

Abstract
Hepatitis C virus (HCV) downregulates the retinoblastoma tumor suppressor protein (Rb), a central cell cycle regulator which is also targeted by oncoproteins expressed by DNA tumor viruses. HCV genome replication is also enhanced in proliferating cells. Thus, it is possible that HCV interactions with host cell cycle regulators, such as Rb, have evolved to modify the intracellular environment to promote viral replication. To test this hypothesis and to determine the impact of viral regulation of Rb on HCV replication, we constructed infectious viral genomes containing mutations in the Rb-binding motif of NS5B which ablate the ability of HCV to regulate Rb. These genomes underwent replication in transfected cells but produced variably reduced virus yields. One mutant, L314A, was severely compromised for replication and rapidly mutated to L314V, thereby restoring both Rb regulation and replication competence. Another mutant, C316A, also failed to downregulate Rb abundance and produced virus yields that were about one-third that of virus with the wild-type (wt) NS5B sequence. Despite this loss of replication competence, purified NS5B-C316A protein was two- to threefold more active than wt NS5B in cell-free polymerase and replicase assays. Although small interfering RNA knockdown of Rb did not rescue the replication fitness of these mutants, we conclude that the defect in replication fitness is not due to defective polymerase or replicase function and is more likely to result from the inability of the mutated NS5B to optimally regulate Rb abundance and thereby modulate host gene expression.
Chronic infection with hepatitis C virus (HCV) is associated with an increased risk for development of hepatocellular carcinoma (11). However, unlike most viruses associated with cancer, HCV has an RNA genome and an exclusively cytoplasmic life cycle. The precise mechanisms by which HCV infection leads to carcinogenesis are unclear. Although chronic inflammation is suspected to play a role, transgenic mice that express a low abundance of the entire HCV polyprotein do not exhibit hepatic inflammation and yet have an increased incidence of hepatocellular carcinoma (10). These data suggest that HCV proteins may have a direct oncogenic effect. Consistent with this, several structural and nonstructural proteins encoded by HCV (core, NS3, NS5A, and NS5B) have been shown to regulate host cell cycle regulators and tumor suppressor proteins, including p53 and the retinoblastoma protein (Rb) (7, 8, 14, 17; for a recent review, see reference 16).
Rb has many important functions in cell cycle control, including the repression of transcription factors needed for the transition from G1/G0 to S phase. Rb is targeted by a number of DNA oncoviruses, including adenovirus (27), simian virus 40 (3), and human papillomavirus (5). These small DNA viruses exploit host cell machinery to facilitate genome replication, and they encode oncoproteins that can sequester Rb or downregulate its abundance. This results in the release of the Rb-imposed repression of various transcription factors that are needed to express genes required for DNA replication, such as proliferating cell nuclear antigen and thymidine kinase (4). In this way, the DNA oncoviruses can induce expression of cellular genes that promote their replication. The regulation of Rb by these DNA viruses is likely to contribute substantially to their oncogenic potential.
Although HCV does not require the host DNA replication machinery to replicate its RNA genome, several in vitro observations suggest that HCV RNA replication and genome accumulation are enhanced in proliferating cells (19, 20, 26). This is an intriguing feature of HCV, since its primary target cell, the hepatocyte, is generally quiescent and, while very active metabolically, demonstrates very slow turnover in the noninfected liver. Our previous studies have shown that HCV has evolved a mechanism to downregulate the Rb protein. The HCV RNA-dependent RNA polymerase (RdRp), NS5B, interacts with Rb, targeting it for E6-associated protein-dependent ubiquitination and proteasomal degradation, thereby promoting cell cycle progression (17, 18). The interaction between NS5B and Rb thus represents an interesting parallel with the oncoproteins of DNA tumor viruses. Like many cellular and viral proteins, HCV NS5B interacts with Rb through a conserved Rb-binding motif (LxC/NxD) homologous to Rb-binding domains found in DNA virus oncoproteins (17, 18). Surprisingly, this Rb-binding motif overlaps with the Gly-Asp-Asp sequence in NS5B that coordinates the binding of divalent metals near the active site of the polymerase.
In order to better understand the role of Rb regulation in the HCV life cycle, we investigated the impact of mutations within the LxC/NxD motif of NS5B on HCV RNA replication and virus production. We show that viruses with mutations in the NS5B Rb-binding site are incapable of regulating Rb abundance and also variably impaired in replication fitness. Additional data suggest that the defect in replication fitness is not due to a direct loss of polymerase or replicase complex activity but may reflect the inability of these viruses to regulate Rb abundance and thereby modulate host gene expression.
MATERIALS AND METHODS
Cells.
Human hepatoma Huh-7 cells and the Huh-7 derivatives Huh-7.5 and FT3-7 were cultured as described previously (28, 29).
Plasmid construction, HCV genome transfection, and virus production.
Plasmid pHJ3-5, previously referred to as pH-NS2/NS3-J(Y361H/Q1251L) (29), carries a chimeric infectious HCV genome consisting of sequence encoding the structural proteins of the genotype 1a H77S virus within the background of the genotype 2a JFH-1 virus. This plasmid contains mutations in E1 (Y361H) and NS3 (Q1251L) that enhance virus production (13). The HCV genome carried by this plasmid was selected as the “wild-type” (wt) HCV for the purpose of this study and will be referred to as HJ3-5 in this study. For mutagenesis of the NS5B sequence, the JFH-1 sequence from nucleotide (nt) 8014 to the end of the genome was released from pHJ3-5 by digestion with HpaI and XbaI and the fragment was inserted into pUC20 at SmaI and XbaI sites in the polylinker. The resulting subclone served as a template for mutagenesis of NS5B using the QuikChange site-directed mutagenesis kit (Stratagene, La Jolla, CA). The mutated NS5B sequence was digested with SnaBI and XbaI and inserted back into similarly digested, full-length pHJ3-5. The sequences of mutated regions in the resulting plasmids were confirmed by direct DNA sequencing. Mutations were named with reference to the amino acid number of the JFH-1 NS5B protein.
HCV infectivity assay.
HCV RNAs were transcribed in vitro using the Megascript T7 kit (Ambion, Austin, TX) and electroporated into cells, and cell culture supernatants containing infectious virus were harvested as described previously (29). The titer of infectious virus released from cells following RNA transfection or virus infection was determined using a focus-forming assay, as described previously (30). Briefly, 100-μl aliquots of cell culture supernatants were used to inoculate naïve Huh-7.5 cells seeded 24 h earlier in eight-well chamber slides at 4 × 104 cells/well. Cells were incubated at 37°C, 5% CO2 and fed with 200 μl medium after 24 h. At 48 h after virus inoculation, cells were washed twice in 1× phosphate-buffered saline (PBS), fixed in methanol-acetone (1:1) for 9 min, and washed again with 1× PBS. Cells were then incubated for 3 h at 37°C with monoclonal antibody to the core protein (C7-50; Affinity Bioreagents, Golden, CO) diluted 1:500 in 3% bovine serum albumin (BSA)-1× PBS. Cells were washed three times in 1× PBS before incubation for 1 h at 37°C with 1:200 dilution of fluorescein isothiocyanate-conjugated goat anti-mouse immunoglobulin G (IgG) antibody (Southern Biotech, Birmingham, AL). Cells were washed three times in 1× PBS and mounted in Vectashield (Vectorlabs, Burlingame, CA).
Analysis of viral RNA replication and sequences of progeny viruses.
Total RNA was extracted from transfected cells using the RNeasy mini kit (Qiagen, Valencia, CA) according to the manufacturer's instructions. Viral RNA abundance was measured by quantitative real-time reverse transcription PCR (qRT-PCR) as previously described (13). Primers DRM59 (GGGCCGTTAACCACATCAAGTCCGTG; corresponding to JFH-1 nt 8007 to 8032) and DRM61 (TGGCGCCCCAAGTTTTCTGAG; nt 9151 to 9131) targeting the NS5B region were used to amplify a fragment of approximately 1.1 kb containing the Rb-binding motif. RT-PCR products were sequenced directly.
Expression and purification of proteins.
wt and mutant JFH-1 NS5B proteins were expressed in Escherichia coli and purified as described previously (24). Briefly, E. coli BL21(DE3) cells transformed with NS5B constructs in pET303 plasmids were grown in LB medium to an optical density at 600 nm of ∼1.0 and induced for expression by treatment with 1 mM IPTG (isopropyl-β-d-thiogalactopyranoside) for 24 h at 16°C. The cells were subsequently pelleted by centrifugation and lysed by sonication in buffer L (50 mM Tris [pH 7.6], 300 mM NaCl, 1 mM β-mercaptoethanol, 10% glycerol, 1 mM phenylmethylsulfonyl fluoride, and 20 mM imidazole). The clarified lysate was applied to a 1-ml His-Trap affinity purification column (GE Healthcare). The column was washed with the sample application buffer, and the bound proteins were eluted in the same buffer containing 500 mM imidazole. The fractions containing the protein were pooled, and the salt concentration was adjusted to 100 mM before applying the sample onto poly(U)-agarose (Sigma, St. Louis, MO). The bound proteins were eluted in buffer A (20 mM Tris [pH 7.6], 10% glycerol, 3 mM dithiothreitol [DTT], and 1 mM phenylmethylsulfonyl fluoride) containing 400 mM NaCl. The proteins were subjected to sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) along with known concentrations of BSA to determine the concentrations.
RdRp activity assays.
RdRp assay mixtures contained 1 pmol of linear template RNA or 30 pmol of circular (C) template RNA and 0.04 μM of NS5B protein (2, 23). The assays were carried out in 20-μl reaction mixtures containing 20 mM sodium glutamate (pH 8.2), 0.5 mM DTT, 4 mM MgCl2, 12, 0.5% (vol/vol) Triton X-100, 200 μM ATP and UTP, and 250 nM [α-32P]CTP (MP Biomedicals). GTP was used at a final concentration of 0.2 mM unless specified. MnCl2 was included in all the assay mixtures at a final concentration of 1 mM. Reaction mixes also contained 20 mM NaCl that came from the protein storage buffer. RNA synthesis reaction mixtures were incubated at 25°C for 60 min. The reactions were stopped by phenol-chloroform extraction, followed by ethanol precipitation of the RNA in the presence of glycogen and 0.5 M ammonium acetate. Products were separated by electrophoresis on denaturing (7.5 M urea) 20% polyacrylamide gels. Gels were wrapped in plastic, and quantification of radiolabeled bands was performed using a PhosphorImager (GE Healthcare).
Circularization of RNA.
The protocol to circularize linear RNA was as described previously (23). Briefly, 2 nmol of a 16-mer linear RNA named “L” was incubated with T4 RNA ligase (Ambion, Austin, TX) according to the manufacturer's specifications for 3 h at 37°C in a 20-μl reaction mixture. An aliquot of the reaction mixture was run on a 20% polyacrylamide-7.5 M urea denaturing gel along with the linear RNA as a control. The gel was stained briefly in toluidine blue, and the RNA was visualized, showing a clear electrophoretic mobility difference between the linear (L) and the circularized (C) molecules (data not shown). The circularized RNA was excised from the gel, eluted using 0.3 M ammonium acetate overnight, and then purified by phenol-chloroform extraction and ethanol precipitation. The concentration of the RNA was measured by its absorbance at 260 nm.
Immunoblot analysis.
Preparation of protein extracts, SDS-PAGE, and subsequent immunoblotting were done as described previously (18) using mouse monoclonal antibodies against β-actin (AC-15; Sigma, St Louis, MO), Rb (G3-245; BD Biosciences, San Jose, CA), and core (C7-50; Affinity BioReagents, Golden, CO); rabbit polyclonal antibodies against NS5B (A266-1 [Virogen, Watertown, MA] or, in replicase assays, ab35586 [Abcam, Cambridge, MA]) and β-tubulin (ab6046; Abcam, Cambridge, MA); and goat polyclonal antibodies against NS3 (ab21124; Abcam). The endoplasmic reticulum marker Rab1b was detected using rabbit polyclonal antibody G-20 (sc-599; Santa Cruz Biotechnology, Santa Cruz, CA). Immunoblots were visualized either by chemiluminescence or by direct detection of infrared fluorescence on an Odyssey infrared imaging system (Li-Cor, Lincoln, NE).
Indirect immunofluorescence microscopy.
Infected or mock-infected cells were seeded onto glass chamber slides at 6 × 104 cells per well and allowed to grow for 24 h. Cells were washed twice with 1× PBS and fixed in methanol-acetone (1:1) at −20°C for 10 min. Fixative was removed and slides air dried in a fume hood for 1 h. For the images shown in Fig. 3C, cells were fixed in 4% paraformaldehyde for 30 min and permeabilized in 0.2% Triton X-100-1× PBS for 12 min. Slides were washed twice in 1× PBS before blocking for 1 h in 10% goat serum (Sigma, St. Louis, MO) in 1× PBS. The slides were then washed twice in 1× PBS and incubated overnight at 4°C with primary antibodies diluted in 1% BSA-1× PBS. A mouse monoclonal antibody was used to detect Rb (G3-245; BD Biosciences, San Jose, CA), and either a human polyclonal anti-HCV serum or a rabbit polyclonal antibody to NS5A (a gift from Craig Cameron) was used for detection of HCV antigens. Slides were washed three times for 10 min each in 1× PBS and incubated in darkness for 1 h at 37°C with secondary antibodies diluted in 1% BSA-1× PBS. Secondary antibodies were Alexa Fluor 488-conjugated goat anti-mouse IgG and Alexa Fluor 594-conjugated goat anti-rabbit IgG (Invitrogen, Carlsbad, CA) or Texas red-conjugated donkey anti-human IgG (Jackson Immunoresearch Laboratories, West Grove, PA). The slides were washed three times for 10 min each in 1× PBS, counterstained with DAPI (4′,6′-diamidino-2-phenylindole), and mounted in Vectashield (Vectorlabs, Burlingame, CA). Fluorescence images were obtained using a Zeiss Axiophot II fluorescence microscope. Confocal fluorescence images were obtained using a Zeiss LSM 510 UV META laser-scanning confocal microscope in the University of Texas Medical Branch Infectious Disease and Toxicology Optical Imaging Core. Recorded digital images and related controls were uniformly enhanced for brightness and contrast using Photoshop CS2 (Adobe Systems, San Jose, CA).
FIG. 3.

Analysis of a partial revertant of the L314A mutant. (A) Virus released into cell culture medium following transfection with HJ3-5, L314A, L314V, C316A, or D318N RNA. Error bars indicate standard deviations. FFU, focus-forming units. (B) Infrared immunoblot analysis of Rb in lysates from cells infected with L314V. Left panel, cells were infected with L314V or mock infected, and lysates collected on the indicated day postinfection were analyzed by immunoblotting using antibodies specific for Rb, NS5B, and β-tubulin (loading control). Right panel, quantitation of the infrared immunoblot, showing the abundance of Rb relative to β-tubulin at the indicated time points. (C) Immunofluorescence analysis of cells infected with HJ3-5, L314A, or L314V. Huh-7.5 cells were infected at an MOI of 0.1 and incubated for 4 days before fixation and staining for Rb or NS5A.
Isolation of heavy membrane fractions containing HCV replication complexes.
Heavy membrane fractions were isolated from cells infected with virus following electroporation of viral RNAs and from stable Huh-7 cell lines containing dicistronic, subgenomic genotype 1a or 2a HCV replicon RNAs. Cells were grown to 70 to 90% confluence, trypsinized, washed twice with PBS, and resuspended in hypotonic lysis buffer containing 10 mM HEPES (pH 7.9), 10 mM KCl, 5 mM DTT, and EDTA-free protease inhibitors (Roche, Mannheim, Germany). Cells were kept at 4°C for 20 min and disrupted by repetitive passage (40 times) through a 25-gauge needle. Nuclei were removed by centrifugation (1,000 × g, 10 min, 4°C). Postnuclear homogenates were centrifuged at 16,000 × g for 30 min at 4°C. Pellets of heavy membranes containing HCV replication complexes (P16) were resuspended in hypotonic buffer containing 10% glycerol and stored at −80°C.
Cell-free HCV RNA synthesis by membrane-bound replicase complexes.
Aliquots of P16 fractions were incubated at 37°C for 3 h in standard transcription mixtures containing 50 mM HEPES (pH 7.9); 5 mM MgCl2; 50 mM KCl; 10 mM DTT; 15 μg/ml actinomycin D; 1 mM spermidine; 800 U RNasin (Promega, Madison, WI) per ml; 1% dimethyl sulfoxide; 1 mM GTP, ATP, and UTP; 10 μM CTP; and 1 mCi of [α-32P]CTP per ml. Total RNA was extracted using the RNeasy mini kit, precipitated with isopropanol, and resolved on a 1% agarose gel containing glyoxal (NorthernMax-Gly; Ambion, Austin, TX).
Gene knockdown by RNA interference.
Small interfering RNAs (siRNAs) were purchased from Dharmacon (Lafayette, CO). An siRNA oligonucleotide SMARTpool, containing four siRNA oligonucleotides specific for the human retinoblastoma gene (L-003296-00), and four individual siRNAs (no. 5, 6, 7, and 9; D-003296-05, -06, -07, and -09, respectively) were used to target Rb. A pool of four siRNAs (D-001206-13) that are not specific for the Rb message and individual control siRNAs 1 and 2 (D-001230-01 and D-001220-01, respectively) were used as negative controls. For knockdown experiments, FT3-7 or Huh-7.5 cells were seeded at 30% confluence in six-well plates and transiently transfected with 50 nM siRNA using Lipofectamine 2000 (Invitrogen, Carlsbad, CA) according to the manufacturer's instructions. Duplicate transfections were used for either HCV infection or supertransfection with DNA plasmids and preparation of protein extracts for immunoblot analysis at 72 to 120 h after transfection.
Plasmid DNA transfection and reporter gene assays.
FT3-7 cells were seeded at 6 × 104 cells per well in 48-well plates at 48 h following siRNA transfection. Cells were transfected with plasmid DNA using Lipofectamine 2000 according to the manufacturer's instructions. The reporter plasmids, pMAD2-Luc, p107-Luc (18), and pIFN-β-Luc (12) (a gift from John Hiscott), were used to transfect cells at 0.2 μg/well, together with pCMV-β-galactosidase at 0.1 μg/well. Cells were harvested at 48 h posttransfection and lysed in reporter lysis buffer (Promega, Madison, WI). Quantification of luciferase and β-galactosidase activities was accomplished with commercial enzyme assay kits according to the manufacturer's instructions (Promega, Madison, WI).
RESULTS
Replication competence of HCV mutants with substitutions in the Rb-binding domain of NS5B.
The downregulation of Rb by HCV is dependent upon the viral RdRp, NS5B, which interacts with Rb through a conserved LxC/NxD motif near the active site of the polymerase (18). Since HCV RNA replication is enhanced in proliferating cells (20, 26), we hypothesized that the NS5B interaction with Rb may have evolved to create an intracellular environment permissive for viral replication. To test this hypothesis, we constructed a series of HCV genomes containing mutations in the conserved Rb-binding domain of NS5B within the background of a chimeric viral genome, HJ3-5 (here considered wt), encoding the nonstructural proteins of JFH-1 virus, which replicates efficiently in cell culture (Fig. 1A). The mutations were selected based on prior studies that demonstrated their ability to ablate NS5B binding to Rb (18). We examined the effect of the mutations on replication competence and infectious virus yield in viral RNA transfection and virus infection experiments. Huh-7 human hepatoma cells were electroporated with synthetic RNAs representing each of the mutated viral genomes. Cells were harvested and the supernatant fluids were collected at 96 h postelectroporation to assess virus yield. Viral RNA abundance in cell lysates was determined by qRT-PCR (Fig. 1B), while the titer of infectious virus released from the cells was determined by a fluorescent-focus assay (Fig. 1C).
FIG. 1.

Mutations in the Rb-binding motif of NS5B reduce RNA replication and replication of HCV. (A) Top, alignments of amino acids 307 to 325 of JFH-1 NS5B (present in the intergenotypic chimeric HJ3-5 virus used as the wt in these studies) and the Rb-binding domains of the E7 protein of human papillomavirus type 18 and pp71 of human cytomegalovirus strain AD169. The Rb-binding motif is shown in bold, while the overlying GDD motif in the NS5B polymerase is shaded. Bottom, amino acid sequences of the NS5B Rb-binding motif mutants used in this study. The mutated residues are underlined. (B) qRT-PCR analysis of HCV RNA accumulated in FT3-7 cells. The cells were transfected with wt HJ3-5 transcript or mutant derivatives as shown and harvested for analysis after 4, 48, 72, 96, and 120 h. Results are shown as the fold increase in viral RNA abundance relative to the replication-incompetent D318N RNA, which was transfected in parallel. (C) Infectious virus released by RNA-transfected FT3-7 cells. Serial dilutions of cell culture supernatant fluids (harvested from FT3-7 cells 96 h after the initial transfection with wt or mutant transcripts) were used to inoculate naïve Huh-7.5 cells. Cells expressing HCV core protein were detected by immunofluorescence analysis, and infectious foci were counted to determine the number of focus-forming units (FFU) per ml. The limit of detection in this assay is 10 FFU/ml. (D) The growth kinetics of mutant C316A were compared to those of the wt in a multicycle infection assay. FT3-7 cells were infected with wt or C316A virus at an MOI of 0.05. Cell culture supernatants were collected every 12 h from 24 to 144 h postinfection. Infectious virus release was measured as described for panel C. Results shown represent the means and standard deviations from three independent infections.
Four of the mutants contained single amino acid substitutions in NS5B, while one contained two substitutions (Fig. 1A). Since the Rb-binding domain overlaps with the divalent metal-coordinating Gly-Asp-Asp residues in the HCV RdRp (18), the mutations within the Rb-binding domain could also negatively influence polymerase activity. Indeed, viral RNA accumulation was reduced in cells infected with each of the five mutants, as determined by the qRT-PCR assay (Fig. 1B). The L314A mutant produced only 1/10 of the RNA abundance achieved by the wt RNA by 120 h after transfection, while replication of the double mutant, L314A/C316A, was severely compromised and resulted in no detectable increase in viral RNA in this assay (Fig. 1B). Consistent with these results, virus production was also reduced with each of the mutants (Fig. 1C), with the most dramatic reduction being observed with the L314A mutant and the double mutant, L314A/C316A. As expected, D318N, which is mutated in the NS5B active site, was unable to produce infectious virus. The C316A mutant, with a substitution in the center of the Rb-binding domain of NS5B, had the least effect on virus production, with virus yields averaging 33% of that from the parental wt RNA. This mutant was further examined in a multicycle infection analysis (Fig. 1D). Virus production by C316A-infected cells was reduced throughout the 144-h assay period compared to that by cells infected in parallel at the same multiplicity of infection (MOI) as the parental wt virus.
Viruses with mutations in the LxCxD domain do not downregulate Rb abundance.
Previously, we have shown that ectopic expression of wt NS5B in human hepatoma cells results in an interaction of NS5B with Rb, targeting Rb for proteasome-dependent degradation in an E6-associated protein-dependent fashion (17, 18). In contrast, ectopic expression of mutant NS5B with substitutions in the LxCxE-like Rb-binding motif does not induce changes in Rb abundance (18). We have also shown that Rb is downregulated in HCV-infected Huh-7 cells (17). It was therefore of interest to determine whether HCVs with mutations in the Rb-binding motif of NS5B are able to downregulate Rb. We initially selected the C316A mutant for study, since it was the least impaired for replication and therefore most likely to be comparable to the parental wt virus in terms of the abundance of viral proteins expressed within infected cells. FT3-7 cells were infected at an MOI of 1 with either wt or C316A virus. After 3 days, the cells were trypsinized and seeded into eight-well chamber slides, and 1 day later they were fixed and immunostained for NS5A and Rb as described in Materials and Methods. NS5A was detected in almost all cells in both the wt- and C316A-infected cultures. Fluorescence microscopy demonstrated that Rb was sharply reduced in abundance in cells infected with wt virus but not in cells infected with the C316A mutant (Fig. 2A).
FIG. 2.

Analysis of Rb abundance in cells infected with either wt or LxCxD domain mutant viruses. (A) Immunofluorescence analysis of cells infected with wt or C316A virus. FT3-7 cells were infected at an MOI of 1 and incubated for 4 days. Infected cells were fixed and immunostained for NS5A and Rb. (B) Laser-scanning confocal microscopy of cells infected with wt, C316A, and L314A viruses. Left panel, cells were labeled with a murine anti-Rb MAb (green) or human polyclonal anti-HCV sera (red). Nuclei were labeled with DAPI. The arrows indicate the presence of cytoplasmic Rb in cells infected with wt virus, a phenomenon not observed in uninfected cells or cells infected with mutant virus. Right panel, expanded view of the wt virus-infected cells showing merged DAPI and Rb images with cytoplasmic Rb. (C) Infrared immunoblot analysis of Rb in lysates from cells infected with wt or C316A virus. Left panel, cells were infected at an MOI of 1 to 2, and lysates collected on the day indicated postinfection were analyzed by immunoblotting using antibodies specific for Rb, NS5B, and β-tubulin (loading control). Right panel, quantitation of the infrared immunoblot, showing the abundance of Rb relative to β-tubulin at various times following infection with the wt or C316A virus.
We also examined cells infected with wt, C316A, or L314A virus by confocal microscopy (Fig. 2B, left panel). Cells were infected at a low MOI (<0.01) and passaged for 8 days prior to fixation and antibody labeling of Rb and HCV antigens. While Rb was found to be predominantly nuclear in its localization in cells infected with each of these viruses, a substantial amount of Rb could be detected within the cytoplasm of many cells infected with the wt virus (Fig. 2B). This finding is consistent with previous observations demonstrating the cytoplasmic accumulation of Rb in cells expressing NS5B, a phenomenon that is enhanced by inhibition of the proteasome and is likely to be due to the trapping of Rb in cytoplasmic complexes with NS5B (17). Importantly, this abnormal, partially cytoplasmic distribution of Rb was never observed in mock-infected cells or in cells infected with the L314A or C316A virus, in which Rb was detected exclusively within the nucleus.
Immunoblots of cell lysates confirmed the downregulation of Rb in cells infected with wt virus and the lack of an effect on Rb abundance in cells infected with the C316A mutant (Fig. 2C). Importantly, the absence of downregulation of Rb in the C316A-infected cells could not be attributed to reduced expression of NS5B, as the abundances of NS5B in the wt and C316A-infected cells were comparable by 120 h after infection (Fig. 2C). Thus, consistent with previous results from experiments involving ectopic expression of NS5B (18), mutations that ablate the Rb-binding motif in NS5B result in infectious viruses that can no longer downregulate Rb abundance.
Stability of the LxCxD domain mutants.
To assess the stability of the C316A mutation, total RNA was isolated from cells at 96 h after transfection with the mutant genome and used as template to amplify a 1.1-kb fragment of the NS5B-coding region (nt 8007 to 9151 of the JFH-1 virus; GenBank accession no. AB047639). The sequence of this fragment retained the engineered mutation and contained no additional changes (data not shown). The stabilities of the other mutations engineered in the Rb-binding domain were also examined over several passages of cell-free virus mutants. With the exception of L314A, each of the mutations was stable for at least three passages after electroporation. In cells infected with the L314A mutant, an Ala-to-Val change was detectable by passage 1 and became present in the majority of the viral sequences examined by passage 2 (data not shown). This new mutant contained a single nucleotide transition (C to T) at nt 8607. In order to determine the effect of the L314V mutation on virus production, it was reconstructed within the wt genome. In FT3-7 cells, the yield of L314V virus was approximately 50% of that of the wt virus, representing a dramatic increase in yield compared to L314A virus (∼1%) (Fig. 3A). The L314V virus yield was consistently greater than that obtained with the C316A virus in three independent experiments, although the differences in the yields of these two mutants were modest. Consistent with these results, L314V RNA accumulation was greater than that of C316A but lower than that of the wt when measured by qRT-PCR (Fig. 1B). These data suggest that the selection of the Ala-to-Val mutation in cells infected with the L314A mutant was due to the enhanced replication efficiency of the L314V virus.
To determine whether the L314V revertant had regained the ability to regulate Rb, we assessed Rb abundance in Huh-7.5 cells at 3 or 4 days after infection with the virus. Compared with that in mock-infected cells, Rb abundance was downregulated in immunoblots of lysates from L314V-infected cells (Fig. 3B). This was confirmed by fluorescence microscopy of cells infected at a low MOI with the L314A mutant, L314V revertant, or wt virus. These studies revealed substantial decreases in nuclear Rb abundance in most cells infected with the wt or L314V virus, while no difference in the nuclear Rb signal was apparent in cells infected with L314A or in noninfected cells adjacent to those infected with wt or L314V virus (Fig. 3C). Some L314V-infected cells showing weak HCV antigen expression appeared to retain a normal nuclear Rb abundance, but in aggregate these data indicate that the L314V revertant had at least partially regained the ability to downregulate Rb.
Mutations that ablate Rb binding do not necessarily impair RdRp activity.
It was important to determine whether the mutations in NS5B affected polymerase activity directly or acted in an indirect fashion to impair replication of the virus through the failure of the LxCxD motif mutants to modulate Rb abundance. To address this question, we purified bacterially expressed wt and mutant NS5B proteins and compared their activities in an in vitro polymerase assay.
The wt JFH-1 NS5B, which is identical to that in the wt HJ3-5 virus, was expressed without the C-terminal 21 residues (named J-Δ21). Four derivatives with mutations as described above, i.e., L314A, L314V, C316A, and the double mutant L314A/C316A, were purified to ∼ 90% homogeneity (Fig. 4A) after separation on an Ni-nitrilotriacetic acid column and a poly(U) column. The L320A mutant could not be purified from the E. coli proteins despite repeated attempts and was not analyzed further. The RNA synthesis activities of the four proteins were first assessed using a 19-nt linear RNA template named LE19. LE19 was designed to report on four distinct activities of the HCV RdRp (Fig. 4B, top): RNA synthesis by de novo initiation (generating a 19-nt RNA product), terminal nucleotide addition (generating 20- and 21-nt RNAs), extension from a primed template (generating a 32-nt RNA that is formed from a partial duplex of two LE19 molecules), and generation of a template switch product (of 38 nt and sometimes longer) (9, 22, 24). J-Δ21 has all four of the activities, which have been previously documented for the genotype 1b RdRp (25) (Fig. 4C, lane 1). In the absence of GTP, the putative initiation nucleotide, the de novo initiation product decreased more severely than the primer extension product, which does not require GTP with the LE19 template (Fig. 4C, lane 3).
FIG. 4.

Effect of mutations in NS5B on RNA synthesis in vitro. (A) SDS-PAGE of the purified proteins. The proteins were expressed in E. coli and purified through two affinity columns (see Materials and Methods). Lane M contains the Benchmark protein molecular weight marker (Invitrogen). (B) Sequence of template LE19, which can direct de novo-initiated RNA synthesis by NS5B. LE19 can also form a partially duplexed dimer that can direct primer extension to result in a 32-nt product. The single-stranded RNA named “L” is the molecule that can be ligated to form the circularized RNA named “C” (see text for additional details on templates). (C) Autoradiograph of the products of an RdRp assay run on a 20% polyacrylamide-7.5 M urea denaturing gel. The templates used are specified above each lane, and the GTP, where present, is at a 0.2 mM final concentration. The sizes of the RNA products are denoted at the left. (D) Quantification of the de novo initiation (19-nt) and primer extension (PE) (32-nt) products of the RdRp assay using LE19 as template. Four replicate reactions were used to quantify the products; results are shown as means ± standard deviations. Both the de novo initiation and primer extension products generated by all the mutant polymerases were normalized to that of the wt, whose activity was set at 100%.
Reproducible results were obtained with the L314A, L314V, C316A, and L314A/C316A mutants in five independent assays (Fig. 4D). The results revealed that the C316A mutant produced higher levels of both the de novo-initiated product and the primer extension product than the wt J-Δ21 protein. The L314A mutant had slightly lower RNA synthetic activity than the wt protein, while the double mutant, L314A/C316A, which demonstrated a severely debilitated replication phenotype (Fig. 1B and C), was severely defective in RNA synthesis in vitro (Fig. 4C, lanes 21 to 23). Thus, while it is apparent that some mutations in the Rb-binding domain cause reduced polymerase activity, this is not true for all mutants. The results obtained with the C316A mutant demonstrate in particular that the Rb-binding function of NS5B is not important for polymerase activity.
We challenged the RdRps with two additional templates to assess whether the mutations may have caused more subtle defects. First, each RdRp was tested with the template LE19p, which has a puromycin covalently linked to the 3′-terminal hydroxyl of LE19, thus rendering it competent for de novo initiation but not primer extension. With the LE19P template, J-Δ21 and each of the mutant RdRps with single amino acid substitutions retained the ability to initiate RNA synthesis by the de novo mechanism (Fig. 4C, lanes 2, 7, 12, and 17). The L314A/C316A mutant was inactive. Next, we assessed the ability of the HCV RdRp to direct RNA synthesis from linear (L) or circular (C) 16-nt templates. Since a circularized template cannot thread a 3′ terminus into the RdRp active site, this template can be recognized only by the RdRp in an open conformation, which then closes for initiation (2). Again, both the wt J-Δ21 and the mutant proteins with single amino acid substitutions retained the ability to generate products, while L314A/C316A was far less active (Fig. 4C, lanes 5, 10, 15, 20, and 25). The products generated from a circularized RNA were different than those from the linear version of the same RNA, demonstrating that the J-Δ21 RdRp can undergo a transition between the open and closed conformations (2). Thus, with respect to each of the NS5B activities that we assessed in these experiments, mutations that disrupt the interaction of NS5B with Rb do not necessarily have a negative effect on RNA synthesis in vitro.
Effects of Rb-binding site mutations on HCV replicase activity.
The finding that the C316A mutant was approximately twice as active in RNA synthesis in vitro was unexpected, given that the virus carrying this mutation is impaired for replication (Fig. 1B) and produces less virus in either RNA-transfected or virus-infected cells (Fig. 1C and D). Therefore, we compared RNA synthesis in cell-free reaction mixtures containing replicase complexes isolated from wt or C316A virus-infected cells. For these experiments, FT3-7 cells were electroporated with wt or C316A genomic RNA and passaged until they were 70 to 90% infected as judged by immunofluorescence detection of core antigen. Cells transfected with the replication-incompetent D318N genomic RNA were passaged in parallel as a control. We sequenced viral RNA extracted from the cells and confirmed that neither the wt nor the C316A virus had acquired mutations in the NS5B-coding region during passage. The cells were then harvested, and heavy membrane fractions containing HCV replicase complexes were isolated in order to measure replicase activity (Fig. 5A), as described in Materials and Methods. In this assay, the viral RNA polymerase uses endogenous viral RNA as template. For comparison between the different heavy membrane preparations, [32P]CTP incorporation into RNA was assessed quantitatively by PhosphorImager analysis (Fig. 5B) and then normalized to NS5B abundance as determined in immunoblots (Fig. 5C). As an additional measure of nonstructural protein abundance in these replicase preparations, we also analyzed immunoblots for NS3 protein (Fig. 5C). In three experiments, the replicase complexes from cells transfected with the C316A mutant synthesized an average of 2.21-fold (±0.48-fold) more labeled product than replicase complexes from the wt RdRp (Fig. 5A and B), consistent with the enhanced in vitro synthetic activity of the recombinant C316A protein described above (Fig. 4C). The increased activity of the C316A mutant relative to the wt was evident across the range of protein concentrations tested in the cell-free reaction (Fig. 5B). As expected, there was no RNA synthesis by heavy membrane fractions prepared from D318N-transfected cells.
FIG. 5.

In vitro RNA synthesis assay from heavy membrane fractions containing functional HCV replication complexes. (A) Heavy membrane fractions isolated from cells infected with wt or C316A virus, or mock-infected cells, were incubated in a transcription reaction mixture containing α-32P-labeled CTP. Heavy membrane fractions from genotype 1a and 2a subgenomic replicon-bearing cell lines (Sg-Rep) were included as positive controls. Labeled products were resolved on a denaturing agarose gel and visualized by autoradiography. (B) Quantification of products from panel A by PhosphorImager analysis. (C) Immunoblot analysis of HCV proteins NS3, NS5B, and core in the heavy membrane fractions used for panel A. The blots were also probed for Rab1b, an endoplasmic reticulum marker, as a loading control for the heavy membrane fractions.
Effects of Rb knockdown on HCV infection.
The results obtained thus far indicated that NS5B mutations that ablate the interaction with Rb do not necessarily reduce the ability of the enzyme to direct HCV RNA synthesis. Given that the same mutations impair HCV RNA replication and virus production, we hypothesized that the lack of regulation of Rb might in some way limit the efficiency of virus replication. To test this, we used RNA silencing to knock down the intracellular abundance of Rb and then examined the impact of this on replication of the virus. Transfection of FT3-7 cells with a pool of siRNAs targeting Rb led to undetectable levels of Rb when assessed by immunoblotting 3 to 5 days later (Fig. 6A). Surprisingly, however, analysis of the culture medium from cells infected with wt or C316A virus (MOI of 2) at 3 days after siRNA knockdown of Rb revealed a modest decrease, and not an increase, in the yields of both wt and C316A viruses compared to cells treated with control siRNAs (Fig. 6B). A similar experiment was performed using four individual siRNAs targeting Rb and two nontargeting control siRNAs. Again, cells transfected with siRNAs targeting Rb demonstrated a negligible abundance of Rb compared to the control siRNA-transfected cells at 4 days posttransfection (Fig. 6C). With three of the four siRNAs, the virus yield was reduced ∼50% compared to that for control siRNA-transfected cells, although one of the Rb-specific siRNAs caused a ∼90% reduction in virus yield (Fig. 6D, no. 9). Similar results were obtained in three independent experiments.
FIG. 6.

Effects of Rb knockdown by siRNA on virus replication. (A) Immunoblot analysis of lysates from FT3-7 cells transfected with a pool of siRNAs targeting Rb or pooled control siRNAs. Rb was efficiently knocked down to undetectable levels at 3, 4, and 5 days post transfection (top). The same blot was also probed for actin as a loading control (bottom). (B) FT3-7 cells transfected with a pool of siRNAs targeting Rb or a similar pool of control siRNAs were infected with either wt or C316A virus at 3 days posttransfection at an MOI of 2. Cell culture supernatants were harvested at 2 days postinfection. Virus released into supernatant fluids of infected cells was determined. Immunoblot analysis of protein from a parallel transfection performed at the same time is shown in panel A. (C) Immunoblot analysis of lysates from FT3-7 cells transfected with individual siRNAs targeting Rb or nonspecific control siRNAs. Lanes: 1, Rb siRNA pool; 2, Rb siRNA no. 5; 3, Rb siRNA no. 6; 4, Rb siRNA no. 7; 5, Rb siRNA no. 9; 6, control siRNA pool; 7, control siRNA no. 1; 8, control siRNA no. 2. (D) FT3-7 cells transfected with various siRNAs for Rb or control siRNAs were infected with wt HJ3-5 virus at 3 days posttransfection at an MOI of 2. Infectious virus in cell culture supernatants was quantitated by focus-forming assay. Immunoblot analysis of protein from a parallel transfection performed at the same time is shown in panel C. (E) FT3-7 cells were infected with either wt, C316A, or L320A virus at an MOI of 0.1. At 6 dpi, cells were seeded into six-well plates and transfected with a pool of siRNAs specific for Rb or pooled control siRNAs. At 10 days postinfection, cell culture supernatants were harvested from the transfected cells and virus titers were determined. Error bars indicate standard deviations. FFU, focus-forming units.
As an alternative experimental strategy, cells were first infected at an MOI of 0.1 with wt, C316A, or L320A virus, and the cells were grown for 7 days prior to transfection with either the pool of siRNAs specific for Rb or the pool of nontargeting control siRNAs. Cell culture supernatants were harvested at 3 days after transfection with siRNA (10 days postinfection), and the titer of virus released from the cells was determined. Immunoblots of cell lysates confirmed efficient knockdown of Rb as before (data not shown). Again, for each of the three viruses, the amount of virus released from cells transfected with Rb-specific siRNAs was lower than that from cells transfected with the nontargeting control siRNAs (Fig. 6E).
To further examine the effect of Rb gene knockdown on HCV replication, we determined the effect of Rb knockdown on the spread of HCV infection in Huh-7.5 cells. Cells were infected with serial dilutions of the wt virus at 3 days after transfection with Rb-specific or control siRNAs and then analyzed 3 days later for the number and size of foci of infected cells identified by core protein-specific immunofluorescence. Immunoblotting confirmed efficient knockdown of Rb (Fig. 7A). Consistent with the results shown in Fig. 6, Rb knockdown resulted in a reduction in the number of infectious foci to less than 50% of that developing in control cells (Fig. 7B). Furthermore, foci of infected cells were approximately 50% smaller in the cells in which Rb expression had been silenced, compared with cells transfected with control siRNA (Fig. 7C).
FIG. 7.
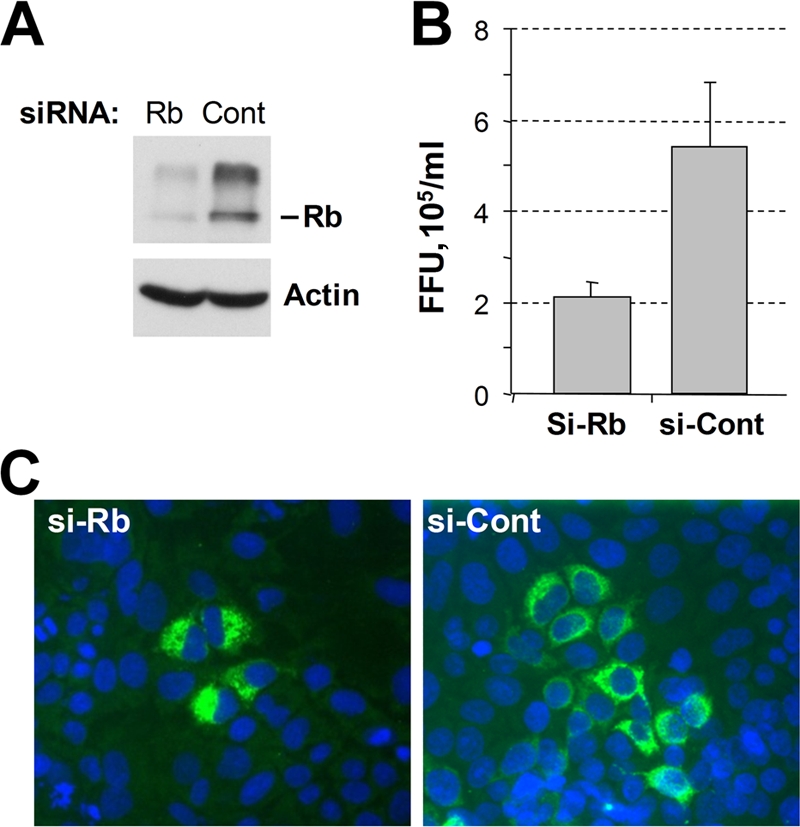
(A) Immunoblot analysis of lysates from Huh-7.5 cells transfected with a pool of siRNAs targeting Rb or pooled control siRNAs. Cells were harvested at 3 days posttransfection. Parallel transfections were trypsinized, seeded into eight-well chamber slides after 2 days (4 × 104 cells per well), and infected with serial dilutions of wt HJ3-5 virus at 3 days posttransfection. Cells were fixed and stained for core antigen at 2 days postinfection. (B) The number of focus-forming units (FFU) counted on cells transfected with control siRNAs was greater than twofold higher than that on cells transfected with Rb-specific siRNAs. Error bars indicate standard deviations. (C) Examples of foci are shown to demonstrate the size of foci.
To confirm that siRNA-mediated Rb knockdown had, as expected, resulted in activation of E2F-responsive promoters in these experiments, we determined the activities of two cellular promoters that are subject to Rb regulation using luciferase reporter assays. The promoters for MAD2 and p107 are both suppressed by Rb through an inhibitory effect of the tumor suppressor on E2F transcription factors (18). Consistent with this, MAD2 and p107 promoter activities were increased by 3.4- and 2-fold, respectively, in cells transfected with Rb-targeting siRNA compared to cells transfected with control siRNA (data not shown). Furthermore, we did not observe an effect on the beta interferon promoter with either siRNA, consistent with the absence of a major effect of Rb on this promoter. These results confirm that the siRNAs used in these experiments were effective in downmodulating both Rb abundance and Rb function.
Overall, these studies indicate that siRNA-mediated Rb knockdown reduces HCV replication in Huh-7 cells, despite the fact that NS5B interacts with Rb and targets it for degradation in infected cells (17, 18). There are several possible explanations that may account for this apparent paradox, as discussed below.
DISCUSSION
The HCV RdRp, NS5B, interacts with and downregulates the cellular abundance of Rb, an important tumor suppressor and cell cycle regulator (18). Here, we have shown that viruses containing mutations in NS5B that disrupt the Rb-binding motif are impaired for replication in cultured Huh-7 hepatoma cells (Fig. 1B and C). Importantly, we have also shown that the loss of fitness was not necessarily accompanied by reduced RdRp function in cell-free RNA synthesis assays. The discordant changes that we observed in the viral fitness and RdRp activities of these NS5B LxCxD domain mutants suggest that the Rb-binding activity of NS5B is not required for polymerase function per se but rather that the regulation of Rb by NS5B may have evolved as a mechanism to orchestrate cellular gene expression in such a manner as to create an intracellular environment that is conducive to HCV replication. We attempted to demonstrate this by assessing the impact of siRNA-mediated knockdown of Rb on the replication of two Rb-binding mutants (C316A and L320A), but we found that this reduced rather than enhanced replication fitness (Fig. 6). Thus, although we believe that a role in modulating cellular gene expression remains the most likely explanation for the regulation of Rb by HCV, there is no simple, straightforward relationship between Rb abundance and viral replication fitness.
Role of the Rb-binding site in NS5B.
Since the LxCxE-like motif overlaps with the GDD active-site motif of NS5B, it was important to examine whether mutations in the LxCxD motif directly affect NS5B activity. To do this, we first measured RNA synthesis in vitro by bacterially expressed wt and mutant NS5B proteins. Although C316A virus was moderately impaired in replication competence (Fig. 1B and C), the NS5B-C316A RdRp was twice as active and retained all of the in vitro activities of the wt NS5B protein (Fig. 4). We also examined RNA synthesis by fully formed membrane-bound replicase complexes extracted from virus-infected cells and confirmed that replicase containing NS5B with the C316A substitution was at least as active (if not more active) for RNA synthesis as the JFH-1 replicase from the parental wt virus used in these studies (Fig. 5). These results confirm that Rb binding is not directly important for NS5B polymerase activity and thus has evolved for other reasons. A very likely scenario is that NS5B may function in two or more populations: one that participates in RNA synthesis as part of the replicase and a second that regulates cellular processes, including the abundance of Rb. This is consistent with estimates from biochemical studies that only a small proportion of NS5B is actively engaged in RNA synthesis in HCV replicon cells (21).
The genomes of each of the LxCxD domain mutants that we constructed contained two or more nucleotide changes from the wt RNA sequence. As a result, they were generally stable and did not revert to the wt sequence on passage. However, the L314A mutant, which was severely handicapped in replication (Fig. 1B and C), underwent additional mutation to L314V on passage, thereby restoring replication fitness to close to that of the wt (Fig. 3A). Interestingly, the increased replication fitness of L314V was associated with restoration of the ability of the virus to downregulate Rb, as assessed by immunoblotting and fluorescence microscopy (Fig. 3B and C). However, the data do not allow us to say whether these two attributes of L314V, enhanced replication fitness and restored ability to regulate Rb, are in any way related to each other. Although the L314V mutation fully corrected the modest defect in function observed with the NS5B L314A mutant in the cell-free polymerase assays (both in de novo initiation and in primer extension) (Fig. 4A), it did not restore viral fitness completely to wt levels (Fig. 1B and 3A).
The Rb-binding motif is LxCxD in most HCV strains and is LxNxD in a subset of genotype 1b viruses (18). Despite the fact that the C316A mutant appears to possess increased polymerase activity compared to the wt protein (Fig. 4C and D), 316A is not found commonly in the RdRps of naturally occurring HCV strains. This suggests that any benefit it might confer upon the enzymatic activity of the NS5B polymerase may be outweighed by the abolition of Rb binding in the context of viral replication. Mutations at L314 and C316 have been found in HCV replicons selected for resistance to a benzofuran inhibitor of NS5B, HCV796, but in agreement with the present study, these mutants were found to have reduced fitness compared to the wt replicon (6). The impact of these resistance mutations on the ability of NS5B to bind Rb is not known.
Rb abundance and optimal HCV replication.
If the ability of NS5B to downregulate Rb has evolved because it facilitates virus replication, it might be expected that HCV replication would be increased in cells in which Rb abundance is reduced by other means. However, downregulation of Rb by siRNA knockdown resulted in no increase, and in fact a modest decrease, in the replication of both wt HCV and mutated HCV lacking a functional Rb-binding domain in NS5B (Fig. 6 and 7). The role of Rb in HCV infection is thus likely to be quite complex, and any effects on the efficiency of HCV replication in Huh-7 cells may be interwoven with other processes regulated by Rb. An important caveat in these experiments is that the only available cell culture systems that are sufficiently robust to study HCV infection in vitro use Huh-7-derived cell lines. Huh-7 cells are known to express high levels of a mutant p53 gene (1) and are thus likely to be defective in other aspects of normal cell cycle regulation. They also contain a constitutively activated NF-κB signaling pathway. Although the expression of wt NS5B, but not NS5B containing Rb-binding motif mutations, was able to downregulate Rb and activate E2F-responsive promoters in Huh-7 cells (18), cell culture systems that more closely mimic primary hepatocytes will be required to fully understand the effects on Rb abundance and cell cycle regulation exerted by HCV. Importantly, our previous studies failed to find any difference in cell cycle regulation in Huh-7-derived cell lines containing replicating, genome-length RNA replicons and their interferon-cured progeny (26). In contrast, we found previously that ectopic expression of NS5B (to an abundance similar to that in replicon cells) stimulates cell proliferation and S-phase entry in U2OS osteosarcoma cells that possess functional Rb and p53 pathways (18).
In vivo, downregulation of Rb might benefit HCV in a number of ways. In addition to its role as a central cell cycle regulator, Rb interacts with several transcription factors. Microarray studies have shown that downregulation of Rb leads to both upregulation of genes required for cell cycle progression and downregulation of genes involved in the modulation of immune functions (15). Over 500 different genes were differentially regulated by the acute loss of Rb in these studies. It is possible that the interaction between NS5B and Rb acts to fine-tune the level of Rb protein in the cell. That is, too much Rb (or too much hypophosphorylated Rb) may be detrimental to HCV infection due to its block in cell cycle progression. However, too little Rb could lead to altered gene expression that also disfavors HCV RNA replication. Given the possibility of such a scenario, it is not surprising to find that RNA silencing of the Rb gene would fail to regulate Rb abundance at the level that is optimal for viral replication. This is particularly so in Huh-7 cells, in which pathways regulating the cell cycle are intrinsically abnormal. Further studies will be needed to sort through these various possibilities and to ascertain the mechanism(s) responsible for the presumed benefit to viral fitness that underlies the evolution of Rb regulation by NS5B.
Acknowledgments
This work was supported in part by grants from the National Institute of Allergy and Infectious Diseases (U19-AI40035 to S.M.L. and RO1-AI073335 to C.C.K.). D.R.M. was a Jeane B. Kempner postdoctoral fellow.
Footnotes
Published ahead of print on 20 May 2009.
REFERENCES
- 1.Bressac, B., K. M. Galvin, T. J. Liang, K. J. Isselbacher, J. R. Wands, and M. Ozturk. 1990. Abnormal structure and expression of p53 gene in human hepatocellular carcinoma. Proc. Natl. Acad. Sci. USA 871973-1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chinnaswamy, S., I. Yarbrough, S. Palaninathan, C. T. Kumar, V. Vijayaraghavan, B. Demeler, S. M. Lemon, J. C. Sacchettini, and C. C. Kao. 2008. A locking mechanism regulates RNA synthesis and host protein interaction by the hepatitis C virus polymerase. J. Biol. Chem. 28320535-20546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.DeCaprio, J. A., J. W. Ludlow, J. Figge, J. Y. Shew, C. M. Huang, W. H. Lee, E. Marsilio, E. Paucha, and D. M. Livingston. 1988. SV40 large tumor antigen forms a specific complex with the product of the retinoblastoma susceptibility gene. Cell 54275-283. [DOI] [PubMed] [Google Scholar]
- 4.DeGregori, J., T. Kowalik, and J. R. Nevins. 1995. Cellular targets for activation by the E2F1 transcription factor include DNA synthesis- and G1/S-regulatory genes. Mol. Cell. Biol. 154215-4224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dyson, N., P. M. Howley, K. Munger, and E. Harlow. 1989. The human papilloma virus-16 E7 oncoprotein is able to bind to the retinoblastoma gene product. Science 243934-937. [DOI] [PubMed] [Google Scholar]
- 6.Howe, A. Y., H. Cheng, S. Johann, S. Mullen, S. K. Chunduru, D. C. Young, J. Bard, R. Chopra, G. Krishnamurthy, T. Mansour, and J. O'Connell. 2008. Molecular mechanism of heaptitis C virus replicon variants with reduced susceptibility to a benzofuran inhibitor, Hcv-796. Antimicrob. Agents Chemother. 523327-3338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ishido, S., and H. Hotta. 1998. Complex formation of the nonstructural protein 3 of hepatitis C virus with the p53 tumor suppressor. FEBS Lett. 438258-262. [DOI] [PubMed] [Google Scholar]
- 8.Kao, C. F., S. Y. Chen, J. Y. Chen, and Y. H. Wu Lee. 2004. Modulation of p53 transcription regulatory activity and post-translational modification by hepatitis C virus core protein. Oncogene 232472-2483. [DOI] [PubMed] [Google Scholar]
- 9.Kim, M. J., and C. Kao. 2001. Factors regulating template switch in vitro by viral RNA-dependent RNA polymerases: implications for RNA-RNA recombination. Proc. Natl. Acad. Sci. USA 984972-4977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lerat, H., M. Honda, M. R. Beard, K. Loesch, J. Sun, Y. Yang, M. Okuda, R. Gosert, S. Y. Xiao, S. A. Weinman, and S. M. Lemon. 2002. Steatosis and liver cancer in transgenic mice expressing the structural and nonstructural proteins of hepatitis C virus. Gastroenterology 122352-365. [DOI] [PubMed] [Google Scholar]
- 11.Levrero, M. 2006. Viral hepatitis and liver cancer: the case of hepatitis C. Oncogene 253834-3847. [DOI] [PubMed] [Google Scholar]
- 12.Lin, R., C. Heylbroeck, P. M. Pitha, and J. Hiscott. 1998. Virus-dependent phosphorylation of the IRF-3 transcription factor regulates nuclear translocation, transactivation potential, and proteasome-mediated degradation. Mol. Cell. Biol. 182986-2996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ma, Y., J. Yates, Y. Liang, S. M. Lemon, and M. Yi. 2008. NS3 helicase domains involved in infectious intracellular hepatitis C virus particle assembly. J. Virol. 827624-7639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Majumder, M., A. K. Ghosh, R. Steele, R. Ray, and R. B. Ray. 2001. Hepatitis C virus NS5A physically associates with p53 and regulates p21/waf1 gene expression in a p53-dependent manner. J. Virol. 751401-1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Markey, M. P., J. Bergseid, E. E. Bosco, K. Stengel, H. Xu, C. N. Mayhew, S. J. Schwemberger, W. A. Braden, Y. Jiang, G. F. Babcock, A. G. Jegga, B. J. Aronow, M. F. Reed, J. Y. Wang, and E. S. Knudsen. 2007. Loss of the retinoblastoma tumor suppressor: differential action on transcriptional programs related to cell cycle control and immune function. Oncogene 266307-6318. [DOI] [PubMed] [Google Scholar]
- 16.McGivern, D. R., and S. M. Lemon. 2008. Tumor suppressors, chromosomal instability, and hepatitis C virus-associated liver cancer. Annu. Rev. Pathol. 4399-415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Munakata, T., Y. Liang, S. Kim, D. R. McGivern, J. M. Huibregtse, A. Nomoto, and S. M. Lemon. 2007. Hepatitis C virus induces E6AP-dependent degradation of the retinoblastoma protein. PLoS. Pathog. 31335-1347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Munakata, T., M. Nakamura, Y. Liang, K. Li, and S. M. Lemon. 2005. Down-regulation of the retinoblastoma tumor suppressor by the hepatitis C virus NS5B RNA-dependent RNA polymerase. Proc. Natl. Acad. Sci. USA 10218159-18164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nelson, H. B., and H. Tang. 2006. Effect of cell growth on hepatitis C virus (HCV) replication and a mechanism of cell confluence-based inhibition of HCV RNA and protein expression. J. Virol. 801181-1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pietschmann, T., V. Lohmann, G. Rutter, K. Kurpanek, and R. Bartenschlager. 2001. Characterization of cell lines carrying self-replicating hepatitis C virus RNAs. J. Virol. 751252-1264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Quinkert, D., R. Bartenschlager, and V. Lohmann. 2005. Quantitative analysis of the hepatitis C virus replication complex. J. Virol. 7913594-13605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ranjith-Kumar, C. T., L. Gutshall, M. J. Kim, R. T. Sarisky, and C. C. Kao. 2002. Requirements for de novo initiation of RNA synthesis by recombinant flaviviral RNA-dependent RNA polymerases. J. Virol. 7612526-12536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ranjith-Kumar, C. T., and C. C. Kao. 2006. Recombinant viral RdRps can initiate RNA synthesis from circular templates. RNA 12303-312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ranjith-Kumar, C. T., Y. C. Kim, L. Gutshall, C. Silverman, S. Khandekar, R. T. Sarisky, and C. C. Kao. 2002. Mechanism of de novo initiation by the hepatitis C virus RNA-dependent RNA polymerase: role of divalent metals. J. Virol. 7612513-12525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ranjith-Kumar, C. T., J. L. Santos, L. L. Gutshall, V. K. Johnston, J. Lin-Goerke, M. J. Kim, D. J. Porter, D. Maley, C. Greenwood, D. L. Earnshaw, A. Baker, B. Gu, C. Silverman, R. T. Sarisky, and C. Kao. 2003. Enzymatic activities of the GB virus-B RNA-dependent RNA polymerase. Virology 312270-280. [DOI] [PubMed] [Google Scholar]
- 26.Scholle, F., K. Li, F. Bodola, M. Ikeda, B. A. Luxon, and S. M. Lemon. 2004. Virus-host cell interactions during hepatitis C virus RNA replication: impact of polyprotein expression on the cellular transcriptome and cell cycle association with viral RNA synthesis. J. Virol. 781513-1524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Whyte, P., K. J. Buchkovich, J. M. Horowitz, S. H. Friend, M. Raybuck, R. A. Weinberg, and E. Harlow. 1988. Association between an oncogene and an anti-oncogene: the adenovirus E1A proteins bind to the retinoblastoma gene product. Nature 334124-129. [DOI] [PubMed] [Google Scholar]
- 28.Yi, M., and S. M. Lemon. 2004. Adaptive mutations producing efficient replication of genotype 1a hepatitis C virus RNA in normal Huh7 cells. J. Virol. 787904-7915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yi, M., Y. Ma, J. Yates, and S. M. Lemon. 2007. Compensatory mutations in E1, p7, NS2, and NS3 enhance yields of cell culture-infectious intergenotypic chimeric hepatitis C virus. J. Virol. 81629-638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yi, M., R. A. Villanueva, D. L. Thomas, T. Wakita, and S. M. Lemon. 2006. Production of infectious genotype 1a hepatitis C virus (Hutchinson strain) in cultured human hepatoma cells. Proc. Natl. Acad. Sci. USA 1032310-2315. [DOI] [PMC free article] [PubMed] [Google Scholar]