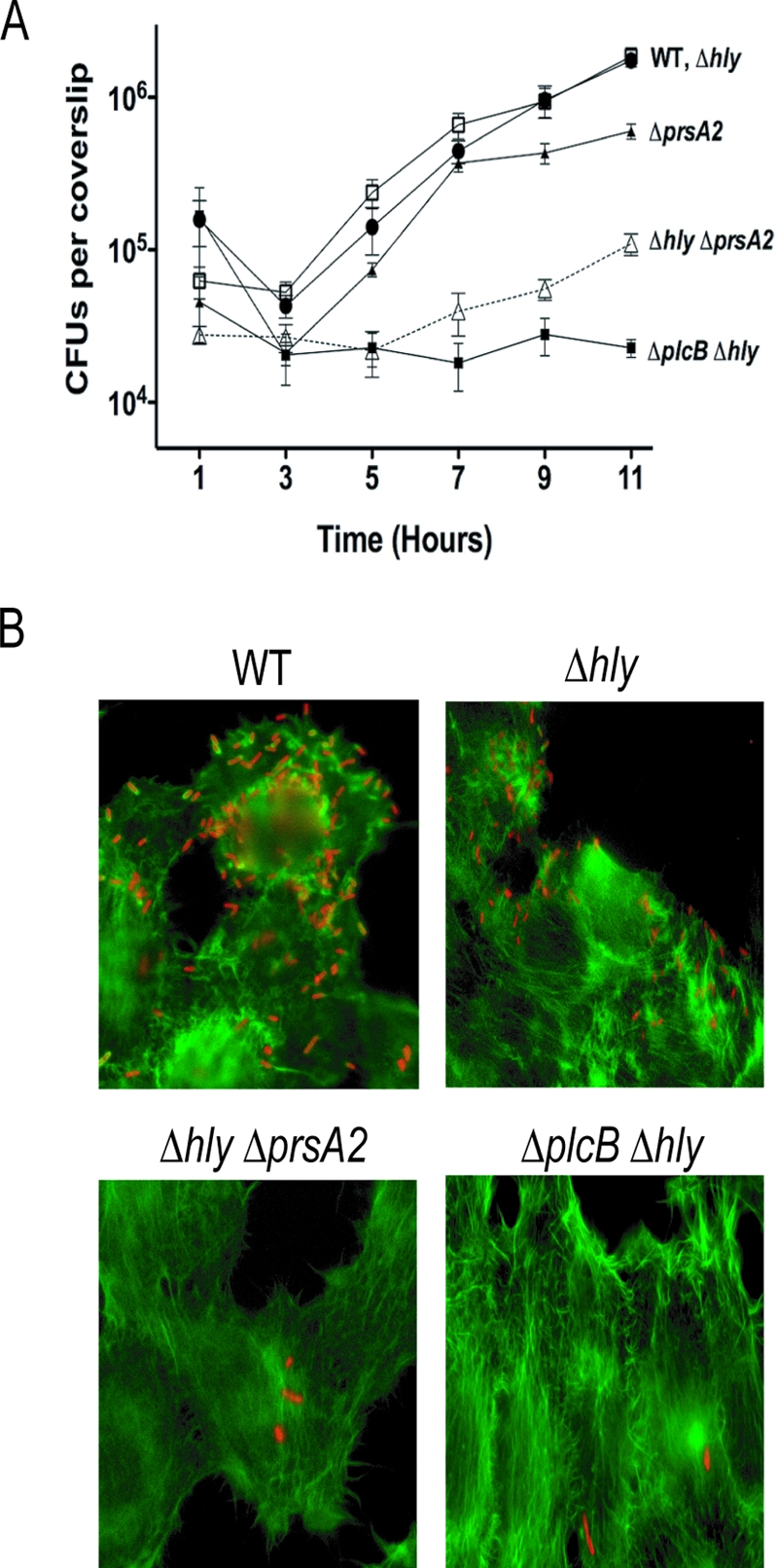
Reduced PC-PLC activity in ΔprsA2::erm strains impairs bacterial vacuolar escape, resulting in reduced intracellular replication. (A) Henle human epithelial cells were infected with the WT or ΔprsA2::erm, Δhly, ΔprsA2::erm Δhly, or ΔplcB Δhly mutant as described in Materials and Methods. After 30 min, infected cells were washed with PBS followed by the addition of gentamicin (50 μg/ml) at 1 h postinfection. The data shown are representative of three independent experiments done in duplicate. □, WT; ▴, ΔprsA2::erm; •, Δhly; ▵, ΔprsA2::erm Δhly double mutant; ▪, ΔplcB Δhly double mutant. (B) Coverslips were also processed for microscopy at 10.5 h postinfection. Cell monolayers were fixed, permeabilized, and stained for L. monocytogenes using an anti-Listeria antibody and tetramethylrhodamine-conjugated goat anti-rabbit secondary antibody to detect bacteria (red) and NBD (7-nitrobanz-2-oxa-1,3-diazole)-phallacidin to detect F-actin (green) at a ×1,000 magnification.
DISCUSSION
L. monocytogenes secretes numerous proteins that enable the organism to invade mammalian cells, gain access to the cytosol, and exploit this protected environment for bacterial replication and systemic spread (55, 71). The expression and activity of secreted virulence factors have been shown to be regulated at multiple steps and include transcriptional, posttranscriptional, and posttranslational regulation (25, 40, 41, 50-53, 58, 77). In this report, we describe the identification of PrsA2, a predicted postsecretion chaperone that is essential for L. monocytogenes pathogenesis. Although L. monocytogenes encodes two highly similar PrsA-like proteins, only PrsA2 was found to influence LLO activity and stability and to promote the processing of pro-PC-PLC to its activated form. Based on our results and on functional analysis of PrsA homologues in other gram-positive bacteria (13, 23, 24, 31, 32, 38, 73, 75), we propose that PrsA2 contributes to L. monocytogenes pathogenesis by promoting the folding, stability, and hence the activity of targeted secreted virulence factors that are essential for bacterial intracellular replication and cell-to-cell spread.
The substantial 5- to 6-log virulence defect observed for mutants lacking prsA2 based upon bacterial recovery from the livers and spleens of infected mice serves to emphasize the essential contributions of PrsA2 to L. monocytogenes virulence. The magnitude of the prsA2 virulence defect is comparable to that observed for strains completely lacking hly or prfA, genes encoding two well-established essential virulence factors (51, 54). This was somewhat surprising given that ΔprsA2::erm strains are not completely lacking in secreted LLO and PC-PLC activity; however, it is probable that PrsA2 contributes to the folding and/or activity of virulence factors in addition to LLO and PC-PLC. Preliminary analysis of secreted and surface-associated proteins from WT and prsA2 mutant strains indicates that a number of proteins appear to be dependent upon PrsA2 activity for secretion and surface association (data not shown). The reduction in bacterial uptake by macrophages (Fig. 3A), also observed for some additional cell lines (data not shown), may indicate a role for PrsA2 in proper folding of proteins required for bacterial adhesion and/or invasion. PrsA2 function may therefore be required for proper function of additional L. monocytogenes-secreted proteins, and it remains to be determined how many of these have functions related to host infection.
Western blot analysis of secreted and cell surface-associated proteins strongly suggests that PrsA2 contributes to LLO stability. PrsA2's role in LLO stability was especially evident in the presence of the mutationally activated prfA(L140F) allele, which increases the expression of L. monocytogenes virulence genes to levels comparable to those observed for cytosolic bacteria (45, 47, 72, 78). Degradation of LLO was evident for both secreted and cell-associated forms of the protein for strains containing WT prsA2 as well as the ΔprsA2::erm mutation; however, strains lacking prsA2 exhibited more extensive degradation (Fig. 6). Changes in protease susceptibility of a given protein are often used to detect changes in protein structure or conformation (22, 45, 66); thus, the appearance of novel degradation products observed in the absence of functional PrsA2 may indicate altered LLO structure or reflect improper folding of the protein. Alternatively, PrsA2 may be required for proper folding of a factor that inhibits LLO degradation. Similarly, PrsA2 may assist in the folding of pro-PC-PLC such that it is more efficiently recognized by the Mpl protease for processing (3, 81), or PrsA2 may be required for full activity of Mpl so that it is capable of pro-PC-PLC processing. Definition of the precise role of PrsA2 with respect to LLO and PC-PLC activity awaits detailed biochemical analyses.
It is worth noting that the virulence defect observed for strains lacking prsA2 was substantially greater than that observed for the prsA2 plasmid insertion mutant (a 5- to 6-log difference versus a 2-log difference in the number of bacterial CFU recovered from the livers of infected mice, respectively) (47). This difference likely reflects the stability of the ΔprsA2::erm mutant versus the temperature-sensitive plasmid insertion. The construction of a stable prsA2 mutation also allowed the detection of the fitness defect conferred by the loss of prsA2 in comparison to WT strains—a defect that was evident in mixed cultures but not in monocultures (see Fig. S1 in the supplemental material). The fitness defect observed for the prsA2 mutants in mixed cultures may reflect a physiological change in the mutant that contributes to the reduction in bacterial virulence observed in infected mice (Fig. 2). Confirmation of this hypothesis awaits a more detailed understanding of PrsA2's role in L. monocytogenes physiology and protein secretion.
It is striking that while L. monocytogenes encodes two PrsA-like proteins, only one is clearly required for bacterial virulence. PrsA1 and PrsA2 share significant amounts of amino acid homology (58% identity and 75% similarity) (Fig. 1A). It is not yet known whether PrsA2 has evolved functional capacities distinct from PrsA1 or whether the failure of prsA1 to compensate for loss of prsA2 is due to a lack of prsA1 expression in the appropriate environment. prsA1, unlike prsA2, does not appear to contain a PrfA binding site within its 5′ upstream region, and little is currently known regarding overall expression patterns for either gene. The construction of L. monocytogenes mutant strains that contain prsA1 coding sequences under the control of the prsA2 promoter should help to clarify the individual roles of PrsA1 and PrsA2. Unlike prsA of B. subtilis, neither prsA1 nor prsA2 appears essential for bacterial viability; however, it is possible that while PrsA1 cannot compensate for the PrsA2 virulence defect, it may share redundant function with respect to an essential role in L. monocytogenes. The PrsA-like protein of L. lactis is not essential for bacterial viability, and Drouault et al. (13) have hypothesized that PrsA-like proteins may be more important for bacteria that are highly dependent on extracellular enzymes for nutrient acquisition, as is the case for soil-dwelling B. subtilis but not for L. lactis grown in milk (13). If the essential nature of PrsA activity is indeed linked to extracellular nutrient acquisition, it will be interesting to determine if L. monocytogenes uses one PrsA-like protein for life in the soil (PrsA1) while having adapted a second PrsA-like protein for life within the host (PrsA2).
PrsA-like proteins have been described in other gram-positive bacteria, including L. lactis (13), Streptococcus pyogenes (38), and Bacillus anthracis (76), and additional homologues to PrsA have been identified in the genomes of both pathogenic and nonpathogenic bacteria (13). Most recognized prsA genes encode a leader peptide followed by a cysteine, indicative of gene products that are lipoproteins. B. subtilis PrsA has been reported to retain activity as a foldase without lipid modification; however, strains with non-lipid-modified PrsA were reduced for the secretion of PrsA-dependent proteins, presumably due to insufficient concentrations of PrsA at the cell surface (36). We first identified PrsA2 as a secreted protein (47); however, preliminary results indicate that a substantial portion of the protein is localized to the cell membrane and cell wall fractions (data not shown). Baumgartner el al (1) have recently confirmed that PrsA2 is indeed lipid modified, although two processed forms of the protein were detected for reasons that were not clear. Some lipoproteins are detectable in secreted protein fractions (1), and it remains to be determined what form of PrsA2 is important for activity and whether PrsA2 activity is restricted to the cell surface, or if secreted PrsA2 fulfills a specific function, perhaps distinct from cell-associated PrsA2. The role of the potential peptidyl-prolyl isomerase activity of PrsA2 also needs to be explored; this activity appears dispensable for the foldase activity of at least some PrsA-like proteins (13, 73).
Recently, Zemansky et al. (82) reported a role for PrsA2 in L. monocytogenes virulence. Their work did not distinguish PrsA1 from PrsA2 but did confirm the attenuation of prsA2 mutants in animal models and the role of PrsA2 in LLO and PC-PLC activity and stability (82). Clearly, PrsA2 is important to the pathogenesis of L. monocytogenes. Its apparently unique role (from PrsA1) in the stability of LLO and in pro-PC-PLC processing is intriguing, and it will be interesting to determine what other secreted factors require PrsA2 for full activity. Clarification of the role of PrsA2 in L. monocytogenes pathogenesis should provide additional insight into the mechanisms by which gram-positive bacterial pathogens use postsecretion chaperones to regulate secretion and promote virulence within the infected host.
Supplementary Material
Acknowledgments
We thank Daniel Portnoy for the L. monocytogenes DP-L937 strain, Helene Marquis for the gift of the PC-PLC antibody, Bobbi Xayarath for assistance with animal studies, Amanda Jones for plasmid pHY304, and members of the Freitag lab for helpful discussions.
This work was supported by Public Health Service grant AI41816 (N.E.F.) from NIAID, a National Science Foundation Graduate Research Fellowship (NSF-GRF) (G.C.P.), and the M. J. Murdock Trust.
The contents of this article are solely the responsibility of the authors and do not necessarily represent the official views of the funding sources.
Editor: A. Camilli
Footnotes
Published ahead of print on 18 May 2009.
Supplemental material for this article may be found at http://iai.asm.org/.
REFERENCES
- 1.Baumgartner, M., U. Kärst, B. Gerstel, M. Loessner, J. Wehland, and L. Jänsch. 2007. Inactivation of Lgt allows systematic characterization of lipoproteins from Listeria monocytogenes. J. Bacteriol. 189313-324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bishop, D. K., and D. J. Hinrichs. 1987. Adoptive transfer of immunity to Listeria monocytogenes: the influence of in vitro stimulation on lymphocyte subset requirements. J. Immunol. 1392005-2009. [PubMed] [Google Scholar]
- 3.Bitar, A. P., M. Cao, and H. Marquis. 2008. The metalloprotease of Listeria monocytogenes is activated by intramolecular autocatalysis. J. Bacteriol. 190107-111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brundage, R. A., G. A. Smith, A. Camilli, J. A. Theriot, and D. A. Portnoy. 1993. Expression and phosphorylation of the Listeria monocytogenes ActA protein in mammalian cells. Proc. Natl. Acad. Sci. USA 9011890-11894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Camilli, A., C. R. Paynton, and D. A. Portnoy. 1989. Intracellular methicillin selection of Listeria monocytogenes mutants unable to replicate in a macrophage cell line. Proc. Natl. Acad. Sci. USA 865522-5526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Camilli, A., L. G. Tilney, and D. A. Portnoy. 1993. Dual roles of plcA in Listeria monocytogenes pathogenesis. Mol. Microbiol. 8143-157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chatterjee, S. S., H. Hossain, S. Otten, C. Kuenne, K. Kuchmina, S. Machata, E. Domann, T. Chakraborty, and T. Hain. 2006. Intracellular gene expression profile of Listeria monocytogenes. Infect. Immun. 741323-1338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen, Y., L. Caruso, B. McClane, D. Fisher, and P. Gupta. 2007. Disruption of a toxin gene by introduction of a foreign gene into the chromosome of Clostridium perfringens using targetron-induced mutagenesis. Plasmid 58182-189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cossart, P., M. F. Vincente, J. Mengaud, F. Baquero, J. C. Perez-Diaz, and P. Berche. 1989. Listeriolysin O is essential for virulence of Listeria monocytogenes: direct evidence obtained by gene complementation. Infect. Immun. 573629-3636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Decatur, A. L., and D. A. Portnoy. 2000. A PEST-like sequence in listeriolysin O essential for Listeria monocytogenes pathogenicity. Science 290992-995. [DOI] [PubMed] [Google Scholar]
- 11.Domann, E., J. Wehland, M. Rohde, S. Pistor, M. Hartl, W. Goebel, M. Leimeister-Wachter, M. Wuenscher, and T. Chakraborty. 1992. A novel bacterial gene in Listeria monocytogenes required for host cell microfilament interaction with homology to the proline-rich region of vinculin. EMBO J. 111981-1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Drevets, D. A., and M. S. Bronze. 2008. Listeria monocytogenes: epidemiology, human disease, and mechanisms of brain invasion. FEMS Immunol. Med. Microbiol. 53151-165. [DOI] [PubMed] [Google Scholar]
- 13.Drouault, S., J. Anba, S. Bonneau, A. Bolotin, S. D. Ehrlich, and P. Renault. 2002. The peptidyl-prolyl isomerase motif is lacking in PmpA, the PrsA-like protein involved in the secretion machinery of Lactococcus lactis. Appl. Environ. Microbiol. 683932-3942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Freitag, N. E. 2000. Genetic tools for use with Listeria monocytogenes, p. 488-498. In V. A. Fischetti, R. P. Novick, J. J. Ferretti, D. A. Portnoy, and J. I. Rood (ed.), Gram-positive pathogens. ASM Press, Washington, DC.
- 15.Freitag, N. E., and D. A. Portnoy. 1994. Dual promoters of the Listeria monocytogenes prfA transcriptional activator appear essential in vitro but are redundant in vivo. Mol. Microbiol. 12845-853. [DOI] [PubMed] [Google Scholar]
- 16.Freitag, N. E., P. Youngman, and D. A. Portnoy. 1992. Transcriptional activation of the Listeria monocytogenes hemolysin gene in Bacillus subtilis. J. Bacteriol. 1741293-1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gaillard, J. L., P. Berche, C. Frehel, E. Gouin, and P. Cossart. 1991. Entry of L. monocytogenes into cells is mediated by internalin, a repeat protein reminiscent of surface antigens from gram-positive cocci. Cell 651127-1141. [DOI] [PubMed] [Google Scholar]
- 18.Gaillard, J.-L., P. Berche, J. Mounier, S. Richard, and P. Sansonetti. 1987. In vitro model of penetration and intracellular growth of Listeria monocytogenes in the human enterocyte-like cell line Caco-2. Infect. Immun. 552822-2829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Glomski, I. J., A. L. Decatur, and D. A. Portnoy. 2003. Listeria monocytogenes mutants that fail to compartmentalize listerolysin O activity are cytotoxic, avirulent, and unable to evade host extracellular defenses. Infect. Immun. 716754-6765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Greiffenberg, L., W. Goebel, K. S. Kim, I. Weiglein, A. Bubert, F. Engelbrecht, M. Stins, and M. Kuhn. 1998. Interaction of Listeria monocytogenes with human brain microvascular endothelial cells: InlB-dependent invasion, long-term intracellular growth, and spread from macrophages to endothelial cells. Infect. Immun. 665260-5267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gründling, A., M. D. Gonzalez, and D. E. Higgins. 2003. Requirement of the Listeria monocytogenes broad-range phospholipase PC-PLC during infection of human epithelial cells. J. Bacteriol. 1856295-6307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Harman, J. G., K. McKenney, and A. Peterkofsky. 1986. Structure-function analysis of three cAMP-independent forms of the cAMP receptor protein. J. Biol. Chem. 26116332-16339. [PubMed] [Google Scholar]
- 23.Hyyrylainen, H. L., M. Sarvas, and V. P. Kontinen. 2005. Transcriptome analysis of the secretion stress response of Bacillus subtilis. Appl. Microbiol. Biotechnol. 67389-396. [DOI] [PubMed] [Google Scholar]
- 24.Jacobs, M., J. B. Andersen, V. Kontinen, and M. Sarvas. 1993. Bacillus subtilis PrsA is required in vivo as an extracytoplasmic chaperone for secretion of active enzymes synthesized either with or without pro-sequences. Mol. Microbiol. 8957-966. [DOI] [PubMed] [Google Scholar]
- 25.Johansson, J., P. Mandin, A. Renzoni, C. Chiaruttini, M. Springer, and P. Cossart. 2002. An RNA thermosensor controls expression of virulence genes in Listeria monocytogenes. Cell 110551. [DOI] [PubMed] [Google Scholar]
- 26.Jones, A. L., R. H. V. Needham, and C. E. Rubens. 2003. The delta subunit of RNA polymerase is required for virulence of Streptococcus agalactiae. Infect. Immun. 714011-4017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jones, S., and D. A. Portnoy. 1994. Characterization of Listeria monocytogenes pathogenesis in a strain expressing perfringolysin O in place of listeriolysin O. Infect. Immun. 625608-5613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kingdon, G. C., and C. P. Sword. 1970. Biochemical and immunological effects of Listeria monocytogenes hemolysin. Infect. Immun. 1363-372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kocks, C., E. Gouin, M. Tabouret, P. Berche, H. Ohayon, and P. Cossart. 1992. L. monocytogenes-induced actin assembly requires the actA gene product, a surface protein. Cell 68521-531. [DOI] [PubMed] [Google Scholar]
- 30.Kocks, C., R. Hellio, P. Gounon, H. Ohayon, and P. Cossart. 1993. Polarized distribution of Listeria monocytogenes surface protein ActA at the site of directional actin assembly. J. Cell Sci. 105699-710. [DOI] [PubMed] [Google Scholar]
- 31.Kontinen, V. P., P. Saris, and M. Sarvas. 1991. A gene (prsA) of Bacillus subtilis involved in a novel, late stage of protein export. Mol. Microbiol. 51273-1283. [DOI] [PubMed] [Google Scholar]
- 32.Kontinen, V. P., and M. Sarvas. 1993. The PrsA lipoprotein is essential for protein secretion in Bacillus subtilis and sets a limit for high-level secretion. Mol. Microbiol. 8727-737. [DOI] [PubMed] [Google Scholar]
- 33.Kuhn, M., S. Kathariou, and W. Goebel. 1988. Hemolysin supports survival but not entry of the intracellular bacterium Listeria monocytogenes. Infect. Immun. 5679-82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lauer, P., M. Y. N. Chow, M. J. Loessner, D. A. Portnoy, and R. Calendar. 2002. Construction, characterization, and use of two Listeria monocytogenes site-specific phage integration vectors. J. Bacteriol. 1844177-4186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Leimeister-Wachter, M., C. Haffner, E. Domann, W. Goebel, and T. Chakraborty. 1990. Identification of a gene that positively regulates expression of listeriolysin, the major virulence factor of Listeria monocytogenes. Proc. Natl. Acad. Sci. USA 878336-8340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Leskela, S., E. Wahlstrom, V. P. Kontinen, and M. Sarvas. 1999. Lipid modification of prelipoproteins is dispensable for growth but essential for efficient protein secretion in Bacillus subtilis: characterization of the Lgt gene. Mol. Microbiol. 311075-1085. [DOI] [PubMed] [Google Scholar]
- 37.Lingnau, A., E. Domann, M. Hudel, M. Bock, T. Nichterlein, J. Wehland, and T. Chakraborty. 1995. Expression of the Listeria monocytogenes EGD inlA and inlB genes, whose products mediate bacterial entry into tissue culture cell lines, by PrfA-dependent and -independent mechanisms. Infect. Immun. 633896-3903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ma, Y., A. E. Bryant, D. B. Salmi, S. M. Hayes-Schroer, E. McIndoo, M. J. Aldape, and D. L. Stevens. 2006. Identification and characterization of bicistronic speB and prsA gene expression in the group A streptococcus. J. Bacteriol. 1887626-7634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Marquis, H., V. Doshi, and D. A. Portnoy. 1995. The broad-range phospholipase C and a metalloprotease mediate listeriolysin O-independent escape of Listeria monocytogenes from a primary vacuole in human epithelial cells. Infect. Immun. 634531-4534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Marquis, H., H. Goldfine, and D. A. Portnoy. 1997. Proteolytic pathways of activation and degradation of a bacterial phospholipase C during intracellular infection by Listeria monocytogenes. J. Cell Biol. 1371381-1392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Marquis, H., and E. J. Hager. 2000. pH-regulated activation and release of a bacteria-associated phospholipase C during intracellular infection by Listeria monocytogenes. Mol. Microbiol. 35289-298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mead, P. S., L. Slutsker, V. Dietz, L. F. McCaig, J. S. Bresee, C. Shapiro, P. M. Griffin, and R. V. Tauxe. 1999. Food-related illness and death in the United States. Emerg. Infect. Dis. 5607-625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mengaud, J., S. Dramsi, E. Gouin, J. A. Vazquez-Boland, G. Milon, and P. Cossart. 1991. Pleiotropic control of Listeria monocytogenes virulence factors by a gene that is autoregulated. Mol. Microbiol. 52273-2283. [DOI] [PubMed] [Google Scholar]
- 44.Milohanic, E., P. Glaser, J. Y. Coppee, L. Frangeul, Y. Vega, J. A. Vazquez-Boland, F. Kunst, P. Cossart, and C. Buchrieser. 2003. Transcriptome analysis of Listeria monocytogenes identifies three groups of genes differently regulated by PrfA. Mol. Microbiol. 471613-1625. [DOI] [PubMed] [Google Scholar]
- 45.Miner, M. D., G. C. Port, and N. E. Freitag. 2008. Functional impact of mutational activation on the Listeria monocytogenes central virulence regulator PrfA. Microbiology 1543579-3589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mueller, K. J., and N. E. Freitag. 2005. Pleiotropic enhancement of bacterial pathogenesis resulting from the constitutive activation of the Listeria monocytogenes regulatory factor PrfA. Infect. Immun. 731917-1926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Port, G. C., and N. E. Freitag. 2007. Identification of novel Listeria monocytogenes secreted virulence factors following mutational activation of the central virulence regulator, PrfA. Infect. Immun. 755886-5897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Portnoy, D. A., P. S. Jacks, and D. J. Hinrichs. 1988. Role of hemolysin for the intracellular growth of Listeria monocytogenes. J. Exp. Med. 1671459-1471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sarvas, M., C. R. Harwood, S. Bron, and J. M. van Dijl. 2004. Post-translocational folding of secretory proteins in Gram-positive bacteria. Biochim. Biophys. Acta 1694311-327. [DOI] [PubMed] [Google Scholar]
- 50.Schnupf, P., J. Hofmann, J. Norseen, I. J. Glomski, H. Schwartzstein, and A. L. Decatur. 2006. Regulated translation of listeriolysin O controls virulence of Listeria monocytogenes. Mol. Microbiol. 61999-1012. [DOI] [PubMed] [Google Scholar]
- 51.Schnupf, P., and D. A. Portnoy. 2007. Listeriolysin O: a phagosome-specific lysin. Microbes Infect. 91176-1187. [DOI] [PubMed] [Google Scholar]
- 52.Schnupf, P., D. A. Portnoy, and A. L. Decatur. 2006. Phosphorylation, ubiquitination and degradation of listeriolysin O in mammalian cells: role of the PEST-like sequence. Cell. Microbiol. 8353-364. [DOI] [PubMed] [Google Scholar]
- 53.Schnupf, P., J. Zhou, A. Varshavsky, and D. A. Portnoy. 2007. Listeriolysin O secreted by Listeria monocytogenes into the host cell cytosol is degraded by the N-end rule pathway. Infect. Immun. 755135-5147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Scortti, M., H. J. Monzo, L. Lacharme-Lora, D. A. Lewis, and J. A. Vazquez-Boland. 2007. The PrfA virulence regulon. Microbes Infect. 91196-1207. [DOI] [PubMed] [Google Scholar]
- 55.Seveau, S., J. Pizarro-Cerda, and P. Cossart. 2007. Molecular mechanisms exploited by Listeria monocytogenes during host cell invasion. Microbes Infect. 91167-1175. [DOI] [PubMed] [Google Scholar]
- 56.Shao, L., S. Hu, Y. Yang, Y. Gu, J. Chen, Y. Yang, W. Jiang, and S. Yang. 2007. Targeted gene disruption by use of a group II intron (targetron) vector in Clostridium acetobutylicum. Cell Res. 17963-965. [DOI] [PubMed] [Google Scholar]
- 57.Sheehan, B., A. Klarsfeld, T. Msadek, and P. Cossart. 1996. A single substitution in the putative helix-turn-helix motif of the pleiotropic activator PrfA attenuates Listeria monocytogenes virulence. Mol. Microbiol. 20785-797. [DOI] [PubMed] [Google Scholar]
- 58.Shen, A., and D. E. Higgins. 2005. The 5′ untranslated region-mediated enhancement of intracellular listeriolysin O production is required for Listeria monocytogenes pathogenicity. Mol. Microbiol. 571460-1473. [DOI] [PubMed] [Google Scholar]
- 59.Shetron-Rama, L. M., H. Marquis, H. G. A. Bouwer, and N. E. Freitag. 2002. Intracellular induction of Listeria monocytogenes actA expression. Infect. Immun. 701087-1096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Shetron-Rama, L. M., K. Mueller, J. M. Bravo, H. G. Bouwer, S. S. Way, and N. E. Freitag. 2003. Isolation of Listeria monocytogenes mutants with high-level in vitro expression of host cytosol-induced gene products. Mol. Microbiol. 481537-1551. [DOI] [PubMed] [Google Scholar]
- 61.Smith, G. A., H. Marquis, S. Jones, N. C. Johnston, D. A. Portnoy, and H. Goldfine. 1995. The two distinct phospholipases C of Listeria monocytogenes have overlapping roles in escape from a vacuole and cell-to-cell spread. Infect. Immun. 634231-4237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Smith, K., and P. Youngman. 1992. Use of a new integrational vector to investigate compartment-specific expression of the Bacillus subtilis spoIIM gene. Biochimie 74705-711. [DOI] [PubMed] [Google Scholar]
- 63.Snyder, A., and H. Marquis. 2003. Restricted translocation across the cell wall regulates secretion of the broad-range phospholipase C of Listeria monocytogenes. J. Bacteriol. 1855953-5958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sun, A. N., A. Camilli, and D. A. Portnoy. 1990. Isolation of Listeria monocytogenes small-plaque mutants defective for intracellular growth and cell-to-cell spread. Infect. Immun. 583770-3778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Swaminathan, B., and P. Gerner-Smidt. 2007. The epidemiology of human listeriosis. Microbes Infect. 91236-1243. [DOI] [PubMed] [Google Scholar]
- 66.Tan, G. S., P. Kelly, J. Kim, and R. M. Wartell. 1991. Comparison of cAMP receptor protein (CRP) and a cAMP-independent form of CRP by Raman spectroscopy and DNA binding. Biochemistry 305076-5080. [DOI] [PubMed] [Google Scholar]
- 67.Theriot, J. A., T. J. Mitchison, L. G. Tilney, and D. A. Portnoy. 1992. The rate of actin-based motility of intracellular Listeria monocytogenes equals the rate of actin polymerization. Nature 357257-260. [DOI] [PubMed] [Google Scholar]
- 68.Tilney, L. G., P. S. Connelly, and D. A. Portnoy. 1990. The nucleation of actin filaments by the bacterial intracellular pathogen, Listeria monocytogenes. J. Cell Biol. 1112979-2988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Tossavainen, H., P. Permi, S. L. Purhonen, M. Sarvas, I. Kilpelainen, and R. Seppala. 2006. NMR solution structure and characterization of substrate binding site of the PPIase domain of PrsA protein from Bacillus subtilis. FEBS Lett. 5801822-1826. [DOI] [PubMed] [Google Scholar]
- 70.Vazquez-Boland, J.-A., C. Kocks, S. Dramsi, H. Ohayon, C. Geoffroy, J. Mengaud, and P. Cossart. 1992. Nucleotide sequence of the lecithinase operon of Listeria monocytogenes and possible role of lecithinase in cell-to-cell spread. Infect. Immun. 60219-230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Vázquez-Boland, J. A., M. Kuhn, P. Berche, T. Chakraborty, G. Dominguez-Bernal, W. Goebel, B. González-Zorn, J. Wehland, and J. Kreft. 2001. Listeria pathogenesis and molecular virulence determinants. Clin. Microbiol. Rev. 14584-640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Vega, Y., M. Rauch, M. J. Banfield, S. Ermolaeva, M. Scortti, W. Goebel, and J. A. Vazquez-Boland. 2004. New Listeria monocytogenes prfA* mutants, transcriptional properties of PrfA* proteins and structure-function of the virulence regulator PrfA. Mol. Microbiol. 521553-1565. [DOI] [PubMed] [Google Scholar]
- 73.Vitikainen, M., I. Lappalainen, R. Seppala, H. Antelmann, H. Boer, S. Taira, H. Savilahti, M. Hecker, M. Vihinen, M. Sarvas, and V. P. Kontinen. 2004. Structure-function analysis of PrsA reveals roles for the parvulin-like and flanking N- and C-terminal domains in protein folding and secretion in Bacillus subtilis. J. Biol. Chem. 27919302-19314. [DOI] [PubMed] [Google Scholar]
- 74.Vitikainen, M., T. Pummi, U. Airaksinen, E. Wahlström, H. Wu, M. Sarvas, and V. P. Kontinen. 2001. Quantitation of the capacity of the secretion apparatus and requirement for PrsA in growth and secretion of α-amylase in Bacillus subtilis. J. Bacteriol. 1831881-1890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Wahlstrom, E., M. Vitikainen, V. P. Kontinen, and M. Sarvas. 2003. The extracytoplasmic folding factor PrsA is required for protein secretion only in the presence of the cell wall in Bacillus subtilis. Microbiology 149569-577. [DOI] [PubMed] [Google Scholar]
- 76.Williams, R. C., M. L. Rees, M. F. Jacobs, Z. Pragai, J. E. Thwaite, L. W. Baillie, P. T. Emmerson, and C. R. Harwood. 2003. Production of Bacillus anthracis protective antigen is dependent on the extracellular chaperone, PrsA. J. Biol. Chem. 27818056-18062. [DOI] [PubMed] [Google Scholar]
- 77.Wong, K. K., H. G. Bouwer, and N. E. Freitag. 2004. Evidence implicating the 5′ untranslated region of Listeria monocytogenes actA in the regulation of bacterial actin-based motility. Cell. Microbiol. 6155-166. [DOI] [PubMed] [Google Scholar]
- 78.Wong, K. K. Y., and N. E. Freitag. 2004. A novel mutation within the central Listeria monocytogenes regulator PrfA that results in constitutive expression of virulence gene products. J. Bacteriol. 1866265-6276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Yao, J., J. Zhong, Y. Fang, E. Geisinger, R. P. Novick, and A. M. Lambowitz. 2006. Use of targetrons to disrupt essential and nonessential genes in Staphylococcus aureus reveals temperature sensitivity of Ll.LtrB group II intron splicing. RNA 121271-1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Yeung, P. S. M., Y. Na, A. J. Kreuder, and H. Marquis. 2007. Compartmentalization of the broad-range phospholipase C activity to the spreading vacuole is critical for Listeria monocytogenes virulence. Infect. Immun. 7544-51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Yeung, P. S. M., N. Zagorski, and H. Marquis. 2005. The metalloprotease of Listeria monocytogenes controls cell wall translocation of the broad-range phospholipase C. J. Bacteriol. 1872601-2608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Zemansky, J., B. C. Kline, J. J. Woodward, J. H. Leber, H. Marquis, and D. A. Portnoy. 17 April 2009. Development of a mariner based transposon and identification of Listeria monocytogenes determinants, including the peptidyl-prolyl isomerase PrsA2, that contribute to its hemolytic phenotype. J. Bacteriol. [Epub ahead of print.] doi: 10.1128/JB.00016-09. [DOI] [PMC free article] [PubMed]
- 83.Zhong, J., M. Karberg, and A. M. Lambowitz. 2003. Targeted and random bacterial gene disruption using a group II intron (targetron) vector containing a retrotransposition-activated selectable marker. Nucleic Acids Res. 311656-1664. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.