

Abstract
Epstein-Barr virus (EBV) is associated with malignant diseases of lymphoid and epithelial cell origin. The tropism of EBV is due to B-cell-restricted expression of CD21, the major receptor molecule for the virus. However, efficient infection of CD21− epithelial cells can be achieved via transfer from EBV-coated B cells. We compare and contrast here the early events following in vitro infection of primary B cells and epithelial cells. Using sensitive, quantitative reverse transcription-PCR assays for several latent and lytic transcripts and two-color immunofluorescence staining to analyze expression at the single cell level, we confirmed and extended previous reports indicating that the two cell types support different patterns of transcription. Furthermore, whereas infection of B cells with one or two copies of EBV resulted in rapid amplification of the viral genome to >20 copies per cell, such amplification was not normally observed after infection of primary epithelial cells or undifferentiated epithelial lines. In epithelial cells, EBNA1 expression was detected in only ca. 40% of EBER+ cells, and the EBV genome was subsequently lost during prolonged culture. One exception was that infection of AGS, a gastric carcinoma line, resulted in maintenance of EBNA1 expression and amplification of the EBV episome. In contrast to B cells, where amplification of the EBV episome occurred even with a replication-defective BZLF1-knockout virus, amplification in AGS cells was dependent upon early lytic cycle gene expression. These data highlight the influence of the host cell on the outcome of EBV infection with regard to genome expression, amplification, and maintenance.
Epstein-Barr virus (EBV) is a gammaherpesvirus that asymptomatically infects the majority of the human population during childhood and then persists for the lifetime of the host. EBV persistence requires the establishment of a latent infection within the resting memory B-cell compartment (1), where the viral gene expression is largely limited to the noncoding EBV-encoded RNA (EBER) and BamA rightward transcript (BART) RNAs, with no expression of protein-coding transcripts; a type of latency sometimes termed latency 0, which allows the virus to evade host immune responses (44). Reactivation from this latent state can be nonproductive (i.e., not producing new virus particles) with the additional expression of up to six nuclear antigens (EBNAs) and a number of membrane proteins (LMP1, LMP2, and vBcl-2), concomitant with growth transformation of the cells. The full range of latent gene expression, termed latency III, can be observed in tonsillar B cells during primary infection manifest clinically as infectious mononucleosis (29) in posttransplant lymphoproliferative disease (67) and in lymphoblastoid cell lines (LCL) established after infection in vitro of primary B cells with EBV (46). Other forms of latency, spanning a spectrum between latency 0 and latency III, are commonly associated with EBV-associated malignancies of both lymphoid and epithelial origin (44).
As with other herpesviruses, EBV-infected cells may in some circumstances reactivate to the lytic virus replication cycle, with the expression of more than 80 viral genes and ultimately the release of infectious virus progeny. In some pathological lesions, lytic infection of B cells (35) or epithelial cells (39, 67) is readily demonstrated. In healthy infected individuals, identification of lytically infected cells has not been definitively demonstrated, although the detection of virus in throat washings and the consistent presence of serum antibodies to lytic cycle antigens are clear indications that reactivation of lytic cycle is a feature of persistent infection in healthy carriers.
The EBV-host cell interactions have been extensively studied in the context of different latency states of the virus in malignant cells, in lymphoproliferative disease of immunosuppressed transplant patients, and in the normal B-cell subsets of infectious mononucleosis patients and healthy infected individuals. Furthermore, the ease with which primary B lymphocytes can be infected with EBV via the EBV-receptor CR2 (CD21) and growth transformed into LCL has provided an amenable model for studying normal EBV-B-cell interactions. LCL exhibit the full latency III program, where all six EBNAs are produced from a large primary transcript driven from one of two upstream promoters, Cp or Wp. However, after infection of B cells in vitro, the first detectable event is expression of EBNA-LP and EBNA2 only from the Wp promoter within hours of infection, which is followed from day 2 onward by a decline in Wp activity and an increase in Cp-initiated transcripts encoding EBNA1, -3A, -3B, and -3C in addition to EBNA-LP and EBNA2. The LMP1, LMP2, and the noncoding EBER transcripts appear relatively late, between 2 and 4 days after infection. The Qp promoter, which is used to transcribe EBNA1 only in the more restricted latency I and latency II states (e.g., in Burkitt's lymphoma and Hodgkin's disease, respectively) is not normally active during the establishment of LCL from normal B cells in culture. Interestingly, after infection in vitro, every B cell that gains a nuclear viral genome not only activates the latency III growth transformation program but then also amplifies the latent genome to an average of 40 to 50 copies per cell in a poorly defined process that is independent of lytic replication (56).
In contrast to B cells, the interaction of EBV with epithelial cells is relatively poorly understood. Early studies showed the presence of the virus in desquamated oropharyngeal epithelial cells of patients with acute infectious mononucleosis (54), which was interpreted to indicate that infection and replication of the virus normally occurs within epithelial cells prior to infection of B cells. Indeed, recent data show the presence of EBV DNA, mRNA, and protein in a small subset of tonsillar epithelial cells from healthy, EBV-positive donors (41). However, some more recent data question the evidence for the replication of EBV, or indeed the presence of the viral genome at all, within the oropharyngeal epithelia of either the asymptomatic host or in patients with infectious mononucleosis (22, 27, 37). Nevertheless, EBV replication has been reported in normal tongue epithelium, suggesting the tongue may be a source of EBV secreted into saliva (60). EBV lytic replication is indisputably observed in epithelial cells in oral hairy leukoplakia, a nonmalignant epithelial lesion occurring in the tongues of individuals infected with human immunodeficiency virus. Oral hairy leukoplakia is characterized by productive EBV replication within the upper-layer epithelial cells undergoing terminal differentiation, apoptosis, and desquamation, strongly suggesting that EBV replication in vivo is dependent upon the differentiation state of the epithelial cell (2, 61, 63, 67).
The evidence to date suggests that true latency in epithelial cells is only observed in EBV-positive epithelial cell tumors such as nasopharyngeal carcinoma (NPC) and a subset of gastric carcinoma (4, 55). NPC is a tumor of undifferentiated epithelial cells, has a 100% association with EBV, and typically displays a latency II pattern of viral gene expression, i.e., expression of EBERs and BARTs RNA, Qp-initiated EBNA1, LMP2, and, in a subset of tumors, LMP1. These tumors contain high numbers (>40 per cell) of latent viral episomes (53); lytic replication rarely seen, only occurs in a small subset of tumor cells, and is usually abortive (9, 17, 33, 38, 68).
Examination of early events following in vitro infection of primary epithelial cells and cell lines has previously been difficult because these CD21− cells are generally refractory to infection with EBV. Gene transfection of CD21 into epithelial lines permits efficient infection of these cells, and apparently results in abortive lytic infection (28, 30). Some rare clones retaining EBV may be isolated with difficulty by limiting dilution cloning (28) or the use of recombinant EBV allows drug resistance selection of EBV-positive clones (25). EBV-positive clones emerging from such experiments typically displayed a latency I or II type gene expression, similar to the EBV expression in the malignant cells of undifferentiated NPC (4).
Recently, we developed a system where EBV is efficiently delivered into epithelial cells by a more physiological process involving transfer from the surface of virus-loaded resting B cells (52). EBV binding to B cells is efficient by virtue of the virally encoded gp350 interacting with cell surface CD21, the EBV receptor. However, binding of EBV to epithelial cells, which lack expression of CD21, is very inefficient. We have previously shown that a considerable enhancement of epithelial infection can be achieved by using a gp350-knockout recombinant virus and that transfer of B-cell-bound EBV to epithelial cells also appears to overcome this barrier. This suggests not only that gp350 inhibits the binding of EBV to epithelial cells but also that the initial binding of EBV to B cells via the gp350-CD21 interaction may expose further ligands on the viral membrane, enabling an increase in EBV binding and entry into epithelial cells. Although a definitive explanation for the phenomenon of transfer infection remains to be resolved, it is likely that it has relevance for infection of epithelial cells in vivo.
The ability to achieve efficient infection of all epithelial cell lines tested allows early infection events to be carefully investigated qualitatively and quantitatively in sufficiently high numbers of cells without reverting to any drug selection to select rare infected clones able to maintain the EBV genome. We report here the results of such an investigation that was designed to address a number of fundamental questions: (i) when EBV enters the epithelial cells, how much reaches the nucleus, and how many of these cells then fire EBV gene expression; (ii) which EBV genes are expressed and with what kinetics; and (iii) how efficient is EBV genome retention in the epithelial cell and does the episomal virus genome amplify as it does in B cells?
MATERIALS AND METHODS
Cells and cell lines.
Primary B cells were isolated from peripheral blood as described previously (51). Cultures of primary tonsillar epithelial cells were generated by finely mincing epithelial cells from freshly excised tonsils and digesting them in a collagenase-dispase solution (20,000 U of collagenase, 120 U of dispase, 1% trypsin inhibitor, and 1.25% bovine serum albumin; Sigma) for 1 h at 37°C with constant agitation. The digested cells were washed twice in phosphate-buffered saline (PBS), and the collagenase-dispase solution was discarded. The cells were resuspended in 10 ml of serum-free keratinocyte growth medium (Invitrogen/Gibco) containing 1% penicillin-streptomycin (Sigma) and 0.2% amphotericin B (Fungizone; Sigma). Cultures of the established epithelial cell lines AdAH, SVK, and HeLa were grown in RPMI 1640 medium supplemented with 10% fetal calf serum. The epithelial cell line AGS was grown in Ham F-12 medium supplemented with 10% fetal calf serum.
Virus preparations and quantitation by EBV genome load assay.
Preparations of wild-type (EBV 2089), EBNA1 knockout (KO) (24), LMP1 KO (12), BZLF1 KO (16), and EBER KO (kindly donated by H.-J. Delecluse) viruses were made from 293 cells carrying a recombinant B95.8 EBV genome (10), with gp110 levels optimized by transfection of a BALF4 expression plasmid (36). Cell-free virus preparations were overlaid on a 50% solution of Optiprep (Axis Shield) buffered with 0.85% (wt/vol) NaCl-60 mM HEPES-NaOH (pH 7.4) and centrifuged in a SW40 rotor at 160,000 × g for 2 h to band the virus sharply at the Optiprep interface. All of the supernatant was removed except for a volume equal to the volume of Optiprep, the contents were then mixed and transferred to a 4.9-ml vertical rotor Beckman tube and centrifuged at 350,000 × g for 2.4 h in a SW60Ti vertical rotor to band the virus in a self-generated gradient of Optiprep, with controlled deceleration. The visible virus band was harvested with a syringe and metal cannula, and the recovered virus was quantitated by a quantitative PCR (Q-PCR) assay amplifying the BALF5 gene (51). The virus recovered comprises of concentrated encapsidated, enveloped virus particles, as observed by electron microscopy (data not shown), and the Q-PCR value was used to determine the multiplicity of infection (MOI).
Transfer infection.
Donor primary B cells were irradiated (5,000 rads) and then incubated with EBV at an MOI of 100 genomes for 3 h at 4°C, and the cells were washed to remove unbound virus and then incubated for 18 h at 37°C to ensure that the B cells were competent to transfer. An aliquot of 106 virus-primed B cells was added to each well of a 24-well plate seeded with 2 × 105 epithelial cells. The plates were gently centrifuged at 130 × g for 2 min to ensure even coverage of the epithelial cells with B cells. After 2 h of coculture, the donor B cells were removed from the acceptor cells by thorough washing and observed microscopically. Transfer infection was assayed 72 h after the initiation of coculture as previously described (52).
Virus transfer and internalization assays.
Virus binding to cells and virus internalized within cells was determined by using a Q-PCR assay of the EBV genome load (51); the assay also quantitates the copies of a unique cellular gene, β2m, and all results are expressed as EBV genome copies per cell. To quantitate virus binding to B cells, freshly isolated B cells were exposed to EBV preparations at an MOI of 50 for 3 h at 4°C; the cells were then washed well to remove unbound virus, and an aliquot of cells immediately subjected to the Q-PCR assay described above. To assay virus internalization, a second aliquot of virus-infected cells was cultured for 16 to 18 h at 37°C and then treated with chymotrypsin to remove all virions from the cell surface (51) before being subjected to Q-PCR assay to determine the mean number of internalized genomes per cell. To quantitate virus binding to epithelial cells, cultures of adherent epithelial cells were exposed for 1 h at 37°C to virus-loaded B cells (see above), and then B cells were washed off as described previously (52) before the epithelial cells were harvested by brief trypsin treatment (which does not to affect surface-bound virions) and washing. Bound and internalized virus was quantitated by Q-PCR as described above for B cells.
FISH.
Viral genomes were detected by fluorescence in situ hybridization (FISH) with an EBV cosmid (cm302-21) probe as described previously (11, 42).
Quantitative reverse transcription-PCR (RT-PCR) for EBV transcripts.
Total RNA was isolated from infected B cells and epithelial cells at 1 to 5 days postinfection, using a Nucleospin II RNA purification kit (Macherey-Nagel) according to the manufacturer's instructions. One microgram of RNA was further digested with DNase (DNA-Free; Ambion) according to the manufacturer's instructions.
For quantitative RT-PCR assay of EBERs, 400 ng of denatured RNA was reverse transcribed in a 20-μl reaction volume with AMT-RT (Roche) for 1 h at 42°C using a combination of 3′ EBER1 (5′-GAC CAC CAG CTG GTA C)-, EBER2 (5′-GGA CAA GCC GAA TAC C)-, and GAPDH (glyceraldehyde-3-phosphate dehydrogenase) (3)-specific primers. The reaction was terminated by heat inactivation. A 25-ng portion of cDNA was added to each quantitative RT-PCR to quantify EBER1 using primers (5′-TGC TAG GGA GGA GAC GTG TGT and 3′-TGA CCG AAG ACG GCA GAA AG) with a 5′FAM and 3′TAMRA-labeled probe (5′-AGA CAA CCA CAG ACA CCG TCC TCA CCA) and to quantify EBER2 using primers (5′-AAC GCT CAG TGC GGT GCT A and 3′-GAA TCC TGA CTT GCA AAT GCT CTA) and probe (5′-CGA CCC GAG GTC AAG TCC CGG) for unspliced transcripts.
Quantitative RT-PCRs for other EBV transcripts were performed using a pool of 3′ EBV gene-specific primers as previously reported (3). Briefly, 20 ng of cDNA was added to each quantitative RT-PCR to quantify Wp- and Cp-initiated EBNA mRNAs; YUK-spliced EBNA1 mRNA; Qp-initiated QUK spliced EBNA1 mRNA; and LMP1, LMP2a, and BZLF1 mRNAs.
EBER ISH.
After transfer infection of epithelial cells (see above) EBER in situ hybridization (ISH) was performed by using an EBER ISH kit (Dako) according to manufacturer's instructions. EBER-positive cells were detected by light microscopy using an alkaline phosphatase-conjugated antibody to an EBER PNA probe in the presence of BCIP (5-bromo-4-chloro-3-indolylphosphate) and nitroblue tetrazolium.
Immunofluorescence detection of EBV proteins.
Epithelial cells were plated onto chamber slides and infected by transfer infection. After 1 to 5 days, the epithelial cells were washed, fixed in 2% paraformaldehyde for 20 min at room temperature, and permeabilized in 0.5% Triton X-100 for 5 min at room temperature. The cells were stained for EBNA1 (R4; kindly donated by Laurie Frappier), LMP1 (CS1-4 [45]), BZLF1 (BZ1 [67]), diffuse early antigen (EA-D; Chemicon), gp350 (72A1 [21]), and VCA (Chemicon). All nonconjugated mouse monoclonal antibodies were labeled with Alexa Fluor 488 or 594 (Zenon kit; Invitrogen) and used immediately postlabeling.
Gardella gel analysis.
Virus particles (5 × 107) from EBV 2089 preparations were precipitated in polyethylene glycol 1500 (PEG), incubated for 3 h at 4°C, and pelleted at 11,000 × g for 20 min, and the virus pellet was resuspended in 10 μl of PBS. One-million green fluorescent protein (GFP)-positive AGS, Raji, and LCL cells were washed and resuspended in 10 μl of PBS. The virus pellet and cells were mixed with 10 μl of 15% Ficoll and then loaded onto a 0.8% Tris-acetate EDTA agarose Gardella gel, lysed “in well” with 20% sodium dodecyl sulfate, and run as previously described for 18 h at 4°C (19). The gel was stained with ethidium bromide for 30 min with gentle rocking and then visualized. The DNA in the gel was depurinated in 0.25 M HCl for 30 min, neutralized in sterile distilled water for 30 min, and then denatured in 0.5 M NaOH-1.5 M NaCl for 30 min. The DNA was then transferred to nitrocellulose membrane (Amersham Hybond N+) by Southern blotting and hybridized in Church buffer. Then, 10 ng of EBV BamW probe was labeled by nick repair with [α-32P]dCTP according to the manufacturer's instructions (Invitrogen) and hybridized to the membrane for 18 h. The membrane was washed five times and exposed to film for 24 h.
RESULTS
Viral entry into epithelial cells.
The first set of experiments sought to compare epithelial and B-cell infections by quantitating the amount of virus binding to the cell surface and its subsequent internalization. The experiments in Fig. 1A show the results of Q-PCR assays ± the standard deviation of four independent epithelial cell experiments and six B-cell experiments measuring the amount of virus initially bound to the cells and the amount of virus that had entered the cells after incubation at 37°C. Thus, B cells exposed to purified virus at an MOI of 50 virus genome copies per cell for 3 h at 4°C bound a mean of about 45 copies per cell (Fig. 1A, left). After 18 h in culture at 37°C, stripping of the virus-loaded B-cell surface by chymotrypsin showed that by that time a mean of 13 virus genomes per cell had been internalized (Fig. 1A, left panel). To analyze infection of epithelial cells, virus-loaded B cells were cocultured for 1 h with either adherent primary epithelial cells or cell lines at a B-cell/epithelial-cell ratio of 5:1, and then the B cells were washed off. In Fig. 1A (middle panel) shows representative results obtained after transfer infection of the AdAH epithelial cell line and primary epithelial cells, which acquired a mean of 15 genomes per cell, of which about 10 genomes per cell had internalized and could not be removed by chymotrypsin. However, many of these genomes did not reach the nucleus, as observed by immunofluorescence for viral capsid (data not shown). Direct exposure of the same epithelial cells to the purified virus preparation alone, again at an MOI of 50 virus genomes per cell, produced low levels of virus binding and internalization, up to a maximum of four particles bound per primary epithelial cell (Fig. 1A, right) and four particles per AdAH cell, although AGS could bind up to twelve particles per cell (data not shown).
FIG. 1.

EBV entry into B cells and primary epithelial cells. Virus binding and internalization assays on primary B cells and on epithelial cells infected by transfer infection and direct infection using purified infectious EBV particles at an MOI of 50. (A) Q-PCR assays were used to measure the amount of virus bound to the cell surface and the amount of virus that had entered after incubation at 37°C for 24 h. The results are the means ± the standard deviations of six independent B-cell infection experiments and four independent primary epithelial cell experiments. (B) Detection of viral genomes by FISH. Primary B cells and primary epithelial cells were exposed to purified EBV as described above, and extracellular or internalized genomes were detected by green fluorescence. Nuclei are identified by DAPI staining. (C) Distribution of EBV genome numbers detectable per primary B-cell, primary epithelial cell, and AdAH epithelial cell line. The results are expressed as the percentage of cells with 1 to 18 genomes per nucleus.
Visualizing cell surface virions by FISH for the viral genome showed that essentially all B cells had bound virus particles after incubation with EBV at an MOI of 50, whereas with epithelial cells ca. 25% of cells typically acquired the genome following transfer infection. Shortly after infection, virus particles were detected by FISH at the cell surface (Fig. 1B, left panel), whereas after incubation of the cells at 37°C, virus genomes were detected intracellularly in the nucleus (Fig. 1B, middle and right panels). Delivery of the virus genome to the nuclei of infected cells at 48 h postinfection was quantified by enumerating FISH signals in individual nuclei from B-cell and epithelial-cell cultures in order to analyze the distribution of viral genome copies at the single-cell level (Fig. 1C). In the primary B-cell, primary epithelial cell, and AdAH cell types, most nuclei had acquired between 1 and 4 genomes, with progressively fewer cells showing higher loads up to a maximum of 18 genomes per nucleus. Similar results were obtained with other epithelial target cells, namely, a simian virus 40-transformed keratinocyte line, SVK, and the semipermissive AGS line. Epithelial cells exposed to virus via coated B cells showed evidence of at least 10-fold greater virus genome entry than cells infected with cell-free virus (data not shown).
Quantitative RT-PCR analysis of EBV gene expression in infected B cells versus infected epithelial cells.
In the next series of experiments, we compared EBV gene expression at the mRNA level in freshly infected B cells and epithelial cells. Freshly isolated primary B cells were infected with wild-type EBV at an MOI of 50; primary epithelial and AdAH cells were infected by transfer infection as in the previous set of experiments. RNA was harvested from the replicate cultures of infected cells at 24-h intervals over 5 days postinfection, and EBV transcripts were measured by using quantitative RT-PCR and normalized against GAPDH transcripts as previously described (3). Figure 2 shows the results from a representative experiment. The first transcripts observed after infection of epithelial cells are the EBERs, which appear about 4 to 6 h postinfection and are transcribed at higher levels than that seen in the primary B cells for the first 3 days postinfection. EBER expression in the B cells is initiated about 24 h postinfection but remains very low until the cells start replicating at 4 to 5 days postinfection. In B cells, the first transcripts are initiated from Wp, followed by Cp-initiated Y-U-K transcripts. In primary and epithelial cell lines, however, very little Wp/Cp-initiated transcription was observed and consequently little EBNA2 or EBNA1 from the Y-U-K splice; EBNA1 was instead transcribed from the Qp-initiated Q-U-K splice by 24 h postinfection and was followed by transcription of LMP1, LMP2a, and BZLF1, which was maximal on average by 2 to 3 days postinfection and gradually dropped over time. The BART transcripts were highly expressed after infection of epithelial cell lines, peaking at days 2 or 3 and then declining to lower levels at days 4 to 5; in contrast, infection of B cells resulted in lower levels of BARTs which peaked later, at day 4 or 5. Interestingly, only very low levels of BARTs were detected in the primary epithelial cells.
FIG. 2.

Kinetics of viral transcription after infection of B cells and epithelial cells. EBV transcripts were assayed by quantitative RT-PCR after infection of primary B cells, primary epithelial cells, and the AdAH epithelial cell line. The results for BARTs, EBERs, Wp, Y-U-K EBNA1, LMP1, and LMP2 transcripts are displayed as gene expression relative to a latency III, Wp-using reference LCL, X50-7; Cp transcripts are displayed relative to a latency III, Cp-using reference LCL, Oku-LCL; Q-U-K EBNA1 transcripts are expressed relative to a reference latency I Burkitt's lymphoma line, Rael; and BZLF1 transcripts are expressed relative to a reference LCL, Chege-LCL, containing ca. 2% cells in lytic cycle.
Viral gene expression at the single cell level.
The quantitative RT-PCR data clearly showed that the pattern of transcription of the EBV genome differed markedly at the population level in a cell-type-dependent manner. We next sought to determine what viral genes are expressed at the single-cell level and whether there is heterogeneity within a culture of infected cells, as there is, for example, in the number of viral genomes in the nucleus (Fig. 1C). We have previously shown that that every primary B cell receiving a viral genome in its nucleus will express EBNA2 and go on to express the full set of growth transforming genes (51). However, we do not know whether every genome within the nucleus of an epithelial cell will fire or what it will express in the first few days of infection.
Figure 3A shows representative photomicrographs from a typical experiment using the AdAH epithelial cell line. Cells were observed for GFP expression as a marker of infection with the recombinant 2089 EBV, and for EBER expression by ISH at 24 h postinfection. Up to 48% of the cells expressed GFP and the EBERs over the first 6 days postinfection. Therefore, most if not all epithelial cells receiving a viral genome in their nucleus fired to express EBERs. This was confirmed by sorting the GFP-expressing cells at 24 h postinfection and then performing ISH for EBERs (data not shown). These cells were also analyzed by immunofluorescence for the expression of the latency genes EBNA1 and LMP1, the immediate-early lytic BZLF1 protein, early antigen (EA), and the late gp350 antigen for 6 days postinfection (Fig. 3B and C). Example photomicrographs are shown to illustrate the results of immunofluorescence staining at 3 days postinfection, when mRNA transcription was maximal. Surprisingly, only 30% of the cells with a viral genome and expressing GFP and the EBERs made EBNA1, the genome maintenance protein, and less than 10% of cells made LMP1. About 15% of the cells entered the lytic cycle, expressing BZLF1 protein, but very few of these progressed to express early or late antigen, showing that the virus lytic cycle is abortive in the majority of BZLF1-expressing cells.
FIG. 3.

Viral gene expression at the single epithelial cell level. (A) Photomicrographs of phase-contrast, GFP fluorescence (upper panels) and EBER ISH (lower panel) of AdAH cultures at 72 h postinfection. GFP and EBER expression were quantified and are represented as the percent expression for an average experiment up to 6 days postinfection. (B) Photomicrographs of phase-contrast and GFP fluorescence of AdAH cells sorted for GFP expression 24 h postinfection. Cells were stained for expression of the latent gene products EBNA1 and LMP1 plus the lytic gene products BZLF1, early antigen EA, and the late antigen gp350. (C) Expression of the latent and lytic gene products was quantitated daily to 6 days postinfection from a population of 100% infected (GFP +ve) cells.
After the EBERs, EBNA1 was the most abundantly expressed of the EBV genes in freshly infected epithelial cell lines. However, EBNA1 was only expressed in 30% of all EBER-positive cells. This raised the question of to what extent EBV latent and lytic proteins were expressed in different subpopulations or were coexpressed in the same cells. Infected epithelial cells were, therefore, double stained to address this question. Epithelial cells were grown on eight-well chamber slides and, on each day for 6 days postinfection, replicate cultures were fixed and stained for the latent gene products EBNA1, LMP1, BZLF1, early antigen (EA-D), and gp350. Figure 4 shows representative photomicrographs of epithelial cells stained at day 3 postinfection. The results show that cells expressing LMP1, BZLF1, EA-D, or gp350 were always coexpressing EBNA1. There was, however, only a partial overlap of LMP1 and BZLF1 expression; this is consistent with the expression of LMP1 as a latent gene in a subpopulation of EBNA1-positive cells and as an early lytic cycle antigen in some BZLF1-positive cells. Together, the results in Fig. 2 to 4 show that by 3 days postinfection of epithelial cells, at least three types of gene expression are observed in different subpopulations: EBER+ only; latency I (EBER and EBNA1); latency II (EBER, EBNA1, and LMP1); and abortive lytic cycle (EBER, EBNA1, BZLF1, ±LMP1, ±EA-D).
FIG. 4.

Heterogeneous viral gene expression at the single-cell level. Phase-contrast and fluorescence images of EBV gene expression show heterogeneous gene expression in different subpopulations of AdAH epithelial cells: EBNA1 alone (latency I); EBNA1 and LMP1 (latency II); and EBNA1, BZLF1, ±LMP1, ±EA (abortive lytic).
Recombinant EBV gene KO-infected AdAH epithelial cells.
The predominance of EBER and EBNA1 expression with differential expression of LMP1 and BZLF1 during the early stages of epithelial cell infection suggests the EBERs or EBNA1 may be key regulators of EBV gene expression in early epithelial cell infection. We therefore used of a panel of recombinant viruses with individual gene knockouts to test whether EBERs or EBNA1 were essential for expression of downstream lytic or latent EBV genes.
Epithelial cells were infected by transfer infection with either wild-type, EBER1/2 KO, EBNA1 KO, LMP1 KO, or BZLF1 KO recombinant viruses. The cells were sorted for GFP expression at 18 h postinfection and then stained daily for EBNA1, LMP1, and BZLF1 for 5 days. Figure 5A shows the percentage of antigen-positive cells for the wild-type and KO virus infections. Infection with the EBER1/2 KO virus showed no difference in the expression of EBNA1, LMP1, or BZLF1 compared to that observed with the wild-type virus. In contrast, infection with the EBNA1 KO virus resulted in a substantial reduction in BZLF1 expression and a small increase in the pool of latent LMP1-positive cells. A similar reduction in the percentage of BZLF1-positive cells, together with a small increase in the percentage of latent EBNA1-positive cells, was also observed when AdAH cells were infected with the LMP1 KO virus. Infection with the BZLF1 KO virus resulted in a slight reduction in EBNA1-positive cells and a smaller reduction in LMP1-positive cells. These data, summarized in the Venn diagrams in Fig. 5B, suggest that the EBERs do not regulate the expression of the other EBV genes in this epithelial cell infection model but that EBNA1 and LMP1 are necessary for efficient entry into lytic cycle.
FIG. 5.

Recombinant EBV gene knockout infected epithelial cells. AdAH epithelial cells were infected with either wild-type, EBER1/2 KO, EBNA1 KO, LMP1 KO, or BZLF1 KO recombinant viruses. The cells were sorted for GFP expression at 18 h postinfection, and the cells were analyzed daily for the viral genome (FISH); for expression of EBER1/2 (ISH); and for expression of EBNA1, LMP1, and BZLF1 (immunofluorescence). The results are displayed as the percent expression of EBNA1, LMP1, and BZLF1 in 100% infected cells (GFP +ve) over 5 days postinfection (A) and as Venn diagrams to identify the subpopulations of cells expressing one or more EBV genes (B).
Maintenance of the viral genome.
After infection of primary resting B cells, every cell in which one viral genome enters the nucleus will fire the Wp promoter to express EBNA2 and EBNA-LP, followed by expression of the full growth transformation program and the generation of a continuously replicating lymphoblastoid cell line without loss of the viral genome. To examine viral genome maintenance in epithelial cells, we sorted GFP-positive (EBV genome-positive) AdAH cells at 24 h postinfection, cultured them for 50 days without hygromycin selection for the recombinant EBV genome, and then analyzed aliquots every few days for GFP expression, viral genome maintenance by FISH, EBER expression by ISH, and EBNA1 expression by immunofluorescence. AdAH cells were used as a model of epithelial cells for this set of experiments, since primary epithelial cells stop growing after a limited number of passages. As initially observed, all cells expressing GFP also contained the viral genome and expressed EBERs, although only 24 to 30% of the cells expressed EBNA1. After serial passage, viral genomes were rapidly lost from the infected cells, concomitant with the loss of GFP and EBERs. More than 80% of the cells lost their EBV genome within the first 15 days postinfection (Fig. 6), but after approximately 20 days the rate of genome, GFP, and EBER loss became much more gradual, with only a 5% loss over the next 30 days. The initial loss of EBNA1 expression was significantly slower than the loss of genome, GFP, and EBERs but, even after 50 days when the virus-positive population appeared to have stabilized, less than half of the genome-positive cells expressed EBNA1.
FIG. 6.
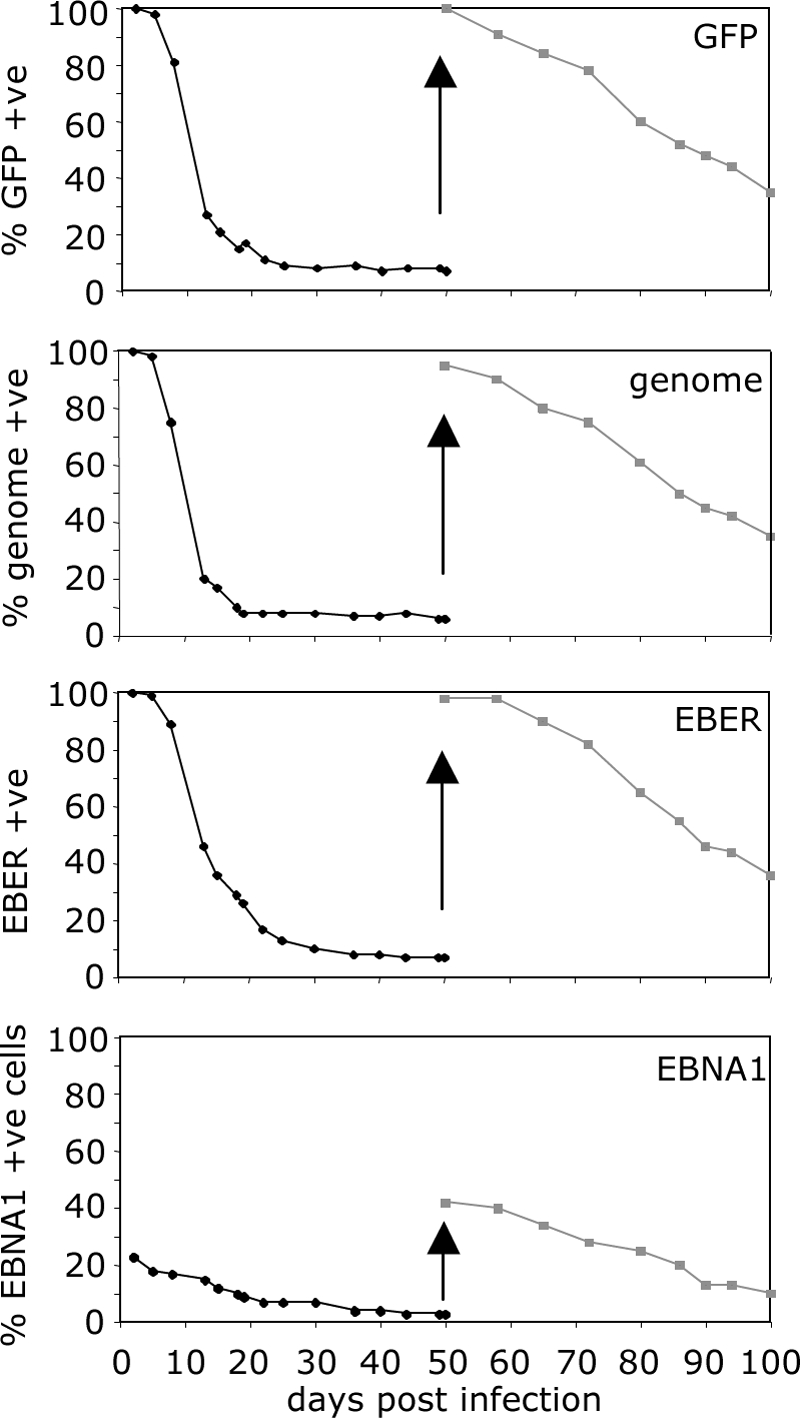
Maintenance of the viral genome in epithelial cells. AdAH epithelial cells were infected with wild-type virus, sorted for GFP expression at 24 h postinfection, and assayed at every passage over 50 days for GFP expression, viral genome (FISH), EBER1/2 expression (ISH), and EBNA1 expression (immunofluorescence). The cells were then resorted for GFP expression at day 50 and assayed for a further 50 days for the above. The results are displayed as the percent expression of each parameter over 100 days.
The GFP-positive cells were resorted at 50 days postinfection and again assayed for GFP, viral genome, EBER, and EBNA1. A similar loss of GFP, genome, EBERs, and EBNA1 was observed. Together, these results suggest that the presence and latent expression of the viral genome in nonpermissive epithelial cells confers no obvious direct growth or survival advantage.
Amplification of the viral genome.
When B cells are infected at a low MOI, so that the majority of cells have one or two genomes per cell after 48 h, an amplification of the episomal copy number subsequently ensues so that by 30 days postinfection the majority of infected B cells have in excess of 20 copies of the EBV episome in each cell (Fig. 7A, first-row histograms). In contrast, infection of primary epithelial or AdAH epithelial cells with similar numbers of viral genomes does not result in genome amplification even when the cultures of AdAH were followed for 100 days (Fig. 7A, second and third rows). Infected cultures of the simian virus 40-transformed keratinocyte line SVK likewise demonstrated an absence of genome amplification. In contrast to these nonpermissive epithelial cells, infection of the semipermissive epithelial cell line, AGS, resulted in the majority of cells maintaining the viral genome and expressing EBNA1. Notably, these cells amplified the virus episome from an average input of two to four genomes to 20 to 40 episomes per cell within 2 to 4 weeks, starting from 4 days postinfection (Fig. 7A, fourth row).
FIG. 7.

Amplification of the viral genome. (A) Primary B cells and primary epithelial cells (upper two panels), AdAH epithelial cells (third panels), and AGS epithelial cells (fourth panels) were infected with wild-type recombinant EBV and sorted for GFP expression at 48 h postinfection. The infected cells were examined immediately or after 30 days for viral genome copy number per cell by FISH. The results are displayed as the numbers of viral genomes per cell at 48 h postinfection (left-hand panels) and at 30 days postinfection (right-hand panels). (B) Detection of viral genomes by FISH. Primary B cells and AGS cells were infected at an MOI of 1 with wild-type or BZLF1 KO viruses. The cells were examined at 40 days postinfection for genome amplification. Viral genomes are detected by green fluorescence, and nuclei are identified by DAPI staining. (C) Detection of circular and linear viral genomes by Gardella gel analysis and Southern blotting. PEG-precipitated viral particles were used as a control for linear viral genomes, Raji cells were used as a control for episomal DNA, and LCLs were used as a control for both episomal and linear DNA. The vast majority of viral DNA in AGS cells with amplified viral genomes corresponds to episomal DNA, with a very small proportion being linear.
The AGS epithelial line differs from the other lines examined in being slightly more differentiated and, subsequently, it is semipermissive for lytic virus production. Typically, 100% of genome-positive cells express EBNA1 and 40% of cells express BZLF1 within 3 days of infection and longer-term cultures show ca. 30% BZLF1-positive cells. Interestingly, fewer than 4% of infected cells progress to late lytic cycle, raising the possibility that abortive lytic cycle might somehow be responsible for genome amplification. In B cells, EBV episomal amplification is independent of lytic replication since LCL established by transforming primary B cells with a recombinant BZLF1-KO EBV (which is unable to enter virus lytic cycle) show genome amplification similar to that seen with LCL transformed with wild-type EBV (Fig. 7B). However, infection of AGS cells with the BZLF1-KO recombinant EBV showed very little or no genome amplification. Gardella gel analysis of AGS cells and primary B cells 4 weeks postinfection showed this that amplification is episomal (Fig. 7C).
DISCUSSION
While EBV infection of primary B cells is readily achieved in vitro and has been well studied, the inability to achieve efficient infection of epithelial cells in vitro has until recently hampered investigations comparing the behavior of virus infection in these two cell types. Now, using our “B-cell transfer” method to infect primary epithelial cells and epithelial lines (52), we have been able to study infection of both B cells and epithelial cells in parallel.
At the population level, infection of primary epithelial cells and transformed lines was distinguished by higher expression of the noncoding EBERs and of LMP1 and LMP2A coding transcripts in the first 48 h postinfection. Furthermore, EBNA1 was exclusively expressed from Qp-derived mRNA. Wp and Cp promoters were essentially silent at all time points after infection of epithelial cells, in contrast to B-cell infections where Wp/Cp-derived mRNA are abundantly expressed and are responsible for the ordered expression of EBNA2 and EBNA-LP and then EBNA1, EBNA3A, EBNA3B, and EBNA3C.
Expression of BZLF1 lytic cycle transcripts was generally more pronounced in epithelial cell lines, although the lytic cycle appears to be abortive. In primary B cells and in primary epithelial cells, the amount of BZLF1 transcripts increased steadily over a 5-day period. In contrast, the BZLF1 transcripts peaked at day 2 in the AdAH epithelial line, a finding reminiscent of what has been observed when the Akata BL line is synchronously induced into lytic cycle (58). It is possible, therefore, that the difference between primary epithelial cells and AdAH with regard to the time course of BZLF1 transcription reflects the heterogeneity of the primary cultures, resulting in asynchronous induction of lytic cycle in different subpopulations at different times postinfection. Another unexpected result was that primary epithelial cells produced virtually no BARTs, as detected by our quantitative RT-PCR assay, relative to infection of transformed epithelial lines or primary B cells. The BARTs comprise a very complex set of transcripts, from which viral micro-RNAs are generated. The exact nature of this altered expression of BARTs in primary epithelial cells, and the consequence for viral micro-RNA expression, is currently under investigation.
Analysis of viral gene expression at the single-cell level showed that every infected B-cell follows an ordered pattern of viral gene expression; EBNA2/EBNA-LP and then EBNA1 and EBNA3A/EBNA3B/EBNA3C. However, EBV gene expression in the epithelial cell lines was very heterogeneous, giving rise to at least three different patterns of gene expression in the same homogeneous culture of cells. Analysis of gene expression at the single cell level showed that LMP1 expression in epithelial cells was detected exclusively in a subpopulation of EBNA1+ cells, and these included both BZLF1+ and BZLF1− cells.
The single-cell analysis also revealed that the EBERs were expressed in all genome-positive cells, whether B cells or epithelial cells. However, in undifferentiated epithelial cells, less than 40% of the EBER+ cells were EBNA1+. Consequently, over a period of culture in which the cells divided many times, the EBV genome was lost from infected epithelial cells. Interestingly, even when EBV+ subpopulations were selected after several weeks in culture, the proportion of EBNA1+ cells remained low, and the genome was again lost at a similar rate as during earlier passages. This suggests that the presence and expression of the EBV genome did not confer a growth advantage over uninfected cells, despite the presence of some cells showing the same latency II pattern of transcription as is found in NPC. In particular, the expression of LMP1 might have been expected to confer some advantage. LMP1 is a potent activator of the NF-κB signaling pathway (15, 23) and other signaling pathways, such as JAK/STAT (20), which together can upregulate in various cell models: antiapoptotic genes such as Bcl-2, A20, and Mcl-1 (18, 47, 62); proinflammatory cytokines such as interleukin-6 (IL-6) and IL-8 (14, 15); and cell surface receptors and adhesion molecules such as EGFR, CD40, and CD54 (5, 34). LMP1 is able to suppress cellular senescence in rat fibroblasts by repressing the expression of the p16INK4a and its downstream mediators, E2F4 and E2F5 (40, 65). However, in vitro infection of primary undifferentiated tonsillar epithelial cells by EBV does not result in the suppression of senescence (C. Shannon-Lowe, unpublished results). In addition, EBNA1 (24), LMP2A (31, 43), EBERs (48, 64), and the BART-5 micro-RNA (8) have all been reported to confer survival and/or proliferative advantage in certain cell models, but clearly their expression with or without LMP1 is insufficient to growth transform primary epithelial cells or to confer a growth advantage to undifferentiated epithelial cell lines after in vitro infection with EBV in our culture system.
A significant feature of EBV-associated lymphoid and epithelial cell tumors is the presence of multiple copies of the EBV genome in every cell, which are maintained as episomes rather than integrated into the host cell chromosomes. NPC is no exception, having at least 40 to 50 copies per cell (53). After infection of primary B cells in vitro using one virus copy per cell, the virus genome amplifies in every infected cell to an average of 40 to 50 copies per cell within the first month after infection (Fig. 7). This amplification is not due to virus lytic replication since it occurs at the same rate and to the same copy number after infection with a recombinant virus defective for lytic replication (ΔBZLF1-EBV). We observed no amplification of the episomal EBV genome after infection of the primary epithelial cells or undifferentiated AdAH or SVK epithelial cells, and these cells progressively lost the viral genomes after multiple passage. The AGS gastric carcinoma line was exceptional in being able to amplify the viral genome within days after infection, resulting in an average of around 40 genomes per cell, the vast majority of which are episomal. Furthermore, as with B cells, the genome-positive AGS cells expressed EBNA1 and were able to maintain the viral genome over serial passage. However, in contrast to B cells, little or no genome amplification was seen in AGS cells infected with the replication-defective ΔBZLF1-EBV, which suggests different mechanisms of genome amplification might operate in these two cell types. It is possible that one or more of the latency-associated growth-transforming viral genes expressed in B cells but not epithelial cells is primarily responsible for episomal genome amplification in B cells and that amplification in epithelial cells is mediated by an early lytic cycle gene. However, the induction of lytic cycle per se is insufficient to initiate episomal genome amplification in epithelial cells, since no such amplification was observed in the AdAH or SVK lines in which a subpopulation of cells entered lytic cycle. This suggests that differentially expressed cellular factors within the epithelial cell lineage might also regulate the ability to achieve episomal genome amplification.
Successful maintenance, duplication, and partitioning of the viral episome within the tumor cells require both EBNA1 and the viral latent origin of plasmid replication (oriP). EBNA1 binds the EBV genome via a DNA-binding domain at specific sites within the oriP, DS, and FR (66) and tethers these genomes to cellular metaphase chromosomes through chromosome-binding domains or AT hooks (32, 49, 50). It is therefore not surprising that, in the absence of significant EBNA1 expression, the viral genome is unable to tether to the host chromosomes and is rapidly lost during cellular replication. Our experiments show that EBNA1 mRNA is transcribed from the Qp promoter after primary infection epithelial cells, similar to what is observed in NPC and gastric carcinomas (3, 57, 59), although the fact that we detect EBNA1 protein in only a subpopulation of EBER+ cells when undifferentiated epithelial cells are infected with EBV suggests that the Qp promoter is firing inefficiently. Qp can, however, be induced by the expression of LMP1 which activates STAT3 and STAT5 and the cytokine IL-6, which in turn upregulate the expression of LMP1 transcripts and also Qp-driven EBNA1 (6, 7, 15). Nevertheless, the expression of LMP1 in infected AdAH or SVK epithelial cells was clearly insufficient to drive EBNA1 expression and maintain the viral episome.
Genome loss after in vitro infection of undifferentiated epithelial cells has been reported previously (28), although the phenomenon is not understood. It has been shown that after rapid loss of the genome from the majority of cells in culture, a small population may remain which retain the episome and still express EBNA1, LMP2a, and the EBERs; these cells can be selected by using a drug selection marker carried by the recombinant EBV used in the experiments (10, 26), but otherwise these cells are lost due to their lack of growth advantage or unstable genome expression. In this context, it is notable that whereas multiple copies of the viral episome are present in every tumor cell of EBV-positive NPC, the episome is rapidly lost upon explantation of NPC tumor cells into culture. It was recently reported (13) that episome loss from the EBV-positive NPC line HONE-1 is biphasic, with two mechanistically distinct phases: the initial phase involves the rapid loss of the entire genome, which is followed by a slow phase where the episome appears to be lost through successive deletion and recombination events, until the essential cis elements oriP, Qp, and EBNA1 are deleted. However, it remains unresolved why the entire genome is lost in the initial phase. One possibility is that it is due to unequal genome segregation, as we have shown to be the case in Burkitt's lymphoma lines that have a propensity to lose the genome (unpublished results).
Whatever the mechanism of genome loss in undifferentiated epithelial cells, the reported observations with NPC tumors suggest that the microenvironment of this tumor, e.g., the reactive T-cell infiltrate, plays a significant role in the maintenance of the EBV genome. To address these issues, further experiments are in progress to examine the possible influence of stroma or lymphoid/monocyte populations on the ability of epithelial cells to maintain the viral genome in vitro.
In summary, examination of the infection of B cells, primary epithelial cells, and epithelial cell lines at the single-cell level has shown substantial differences in viral gene expression, genome amplification, and genome loss. A key distinguishing feature identified in the present study is the ability to amplify the viral genome once the virus enters the cell. Whereas viral genomes that enter a primary B-cell fire to express EBNA2/LP, followed by a highly ordered pattern of viral gene expression resulting in the growth transformation of the B-cell and lytic cycle-independent amplification of the viral genomes, the more restricted pattern of gene expression and the cellular phenotype of epithelial cells appears to be generally incompatible with viral genome amplification in vitro. When amplification was observed in a more differentiated epithelial line (AGS), the mechanism clearly differed from that in B cells since it was dependent upon early lytic gene expression. These observations have implications for the pathogenesis of epithelial cell tumors. One essential step must include factors that promote viral genome amplification, followed by conditions that allow expression of EBNA1 and maintenance of the genomes. Given that our data suggest that episomal genome amplification in epithelial cells requires expression of lytic cycle genes, it is possible that the characteristically elevated titers of antibodies to lytic cycle proteins in NPC patients, where the majority of the established tumor cells are nonlytic, might reflect conditions that predispose to episomal genome amplification in premalignant epithelial cells.
Acknowledgments
This study was funded by the Association for International Cancer Research, Edinburgh, United Kingdom. Additional support was provided to M.R., A.B.R., and A.I.B. by the Cancer Research-UK and to E.A., C.S.-L., and M.R. by the Leukemia Research Fund, London, United Kingdom.
Footnotes
Published ahead of print on 13 May 2009.
REFERENCES
- 1.Babcock, G. J., L. L. Decker, M. Volk, and D. A. Thorley-Lawson. 1998. EBV persistence in memory B cells in vivo. Immunity 9395-404. [DOI] [PubMed] [Google Scholar]
- 2.Becker, J., U. Leser, M. Marschall, A. Langford, W. Jilg, H. Gelderblom, P. Reichart, and H. Wolf. 1991. Expression of proteins encoded by Epstein-Barr virus transactivator genes depends on the differentiation of epithelial cells in oral hairy leukoplakia. Proc. Natl. Acad. Sci. USA 888332-8336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bell, A. I., K. Groves, G. L. Kelly, D. Croom-Carter, E. Hui, A. T. Chan, and A. B. Rickinson. 2006. Analysis of Epstein-Barr virus latent gene expression in endemic Burkitt's lymphoma and nasopharyngeal carcinoma tumour cells by using quantitative real-time PCR assays. J. Gen. Virol. 872885-2890. [DOI] [PubMed] [Google Scholar]
- 4.Brooks, L., Q. Y. Yao, A. B. Rickinson, and L. S. Young. 1992. Epstein-Barr virus latent gene transcription in nasopharyngeal carcinoma cells: coexpression of EBNA1, LMP1, and LMP2 transcripts. J. Virol. 662689-2697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Busson, P., Q. Zhang, J. M. Guillon, C. D. Gregory, L. S. Young, B. Clausse, M. Lipinski, A. B. Rickinson, and T. Tursz. 1992. Elevated expression of ICAM1 (CD54) and minimal expression of LFA3 (CD58) in Epstein-Barr-virus-positive nasopharyngeal carcinoma cells. Int. J. Cancer 50863-867. [DOI] [PubMed] [Google Scholar]
- 6.Chen, H., L. Hutt-Fletcher, L. Cao, and S. D. Hayward. 2003. A positive autoregulatory loop of LMP1 expression and STAT activation in epithelial cells latently infected with Epstein-Barr virus. J. Virol. 774139-4148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chen, H., J. M. Lee, Y. Wang, D. P. Huang, R. F. Ambinder, and S. D. Hayward. 1999. The Epstein-Barr virus latency BamHI-Q promoter is positively regulated by STATs and Zta interference with JAK/STAT activation leads to loss of BamHI-Q promoter activity. Proc. Natl. Acad. Sci. USA 969339-9344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Choy, E. Y., K. L. Siu, K. H. Kok, R. W. Lung, C. M. Tsang, K. F. To, D. L. Kwong, S. W. Tsao, and D. Y. Jin. 2008. An Epstein-Barr virus-encoded microRNA targets PUMA to promote host cell survival. J. Exp. Med. 2052551-2560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cochet, C., D. Martel-Renoir, V. Grunewald, J. Bosq, G. Cochet, G. Schwaab, J. F. Bernaudin, and I. Joab. 1993. Expression of the Epstein-Barr virus immediate-early gene, BZLF1, in nasopharyngeal carcinoma tumor cells. Virology 197358-365. [DOI] [PubMed] [Google Scholar]
- 10.Delecluse, H. J., T. Hilsendegen, D. Pich, R. Zeidler, and W. Hammerschmidt. 1998. Propagation and recovery of intact, infectious Epstein-Barr virus from prokaryotic to human cells. Proc. Natl. Acad. Sci. USA 958245-8250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Delecluse, H. J., S. Schuller, and W. Hammerschmidt. 1993. Latent Marek's disease virus can be activated from its chromosomally integrated state in herpesvirus-transformed lymphoma cells. EMBO J. 123277-3286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dirmeier, U., B. Neuhierl, E. Kilger, G. Reisbach, M. L. Sandberg, and W. Hammerschmidt. 2003. Latent membrane protein 1 is critical for efficient growth transformation of human B cells by Epstein-Barr virus. Cancer Res. 632982-2989. [PubMed] [Google Scholar]
- 13.Dittmer, D. P., C. J. Hilscher, M. L. Gulley, E. V. Yang, M. Chen, and R. Glaser. 2008. Multiple pathways for Epstein-Barr virus episome loss from nasopharyngeal carcinoma. Int. J. Cancer 1232105-2112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Eliopoulos, A. G., N. J. Gallagher, S. M. Blake, C. W. Dawson, and L. S. Young. 1999. Activation of the p38 mitogen-activated protein kinase pathway by Epstein-Barr virus-encoded latent membrane protein 1 coregulates interleukin-6 and interleukin-8 production. J. Biol. Chem. 27416085-16096. [DOI] [PubMed] [Google Scholar]
- 15.Eliopoulos, A. G., M. Stack, C. W. Dawson, K. M. Kaye, L. Hodgkin, S. Sihota, M. Rowe, and L. S. Young. 1997. Epstein-Barr virus-encoded LMP1 and CD40 mediate IL-6 production in epithelial cells via an NF-κB pathway involving TNF receptor-associated factors. Oncogene 142899-2916. [DOI] [PubMed] [Google Scholar]
- 16.Feederle, R., M. Kost, M. Baumann, A. Janz, E. Drouet, W. Hammerschmidt, and H. J. Delecluse. 2000. The Epstein-Barr virus lytic program is controlled by the co-operative functions of two transactivators. EMBO J. 193080-3089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Feng, P., E. C. Ren, D. Liu, S. H. Chan, and H. Hu. 2000. Expression of Epstein-Barr virus lytic gene BRLF1 in nasopharyngeal carcinoma: potential use in diagnosis. J. Gen. Virol. 812417-2423. [DOI] [PubMed] [Google Scholar]
- 18.Fries, K. L., W. E. Miller, and N. Raab-Traub. 1996. Epstein-Barr virus latent membrane protein 1 blocks p53-mediated apoptosis through the induction of the A20 gene. J. Virol. 708653-8659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gardella, T., P. Medveczky, T. Sairenji, and C. Mulder. 1984. Detection of circular and linear herpesvirus DNA molecules in mammalian cells by gel electrophoresis. J. Virol. 50248-254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gires, O., F. Kohlhuber, E. Kilger, M. Baumann, A. Kieser, C. Kaiser, R. Zeidler, B. Scheffer, M. Ueffing, and W. Hammerschmidt. 1999. Latent membrane protein 1 of Epstein-Barr virus interacts with JAK3 and activates STAT proteins. EMBO J. 183064-3073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hoffman, G. J., S. G. Lazarowitz, and S. D. Hayward. 1980. Monoclonal antibody against a 250,000-dalton glycoprotein of Epstein-Barr virus identifies a membrane antigen and a neutralizing antigen. Proc. Natl. Acad. Sci. USA 772979-2983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hudnall, S. D., Y. Ge, L. Wei, N. P. Yang, H. Q. Wang, and T. Chen. 2005. Distribution and phenotype of Epstein-Barr virus-infected cells in human pharyngeal tonsils. Mod. Pathol. 18519-527. [DOI] [PubMed] [Google Scholar]
- 23.Huen, D. S., S. A. Henderson, D. Croom-Carter, and M. Rowe. 1995. The Epstein-Barr virus latent membrane protein-1 (LMP1) mediates activation of NF-κB and cell surface phenotype via two effector regions in its carboxy-terminal cytoplasmic domain. Oncogene 10549-560. [PubMed] [Google Scholar]
- 24.Humme, S., G. Reisbach, R. Feederle, H. J. Delecluse, K. Bousset, W. Hammerschmidt, and A. Schepers. 2003. The EBV nuclear antigen 1 (EBNA1) enhances B-cell immortalization several thousandfold. Proc. Natl. Acad. Sci. USA 10010989-10994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Imai, S., J. Nishikawa, and K. Takada. 1998. Cell-to-cell contact as an efficient mode of Epstein-Barr virus infection of diverse human epithelial cells. J. Virol. 724371-4378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kanda, T., M. Yajima, N. Ahsan, M. Tanaka, and K. Takada. 2004. Production of high-titer Epstein-Barr virus recombinants derived from Akata cells by using a bacterial artificial chromosome system. J. Virol. 787004-7015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Karajannis, M. A., M. Hummel, I. Anagnostopoulos, and H. Stein. 1997. Strict lymphotropism of Epstein-Barr virus during acute infectious mononucleosis in nonimmunocompromised individuals. Blood 892856-2862. [PubMed] [Google Scholar]
- 28.Knox, P. G., Q. X. Li, A. B. Rickinson, and L. S. Young. 1996. In vitro production of stable Epstein-Barr virus-positive epithelial cell clones which resemble the virus-cell interaction observed in nasopharyngeal carcinoma. Virology 21540-50. [DOI] [PubMed] [Google Scholar]
- 29.Laytragoon-Lewin, N., F. Chen, J. Avila-Carino, G. Klein, and H. Mellstedt. 1997. Epstein-Barr virus (EBV) gene expression in lymphoid B cells during acute infectious mononucleosis (IM) and clonality of the directly growing cell lines. Int. J. Cancer 71345-349. [DOI] [PubMed] [Google Scholar]
- 30.Li, Q. X., L. S. Young, G. Niedobitek, C. W. Dawson, M. Birkenbach, F. Wang, and A. B. Rickinson. 1992. Epstein-Barr virus infection and replication in a human epithelial cell system. Nature 356347-350. [DOI] [PubMed] [Google Scholar]
- 31.Mancao, C., and W. Hammerschmidt. 2007. Epstein-Barr virus latent membrane protein 2A is a B-cell receptor mimic and essential for B-cell survival. Blood 1103715-3721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Marechal, V., A. Dehee, R. Chikhi-Brachet, T. Piolot, M. Coppey-Moisan, and J. C. Nicolas. 1999. Mapping EBNA-1 domains involved in binding to metaphase chromosomes. J. Virol. 734385-4392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Martel-Renoir, D., V. Grunewald, R. Touitou, G. Schwaab, and I. Joab. 1995. Qualitative analysis of the expression of Epstein-Barr virus lytic genes in nasopharyngeal carcinoma biopsies. J. Gen. Virol. 76(Pt. 6)1401-1408. [DOI] [PubMed] [Google Scholar]
- 34.Miller, W. E., H. S. Earp, and N. Raab-Traub. 1995. The Epstein-Barr virus latent membrane protein 1 induces expression of the epidermal growth factor receptor. J. Virol. 694390-4398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Montone, K. T., R. L. Hodinka, K. E. Salhany, E. Lavi, A. Rostami, and J. E. Tomaszewski. 1996. Identification of Epstein-Barr virus lytic activity in posttransplantation lymphoproliferative disease. Mod Pathol. 9621-630. [PubMed] [Google Scholar]
- 36.Neuhierl, B., R. Feederle, W. Hammerschmidt, and H. J. Delecluse. 2002. Glycoprotein gp110 of Epstein-Barr virus determines viral tropism and efficiency of infection. Proc. Natl. Acad. Sci. USA 9915036-15041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Niedobitek, G., A. Agathanggelou, H. Herbst, L. Whitehead, D. H. Wright, and L. S. Young. 1997. Epstein-Barr virus (EBV) infection in infectious mononucleosis: virus latency, replication and phenotype of EBV-infected cells. J. Pathol. 182151-159. [DOI] [PubMed] [Google Scholar]
- 38.Niedobitek, G., H. Herbst, L. S. Young, M. Rowe, D. Dienemann, C. Germer, and H. Stein. 1992. Epstein-Barr virus and carcinomas. Expression of the viral genome in an undifferentiated gastric carcinoma. Diagn. Mol. Pathol. 1103-108. [PubMed] [Google Scholar]
- 39.Niedobitek, G., L. S. Young, R. Lau, L. Brooks, D. Greenspan, J. S. Greenspan, and A. B. Rickinson. 1991. Epstein-Barr virus infection in oral hairy leukoplakia: virus replication in the absence of a detectable latent phase. J. Gen. Virol. 72(Pt. 12)3035-3046. [DOI] [PubMed] [Google Scholar]
- 40.Ohtani, N., P. Brennan, S. Gaubatz, E. Sanij, P. Hertzog, E. Wolvetang, J. Ghysdael, M. Rowe, and E. Hara. 2003. Epstein-Barr virus LMP1 blocks the p16INK4a-RB pathway by promoting nuclear export of E2F4/5. J. Cell Biol. 162173-183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pegtel, D. M., J. Middeldorp, and D. A. Thorley-Lawson. 2004. Epstein-Barr virus infection in ex vivo tonsil epithelial cell cultures of asymptomatic carriers. J. Virol. 7812613-12624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Polack, A., G. Hartl, U. Zimber, U. K. Freese, G. Laux, K. Takaki, B. Hohn, L. Gissmann, and G. W. Bornkamm. 1984. A complete set of overlapping cosmid clones of M-ABA virus derived from nasopharyngeal carcinoma and its similarity to other Epstein-Barr virus isolates. Gene 27279-288. [DOI] [PubMed] [Google Scholar]
- 43.Portis, T., and R. Longnecker. 2004. Epstein-Barr virus (EBV) LMP2A mediates B-lymphocyte survival through constitutive activation of the Ras/PI3K/Akt pathway. Oncogene 238619-8628. [DOI] [PubMed] [Google Scholar]
- 44.Rickinson, A. B., and E. Kieff. 2007. Epstein-Barr virus, p. 2655-2700. In D. M. Knipe, P. M. Howley, D. E. Griffin, R. A. Lamb, M. A. Martin, B. Roizman, and S. E. Straus (ed.), Fields virology, 5th ed, vol. 2. Lippincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 45.Rowe, M., H. S. Evans, L. S. Young, K. Hennessy, E. Kieff, and A. B. Rickinson. 1987. Monoclonal antibodies to the latent membrane protein of Epstein-Barr virus reveal heterogeneity of the protein and inducible expression in virus-transformed cells. J. Gen. Virol. 68(Pt. 6)1575-1586. [DOI] [PubMed] [Google Scholar]
- 46.Rowe, M., A. L. Lear, D. Croom-Carter, A. H. Davies, and A. B. Rickinson. 1992. Three pathways of Epstein-Barr virus gene activation from EBNA1-positive latency in B lymphocytes. J. Virol. 66122-131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rowe, M., M. Peng-Pilon, D. S. Huen, R. Hardy, D. Croom-Carter, E. Lundgren, and A. B. Rickinson. 1994. Upregulation of bcl-2 by the Epstein-Barr virus latent membrane protein LMP1: a B-cell-specific response that is delayed relative to NF-κB activation and to induction of cell surface markers. J. Virol. 685602-5612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ruf, I. K., P. W. Rhyne, C. Yang, J. L. Cleveland, and J. T. Sample. 2000. Epstein-Barr virus small RNAs potentiate tumorigenicity of Burkitt lymphoma cells independently of an effect on apoptosis. J. Virol. 7410223-10228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sears, J., J. Kolman, G. M. Wahl, and A. Aiyar. 2003. Metaphase chromosome tethering is necessary for the DNA synthesis and maintenance of oriP plasmids but is insufficient for transcription activation by Epstein-Barr nuclear antigen 1. J. Virol. 7711767-11780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sears, J., M. Ujihara, S. Wong, C. Ott, J. Middeldorp, and A. Aiyar. 2004. The amino terminus of Epstein-Barr virus (EBV) nuclear antigen 1 contains AT hooks that facilitate the replication and partitioning of latent EBV genomes by tethering them to cellular chromosomes. J. Virol. 7811487-11505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Shannon-Lowe, C., G. Baldwin, R. Feederle, A. Bell, A. Rickinson, and H. J. Delecluse. 2005. Epstein-Barr virus-induced B-cell transformation: quantitating events from virus binding to cell outgrowth. J. Gen. Virol. 863009-3019. [DOI] [PubMed] [Google Scholar]
- 52.Shannon-Lowe, C. D., B. Neuhierl, G. Baldwin, A. B. Rickinson, and H. J. Delecluse. 2006. Resting B cells as a transfer vehicle for Epstein-Barr virus infection of epithelial cells. Proc. Natl. Acad. Sci. USA 1037065-7070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Shao, J. Y., Y. H. Li, H. Y. Gao, Q. L. Wu, N. J. Cui, L. Zhang, G. Cheng, L. F. Hu, I. Ernberg, and Y. X. Zeng. 2004. Comparison of plasma Epstein-Barr virus (EBV) DNA levels and serum EBV immunoglobulin A/virus capsid antigen antibody titers in patients with nasopharyngeal carcinoma. Cancer 1001162-1170. [DOI] [PubMed] [Google Scholar]
- 54.Sixbey, J. W., J. G. Nedrud, N. Raab-Traub, R. A. Hanes, and J. S. Pagano. 1984. Epstein-Barr virus replication in oropharyngeal epithelial cells. N. Engl. J. Med. 3101225-1230. [DOI] [PubMed] [Google Scholar]
- 55.Smith, P. R., and B. E. Griffin. 1992. Transcription of the Epstein-Barr virus gene EBNA-1 from different promoters in nasopharyngeal carcinoma and B-lymphoblastoid cells. J. Virol. 66706-714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sternas, L., T. Middleton, and B. Sugden. 1990. The average number of molecules of Epstein-Barr nuclear antigen 1 per cell does not correlate with the average number of Epstein-Barr virus (EBV) DNA molecules per cell among different clones of EBV-immortalized cells. J. Virol. 642407-2410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sugiura, M., S. Imai, M. Tokunaga, S. Koizumi, M. Uchizawa, K. Okamoto, and T. Osato. 1996. Transcriptional analysis of Epstein-Barr virus gene expression in EBV-positive gastric carcinoma: unique viral latency in the tumour cells. Br. J. Cancer 74625-631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Takada, K., and Y. Ono. 1989. Synchronous and sequential activation of latently infected Epstein-Barr virus genomes. J. Virol. 63445-449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tsai, C. N., S. T. Liu, and Y. S. Chang. 1995. Identification of a novel promoter located within the BamHI-Q region of the Epstein-Barr virus genome for the EBNA 1 gene. DNA Cell Biol. 14767-776. [DOI] [PubMed] [Google Scholar]
- 60.Walling, D. M., C. M. Flaitz, C. M. Nichols, S. D. Hudnall, and K. Adler-Storthz. 2001. Persistent productive Epstein-Barr virus replication in normal epithelial cells in vivo. J. Infect. Dis. 1841499-1507. [DOI] [PubMed] [Google Scholar]
- 61.Walling, D. M., P. D. Ling, A. V. Gordadze, M. Montes-Walters, C. M. Flaitz, and C. M. Nichols. 2004. Expression of Epstein-Barr virus latent genes in oral epithelium: determinants of the pathogenesis of oral hairy leukoplakia. J. Infect. Dis. 190396-399. [DOI] [PubMed] [Google Scholar]
- 62.Wang, S., M. Rowe, and E. Lundgren. 1996. Expression of the Epstein Barr virus transforming protein LMP1 causes a rapid and transient stimulation of the Bcl-2 homologue Mcl-1 levels in B-cell lines. Cancer Res. 564610-4613. [PubMed] [Google Scholar]
- 63.Webster-Cyriaque, J., J. Middeldorp, and N. Raab-Traub. 2000. Hairy leukoplakia: an unusual combination of transforming and permissive Epstein-Barr virus infections. J. Virol. 747610-7618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Yajima, M., T. Kanda, and K. Takada. 2005. Critical role of Epstein-Barr virus (EBV)-encoded RNA in efficient EBV-induced B-lymphocyte growth transformation. J. Virol. 794298-4307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Yang, X., Z. He, B. Xin, and L. Cao. 2000. LMP1 of Epstein-Barr virus suppresses cellular senescence associated with the inhibition of p16INK4a expression. Oncogene 192002-2013. [DOI] [PubMed] [Google Scholar]
- 66.Yates, J., N. Warren, D. Reisman, and B. Sugden. 1984. A cis-acting element from the Epstein-Barr viral genome that permits stable replication of recombinant plasmids in latently infected cells. Proc. Natl. Acad. Sci. USA 813806-3810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Young, L. S., R. Lau, M. Rowe, G. Niedobitek, G. Packham, F. Shanahan, D. T. Rowe, D. Greenspan, J. S. Greenspan, A. B. Rickinson, et al. 1991. Differentiation-associated expression of the Epstein-Barr virus BZLF1 transactivator protein in oral hairy leukoplakia. J. Virol. 652868-2874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zhang, J. X., H. L. Chen, Y. S. Zong, K. H. Chan, J. Nicholls, J. M. Middeldorp, J. S. Sham, B. E. Griffin, and M. H. Ng. 1998. Epstein-Barr virus expression within keratinizing nasopharyngeal carcinoma. J. Med. Virol. 55227-233. [DOI] [PubMed] [Google Scholar]